
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A tumor-targeted polymer theranostics platform for positron emission tomography and fluorescence imaging†
Eva
Koziolová‡
a,
Shreya
Goel‡
b,
Petr
Chytil
a,
Olga
Janoušková
a,
Todd E.
Barnhart
c,
Weibo
Cai
 bcd and
Tomáš
Etrych
*a
bcd and
Tomáš
Etrych
*a
aInstitute of Macromolecular Chemistry, Czech Academy of Sciences, Heyrovsky Sq. 2, Prague 6, 162 06, Czech Republic. E-mail: etrych@imc.cas.cz; Fax: +420-296809 410; Tel: +420-296 809 231
bMaterials Science Program, University of Wisconsin-Madison, Madison, Wisconsin, USA
cDepartments of Radiology and Medical Physics, University of Wisconsin-Madison, Madison, Wisconsin, USA
dUniversity of Wisconsin Carbone Cancer Center, Madison, Wisconsin, USA
First published on 4th July 2017
Abstract
Here, we describe a novel polymer platform suitable for efficient diagnostics and potential theranostics based on 89Zr-labeled N-(2-hydroxypropyl)methacrylamide (HPMA)-based copolymer conjugates. A set of polymers differing in molecular weight with either low dispersity or high dispersity were designed and synthesized and their biodistribution in vivo was successfully and precisely observed over 72 h. Moreover, the feasibility of two imaging techniques, fluorescence imaging (FI) and positron emission tomography (PET), was compared using labeled polymer conjugates. Both methods gave comparable results thus showing the enhanced diagnostic potential of the prepared polymer–dye or polymer–chelator–89Zr constructs. The in vivo and ex vivo PET/FI studies indicated that the dispersity and molecular weight of the linear HPMA polymers have a significant influence on the pharmacokinetics of the polymer conjugates. The higher molecular weight and narrower distribution of molecular weights of the polymer carriers improve their pharmacokinetic profile for highly prolonged blood circulation and enhanced tumor uptake. Moreover, the same polymer carrier with the anticancer drug doxorubicin bound by a pH-sensitive hydrazone bond showed higher cytotoxicity and cellular uptake in vitro. Therefore, HPMA copolymers with low dispersity and a molecular weight near the limit of renal filtration can be used as highly efficient polymer carriers of tumor-targeted therapeutics or for theranostics with minimal side effects.
Introduction
Despite notable advances in healthcare provided to clinical patients, early and accurate tumor diagnosis, together with efficient and safe cancer therapy, remains one of the biggest medical challenges of the 21st century. Cancer treatment with conventional cytostatics often suffers from low efficiency and occurrence of severe side effects. Recently, numerous nanosized delivery systems of anticancer agents have been designed and tested, among which N-(2-hydroxypropyl)methacrylamide (HPMA) copolymer conjugates with anthracyclines, doxorubicin (Dox) or pirarubicin, with controlled tumor-oriented drug release exhibited high anti-tumor activity with reduced side effects in humans.1,2 The attachment of cytostatics to HPMA copolymers prolongs their blood circulation, and due to the enhanced permeability and retention (EPR) effect, the polymer–drug conjugates accumulate predominantly in solid tumors.3 The widely studied pH-sensitive hydrazone bond between the drug and the HPMA copolymer backbone is relatively stable at blood pH (pH ∼ 7.4) and the hydrolysis rate increases with decreasing pH, therefore the drug is mainly released within the tumor tissue (pH ≈ 6) and even faster after the cellular uptake in the endosomes or lysosomes (pH 5) of tumor cells, thus significantly reducing the side effects of chemotherapeutics.4The tumor accumulation rate and blood circulation time of polymer carriers and drug conjugates increase with their growing molecular weight.5 Nevertheless, the polymer carrier should be entirely excreted after drug delivery to solid tumors. Therefore, HPMA copolymers with the molecular weight, Mw, close to the renal threshold (70 kg mol−1 for linear HPMA copolymers6) and with low dispersity are expected to be highly efficient in tumor treatment without undesired polymer persistence in the body compartments.
The development of the controlled radical Reversible Addition Fragmentation Chain Transfer (RAFT) polymerization technique enabled the synthesis of HPMA copolymers with dispersity close to one.7–11 We have recently shown that HPMA copolymer–Dox conjugates with low dispersity prepared by RAFT copolymerization exhibited higher tumor accumulation and enhanced therapeutic activity in vivo compared to the corresponding conjugates of polymers with high dispersity prepared by free radical polymerization with a similar molecular weight.12
Not only the verification of therapeutic activity and efficiency, but also a detailed study of polymer carrier biodistribution and its clearance, is essential for its potential introduction in clinical practice. Single photon emission computed tomography was exploited for in vivo observing the fate of various HPMA copolymers varying in molecular weight or type and content of side chain functional groups.13 Optical imaging has been recently utilized to observe the in vitro and in vivo fate of dual fluorescently-labeled HPMA copolymers with high dispersity.14,15 Despite high resolution, multicolor imaging capacity and ease of operation, fluorescence imaging (FI) suffers from poor tissue depth penetration, low sensitivity and semi-quantitative nature.16 In comparison, tumor imaging utilizing positron emission tomography (PET) is more sensitive and quantitative with unlimited tissue penetration, but its application is hampered by poor resolution and lack of anatomical information.16,17 Thus, a dual-modality PET/optical probe can provide synergistic advantages over a single modality alone.16 The versatility and multivalence of HPMA copolymers enable the attachment of fluorescent dyes, radiotracers and drugs allowing simultaneous therapy and diagnostics, i.e. the theranostics of tumors.
In this work, the influence of molecular weight and dispersity of HPMA copolymers on their biodistribution has been studied using two complementary imaging methods – PET and FI. HPMA copolymers differing in molecular weight were synthesized either by free radical polymerization (FRP) or by controlled radical RAFT polymerization and functionalized with a well-known chelator, deferoxamine (Def), for further radiolabeling with zirconium-89 (89Zr; t1/2 = 78.4 h) prior to in vivo and ex vivo studies. To demonstrate their multifunctionality, the polymers were labeled with fluorescent dyes, Dy633 or Dy676, and their in vitro cellular uptake or in vivo biodistribution was studied via optical imaging.
In order to prove the potential of the HPMA copolymer conjugates for tumor-targeted theranostics with controlled drug release at the site of interest, the cytostatic drug, Dox, was attached to the fluorescently-labeled copolymer by a pH-sensitive hydrazone bond and the influence of molecular weight and dispersity of the polymer–Dox conjugates was studied using various tumor cell lines.
Experimental
Chemicals
1-Aminopropan-2-ol, methacryloyl chloride, 2,2′-azobis(isobutyronitrile) (AIBN), 6-aminohexanoic acid, 4-(dimethylamino)pyridine, N,N′-dicyclohexylcarbodiimide, 4,5-dihydrothiazole-2-thiol, tert-butylsemicarbazate, 2-cyanopropan-2-yl benzodithioate, N,N-diisopropylethylamine (DIPEA), dimethyl sulfoxide (DMSO), tert-butyl alcohol, 2,4,6-trinitrobenzene-1-sulfonic acid (TNBSA), Chelex 100 resin (50–100 mesh) and deferoxamine mesylate were purchased from Sigma-Aldrich. Doxorubicin hydrochloride was purchased from Meiji Seika, Japan. DY-633-NHS-ester and DY-676-NHS-ester fluorescent dyes were purchased from Dyomics GmbH, Germany. LH-20, Sephadex G-25 and PD-10 desalting columns were purchased from GE Healthcare.Water and all buffers were of Millipore grade and pre-treated with Chelex 100 resin to ensure that the aqueous solution was heavy metal-free. All other chemicals and solvents were of analytical grade. The solvents were dried and purified by conventional procedures.
Synthesis of the monomers
The synthesis and characterization of N-(2-hydroxypropyl)methacrylamide (HPMA), (6-methacrylamido hexanoyl)hydrazide (MA-Ahex-NHNH2), N-(tert-butoxycarbonyl)-2-(6-methacrylamido hexanoyl)hydrazine (MA-Ahex-NHNH-Boc) and 3-(3-methacrylamidopropanoyl)thiazolidine-2-thione (MA-APr-TT) monomers have been already published.4,18–20Synthesis of the polymer precursors
The random copolymer poly(HPMA-co-MA-Ahex-NHNH2) (HD-P(hyd)) was prepared by a solution FRP of HPMA with MA-Ahex-NHNH2 using AIBN as an initiator carried out in methanol at 60 °C for 17 h as described previously.4 The random copolymer poly(HPMA-co-MA-APr-TT) (HD-P(TT)) was prepared by a solution FRP of HPMA with MA-APr-TT using AIBN as an initiator performed in DMSO at 60 °C for 6 h according our previous work.18The random copolymers poly(HPMA-co-MA-Ahex-NHNH2) with a narrow distribution of molecular weight (LD-P(hyd)-30 and LD-P(hyd)-45) were prepared by the RAFT polymerization of HPMA with MA-Ahex-NHNH-Boc, followed by dithiobenzoate end group removal and deprotection of hydrazide groups. The random copolymers poly(HPMA-co-MA-APr-TT) with a narrow distribution of molecular weight (LD-P(TT)-30 and LD-P(TT)-45) were prepared by the RAFT polymerization of HPMA with MA-APr-TT, followed by dithiobenzoate end group removal. Polymerizations were adapted from previous studies19,21 using 2-cyanopropan-2-yl benzodithioate as a chain transfer agent (CTA) and AIBN as an initiator at 70 °C for 16 h in a mixture of 85% tert-butyl alcohol and 15% DMSO. The molar ratios of monomers![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CTA:initiator were 400:2:1 for LD-P(hyd)-30, 600:2:1 for LD-P(hyd)-45, 550:2:1 for LD-P(TT)-30 and 700:2:1 for LD-P(TT)-45. The molar ratio of HPMA to MA-Ahex-NHNH-Boc in the reaction mixture was 9:1; the molar ratio of HPMA to MA-APr-TT in the reaction mixture was 24:1. The dithiobenzoate ω-end group was removed using AIBN as described in the literature.22 The Boc protecting groups were removed in the presence of TFA according to ref. 19.
:CTA:initiator were 400:2:1 for LD-P(hyd)-30, 600:2:1 for LD-P(hyd)-45, 550:2:1 for LD-P(TT)-30 and 700:2:1 for LD-P(TT)-45. The molar ratio of HPMA to MA-Ahex-NHNH-Boc in the reaction mixture was 9:1; the molar ratio of HPMA to MA-APr-TT in the reaction mixture was 24:1. The dithiobenzoate ω-end group was removed using AIBN as described in the literature.22 The Boc protecting groups were removed in the presence of TFA according to ref. 19.
Synthesis of the polymer conjugates with fluorescent dyes or drug
The fluorescently labeled copolymers were prepared by a reaction of DY-633-NHS-ester or DY-676-NHS-ester with hydrazide groups of the respective polymer precursors (HD-P(hyd), LD-P(hyd)-30 and LD-P(hyd)-45), forming a physiologically stable hydrazide bond. An example of HD-P + DY-676 synthesis: DY-676-NHS-ester (0.5 mg) dissolved in 0.1 mL of methanol was added into a stirred solution of 30 mg of HD-P(hyd) dissolved in 0.2 mL of methanol. After 20 h of stirring at room temperature, the unbound dye was purified by gel filtration on a Sephadex LH-20 column in methanol. The polymer–dye conjugate was isolated by precipitation in ethyl acetate.Polymer conjugates with doxorubicin (Dox) were prepared by a reaction of the C13 keto group of Dox with hydrazide groups of the respective polymer precursors (HD-P(hyd), LD-P(hyd)-30 and LD-P(hyd)-45) or copolymers labeled by DY-633 (HD-P + DY-633, LD-P-30 + DY-633 and LD-P-45 + DY-633), forming a pH-sensitive hydrazone bond according to our previous work.23 After 17 h of stirring at room temperature, the polymer–drug conjugates were purified from the unbound drug by gel filtration, using a Sephadex LH-20 column and methanol as the eluent followed by precipitation in ethyl acetate.
Synthesis of the polymer conjugates with a chelator
The polymer–chelator conjugates were prepared by aminolysis of the thiazolidine-2-thione groups of the respective copolymers (HD-P(TT), LD-P(TT)-30 and LD-P(TT)-45) by the chelator, deferoxamine (Def), forming a physiologically stable amide bond. An example of HD-P + Def synthesis: the polymer precursor HD-P(TT) (200 mg, 1.2 × 10−5 mol of TT groups) was dissolved in 1 mL of methanol and 20 μL of DIPEA (1.2 × 10−4 mol) was added. Deferoxamine mesylate (79 mg, 1.2 × 10−4 mol) dissolved in 1 mL of methanol was stirred while the polymer solution was added dropwise. The reaction was carried out at room temperature for 1 h. The crude polymer–deferoxamine conjugate was isolated by precipitation in diethyl ether and purified by subsequent gel filtration on a Sephadex G-25 column in water. The polymer–chelator conjugate was isolated by freeze-drying.Characterization of the conjugates
Determination of the molecular weight and dispersity of the copolymers was performed with an HPLC Shimadzu system equipped with a size exclusion chromatography (SEC) column (TSKgel G3000SWx (300 × 7.8 mm; 5 μm), a photo-diode array (PDA, Shimadzu SPD-M20A), refractive index (RI) Optilab®-rEX and multiangle light scattering (MALS) DAWN HELEOS II (Wyatt Technology Co., USA) detectors, using a methanol–sodium acetate buffer (0.3 M; pH 6.5) mixture (80:20 vol%; flow rate 0.3 mL min−1)). The Mw, Mn and Đ were calculated using ASTRA VI software and a refractive increment index dn/dc between 0.167 and 0.175 mL g−1 was used. The dn/dc values were determined by two methods, using the “100% recovery procedure” within the MALS-GPC instrument or by the measurement of the RI of several concentrations of polymer solution in a given solvent with extrapolation to zero concentration.
The content of the TT groups in HD-P(TT), LD-P(TT)-30 and LD-P(TT)-45 was determined by UV/Vis spectrophotometry in methanol using the molar absorption coefficient ε305 = 10000 L mol−1 cm−1 estimated for MA-APr-TT.
The content of the hydrazide groups in HD-P(hyd), LD-P(hyd)-30 and LD-P(hyd)-45 was determined by a modified TNBSA assay.4
The content of Dox in non-labeled polymer conjugates was measured by UV spectrophotometry in water.4 The content of Dox in fluorescently labeled polymer conjugates was determined by HPLC (using a reverse-phase column Chromolith Performance RP-18e 100-4.6 with PDA detection; eluent water–acetonitrile with acetonitrile gradient 0–100 vol%; flow rate, 5 mL min−1; fluorescence detection, excitation at 488 nm and emission at 560 nm) as its aglycone after acidic hydrolysis in 1 M HCl (1 h at 50 °C) followed by extraction in chloroform.
The content of fluorescent dyes DY-633 or DY-676 was measured by UV spectrophotometry in methanol using the molar absorption coefficient ε637 = 200000 L mol−1 cm−1 for DY-633 and ε671 = 219000 L mol−1 cm−1 for DY-676. The purity of the polymer conjugates (content or absence of unbound drug or dye) was tested by HPLC using a PDA detector (Shimadzu SPD-M20A) with the same SEC column, mobile phase and flow rate as mentioned above.
The content of Def was determined by 1H-NMR spectroscopy (Bruker spectrometer, 300 MHz) in methanol-d4 by comparison of the integral intensities δ (ppm) = 3.87 (1 H, br, CH–OH) of the HPMA monomer unit and integral intensities of δ (ppm) = 3.85 (6 H, m, CH2–N(OH)–CO) of deferoxamine.
89Zr-Labeling of Def-conjugated polymers
89Zr-Oxalate was produced according to the previously reported procedures with minor modifications.24 Briefly, natural yttrium foil (250 μm, 99.9%) was irradiated with a proton beam to create 89Zr via the 89Y(p,n)89Zr reaction. 89Zr was separated from the target material using a hydroxamate-functionalized column and then eluted in 1 M oxalic acid. Typically >90% of the radioactivity was eluted in 1 mL with an end-of-bombardment (EOB) specific activity of ∼111 GBq μmol−1.For radiolabeling, 37–74 MBq of 89Zr-oxalate was diluted in 300 μL of 0.5 M HEPES buffer, neutralized with a 2 M Na2CO3 solution and added to aqueous solutions of Def-conjugated polymers HD-P + Def, LD-P-30 + Def and LD-P-45 + Def, where 1.0–1.5 mg of Def polymer conjugate was used per 37 MBq of 89Zr. The mixture was incubated for 2 h at 37 °C with constant shaking. 89Zr-Labeled polymers were purified on PD-10 columns, using PBS as the mobile phase. Radioactive fractions containing 89Zr-Def–polymer conjugates were collected for further in vivo experiments. Radiochemical yields were determined via instant thin layer chromatography using silica backed iTLC plates and 50 mM EDTA as the running buffer.
Cell lines and animal models
The Jurkat human T-lymphocyte cell line (ATCC, LGC Standards Sp. z.o.o., Poland) was cultivated in RPMI medium, supplemented with heat inactivated 10% fetal calf serum (FCS), 100 U mL−1 penicillin, and 100 μg mL−1 streptomycin (Thermo Scientific, Czech Republic). The human and mouse cell lines, for in vitro study, isolated from the mammary gland, MCF-7 and 4T1 respectively (MCF-7 were kindly provided by Prof. Neužil, Institute of Biotechnology AS CR and 4T1 were kindly provided by Prof. Ražka, First Medical Faculty Charles University) were cultivated in DMEM medium with heat inactivated 10% fetal calf serum (FCS), penicillin (100 U mL−1), and streptomycin (100 μg mL−1) (Thermo Scientific, Czech Republic). The 4T1 murine breast cancer cell line for in vivo studies was purchased from the American Type Culture Collection (ATCC, Manassas, VA). The cells were cultured in RPMI 1640 medium (Invitrogen, Carlsbad, CA) with 10% fetal bovine serum and incubated at 37 °C with 5% CO2 and when they reached 75% confluence.All animal studies were conducted under a protocol approved by the University of Wisconsin Institutional Animal Care and Use Committee. For the 4T1 tumor model, four- to five-week-old female Balb/c mice were purchased from Envigo (Indianapolis, IN) and tumors were established by subcutaneously injecting 2 × 106 cells, suspended in 50 μL of PBS, into the front or hind flanks of the mice. Tumor sizes were monitored every other day and the mice were used for in vivo experiments when the diameter of the tumors reached 5–8 mm (typically 1–2 weeks after inoculation).
In vitro cytotoxicity
5 × 104 cells were seeded in 100 μL of media in 96-well flat-bottom plates (TPP, Czech Republic) 24 h before adding the polymer–Dox conjugates or the Dox itself. Their concentrations varied in the range 0.1–500 μg mL−1 for conjugates or 0.002–5 μg mL−1 for Dox. The cells were cultivated for 72 h in 5% CO2 at 37 °C. Then 10 μL of alamarBlue® cell viability reagent (Thermo Scientific, Czech Republic) was added to each well and incubated for 4 h at 37 °C. The active component of the alamarBlue reagent resazurin was reduced to the highly fluorescent compound resorufin only in viable cells. Fluorescence was detected using the plate reader Synergy Neo (Bio-Tek, Czech Republic) with excitation at 570 nm and emission at 600 nm. Cells cultivated in medium without Dox or the polymer–Dox conjugates were employed as controls. Three wells were used for each concentration. The assay was repeated two times in triplicate.In vitro cellular uptake
The mouse cell lines 4T1 isolated from the mammary gland were used for the evaluation of polymer conjugate uptake using confocal laser scanning microscopy. Fluorescently labeled polymers, HD-P + Dy-633, LD-P-30 + Dy-633 and LD-P-45 + Dy-633, and polymer–drug conjugates, HD-P + Dox/Dy-633, LD-P-30 + Dox/Dy-633 and LD-P-45 + Dox/Dy-633, all with covalently bound Dyomics-633 were incubated with 4T1 cells for 24 h in a 5% CO2 atmosphere at 37 °C. The amount of the polymer conjugates added to the cell suspensions was normalized to the Dyomics-633 content (2 μg mL−1). The normalization to the dye was used to evaluate the difference in the internalization rate between the polymers and polymer conjugates. After incubation, the cells were washed two times with PBS and the cells were labeled with Hoechst 33342 in PBS (5 μg mL−1). The polymer-bound dye Dyomics-633 was excited at 647 nm and the emitted light was detected through a 650–750 nm filter. The Hoechst 33342 dye labelling the nuclei was excited at 405 nm and the emitted light was detected through a 425–500 nm filter. The drug Dox was excited at 488 nm and the emitted light was detected through a 500–600 nm filter. The laser scanning confocal microscope (Olympus IX83) with FV10-ASW software (Olympus, Czech Republic) was used to observe the fluorescence and transmitted light. The samples were scanned with a 60× oil immersion objective Plan ApoN (1.42 numerical aperture).PET imaging and biodistribution studies
Small animal PET scans were performed using an Inveon microPET/microCT rodent model scanner (Siemens Medical Solutions USA, Inc.). Each 4T1 tumor-bearing mouse was injected intravenously with 5–10 MBq of the radio-labeled polymer conjugate and static PET scans were performed at 0.5, 3, 6, 20, 48 and 72 h post-injection (p.i.). The images were reconstructed using a maximum a posteriori (MAP) algorithm, with no attenuation or scatter correction, and are presented as maximum intensity projections (MIPs). For each microPET scan, three-dimensional (3D) regions-of-interest (ROIs) were drawn over the tumor and major organs using vendor software (Inveon Research Workplace) on decay-corrected whole-body images, and are presented as percentage of injected dose per gram (%ID g−1). The data obtained from the ROI analysis were used to construct the time–activity curves (TAC) for the tumor and other major organs.Ex vivo biodistribution studies were carried out after the final PET scan at 72 h p.i. to confirm that the quantitative values based on PET imaging truly represented the radioactivity distribution in tumor-bearing mice. The mice were euthanized and blood, 4T1 tumors, and major organs/tissues were collected and wet-weighed. The radioactivity in the tissue was measured using a gamma-counter (PerkinElmer) and presented as %ID g−1 (mean ± SD).
In vivo and ex vivo fluorescence imaging
For all in vivo fluorescence studies 38 μg of Dy676 conjugated polymers HD-P + DY-676 and LD-P-45 + DY-676 (∼1.9 mg kg−1 dose per mouse; corresponding to a dose of ∼120 pmol of Dy-676) were injected into the tail vein of 4T1 tumor-bearing mice. The mice (n = 3 per group) were imaged at 0.5, 3, 24 and 48 h p.i. on a PerkinElmer IVIS system using 675/720 nm excitation/emission filters. After the last scan at 48 h p.i., the mice were euthanized and 4T1 tumors and other major organs were harvested and imaged again ex vivo to validate the in vivo findings. Using vendor software, ROIs were drawn on the explanted tumor and organs and total signal intensity (presented in units of radiant efficiency (p s−1 cm−2 sr−1)/(μW cm−2)) within the ROI was used for subsequent semi-quantitative analysis.Results and discussion
We have investigated various water-soluble HPMA copolymer carriers in terms of their in vivo suitability as efficient and safe drug delivery carriers. The aim of this study was to determine the influence of the dispersity and molecular weight of polymers on their biological behavior in vitro and in vivo, especially on biodistribution, tumor accumulation and elimination from the body, as well as on the cellular uptake and cytotoxicity of the corresponding polymer carriers and conjugates of Dox.Synthesis of HPMA-based polymer precursors
The properties of polymer precursors with high dispersity (HD) prepared by free radical polymerization (HD-P(hyd) or HD-P(TT)) were compared with the properties of polymer precursors with low dispersity (LD), prepared by controlled radical RAFT polymerization, either with Mw values similar to HD precursors (LD-P(hyd)-30 or LD-P(TT)-30) or with Mw closely below the limit of renal filtration (LD-P(hyd)-45 or LD-P(TT)-45) (the reaction schemes and characteristics are given in Fig. S1† and Table 1, respectively). The polymerization conditions were adapted from our previous studies.19,21 The content of the hydrazide or TT groups in the polymer precursors was sufficient for further attachment the fluorescent dyes, drug or chelator.| Copolymer no. | Polymerization type |
M
wa (kg mol−1) |
Đ | Content of functional groupsb (mol%) | |
|---|---|---|---|---|---|
| Hydrazides | TT | ||||
| a The molecular weights (Mw) and dispersity (Đ) were determined by SEC using RI and MALS detection. b The content of hydrazide or TT groups was determined spectrophotometrically. | |||||
| HD-P(hyd) | FRP | 27.8 | 1.74 | 5.1 | — |
| HD-P(TT) | FRP | 25.6 | 1.54 | — | 1.7 |
| LD-P(hyd)-30 | RAFT | 32.6 | 1.12 | 7.7 | — |
| LD-P(TT)-30 | RAFT | 33.3 | 1.06 | — | 1.7 |
| LD-P(hyd)-45 | RAFT | 45.2 | 1.13 | 6.9 | |
| LD-P(TT)-45 | RAFT | 43.3 | 1.07 | — | 3.6 |
Due to rather high dispersity both HD-polymer precursors with Mw about 27 kg mol−1 contained 25% of polymer chains (lower quartile, Q1) with Mw below 12 kg mol−1 and 25% of polymer chains (upper quartile, Q3) with Mw above 30 kg mol−1 (Fig. 1), thus a significant portion of polymer chains had very low molecular weight, predisposing them to fast excretion and low tumor accumulation due to the EPR effect. Due to the RAFT polymerization technique, a low dispersity of LD-copolymers (e.g., Đ = 1.12 for LD-P(hyd)-30 and Q1 = 26 kg mol−1 and Q3 = 37 kg mol−1) was achieved compared with the high dispersity of HD-copolymers (e.g., Đ = 1.87 for HD-P(hyd)) prepared by FRP, thus the portion of polymers with a molecular weight under 15 kg mol−1 is highly reduced and its interquartile range (Q3 − Q1) is much lower (compare Q3 − Q1 = 10 kg mol−1 for LD precursors and 18 kg mol−1 for HD precursors) (Fig. 1). By increasing the ratio between the monomers and CTA, copolymers with the hydrazide (LD-P(hyd)-45) or TT (LD-P(TT)-45) groups with higher molecular weight (Mw about 44 kg mol−1) but with the same dispersity (Đ about 1.1) could be synthesized. As expected, the molecular weight of the LD-45 copolymers in both quartiles (e.g., Q1 = 34 kg mol−1 and Q3 = 47 kg mol−1 for LD-P(TT)-45) was higher, while the interquartile range remained nearly unchanged. Thus, the majority of LD polymer carrier chains should exhibit prolonged blood circulation and enhanced passive tumor accumulation.
 | ||
| Fig. 1 The cumulative weight molar mass of polymer precursors HD-P(TT) (red line), LD-P(TT)-30 (green line) and LD-P(TT)-45 (blue line) determined by SEC using MALS detection. Determined quartiles Q1 and Q3 are depicted by the same colors. | ||
A highly effective, and at the same time safe, drug delivery system designed for use in medicine should be excreted from the body within a reasonable time after fulfilling its role as a drug carrier. We have investigated this issue in detail and compared the quantiles Q0.9 and Q0.95, i.e. 90% or 95%, of polymer chains having Mw under this value, for all the synthesized polymers. As seen in Fig. S2,†Q0.9 is the lowest for LD-P(TT)-30 and it increases for HD-P(TT) and LD-P(TT)-45. Nevertheless, their Q0.9 values are still in the range of renally eliminable polymers (Q0.9 for HD-P(TT) reached 50 kg mol−1, and for LD-P(TT)-45, ∼56 kg mol−1). Furthermore, Q0.95 values (Fig. 2) definitely proved the safe excretion profiles of all the studied polymers, as the value for both HD-P(TT) and LD-P(TT)-45 reached 65 kg mol−1, which is still below the renal threshold for HPMA copolymers.6 Thus, the Q0.95 values for HD-P(TT) and LD-P(TT)-45 polymers confirmed the safe in vivo applicability in terms of polymer carrier elimination from the body, moreover, they unequivocally showed the potential of low-dispersity polymers prepared by the RAFT technique. Such polymers offer application of the higher Mw of polymer carriers while ensuring their safe elimination after fulfilment of their role in vivo.
 | ||
| Fig. 2 The cumulative weight molar mass and RI and LS chromatograms of polymer precursors HD-P(TT) (red line), LD-P(TT)-30 (green line) and LD-P(TT)-45 (blue line) determined by SEC using RI and LS detection. Determined quantiles Q0.95 are depicted by the same colors. Dashed areas in chromatograms represent 5% of cumulative weight fraction. | ||
Synthesis of the polymer conjugates with the imaging agents and drug
The chemical structures and the characteristics of the polymer conjugates with imaging agents and drug are displayed in Fig. 3 and Table 2, respectively.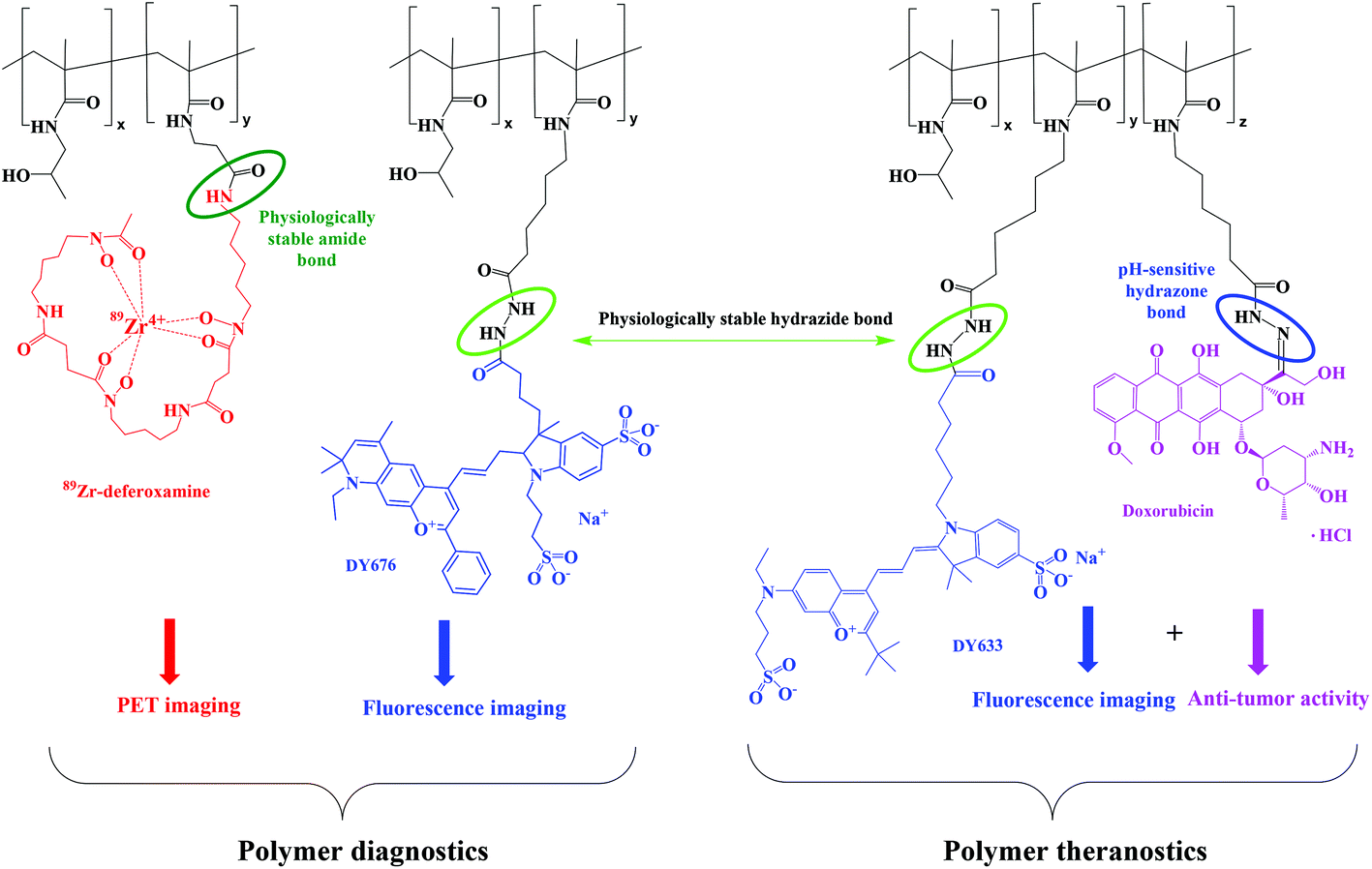 | ||
| Fig. 3 The chemical structure of polymer conjugates with imaging agents and/or Dox. | ||
| Sample name | Prepared from | Def contenta (wt%/mol%) | Dye contentb (wt%/mol%) | Dox contentb (wt%/mol%) |
|---|---|---|---|---|
| a Deferoxamine content was determined by 1H-NMR spectroscopy. b Dye or Dox content was determined spectrophotometrically. | ||||
| HD-P + DY-633 | HD-P(hyd) | — | 2.7/0.5 | — |
| LD-P-30 + DY-633 | LD-P(hyd)-30 | — | 1.4/0.3 | — |
| LD-P-45 + DY-633 | LD-P(hyd)-45 | — | 2.3/0.4 | — |
| HD-P + Dox | HD-P(hyd) | — | — | 9.8/2.7 |
| LD-P-30 + Dox | LD-P(hyd)-30 | — | — | 9.4/2.6 |
| LD-P-45 + Dox | LD-P(hyd)-45 | — | — | 9.6/2.6 |
| HD-P + Dox/DY-633 | HD-P + DY-633 | — | 2.7/0.5 | 9.4/2.6 |
| LD-P-30 + Dox/DY-633 | LD-P-30 + DY-633 | — | 1.4/0.3 | 9.6/2.6 |
| LD-P-45 + Dox/DY-633 | LD-P-45 + DY-633 | — | 2.3/0.4 | 9.8/2.7 |
| HD-P + DY-676 | HD-P(hyd) | — | 0.9/0.2 | — |
| LD-P-45 + DY-676 | LD-P(hyd)-45 | — | 1.1/0.2 | — |
| HD-P + Def | HD-P(TT) | 5.8/1.7 | — | — |
| LD-P-30 + Def | LD-P(TT)-30 | 5.8/1.7 | — | — |
| LD-P-45 + Def | LD-P(hyd)-45 | 10.3/3.2 | — | — |
The fluorescently-labeled copolymers HD-P + DY-633, LD-P-30 + DY-633 and LD-P-45 + DY-633 or HD-P + DY-676 and LD-P-45 + DY-676 were prepared by the reaction of hydrazide groups of the respective polymer precursors with the NHS-ester of the fluorescent dyes DY-676 or DY-633 forming a hydrazide bond. The dye attachment did not influence the dispersity of the polymer–dye conjugates compared to their precursors. The amount of fluorescent dye (approximately 1–2.5 wt%) was sufficient for successful imaging in vitro and in vivo.
The polymer–doxorubicin conjugates HD-P + Dox, LD-P-30 + Dox and LD-P-45 + Dox or their fluorescently-labeled analogues HD-P + Dox/DY-633, LD-P-30 + Dox/DY-633 and LD-P-45 + Dox/DY-633 were prepared by the reaction of Dox·HCl with hydrazide groups of the respective polymer precursors forming a hydrazone bond. The molecular weights of the conjugates slightly increased compared with those of the copolymer precursors; however, their dispersity did not change significantly. As expected, no differences in reactivity during conjugation reaction or in physico-chemical properties of the prepared polymer–Dox conjugates were observed as previously described.12,19 Thus, the utilization of the HPMA copolymer enabled the simultaneous attachment of an imaging agent and the anticancer drug, Dox.
GPC profiles of the fluorescently labeled polymers corresponded with those obtained for polymer precursors; nevertheless, the precise calculation of Mw could not be executed due to the interaction of the LS detector laser with the fluorescent dye.
The polymer–deferoxamine conjugates HD-P + Def, LD-P-30 + Def and LD-P-45 + Def were prepared by reaction of the amino group of deferoxamine mesylate with the TT group of the respective polymer precursors forming an amide bond. In order to reduce the side reaction of the hydroxylamine group of deferoxamine with the TT groups of the polymer backbone causing potential crosslinking of the polymer conjugate, the polymer precursor solution was added dropwise to the solution of ten molar excess of deferoxamine mesylate with DIPEA. Under these conditions, the reaction yield reached nearly 100% and the attachment of deferoxamine had no significant influence on the molecular weight and dispersity of the polymer conjugate; Mw = 27.1 kg mol−1 and Đ = 1.63 for HD-P + Def; Mw = 35.0 kg mol−1 and Đ = 1.13 for LD-P-30 + Def; Mw = 50.5 kg mol−1 and Đ = 1.21 for LD-P-45 + Def.
In vitro cytotoxicity of polymer conjugates
The cytostatic activities of the free Dox and polymer conjugates HD-P + Dox, LD-P-30 + Dox and LD-P-45 + Dox were evaluated on three different cancer cell lines, including 4T1 murine breast cancer cells, MCF-7 human breast cancer cells and Jurkat human T-lymphocyte cells, where the Jurkat cell line is much more sensitive to Dox compared with the other two cell lines. As mentioned in our several recent reports, the cytotoxicity of the polymer conjugates in vitro is always lower than the cytotoxicity of the free drug. In contrast to the low molecular weight drug, which enters the cells quite rapidly by diffusion, the polymer conjugates reach cells via the slower process of endocytosis. Similar observations were also seen in the case of the described HD and LD polymers, as the cytotoxicities of the polymer conjugates were always lower than that of free Dox. The values of IC50, i.e. the concentration of the drug that inhibits cell proliferation by 50%, for all cell lines and conjugates are summarized in Table 3. We observed a small difference in the cytotoxicity of the polymers; the cytotoxicity slightly increased with the decreased dispersity and increased molecular weight of the polymer–Dox conjugates. We hypothesize that the slight increase in the cytotoxicity of the less dispersed LD-P-45 + Dox and LD-P-30 + Dox polymer conjugates could be explained by the higher number of polymer chains with a higher molecular weight, which enables the transfer into the endosomal compartment, which is usually 50 nm in diameter, of a higher amount of drug per endosome. Moreover, no significant influence of fluorophores or Def was found in the in vitro cytotoxicity tests. Thus, we can summarize that the studied low-dispersed polymers are suitable candidates for further pharmacological development as highly potent stimuli-sensitive drug delivery systems.| Sample | Jurkat (μg ml−1) | MCF-7 (μg ml−1) | 4T1 (μg ml−1) |
|---|---|---|---|
| HD-P + Dox | 0.37 ± 0.09 | 1.94 ± 0.75 | 20.72 ± 2.14 |
| LD-P-30 + Dox | 0.32 ± 0.13 | 1.90 ± 0.42 | 19.55 ± 4.59 |
| LD-P-45 + Dox | 0.29 ± 0.08 | 1.44 ± 0.11 | 16.66 ± 3.91 |
| Dox | 0.03 ± 0.01 | 0.09 ± 0.03 | 0.70 ± 0.33 |
Cellular uptake of fluorescently labeled polymer conjugates in vitro
To further clarify the slightly increased cytotoxicity of sparsely dispersed polymers, LD-P-30 + Dox and LD-P-45 + Dox compared with HD-P + Dox, we evaluated the uptake of the fluorescently labeled polymers and polymer–Dox conjugates in the murine cancer cell line 4T1 using confocal laser scanning microscopy. We observed a higher uptake for both sparsely dispersed polymers and polymer–drug conjugates, and Dy633 labeled systems than for the Dyomics-633 labeled HD polymer or polymer–Dox conjugates (Fig. 4). Using the region of interest (ROI) analysis we have calculated that the uptake of LD-P-30 + Dox and LD-P-45 + Dox compared to HD-P + Dox was 3.85 times (±1.69) and 2.89 times (±0.68) respectively, higher. Similarly, the ROI analysis proved the same trend in the uptake of polymers without Dox, the uptake of LD-P-30 + Dy-633 and LD-P-45 + Dy-633 compared to HD-P + Dy-633 was 3.39 times (±1.03) and 2.93 times (±1.57) higher, respectively. Thus, LD polymers or conjugates are capable of increasing their uptake into cancer cell lines in concordance with the cytotoxicity study presented above. | ||
| Fig. 4 Uptake of polymers (top row) and polymer–Dox conjugates (bottom row) labeled by fluorescent dye Dyomics-633 (coloured in orange), the nucleus stained with Hoechst 33342 (coloured in blue), after incubation of the parent murine mammary 4T1 cell line for 24 h with fluorescently labeled polymers (A) HD-P + Dy-633, (B) LD-P-30 + Dy-633 and (C) LD-P-45 + Dy-633 and polymer–drug conjugates (D) HD-P + Dox/Dy-633, (E) LD-P-30 + Dox/Dy-633 and (F) LD-P-45 + Dox/Dy-633 at 37 °C and 5% CO2. | ||
Moreover, except for the polymer uptake study we have also evaluated the cell uptake of Dox carried out on polymer conjugates (Fig. 5). Similarly to the polymer uptake studies we also found an increased uptake of Dox in the case of LD-P-30 + Dox/Dy-633 and LD-P-45 + Dox/Dy-633 polymers with low dispersity. The Dox signal was much more profound within the cells than for polymer HD-P + Dox/Dy-633. Altogether, both uptake studies unequivocally proved the enhanced uptake by the cancer cell line of LD polymers with higher molecular weight, thus validating the great potential of novel LD polymer carriers for targeted cancer therapy. Additionally, co-localization of the polymer label Dyomics-633 and the Dox signal (Fig. 5) proved univocally the pH-sensitive release mechanism of the hydrazone bond containing polymers. The green signal of the released Dox was present in the cells after 24 h incubation – near to the yellow spots of the co-localized polymer conjugates with attached Dox.
 | ||
| Fig. 5 Uptake of Dox from polymer–Dox conjugates (top row) and co-localization of Dox and polymer carriers labeled with Dyomics-633 (bottom row). Fluorescent dye Dyomics-633 colored in red, Dox colored in green, the nucleus stained with Hoechst 33342 colored in blue, colocalization of Dox and polymer colored in yellow, colocalization of Dox Hoechst 33342 in the nucleus is colored in light blue. Pictures determined after incubation of the parent murine mammary 4T1 cell line for 24 hours with fluorescently labeled polymer–drug conjugates (A) HD-P + Dox/Dy-633, (B), (D) LD-P-30 + Dox/Dy-633 and (C), (E) LD-P-45 + Dox/Dy-633 at 37 °C and 5% CO2. | ||
In vivo and ex vivo PET imaging with 89Zr-labeled Def polymer conjugates
The relatively low positron energy (β+avg = 395.5 keV) and long half-life of 89Zr allow long-term monitoring of 89Zr-labeled radiotracers.25 Therefore, the 89Zr-labeling of Def–polymer conjugates enables the observation of their biodistribution for up to 72 h. The cytotoxicity of Dox conjugates in vitro was not influenced by the presence of Def or fluorophores, nor the molecular weight or dispersity of polymer precursors. However, their anti-tumor activity in vivo is expected to depend not only on direct cell cytotoxicity but also on their biodistribution and the rate of body clearance.Allthe Def–polymer conjugates demonstrated high radiolabeling yields (>80%), when incubated with 89Zr in 0.5 M HEPES buffer for 2 h. MicroPET was used to assess the in vivo biodistribution of 89Zr-HD-P + Def, 89Zr-LD-P-30 + Def and 89Zr-LD-P-45 + Def after i.v. injection into 4T1-tumor bearing mice (n = 3 per group) at 0.5, 3, 6, 20, 48 and 72 h p.i. The initial blood pool activity for 89Zr-HD-P + Def was considerably lower than for 89Zr-LD-P-30 + Def; 12.3 ± 2.0 compared with 21.7 ± 3.5% ID g−1 at 0.5 h p.i.; whereas a comparable amount of both polymers remained in circulation after 6 h p.i. (6.3 ± 0.9 and 8.4 ± 0.6% ID g−1 for 89Zr-HD-P + Def and 89Zr-LD-P-30 + Def, respectively) (Fig. 6). As expected, 89Zr-LD-P-45 + Def demonstrated significantly longer blood circulation and more pronounced tumor uptake than its counterparts 89Zr-HD-P + Def, 89Zr-LD-P-30 + Def (Fig. 6(a), bottom panel). During the first scan, at a 0.5 h time-point, the blood pool showed a high concentration of 89Zr-LD-P-45 + Def (26.4 ± 2.7% ID g−1) which decreased slowly over time, with 3.8 ± 3.0% ID g−1 remaining in the circulation on day 3 p.i. In terms of polymer carrier elimination from the body, both the detailed analysis of the Mw distribution of the studied polymers (Q0.9 and Q0.95 values) as well as thorough in vivo study confirmed that sparsely dispersed polymers with higher Mw, prepared by RAFT polymerization can be administered to the body while guaranteeing their safe elimination after fulfilling their role.
 | ||
| Fig. 6 In vivo PET imaging and biodistribution study: (a) serial maximum intensity projection (MIP) images, (b) time–activity curves, (c) and comparison of radioactivity retained in the liver, blood, tumor and muscle of 89Zr-HD-P + Def, 89Zr-LD-P-30 + Def and 89Zr-LD-P-45 + Def. | ||
The gradual decrease in plasma activity was accompanied by a concomitant increase in the accumulation of the tracers at the tumor site. As can be clearly seen in the MIPs presented in Fig. 6(a)89Zr-HD-P + Def showed a rapid and persistent uptake at the 4T1 tumor site (2.6 ± 1.3 at 0.5 h p.i.), which increased to a maximum of 3.7 ± 0.3 at 24 h p.i. and remained constant thereafter (up to 72 h). Compared with 89Zr-HD-P + Def, 89Zr-LD-P-30 + Def displayed longer circulation and enhanced tumor tropism (3.3 ± 1.0% ID g−1 accumulation within 0.5 h p.i., reaching a maximum of 5.7 ± 0.6% ID g−1 at 48 h p.i.), owing to lower dispersity, resulting in greater accumulation of polymers with a higher molecular weight at the tumor site (see Fig. 1) due to a more effective enhanced permeability and retention (EPR) effect (Fig. 6(b)). By increasing the Mw of 89Zr-LD-P-45 + Def close to the renal threshold, slower plasma clearance of 89Zr-LD-P-45 + Def and considerably enhanced 4T1 tumor uptake (11.6 ± 1.8% ID g−1 at 72 h p.i., respectively) were observed (Fig. 6(c)).
Slow clearance and prolonged accumulation of nanomaterials in off-target organs like the liver and spleen raise concerns over their potential systemic toxicity, and have posed a hurdle towards their successful transfer to the clinic. It is worth mentioning that all the mice injected with labeled polymers, either 89Zr-Def or fluorophore, did not show any sign of toxicity as their body weight remain unchanged after in vivo administration. Notably, in addition to appropriate blood circulation and tumor homing properties, the polymer conjugates also exhibited rapid systemic clearance, resulting in excellent tumor to normal tissue contrast. Clearance from the blood was accompanied by a rapid increase in the signal from the bladder within 30 min p.i., which seems to peak at 3 h p.i., tapering off thereafter. The signal from the liver was found to be the highest just after administration (5.3 ± 0.7, 8.6 ± 1.6 and 10.3 ± 0.3% ID g−1 at 0.5 h for 89Zr-HD-P + Def, 89Zr-LD-P-30 + Def and 89Zr-LD-P-45 + Def, respectively), decreasing continuously thereafter to final values of 2.0 ± 0.2, 2.5 ± 0.1 and 6.5 ± 1.5% ID g−1 at day 3 p.i., indicating initially rapid and afterward slower, but progressive clearance of these tracers via the renal and hepatic pathways. Although all three polymer conjugates show similar biodistribution profiles, the uptake and retention in the tumor and other major organs follows the order: 89Zr-HD-P + Def < 89Zr-LD-P-30 + Def < 89Zr-LD-P-45 + Def, corresponding to their respective dispersity and molecular weights (Fig. 6 and Fig. S3†). The higher molecular weight and narrower distribution of the molecular weights of polymer carriers improve their pharmacokinetic profile with prolonged blood circulation and enhanced tumor uptake.
Also noteworthy is the absence of signals from the bones and joints (89Zr is a well-known osteophile),26 even at day 3 p.i. indicating the excellent radiostability of 89Zr-HD-P + Def, 89Zr-LD-P-30 + Def and 89Zr-LD-P-45 + Def. Since PET can only detect the radioisotope, which in many cases follows a completely different trajectory than the parent tracers, the high integrity of the radioisotope: tracer complex in vivo is of utmost importance for accurate prediction of tracer kinetics and reliable disease diagnoses.
After the final scans at 72 h p.i., the mice from all the groups were euthanized and the tumors and other major organs were harvested. Ex vivo longitudinal PET (Fig. 7(a–c)) and gamma counting (Fig. 7(d)) of the explanted tumors and organs validated the in vivo findings. In addition, minimal retention was observed in off-target organs like skin, muscle, lungs and brain tissue (Fig. 7), further documenting the efficacy, specificity and safety of the reported polymers and their potential to serve as efficient tumor-targeted polymer therapeutic carriers (polymer carriers with attached anticancer agents) or even for polymer theranostics (polymer carriers with attached anticancer agents and imaging agents). The slightly elevated retention of 89Zr-LD-P-45 + Def in the liver and spleen after 72 h p.i. is strongly connected with the increased blood circulation and is continuously decreasing as evident from Fig. 6.
 | ||
| Fig. 7 Ex vivo study: longitudinal PET (a–c) and gamma counting (d) of the explanted tumor and organs, 72 h after the intravenous administration of 89Zr-HD-P + Def, 89Zr-LD-P-30 + Def and 89Zr-LD-P-45 + Def. | ||
In vivo and ex vivo fluorescence imaging with DY676-labeled polymer conjugates
By simply substituting the chelator Def with a fluorescent dye DY-676 (Ex: 677 nm; Em: 700 nm) on the HPMA copolymer backbone, the fluorescently-labeled HPMA copolymers could be utilized for optical imaging, thereby validating their potential for versatile multimodal molecular imaging. In order to compare these two imaging techniques – PET and fluorescence, polymers HD-P + DY-676 and LD-P-45 + DY-676 were chosen for this proof-of-concept study. After i.v. administration of HD-P + DY-676 and LD-P-45 + DY-676 a distinct fluorescence signal from the 4T1 tumors was visible as early as 0.5 h p.i. (Fig. 8(a)). The intensity of the signal from the tumor increased over time up to 24 h p.i., corresponding to the increasing accumulation of the polymers at the target site, after which it decreased slightly. Although both polymer conjugates show similar behavior regarding enhanced tumor accumulation, the signal in the tumor was much higher from LD-P-45 + DY-676 compared with HD-P + DY-676, corresponding to their respective dispersity and molecular weights (Fig. 8a and Table 3). In contrast, fluorescence signals from the non-target tissues remained negligible. Owing to the limited depth of tissue penetration, fluorescence signals from the liver and spleen could not be visualized in vivo, warranting further ex vivo analysis. | ||
| Fig. 8 In vivo and ex vivo optical imaging. (a) Serial in vivo optical imaging of 4T1 tumor-bearing mice injected with HD-P + DY-676 (top panels) and LD-P-45 + DY-676 (bottom panels) at 0.5 h, 3 h, 24 h and 48 h p.i. (b) Ex vivo images of tumor and major organs 48 h p.i. of the tracers. Images are representative of 3 mice per group. Green arrowheads indicate the position of the 4T1 tumor. | ||
After the final scan at 48 h p.i., the mice from both the groups were euthanized and 4T1 tumors and other major organs were harvested and imaged ex vivo. The tumor was the dominant site of uptake for both polymers HD-P + DY-676 and LD-P-45 + DY-676, while the clearance organs like liver, spleen and kidney showed attenuated signals indicating a successful clearance of the polymers via both the renal and hepatic pathways (Fig. 8(b)). The semi-quantitative analysis of the explanted tumors and major organs proved observable differences in the in vivo fate of the two polymers (Table S1†). LD-P-45 + DY-676 demonstrated higher tumor accumulation (1.6 times) and longer in vivo residence, when compared with that of HD-P + DY-676 (Fig. 7 and Table S1†), which is in excellent agreement with the PET observations (2.7 times higher tumor accumulation of 89Zr-LD-P-45 + Def compared with that of 89Zr-HD-P + Def). Similar agreement between PET and FI data were found for other observed tissues. Slightly higher accumulation was found for the liver, lung and spleen, owing to longer blood circulation and higher molecular weight of the polymer. To sum up, we have proved the applicability of PET and FI imaging for a HPMA copolymer based platform for theranostics with high tumor accumulation.
Conclusion
Novel polymer diagnostics and potential therapeutics based on 89Zr- or fluorescently-labeled HPMA-copolymer conjugates differing in molecular weight with either low dispersity or high dispersity were designed, synthesized, and in vitro characterized and their biodistribution in vivo was successfully observed within 72 h. The in vitro cellular uptake and cytotoxicity study of Dox-conjugates showed increased cellular uptake and cytotoxicity to 4T1 cancer cells for LD polymers with increased molecular weight or lower dispersity of polymer carriers. Moreover, our in vivo and ex vivo PET/optical imaging studies indicated that the dispersity and molecular weight of the linear HPMA polymer carriers have a significant influence on the in vivo fate of the polymer conjugates. In addition, HPMA polymer conjugates can be successfully utilized as dual modality PET/FI diagnostics with unprecedented tumour to normal tissue contrast and favorable pharmacokinetic profiles. Moreover, the feasibility of the two imaging techniques, FI and PET, was successfully evaluated and compared proving the potential of the described polymer carriers in tumor diagnostics. Finally, the HPMA copolymers prepared with low dispersity (Đ = 1.1) and molecular weight close to the renal threshold (Mw ≈ 45 kg mol−1) prepared by controlled radical RAFT polymerization are very promising carriers of imaging agents and/or anticancer drugs for highly efficient tumor treatment and diagnostics with minimal side effects.Acknowledgements
This work was supported by the Czech Science Foundation (15-02986S), the Ministry of Health of the Czech Republic (16-28594A), the National Sustainability Program I (POLYMAT LO1507), the National Institutes of Health (R01CA169365 and P30CA014520), and the American Chemical Society (125246-RSG-13-099-01-CCE).References
- H. Dozono, S. Yanazume, H. Nakamura, T. Etrych, P. Chytil, K. Ulbrich, J. Fang, T. Arimura, T. Douchi, H. Kobayashi, M. Ikoma and H. Maeda, HPMA Copolymer-Conjugated Pirarubicin in Multimodal Treatment of a Patient with Stage IV Prostate Cancer and Extensive Lung and Bone Metastases, Targeted Oncol., 2016, 11, 101–106 CrossRef PubMed.
- L. W. Seymour, D. R. Ferry, D. J. Kerr, D. Rea, M. Whitlock, R. Poyner, C. Boivin, S. Hesslewood, C. Twelves, R. Blackie, A. Schatzlein, D. Jodrell, D. Bissett, H. Calvert, M. Lind, A. Robbins, S. Burtles, R. Duncan and J. Cassidy, Phase II studies of polymer-doxorubicin (PK1, FCE28068) in the treatment of breast, lung and colorectal cancer, Int. J. Oncol., 2009, 34, 1629–1636 CrossRef CAS PubMed.
- H. Maeda and Y. Matsumura, Tumoritropic and lymphotropic principles of macromolecular drugs, Crit. Rev. Ther. Drug Carrier Syst., 1989, 6, 193–210 CAS.
- T. Etrych, T. Mrkvan, P. Chytil, Č. Koňák, B. Říhová and K. Ulbrich, N-(2-hydroxypropyl)methacrylamide-based polymer conjugates with pH-controlled activation of doxorubicin. I. New synthesis, physicochemical characterization and preliminary biological evaluation, J. Appl. Polym. Sci., 2008, 109, 3050–3061 CrossRef CAS.
- T. Etrych, L. Kovář, J. Strohalm, P. Chytil, B. Říhová and K. Ulbrich, Biodegradable star HPMA polymer-drug conjugates: Biodegradability, distribution and anti-tumor efficacy, J. Controlled Release, 2011, 154, 241–248 CrossRef CAS PubMed.
- T. Etrych, V. Šubr, J. Strohalm, M. Šírová, B. Říhová and K. Ulbrich, HPMA copolymer-doxorubicin conjugates: The effects of molecular weight and architecture on biodistribution and in vivo activity, J. Controlled Release, 2012, 164, 346–354 CrossRef CAS PubMed.
- C. W. Scales, Y. A. Vasilieva, A. J. Convertine, A. B. Lowe and C. L. McCormick, Direct, Controlled Synthesis of the Nonimmunogenic, Hydrophilic Polymer, Poly(N-(2-hydroxypropyl)methacrylamide) via RAFT in Aqueous Media, Biomacromolecules, 2005, 6, 1846–1850 CrossRef CAS PubMed.
- M. J. Yanjarappa, K. V. Gujraty, A. Joshi, A. Saraph and R. S. Kane, Synthesis of copolymers containing an active ester of methacrylic acid by RAFT: controlled molecular weight scaffolds for biofunctionalization, Biomacromolecules, 2006, 7, 1665–1670 CrossRef CAS PubMed.
- B. S. Tucker and B. S. Sumerlin, Poly(N-(2-hydroxypropyl) methacrylamide)-based nanotherapeutics, Polym. Chem., 2014, 5, 1566–1572 RSC.
- J. Yang and J. Kopeček, Macromolecular Therapeutics, J. Controlled Release, 2014, 288–303 CrossRef PubMed.
- A. Gregory and M. H. Stenzel, Complex polymer architectures via RAFT polymerization: From fundamental process to extending the scope using click chemistry and nature's building blocks, Prog. Polym. Sci., 2012, 37, 38–105 CrossRef CAS.
- P. Chytil, M. Šírová, E. Koziolová, K. Ulbrich, B. Říhová and T. Etrych, The comparison of in vivo properties of water-soluble HPMA-based polymer conjugates with doxorubicin prepared by controlled RAFT or free radical polymerization, Physiol. Res., 2015, 64, 41–49 Search PubMed.
- T. Lammers, R. Kuhnlein, M. Kissel, V. Šubr, T. Etrych, R. Pola, M. Pechar, K. Ulbrich, G. Storm, P. Huber and P. Peschke, Effect of physicochemical modification on the biodistribution and tumor accumulation of HPMA copolymers, J. Controlled Release, 2005, 110, 103–118 CrossRef CAS PubMed.
- S. Hoffmann, L. Vystrčilová, K. Ulbrich, T. Etrych, H. Caysa, T. Mueller and K. Mäder, Dual Fluorescent HPMA Copolymers for Passive Tumor Targeting with pH-Sensitive Drug Release: Synthesis and Characterization of Distribution and Tumor Accumulation in Mice by Noninvasive Multispectral Optical Imaging, Biomacromolecules, 2012, 13, 652–663 CrossRef CAS PubMed.
- P. Chytil, E. Koziolova, O. Janouskova, L. Kostka, K. Ulbrich and T. Etrych, Synthesis and Properties of Star HPMA Copolymer Nanocarriers Synthesised by RAFT Polymerisation Designed for Selective Anticancer Drug Delivery and Imaging, Macromol. Biosci., 2015, 15, 839–850 CrossRef CAS PubMed.
- W. Cai and X. Chen, Multimodality molecular imaging of tumor angiogenesis, J. Nucl. Med., 2008, 49(suppl 2), 113–128 CrossRef PubMed.
- S. Goel, C. G. England, F. Chen and W. Cai, Positron emission tomography and nanotechnology: A dynamic duo for cancer theranostics, Adv. Drug Delivery Rev., 2016 DOI:10.1016/j.addr.2016.08.001.
- V. Šubr and K. Ulbrich, Synthesis and properties of new N,-(2-hydroxypropyl)methacrylamide copolymers containing thiazolidine-2-thione reactive groups, React. Funct. Polym., 2006, 66, 1525–1538 CrossRef.
- P. Chytil, T. Etrych, J. Kříž, V. Šubr and K. Ulbrich, N-(2-Hydroxypropyl)methacrylamide-based polymer conjugates with pH-controlled activation of doxorubicin for cell-specific or passive tumour targeting. Synthesis by RAFT polymerisation and physicochemical characterisation, Eur. J. Pharm. Sci., 2010, 41, 473–482 CrossRef CAS PubMed.
- K. Ulbrich, T. Etrych, P. Chytil, M. Jelínková and B. Říhová, Antibody-targeted polymer-doxorubicin conjugates with pH-controlled activation, J. Drug Targeting, 2004, 12, 477–489 CrossRef CAS PubMed.
- P. Šácha, T. Knedlík, J. Schimer, J. Tykvart, J. Parolek, V. Navrátil, P. Dvořáková, F. Sedlák, K. Ulbrich, J. Strohalm, P. Majer, V. Šubr and J. Konvalinka, iBodies: Modular Synthetic Antibody Mimetics Based on Hydrophilic Polymers Decorated with Functional Moieties, Angew. Chem., Int. Ed., 2016, 55, 2356–2360 CrossRef PubMed.
- S. Perrier, P. Takolpuckdee and C. A. Mars, Reversible addition-fragmentation chain transfer polymerization: End group modification for functionalized polymers and chain transfer agent recovery, Macromolecules, 2005, 38, 2033–2036 CrossRef CAS.
- T. Etrych, P. Chytil, M. Jelínková, B. Říhová and K. Ulbrich, Synthesis of HPMA Copolymers Containing Doxorubicin Bound via a Hydrazone Linkage. Effect of Spacer on Drug Release and in vitro Cytotoxicity, Macromol. Biosci., 2002, 2, 43–52 CrossRef CAS.
- H. Hong, G. W. Severin, Y. Yang, J. W. Engle, Y. Zhang, T. E. Barnhart, G. Liu, B. R. Leigh, R. J. Nickles and W. Cai, Positron emission tomography imaging of CD105 expression with 89Zr-Df-TRC105, Eur. J. Nucl. Med. Mol. Imaging, 2012, 39, 138–148 CrossRef CAS PubMed.
- F. Chen, S. Goel, H. F. Valdovinos, H. Luo, R. Hernandez, T. E. Barnhart and W. Cai, In Vivo Integrity and Biological Fate of Chelator-Free Zirconium-89-Labeled Mesoporous Silica Nanoparticles, ACS Nano, 2015, 9, 7950–7959 CrossRef CAS PubMed.
- M. A. Deri, B. M. Zeglis, L. C. Francesconi and J. S. Lewis, PET Imaging with (89)Zr: From Radiochemistry to the Clinic, Nucl. Med. Biol., 2013, 40, 3–14 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7nr03306k |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2017 |