
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reducing hole transporter use and increasing perovskite solar cell stability with dual-role polystyrene microgel particles†
Mu
Chen
a,
Muhamad Z.
Mokhtar
a,
Eric
Whittaker
b,
Qing
Lian
a,
Bruce
Hamilton
b,
Paul
O'Brien
 ac,
Mingning
Zhu
a,
Zhengxing
Cui
a,
Saif A.
Haque
d and
Brian R.
Saunders
*a
ac,
Mingning
Zhu
a,
Zhengxing
Cui
a,
Saif A.
Haque
d and
Brian R.
Saunders
*a
aSchool of Materials, University of Manchester, MSS Tower, Manchester, M13 9PL, UK. E-mail: brian.saunders@manchester.ac.uk
bPhoton Science Institute, University of Manchester, Alan Turing Building, Oxford Road, Manchester, M13 9PL, UK
cSchool of Chemistry, University of Manchester, Manchester, M13 9PL, UK
dDepartment of Chemistry and Centre for Plastic Electronics, Imperial College London, South Kensington Campus, SW7 2AZ, UK
First published on 30th June 2017
Abstract
Perovskite solar cells (PSCs) are a disruptive technology that continues to attract considerable attention due to their remarkable and sustained power conversion efficiency increase. Improving PSC stability and reducing expensive hole transport material (HTM) usage are two aspects that are gaining increased attention. In a new approach, we investigate the ability of insulating polystyrene microgel particles (MGs) to increase PSC stability and replace the majority of the HTM phase. MGs are sub-micrometre crosslinked polymer particles that swell in a good solvent. The MGs were prepared using a scalable emulsion polymerisation method. Mixed HTM/MG dispersions were subsequently spin-coated onto PSCs and formed composite HTM-MG layers. The HTMs employed were poly(triaryl amine) (PTAA), poly(3-hexylthiophene) (P3HT) and Spiro-MeOTAD (Spiro). The MGs formed mechanically robust composite HTMs with PTAA and P3HT. In contrast, Spiro-MG composites contained micro-cracks due the inability of the relatively small Spiro molecules to interdigitate. The efficiencies for the PSCs containing PTAA-MG and P3HT-MG decreased by only ∼20% compared to control PSCs despite PTAA and P3HT being the minority phases. They occupied only ∼35 vol% of the composite HTMs. An unexpected finding from the study was that the MGs dispersed well within the PTAA matrix. This morphology aided strong quenching of the CH3NH3PbI3−xClx fluorescence. In addition, the open circuit voltages for the PSCs prepared using P3HT-MG increased by ∼170 mV compared to control PSCs. To demonstrate their versatility the MGs were also used to encapsulate P3HT-based PSCs. Solar cell stability data for the latter as well as those for PSCs containing composite HTM-MG were both far superior compared to data measured for a control PSC. Since MGs can reduce conjugated polymer use and increase stability they have good potential as dual-role PSC additives.
Introduction
Perovskite solar cells (PSCs) have been very widely studied in recent years due in large part to the remarkable increases in their power conversion efficiency (PCE).1–3 The maximum certified PCE reported to date is now an impressive 22.1%4 for single junction devices and they have achieved values of more than 25% in tandem cells with silicon.5 The hole transport materials (HTM) employed in PSC fabrication6,7 play important roles in determining device performance with Spiro-MeOTAD (Spiro) being widely used. Other widely used conjugated polymer HTMs include poly(3-hexylthiophene) (P3HT)8 and poly(triaryl amine) (PTAA).9 For longer term PSC scale-up P3HT is a strong HTM candidate with two roll-to-roll proof-of-concept studies already reported.10,11 However, the relatively high cost of these HTMs drives research for reducing their use or replacing them. An important concern for PSCs is their relatively poor stability.12–15 PSC performance is degraded by moisture, light, oxygen and elevated temperature.16–18 The HTM provides an important opportunity to design a barrier to protect the underlying perovskite from the atmosphere which is explored in the present study. Microgels (MGs) are sub-micrometre crosslinked polymer particles that swell in a thermodynamically good solvent.19 Here, the use of MGs as HTM-MG composites that not only reduce HTM use but increase PSC stability is studied. We also study the use of MGs as an encapsulating layer for PSCs.Whilst insulating MGs have been extensively studied as model colloids20 and surface coatings21 there are no reported studies of their use within solar cells. Polystyrene (PS) MGs were first reported more than 50 years ago.22 The PS MGs used here (Fig. 1, lower centre) were crosslinked with divinylbenzene (DVB) and were prepared using emulsion polymerisation which can be conducted on a multi-tonne scale. MGs are well suited for use with PSCs for several reasons. PS MGs increase structural order and P3HT light absorption within composite P3HT-MG films.23 The MGs force P3HT to percolate around the particles which preserves conductivity.23 Here, we greatly extend our earlier approach to include PTAA and Spiro in the context of composite HTMs for PSCs. MG dispersions also have excellent colloid stability20 and good film forming properties.23–26 PS MGs are hydrophobic which offers potential for water barrier formation. We hypothesised that MGs could be blended with HTMs to provide improved device stability without substantially compromising device performance. In addition, we hypothesised that MGs would provide a hydrophobic barrier and thereby improve PSC stability. The results of this study support these hypotheses. However, several unexpected results were also found and these are discussed.
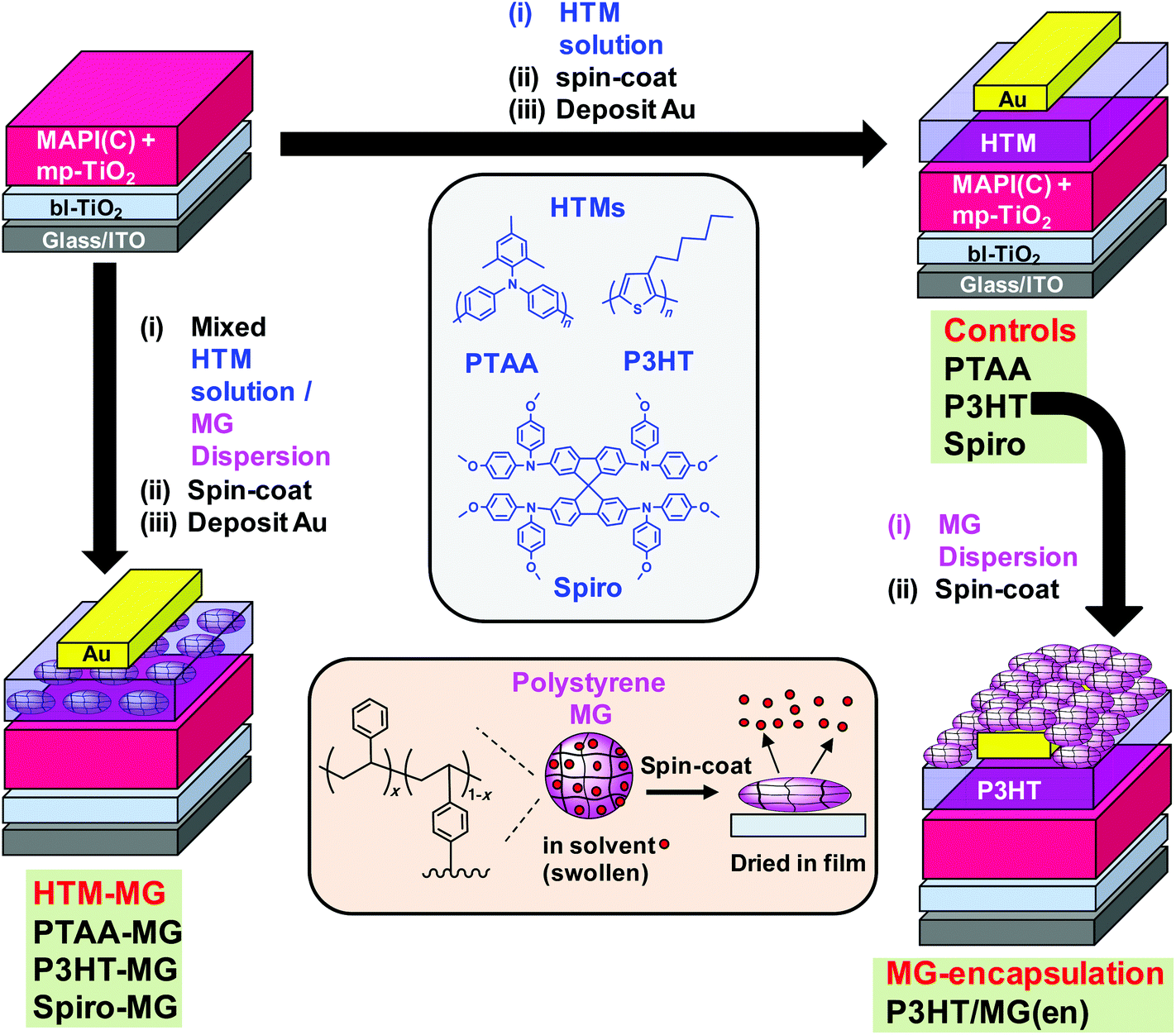 | ||
| Fig. 1 Schematic depicting the two complementary approaches for using microgels (MGs) to improve MAPbI3−xClx (MAPI(C)) solar cell performance. Left hand side: HTM species (PTAA, P3HT or Spiro) were mixed with MGs and spin-coated to form composite hole transport layers (HTM-MG). Right-hand side: starting from the unmodified PSCs (controls), MG particles were deposited as an encapsulating layer for the P3HT-based PSCs (MG-encapsulation). The codes used for the PSCs and their geometries are shown. Lower centre: depiction of polystyrene MGs as swollen spheres when dispersed in a good solvent that become flattened when spin-coated. | ||
A number of studies have investigated including additives within PSC HTM layers. In a seminal paper, Habisreutinger et al. used a composite HTM comprised of single-walled carbon nanotubes (SWNTs) embedded within an insulating polymer.27 Those PSCs showed excellent stability. Gatti et al.28 doped P3HT with SWNTs and reduced graphene oxide and reported improvements of the PCE and stability. Aitola et al. demonstrated that PSCs containing SWNT-based HTM, which also functioned as the hole contact, had improved stabilities compared to devices containing Spiro and a gold back contact.29 Wang et al.30 incorporated gold nanoparticles within a P3HT matrix and also reported an improved PCE. Xiao et al. included graphdiyne into the P3HT layers and reported an improved hole extraction.31 Whilst each study provided improvements in PCE. Stability or charge extraction, they all used conductive additives that were high cost and are not well suited to large scale preparation. In contrast, we use very low cost (insulating) PS MG particles that are highly scalable.
A major source of instability for perovskite is reaction with water vapour32 which has led to encapsulation of PSCs becoming an important area of research. Chang et al.33 used atomic layer deposition to encapsulate PSCs with Al2O3. Whilst the stability of their PSCs was improved, their approach required high vacuum and specialist equipment. Hwang et al. encapsulated PSCs using Teflon which was cast from solution and improved device stability due to hydrophobic passivation. In the context of future scale up, Teflon is a more expensive polymer than PS. Liu et al.34 encapsulated their PSCs with poly(dimethylsiloxane) and reported improved efficiencies. That approach required in situ chemical crosslinking. Here, we used a film-forming dispersion of hydrophobic PS MG particles (Fig. 1) which offers a much simpler, low-cost PSC encapsulation approach which does not involve a chemical reaction. Robust physically interlinked MG films form due to interpenetration of neighbouring MGs.
The approach used in this study is depicted in Fig. 1. MAPbI3−xClx (MAPI(C)) was the perovskite employed. In the first approach (left hand side) binary mixtures of dissolved HTM and dispersed MGs were prepared and spin-coated to form composite HTM-MG layers. Because the MGs were deformable in the swollen state they flattened during spin-coating and solvent evaporation (lower centre of Fig. 1). We previously reported that mixed MG/P3HT dispersions formed electrically conducting composite P3HT-MG films.23 Here, we studied PSCs coated with HTM-MG films containing PTAA, P3HT and Spiro and compared their properties. In the second approach, MG films were spin-coated onto a pure P3HT HTM as an encapsulating layer from MG dispersions to give P3HT/MG(en) PSCs (Fig. 1, right hand side).
The study begins by characterising the MGs and investigating the morphologies of the composite films formed with PTAA, P3HT and Spiro. Performance data for HTM-MG devices are then considered and the results compared. The use of MGs enabled a dramatic decrease in the conjugated polymer that could be used whilst obtaining good PCE values. The optoelectronic properties of the MAPI(C)/HTM-MG films are investigated to gain insight into charge transfer. Unexpectedly, the MGs were found to disperse well in PTAA and enabled very efficient fluorescence quenching. They also increased Voc for the P3HT-MG PSCs. Device stability data are then presented for the P3HT-MG and P3HT/MG(en) PSCs which demonstrate substantial improvements compared to control data. The results of this study show that PS MGs should enable future PSCs to be prepared at any scale with good PCEs and improved stability whilst using much less conjugated polymer.
Experimental details
Materials
P3HT had a regioregularity of greater than 95% and a number-average molecular weight range of 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–35000 g mol−1 (polydispersity <2) and was used as received (Aldrich). Spiro (N2,N2,N2′,N2′,N7,N7,N7′,N7′-octakis(4-methoxyphenyl)-9,9′-spirobi[9H-fluorene]-2,2′,7,7′-tetramine, Fenglin Chemicals, 99.5%) was also used as received. PTAA was purchased from Aldrich and had a number-average molecular weight of 7000–10000 g mol−1 with a polydispersity of 2.0–2.2 and used as received. Styrene (≥99%), divinylbenzene (DVB, 80%) 4,4′-azobis(4-cyanovaleric acid) (ACVA, 98%), toluene (99.8%), chlorobenzene (CBZ, 99.8%), isopropanol (IPA, anhydrous, 99.5%), 4-tert-butylpyridine (TBP, 96%), lithium bistrifluoromethanesulfonimidate (LiTFSI, 99.95%) were all purchased from Sigma-Aldrich and used as received. Methyl amine solution (33 wt% in absolute ethanol) and hydriodic acid (57 wt%), titanium diisopropoxide bis(acetylacetonate) (TDB, 75 wt% in IPA) and PbCl2 (98%), anhydrous N,N-dimethylformamide (DMF, 99.8%) were also purchased from Aldrich and used as received. Methylammonium iodide was synthesised and purified using the method previously reported.35 Titania paste (TiO2, 18 NRT) was purchased from Dyesol and used as received. Water was of ultrahigh purity and de-ionised.
000–35000 g mol−1 (polydispersity <2) and was used as received (Aldrich). Spiro (N2,N2,N2′,N2′,N7,N7,N7′,N7′-octakis(4-methoxyphenyl)-9,9′-spirobi[9H-fluorene]-2,2′,7,7′-tetramine, Fenglin Chemicals, 99.5%) was also used as received. PTAA was purchased from Aldrich and had a number-average molecular weight of 7000–10000 g mol−1 with a polydispersity of 2.0–2.2 and used as received. Styrene (≥99%), divinylbenzene (DVB, 80%) 4,4′-azobis(4-cyanovaleric acid) (ACVA, 98%), toluene (99.8%), chlorobenzene (CBZ, 99.8%), isopropanol (IPA, anhydrous, 99.5%), 4-tert-butylpyridine (TBP, 96%), lithium bistrifluoromethanesulfonimidate (LiTFSI, 99.95%) were all purchased from Sigma-Aldrich and used as received. Methyl amine solution (33 wt% in absolute ethanol) and hydriodic acid (57 wt%), titanium diisopropoxide bis(acetylacetonate) (TDB, 75 wt% in IPA) and PbCl2 (98%), anhydrous N,N-dimethylformamide (DMF, 99.8%) were also purchased from Aldrich and used as received. Methylammonium iodide was synthesised and purified using the method previously reported.35 Titania paste (TiO2, 18 NRT) was purchased from Dyesol and used as received. Water was of ultrahigh purity and de-ionised.
Polystyrene microgel preparation
PS MG was prepared using surfactant-free emulsion polymerisation following the method reported earlier.23 Briefly, water (265 ml) was adjusted to pH of 9.0 using NaOH solution and then added to a 500 ml reaction vessel and stirred at 200 rpm at 70 °C and deoxygenated. ACVA (0.244 g, 0.871 mmol) was dissolved in water (7.0 ml) and adjusted to pH of 11.0 using aqueous NaOH solution. DVB (0.086 g, 0.661 mmol) was mixed with styrene (28.6 g, 0.275 mol) and added to the vessel. The ACVA solution was then quickly added under a N2 atmosphere and the mixture stirred for 16 h. The particle dispersion (in collapsed, latex, form) was purified using repeated centrifugation and re-dispersion in water. After purification, the dispersion was freeze-dried and the powder re-dispersed in toluene or CBZ. To aid re-dispersion a few drops of methanol were added prior to solvent addition. The MG had a nominal composition of 99.7 wt% styrene and 0.3 wt% DVB.Perovskite film fabrication
All solution and film preparation steps (including annealing) were conducted in a glove box (humidity ∼2%). Laser-patterned, ITO-coated glass substrates (20 Ω sq−1) were cleaned by ultrasonication in a 2% Hellmanex solution, rinsed with water and IPA, and dried. A TiO2 hole-blocking layer (bl-TiO2) (48 nm) was spin-coated at 2000 rpm for 60 s onto the ITO using TDP solution in 1-butanol (0.15 M) and subsequent heating at 125 °C for 5 min. The procedure was repeated using TDB solution (0.30 M). Titania paste (70 μL, 1:5 in ethanol) was spin-coated onto the cleaned glass slides at 5000 rpm for 30 s to form a mesoporous scaffold (mp-TiO2). The films were then annealed at 500 °C for 30 min and had an average thickness of 250 nm. After cooling to room temperature a MAPI(C) precursor solution was spin-coated onto ITO/bl-TiO2/mp-TiO2 substrate at 2000 rpm for 60 s. The precursor solution contained MAI and PbCl2 (3:1 molar ratio) in DMF (30 wt%). The film was dried at 100 °C for 45 min. The capping layer thickness was ∼250 nm. All films were stored in a desiccator over P2O5 in the dark until investigation.
Perovskite solar cell fabrication
All stages of device preparation were conducted in the glove box. The procedure to form the ITO/bl-TiO2/mp-TiO2/MAPI(C) films was described above. PTAA solution was prepared by adding PTAA (15 mg) to CBZ (0.865 g) at room temperature. LiTFSI (15 μl, 170 mg ml−1) and t-BP (7.5 μl) were also added.36 The PTAA solution was spin-coated onto the MAPI(C) layer at 4000 rpm for 30 s to give an average PTAA thickness of 120 nm. PTAA-MG composite HTMs were prepared using mixed dispersions containing MG particles and PTAA. The MG dispersion was prepared using MG (64 mg) in CBZ at room temperature with a total dispersion weight of 1.0 g. PTAA solution (3.0 wt%) was prepared by adding PTAA (30 mg) to CBZ (total weight 1.0 g) at room temperature. The MG dispersion and PTAA solution were then mixed at a weight ratio 0.47:0.53. LiTFSI (15 μl, 170 mg ml−1) and t-BP (7.5 μl) were added. The PTAA-MG films were formed by spin-coating at 4000 rpm for 30 s and had an average thickness of 180 nm.
Similar procedures to those described above were used for the devices containing P3HT and P3HT-MG. The solvent used in these cases was toluene at 70 °C. For the P3HT systems, LiTFSI (10 μL) and TBP (10 μL) were added.37 The average film thicknesses for the P3HT and P3HT-MG HTMs were 100 nm and 160 nm, respectively. For the Spiro-containing films, CBZ was used as the solvent at room temperature; LiTFSI (4.8 μl, 520 mg ml−1) and TBP (8.0 μl) were also added.38 The average thicknesses for the Spiro and Spiro-MG films were 450 nm and 160 nm, respectively.
For all devices, a gold layer (80 nm) was deposited by thermal evaporation onto the HTMs and HTM-MG composite layers. In the case of MG encapsulation the MG particles were dispersed in toluene at a concentration (CMG) of 3.0 wt%. The MG dispersions were spin-coated at 3000 rpm for 30 s at room temperature. The thickness of the encapsulating MG layer was 145 nm.
Physical measurements
Dynamic light scattering (DLS) measurements were conducted using a 50 mW He/Ne laser operated at 633 nm with a standard avalanche photodiode (APD) and 90° detection optics connected to a Malvern Zetasizer Nano ZS100 autocorrelator. XRD patterns were obtained using a Bruker D8 Advance diffractometer (Cu-Kα). Films were scanned between 10 and 50° with a step size of 0.05. The films were prepared and measured under a nitrogen atmosphere. Atomic force microscopy (AFM) images were obtained using an Asylum Research MFP-3D operating in AC (“tapping”) mode. UV-visible spectra were obtained using a Hitachi U-1800 spectrophotometer. Film thickness measurements were conducted using a Dektak 8 Stylus Profilometer (Bruker). Contact angle measurements were performed using water and a Krüss Drop Shape Analysis (DSA100). Photoluminescence (PL) spectra were obtained using an Edinburgh Instruments FLS1080 spectrometer. The beam was incident on the film surface side and an excitation wavelength of 480 nm was used. The substrates were ITO/bl-TiO2/mp-TiO2 for the films containing MAPI(C) or glass for the samples that did not contain MAPI(C).Device measurements
The current density–voltage (J–V) curves were measured using a Keithley 2420 sourcemeter, 100 mW cm−2 illumination (AM 1.5G) and a calibrated Oriel Si-reference cell that had been certified by NREL. An Oriel solar simulator (SOL3A) was used for these measurements. The active area of the devices (0.025 cm2) was defined using a square aperture within a mask. The data shown are from the reverse scan unless otherwise stated (Voc to Jsc) and the sweep rate was 100 mV s−1.Results and discussion
Morphologies of composite HTM-MG films
The PS MGs used in this study are depicted in Fig. 1 (lower centre). SEM and DLS characterisation data for the MGs are presented in Fig. S1 and discussed in the ESI.† Briefly, the SEM data (Fig. S1, ESI†) showed that the MGs were monodisperse. DLS data revealed that the particles had a collapsed z-average diameter (dz) of 281 nm when dispersed in water (a poor solvent). The particles had dz values of 529 and 712, nm, respectively, when dispersed in toluene or CBZ and were in their (swollen) MG state. CBZ was the solvent used to spin-coat the PTAA- and Spiro-based films; whereas, toluene was used for the P3HT-based films (Fig. 1).Composite PTAA-MG, P3HT-MG and Spiro-MG films were investigated in this study for their ability to provide efficient PSCs whilst using much lower HTM concentrations. All of the composite films contained only 35 wt% of conjugated material and 65 wt% MG. Therefore, the MGs were the majority phase. We selected this composition because previous work on related P3HT-MG films deposited from ethylbenzene established that those films had similar conductivities to that of P3HT.23 An AFM image and line profile for the PTAA-MG film are shown in Fig. 2a. The morphology is remarkable because the MG particles were very well dispersed within the PTAA matrix. (A larger area image is shown in Fig. S2a.†) This well dispersed morphology is unexpected for a composite film prepared from a mixture of polymer colloid particles and semiconducting polymer and has not been reported before. We attribute the excellent dispersion of MGs to the similar structures of PS and PTAA (Fig. 1). This meant that the segment-segment interactions were similar between and within each polymer. The RMS roughness was 19.0 nm and relatively low. An expanded view of a representative particle is shown in above Fig. 2 which reveals that the MGs were flattened. MG particles flatten during deposition occurs because of their swollen nature.39,40 The good dispersion of the MG particles is supported by an optical micrograph of the PTAA-MG film which was relatively featureless (Fig. 2d). Additional optical micrographs are shown in Fig. S2d.†
 | ||
| Fig. 2 Morphology of PTAA-MG (left column), P3HT-MG (middle column) and Spiro-MG (right column) composite films. AFM tapping mode images are shown in (a) to (c). Expanded views and line profiles are also shown. The top inset shows a line profile for a representative particle using the same scale for the height and distance axes. The yellow arrows in (b) highlight P3HT-rich regions. Optical micrographs for the films are shown in (d) to (f). Micro-cracks were present for the Spiro-MG film (f) and some of these are highlighted with red arrows. See Fig. S2† for more images. | ||
In previous work ethylbenzene was used to prepare P3HT-MG composite films.23 In this study toluene was used because it is a non-solvent for MAPbI341 and did not dissolve MAPI(C) during HTM spin-coating. AFM images (Fig. 2b and Fig. S2b†) for the P3HT-MG film showed close packed particles and regions between them that were P3HT-rich (yellow arrows). The RMS roughness was 24.0 nm, which was slightly greater than that observed for the PTAA-MG film. Whilst this non-uniform morphology is likely due decreased miscibility between P3HT and PS,42 it was not detrimental to the composite film mechanical or electrical properties. The optical micrographs for the film (Fig. 2e and Fig. S2e†) showed a textured appearance. The P3HT-MG films were hydrophobic as judged by the contact angle for water (95°, Fig. S3a†) which was slightly higher than the value of 93° measured for P3HT (Fig. S3b†) but less than the value of 102° measured for the MG film (Fig. S9b†). The contact angle measured for P3HT compares well to the value of 91° reported for P3HT by Fang et al.43 It follows that P3HT was on the top surface, which was important for achieving a good electrical contact with Au for the P3HT-MG PSCs (Fig. 1).
AFM images and a line profile are shown for the Spiro-MG film in Fig. 2c and Fig. S2c.† The MGs were irregularly arranged and aggregated. The line profile (Fig. 2c) shows the surfaces of these films were the most rough of all three HTM-MG composites with a RMS roughness value of 31 nm. Interestingly, the optical micrographs for Spiro-MG (Fig. 2f and Fig. S2f†) revealed the presence of abundant micro-cracks. We stress that micro-cracks were absent for the PTAA-MG and P3HT-MG films. The micro-cracks were therefore due to the properties of Spiro. We propose that the micro-cracks formed because of the inability of the relatively small Spiro molecules (Fig. 1) to interdigitate.32 In comparison, relatively long polymer macromolecules (e.g., PTAA and P3HT) were able to interdigitate. The polymer chains formed entanglements and physical crosslinks that generated elastic (and mechanically tougher) films that were able to tolerate the local strain imbalances caused by the embedded MGs.
Perovskite solar cells prepared using HTM-MG blends
Representative J–V curves are shown in Fig. 3a–c for devices containing PTAA-MG, P3HT-MG, and Spiro-MG composite HTMs. Control devices were also prepared using PTAA, P3HT and Spiro. The solar cell metric data are shown in Fig. 3d and Table S1.† The PCEs for the PTAA (11.23%), P3HT (6.64%) and Spiro (8.62%) control devices were all similar to reported values for devices prepared using simple MAPI processing conditions.8,44–46 Higher PCE values should be obtainable using processing enhancements designed to increase the PCE. For example, MAPI/Spiro PCSs with efficiency values as high as 16.27%, 15.76 and 16.27% have been reported using extra-dry isopropanol,47 consecutive compact and mesoporous TiO248 or anti-solvents,41 respectively. In this first study we used simple MAPI(C) processing conditions (with MGs included in the HTM) to establish a reliable procedure that should be suitable for processing enhancement in the future.
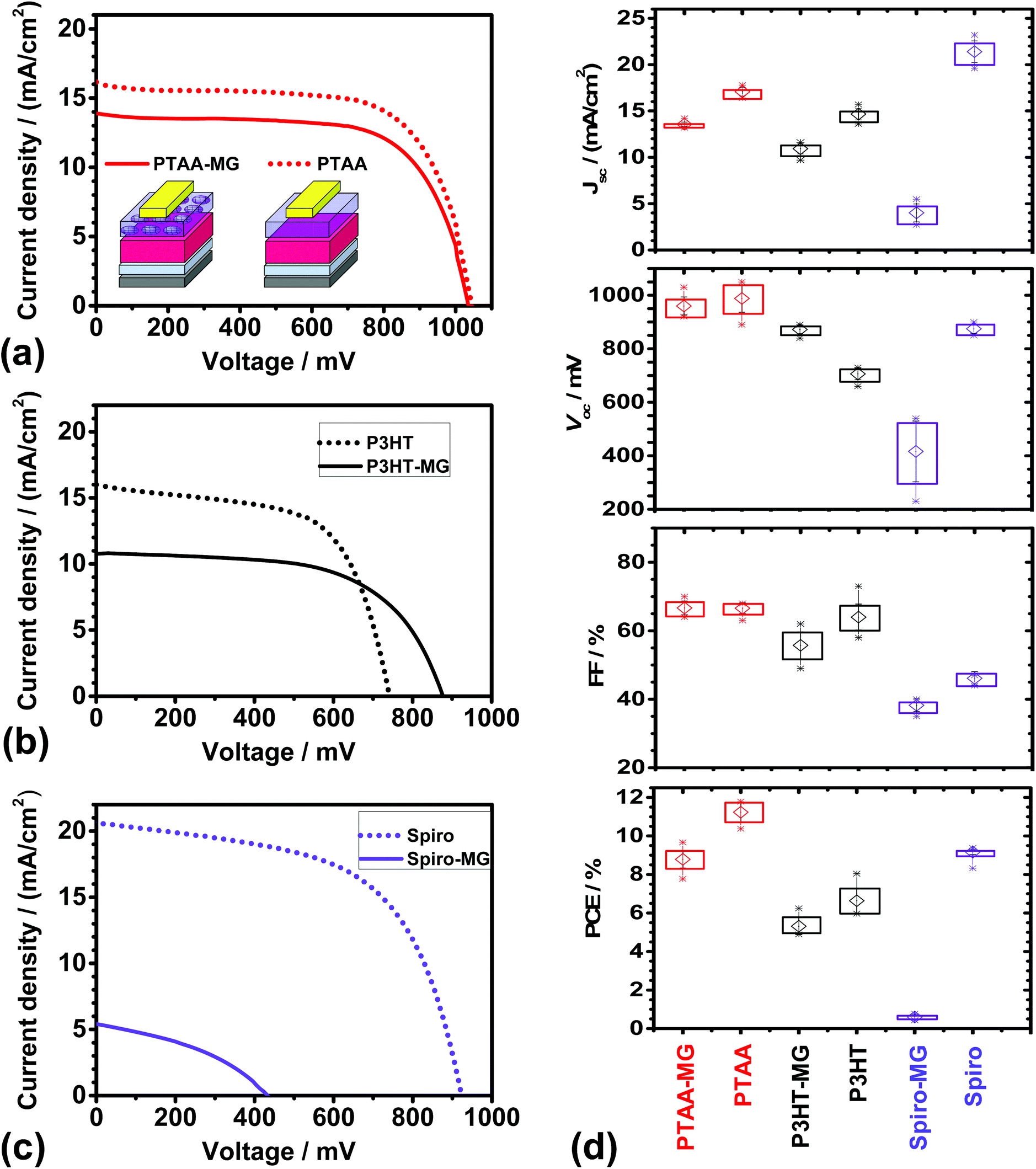 | ||
| Fig. 3 J–V curves for PSCs with HTMs of (a) PTAA-MG or PTAA, (b) P3HT-MG or P3HT and (b) Spiro-MG or Spiro. (d) Solar cell metrics for the various PSCs. The average values are shown by the open symbols. The rectangles show the upper and lower values for the average ± the standard deviation. The maximum and minimum values measured for each PSC are shown by the stars. | ||
Incorporating MGs into the HTM layer had different effects for the three conjugated materials studied. The PSCs prepared using PTAA-MG and P3HT-MG had good performance as judged by the PCE values. The devices containing Spiro-MG had an average PCE value of only 0.60%, which was much lower than for the control Spiro PSC. The Spiro-MG films had a disordered morphology (Fig. 2c) and the film integrity was compromised by micro-cracks (Fig. 2f and Fig. S2f†). The latter would have allowed Au to come into contact with MAPI(C). These effects are proposed to account for the low Jsc, Voc, FF and hence PCE values for the Spiro-MG PSCs (Fig. 3d). It follows that the MG particles are well suited to blending with polymeric HTMs, but not small-molecule HTMs.
The average PCE values for the PTAA-MG and P3HT-MG PSCs were 8.79 and 5.31%, respectively (Table S1, ESI†). These values were only 22% and 20%, respectively, lower than those for the control PSCs. The decreased PCE values were mainly due to lower Jsc values (Fig. 3d). This decrease is attributed to the greater average distance photo-generated holes had to travel to reach the Au contact. We stress that the weight fraction of PTAA and P3HT had decreased by 65% for both the PTAA-MG and P3HT-MG PSCs and the MGs were the majority phase.
The cost of energy production will be important for future PSC deployment. The future cost of electricity produced by PSCs should be an increasing function of the weight fraction of conjugated polymer in the HTM (WCP) to PCE ratio (i.e., WCP/PCE). High and low WCP/PCE values imply, respectively, higher and lower energy cost. The MG-containing and control (pure conjugated polymer) HTMs had WCP values of 35% and 100%, respectively. From these values and the PCE values in Table S1,† the calculated WCP/PCE values for the PTAA-MG and PTAA devices were 4.0 and 8.9, respectively. The WCP/PCE values for the P3HT-MG and P3HT devices were 6.6 and 15.1, respectively. It is noteworthy that the WCP/PCE values for the PTAA-MG and P3HT-MG PSCs were less than half of those for the respective control devices.
An unexpected result from this study is that the Vocvalue for the P3HT-MG devices was ∼170 mV higher than that for the P3HT device (Fig. 3d). The Voc increase may be due to formation of MG/MAPI(C) contacts which decreased the extent of P3HT/mp-TiO2 contacts which, in turn, decreased shunting. Related effects have been reported to increase Voc for PSCs.49 To further elaborate this hypothesis the dark J–V curves for the P3HT-MG and P3HT devices were investigated. The dark J–V curves for the devices shown in Fig. 3b appear in Fig. S4a.† The shunt resistance was obtained from the reciprocal of the gradient at zero bias (see Fig. S4b†).50 Analysis of all of the dark J–V curves for the P3HT-MG and P3HT devices gave average shunt resistances of 12600 ± 2900 and 5730 ± 810 Ω cm2, respectively. The latter value is comparable to values reported for MAPI PSCs by Tu et al.51 Here, the MGs increased the shunt resistance by a factor of ∼2, which supports our hypothesis that the MGs decreased shunting.
In contrast to the P3HT-based devices discussed above the Voc values for the PTAA-MG and PTAA devices were not significantly different (Table S1†) and were relatively high. It is noted that Voc values in the range of 0.96 to 1.1 V have been reported for MAPI solar cells with PTAA HTMs44,52,53 and this range covers the Voc values reported here. Hence, the Voc values for our devices were most likely limited by the perovskite. Whilst one may speculate that good dispersion of the MGs within the PTAA phase enabled the high Voc value for the MG-PTAA device to be achieved, it is not possible to confirm this conjecture from the data available.
Current–voltage hysteresis for PSCs is known to be influenced by morphology,35,54 device geometry55 as well as nature of the electron56 and hole57 transport layers and ion transport.58 Recent reports have also established a link between ion migration and recombination near the perovskite-contact interfaces.59 We found that the PTAA-MG and PTAA devices had the strongest current–voltage hysteresis; whereas, hysteresis was much less for the P3HT-MG, P3HT, Spiro-MG and Spiro devices (Fig. S5†). The presence of the MGs within the HTMs did not significantly affect hysteresis. It appears that the MGs did not affect recombination at the HTM/MAPI(C) interface.
Photoluminescence quenching in perovskite/composite HTM-MG films
PL spectra were measured to probe the possible influence of the MGs in the charge transfer between the conjugated polymers and MAPI(C). The PL spectra for MAPI(C) (Fig. 4a) had a λmax value of 765 ± 1 nm, which is similar to that reported for related perovskites.60 The presence of PTAA or PTAA-MG layers caused very strong quenching of the MAPI(C) PL signal (see inset). The UV-visible spectra (Fig. 4b) showed that absorption was dominated by the MAPI(C) because PTAA did not absorb light over the wavelength range studied. The PL quenching efficiencies (Q) were calculated using61Q = 100(y − x)/y, where y is the PL intensity for MAPI(C) and x is the PL intensity for the sample. The Q values for PTAA-MG and PTAA were 97.5 and 98.0%, respectively. These high Q values are attributed to efficient transfer of photogenerated holes from the valence band of MAPI(C) to the HOMO of the conjugated polymer.8,62 It is remarkable that high quenching efficiency was achieved when PTAA-MG contained 65 wt% of MG particles. Whilst the AFM data showed that individual MG particles were uniformly distributed within the PTAA matrix (Fig. 2a), the PL data demonstrate that these relatively large, insulating, particles did not interfere significantly with hole transfer and hence a key function of the HTM. The uniform dispersion of particles enabled a continuous, conductive, pathway of PTAA.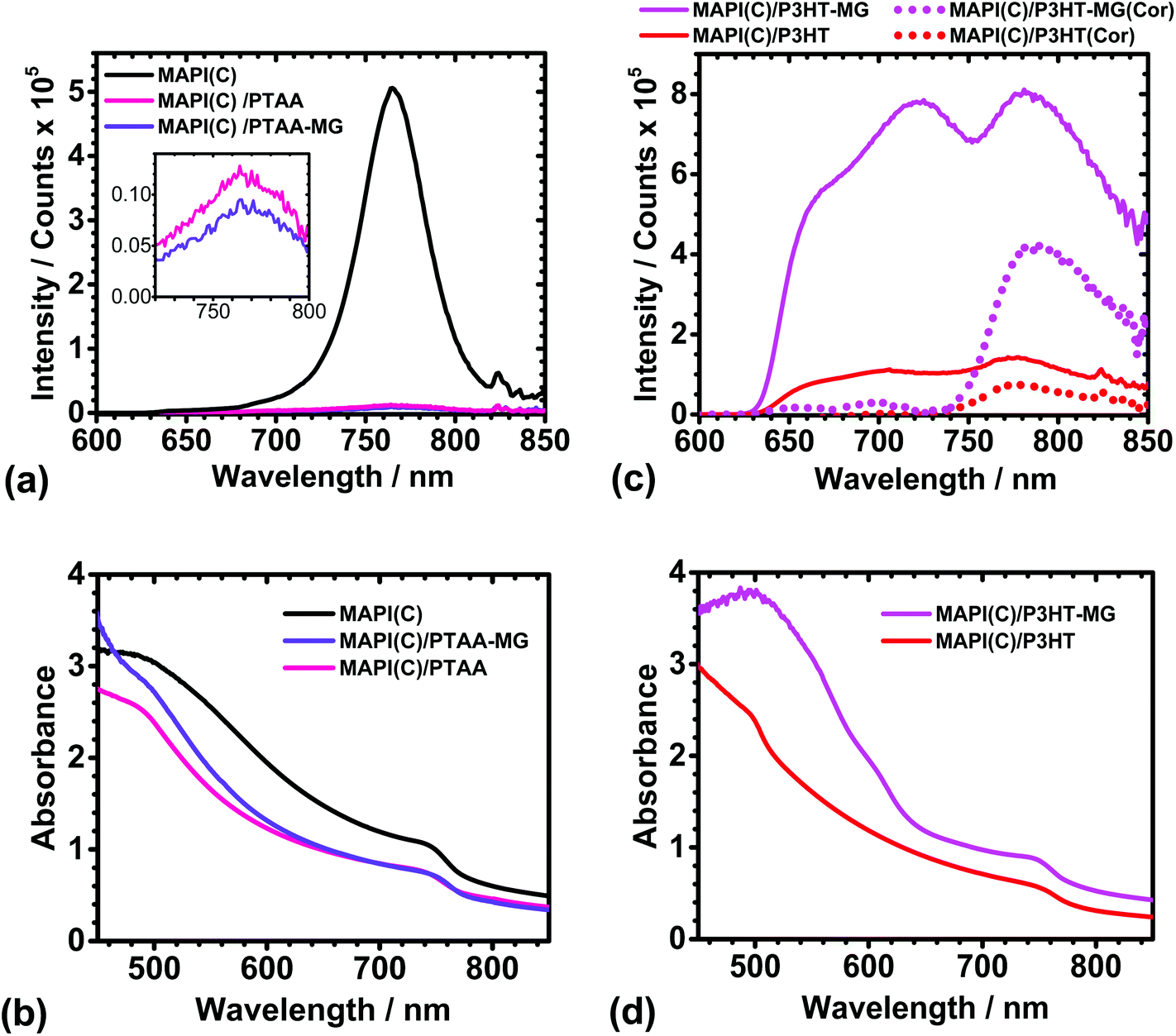 | ||
| Fig. 4 PL spectra (a and c) and UV-visible spectra (b and d) for MAPI(C), MAPI(C)/PTAA, MAPI(C)/PTAA-MG, MAPI(C)/P3HT and MAPI(C)/P3HT-MG. The inset for (a) shows the weak MAPI(C) signals due to quenching. PL spectra that have been corrected by subtracting the PL spectra for the respective polymers (Fig. S6a†) multiplied by 0.48 are also shown in (c) as the dotted lines (see text). | ||
The PL spectra for the MAPI(C) films spin-coated with P3HT or P3HT-MG are shown in Fig. 4c. The PL spectra for the MAPI(C)/P3HT film shows signals due to both P3HT and MAPI(C) at ∼720 and 780 nm, respectively. The PL signal from MAPI(C) was superimposed on the signal from P3HT. To separate the signals we used the PL spectrum measured for P3HT on glass (Fig. S6a†) and then subtracted a reduced spectrum which had been multiplied by a constant (of 0.48) which gave a good fit to the P3HT maximum present within the MAPI(C)/P3HT spectrum. (The fitting procedure is described and the fits are shown in Fig. S7.†) The latter spectrum was subtracted from the measured MAPI(C)/P3HT spectrum to give the corrected PL spectrum (red dotted curve in Fig. 4c). This spectrum had a maximum that was due to MAPI(C). The latter signal had a much lower intensity compared to the parent MAPI(C) signal (Fig. 4a) and a Q value of 85.0% was calculated. This value indicates less efficient quenching occurred for MAPI(C)/P3HT compared to the MAPI(C)/PTAA films. PTAA has a high hole mobility and has been suggested to interact more strongly with perovskites than P3HT.45
The PL spectra for MAPI(C)/P3HT-MG was relatively intense (Fig. 4c), which was also the case for P3HT-MG on glass (Fig. S6a†). The UV-visible spectrum for the former (Fig. 4d) shows strong absorption in the region of 480 nm, which is associated with the enhanced absorption of the P3HT-MG.23 We performed the same fitting procedure as described above to obtain a corrected MAPI(C)/P3HT-MG spectrum (see Fig. S7b†). The corrected spectrum showing the MAPI(C) signal is shown in Fig. 4c (pink dots). The intensity was less than that measured for MAPI(C) (Fig. 4a) and corresponded to Q = 17.0%. This value is less than that apparent for the MAPI(C)/P3HT system. This effect can be explained by the presence of close packed MG particles (Fig. 2b), which restricted pathways for photo-generated holes to move from MAPI(C) to the P3HT matrix. The latter conclusion is consistent with the lower Jsc (Table S1†) and higher shunt resistance found for the PHT-MG devices compared to the P3HT devices (discussed above). The PL data discussed above have shown for the first time that the extent of MG dispersion within the conjugated polymer matrix significantly affects quenching. Quenching is most efficient when the MGs are well dispersed within the HTM matrix.
An interesting question concerning the MAPI(C)/P3HT-MG and MAPI(C)/P3HT films is why the PL and UV-visible spectra (Fig. 4c and d) were so different. The PL intensities and the absorbance values were much higher for the P3HT-MG HTMs. Indeed, the same general trend can be seen from the PL and UV-visible spectra for P3HT-MG and P3HT films (Fig. S6a and b†). The refractive indexes of P3HT (1.8763) and PS (1.5964) differ considerably which would favour light scattering for P3HT-MG films. In addition, the MG particles have a size (Fig. S1†) that is comparable to the wavelength of light, which would favour light scattering. We reported earlier that light absorption by P3HT within P3HT-MG films increased strongly when the MG concentration increased23 as a result of light scattering. Related studies have also shown that PL and light absorption by P3HT can be greatly enhanced by light scattering species.65,66 Hence, the relatively high PL and UV-visible values for the P3HT-MG HTM (Fig. 4c and d) and P3HT-MG film (Fig. S6a and b†) originate from the longer pathlengths travelled due to enhanced light scattering caused by the MG particles.
In summary, the PL data point to differences in the MG roles in influencing hole transport which can be rationalised in terms of MG distribution. For the PTAA-MG films hole transport from the perovskite to the PTAA phase appeared to be efficient (as indicated by strong quenching) and this was attributed to the MGs being well dispersed in the PTAA matrix. In the case of P3HT-MG films the less efficient quenching was attributed to the MGs forming close-packed clusters near the mp-TiO2/P3HT interface.
Using microgel particles to improve solar cell stability
The ability of MG particles to form an encapsulation layer to improve PSC stability was investigated. A preliminary study was conducted where MG dispersed in toluene were spin-coated onto glass (Fig. S8†). The fractional coverage data obtained from optical micrographs showed that the MG dispersions formed a uniform film when the MG concentration (CMG) reached 3.0%. AFM data showed the MG films consisted of close packed (flattened) particles (Fig. S8f†) with a low RMS roughness of 16.0 nm. To probe the optical clarity of the films a UV-visible spectrum was measured (Fig. S9a†). The absorbance was low at wavelengths greater than 350 nm and increased in the UV-region, which may be useful in the future because UV protection has been found to enhance the stability of PSCs.12 The MG films had a contact angle of 102° (Fig. S9b†) and were more hydrophobic than atactic polystyrene which has a contact angle of67 90°.The ability of MGs to encapsulate MAPI(C) surfaces was investigated. Fig. 5a shows an AFM image for a MAPI(C) film covered with an MG film (MAPI(C)/MG(en)). The MG particles formed a well ordered film with closely packed particles. The RMS average roughness for the encapsulated film was 22 nm. In contrast, an AFM image for an unmodified control MAPI(C) film is shown in Fig. 5b. The film consisted of MAPI(C) nanocrystals and was relatively rough with an RMS average roughness of 50 nm. Comparison of the AFM images (Fig. 5a and b) shows that the MG particles were able to coat MAPI(C) and appeared to fill large voids, which demonstrates good film forming ability. The UV-visible and PL spectra (Fig. 5c and d) as well as the X-ray diffractograms for the MAPI(C)/MG(en) and MAPI(C) films were similar and had no evidence of residual PbI2.
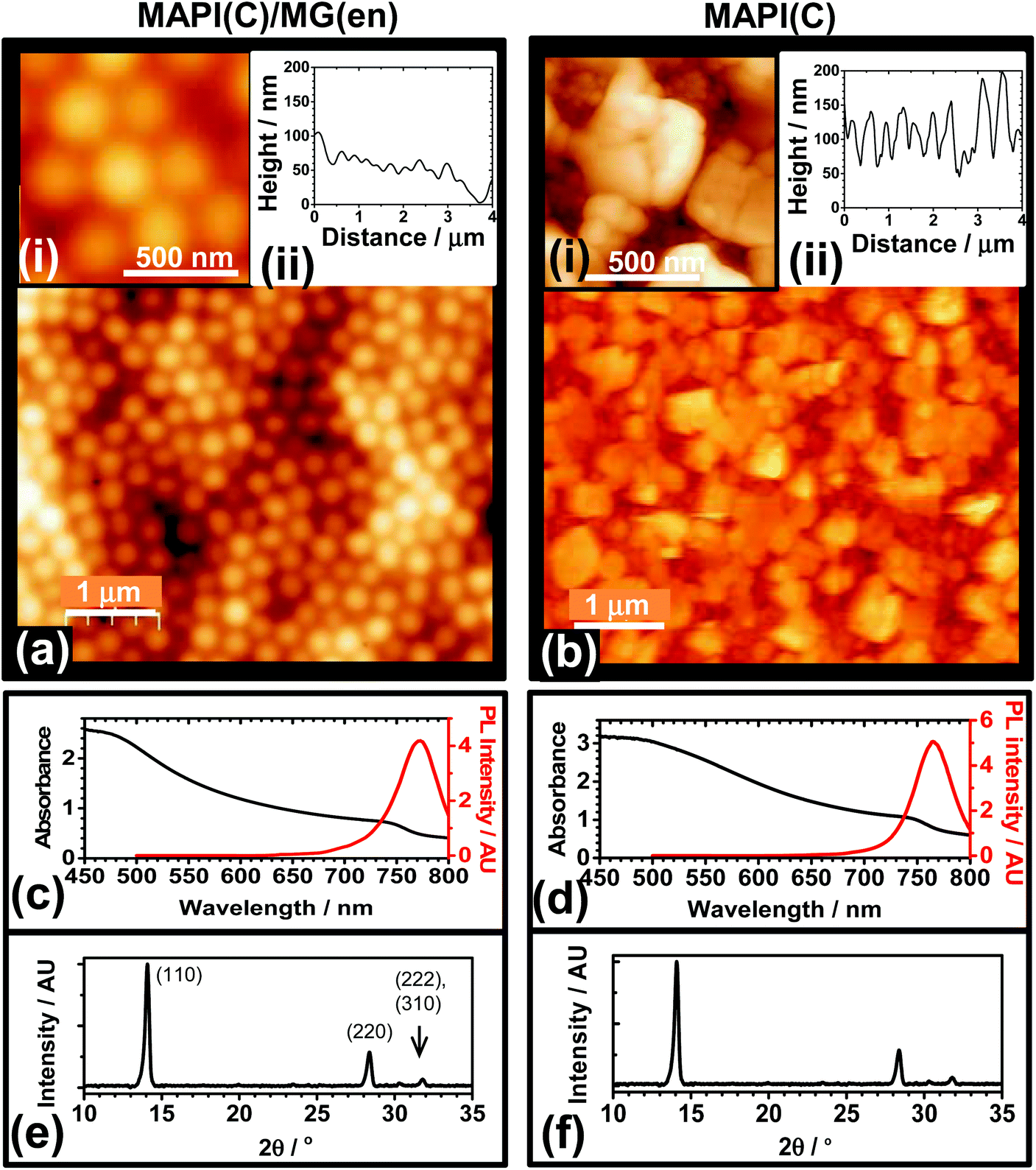 | ||
| Fig. 5 AFM images and line-traces (a and b), UV-visible and PL spectra (c and d) and XRD data (e and f) for MAPI(C) encapsulated by an MG film (MAPI(C)/MG(en), CMG = 4.6%) and an unmodified MAPI(C) film. The films were deposited on ITO/bl-TiO2. | ||
We conducted an investigation of the stability P3HT-containing devices because PSCs containing P3HT as the HTM are a plausible candidate for scale-up.10 P3HT devices were constructed that were encapsulated using a MG layer and J–V data measured (Fig. S10a†). The average PCE was 5.06% (Table S1†). This value was lower than that for the control P3HT device due to a combination of slight decreases in each of Jsc, Voc and FF. A device cross-section for the P3HT/MG(en) device (Fig. S10b†) showed flattened MG particles at the surface of the encapsulating layer. The PSC performance was measured at room temperature with an average humidity of 40% at regular time intervals. For comparison, we also measured the performance of P3HT-MG and control P3HT PSCs (See Fig. 6a–d). It should be noted that P3HT-MG contained MG only within the HTM phase and did not contain an encapsulating layer. The P3HT PSC showed rapid degradation in performance with device measurements no longer possible after 1 day. The PCE for the P3HT-MG device decreased at a more gradual, but relatively slow, rate over the entire experimental period. By contrast, the PCE for the P3HT/MG(en) device did not decrease significantly for 28 days. This result agrees favourably with the those of Hwang et al. where a Teflon coating was used to encapsulate PSCs.68
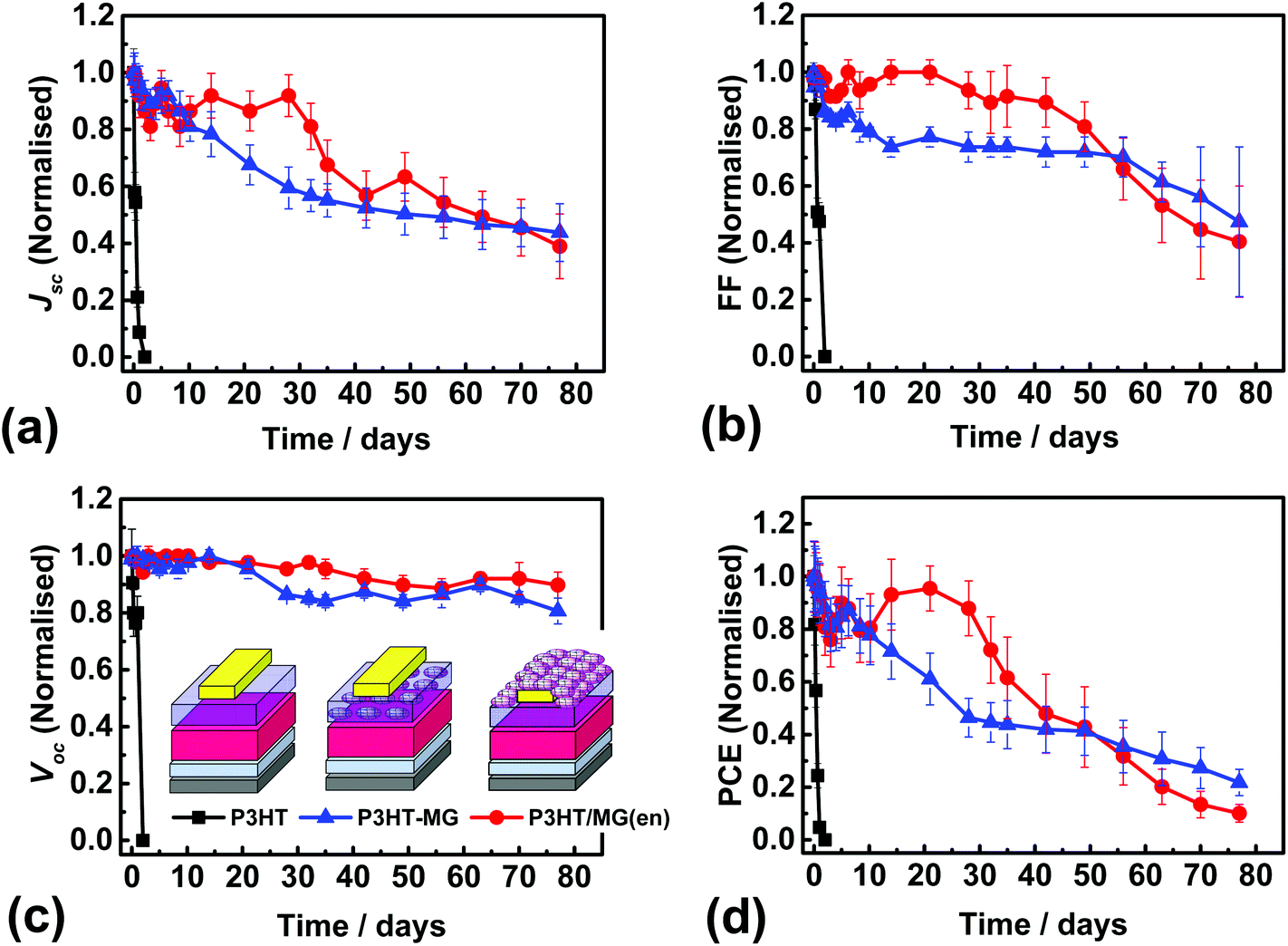 | ||
| Fig. 6 (a) to (d) Normalised solar cell figures of merit for P3HT/MG(en) (CMG = 3.0%), P3HT-MG and control P3HT PSCs exposed to 40% humidity at room temperature. The PSCs were stored in the dark between measurements. The legend from (c) also applies to (a), (b) and (d). | ||
The stability of a PSC can be quantified using the half-life (t1/2), which is the time taken for the PCE to reach half the initial value. Accordingly, the t1/2 values for the devices containing P3HT/MG(en), P3HT-MG and P3HT were 41, 26 and 0.5 days, respectively. These results demonstrate that MGs strongly enhanced the stability of the PSCs whether included as an encapsulating layer or as a HTM-MG composite layer. We attribute this effect to hydrophobic passivation which opposed water vapour ingress. The P3HT-MG PSC stability enhancement was remarkable because it demonstrates that the MGS were able to play a dual role of HTM extender and stability enhancer.
Conclusions
In this study we investigated MAPI(C) PSCs that contained PS MGs for the first time. The MGs were incorporated within the HTM phase as a composite or as an encapsulating layer. The MGs were successfully used with PTAA and P3HT which was attributed to the ability of these polymer HTMs to withstand strain imbalances imposed by the MGs. The PCE values for devices containing MG-PTAA or MG-P3HT decreased only by about 20% compared to control PSCs. This is despite the fact that the conjugated polymer concentration decreased by 65%. MGs were also shown to be effective at increasing the PSC stability when used as an encapsulating layer. The work indicates that introduction of MGs (which could be made on the tonne scale) may increase the cost-effectiveness of conjugated polymer use and increase performance stability for future PSCs.It is useful to summarise the role of the MGs within the PTAA and P3HT HTMs. For the PTAA-MG films the MGs were well dispersed and did not significantly interfere with hole transport from the perovskite to the PTAA phase as judged by the PL data. Hence, their primary role was that of a HTM extender, which allowed much less PTAA to be used to prepare efficient devices. In the case of P3HT-MG films the MGs also acted as a HTM extender. Additionally, the device stability studies (Fig. 6) showed that MGs present within the HTM increased device stability. Hence, for the P3HT-MG HTMs the MGs were shown to have a second role of stability enhancer.
A number of unexpected results were found from this study. Firstly, the MGs dispersed remarkably well within the PTAA matrix. Secondly, the Voc value increased by ∼170 mV for the MG-P3HT devices. Thirdly, the PTAA-MG films were highly efficient at quenching MAPI(C) fluorescence, despite their majority MG content. Finally, MG dispersed within the P3HT layer provided a surprisingly effective stability improvement. Consequently, MGs had dual roles. They more than halved the conjugated polymer weight fraction/PCE ratio for the PSCs, provided increased stability and also augmented Voc. Because MGs can be readily functionalised20 they are an excellent vehicle for introducing additional versatility into future composite HTMs for PSCs and would seem to have good potential for study in other PSCs and eventual application.
Acknowledgements
BRS would like to thank the EPSRC for funding (EP/K010298/1). We thank one of the referees for their helpful suggestions.References
- T. A. Berhe, W.-N. Su, C.-H. Chen, C.-J. Pan, J.-H. Cheng, H.-M. Chen, M.-C. Tsai, L.-Y. Chen, A. A. Dubale and B.-J. Hwang, Energy Environ. Sci., 2016, 9, 323–356 CAS.
- M. M. Lee, J. Teuscher, T. Miyasaka, T. N. Murakami and H. J. Snaith, Science, 2016, 338, 5 Search PubMed.
- J. M. Ball, M. M. Lee, A. Hey and H. J. Snaith, Energy Environ. Sci., 2013, 6, 1739 CAS.
- National Renewable Energy Laboratory, Best Research-Cell Efficiencies chart; http://www.nrel.gov/ncpv/images/efficiency_chart.jpg.
- D. P. McMeekin, G. Sadoughi, W. Rehman, G. E. Eperon, M. Saliba, M. T. Hörantner, A. Haghighirad, N. Sakai, L. Korte, B. Rech, M. B. Johnston, L. M. Herz and H. J. Snaith, Science, 2016, 351, 151–155 CrossRef CAS PubMed.
- M. Maciejczyk, A. Ivaturi and N. Robertson, J. Mater. Chem. A, 2016, 4, 4855–4863 CAS.
- S. Ameen, M. A. Rub, S. A. Kosa, K. A. Alamry, M. S. Akhtar, H.-S. Shin, H.-K. Seo, A. M. Asiri and M. K. Nazeeruddin, ChemSusChem, 2016, 9, 10–27 CrossRef CAS PubMed.
- Y. Zhang, W. Liu, F. Tan and Y. Gu, J. Power Sources, 2015, 274, 1224–1230 CrossRef CAS.
- N. J. Jeon, J. H. Noh, W. S. Yang, Y. C. Kim, S. Ryu, J. Seo and S. I. Seok, Nature, 2015, 517, 476–480 CrossRef CAS PubMed.
- K. Hwang, Y.-S. Jung, Y.-J. Heo, F. H. Scholes, S. E. Watkins, J. Subbiah, D. J. Jones, D.-Y. Kim and D. Vak, Adv. Mater., 2015, 27, 1241–1247 CrossRef CAS PubMed.
- F. Matteocci, S. Razza, F. Di Giacomo, S. Casaluci, G. Mincuzzi, T. M. Brown, A. D'Epifanio, S. Licoccia and A. Di Carlo, Phys. Chem. Chem. Phys., 2014, 16, 3918–3923 RSC.
- F. Bella, G. Griffini, J.-P. Correa-Baena, G. Saracco, M. Grätzel, A. Hagfeldt, S. Turri and C. Gerbaldi, Science, 2016, 203–206 Search PubMed.
- L. N. Quan, M. Yuan, R. Comin, O. Voznyy, E. M. Beauregard, S. Hoogland, A. Buin, A. R. Kirmani, K. Zhao, A. Amassian, D. H. Kim and E. H. Sargent, J. Am. Chem. Soc., 2016, 138, 2649–2655 CrossRef CAS PubMed.
- H. Tan, A. Jain, O. Voznyy, X. Lan, F. P. García de Arquer, J. Z. Fan, R. Quintero-Bermudez, M. Yuan, B. Zhang, Y. Zhao, F. Fan, P. Li, L. N. Quan, Y. Zhao, Z.-H. Lu, Z. Yang, S. Hoogland and E. H. Sargent, Science, 2017 DOI:10.1126/science.aai9081.
- Z. Wang, D. P. McMeekin, N. Sakai, S. van Reenen, K. Wojciechowski, J. B. Patel, M. B. Johnston and H. J. Snaith, Adv. Mater., 2017, 29, 9986–9992 Search PubMed.
- N. Aristidou, I. Sanchez-Molina, T. Chotchuangchutchaval, M. Brown, L. Martinez, T. Rath and S. A. Haque, Angew. Chem., Int. Ed., 2015, 54, 8208–8212 CrossRef CAS PubMed.
- G. Niu, X. Guo and L. Wang, J. Mater. Chem. A, 2015, 3, 8970–8980 CAS.
- H. J. Snaith, J. Phys. Chem. Lett., 2013, 4, 3623–3630 CrossRef CAS.
- B. R. Saunders and B. Vincent, Adv. Colloid Interface Sci., 1999, 80, 25 CrossRef.
- B. R. Saunders, N. Laajam, E. Daly, S. Teow, X. Hu and R. Stepto, Adv. Colloid Interface Sci., 2009, 147–148, 251–262 CrossRef CAS PubMed.
- M. Nair, Prog. Org. Coat., 1992, 20, 53–61 CrossRef CAS.
- C. L. Sieglaff, J. Polym. Sci., 1959, 41, 319–326 CrossRef CAS.
- M. Chen, Z. Cui, S. Edmondson, N. Hodson, M. Zhou, J. Yan, P. O'Brien and B. R. Saunders, Soft Matter, 2015, 11, 8322–8332 RSC.
- Y. Guan and Y. Zhang, Soft Matter, 2011, 7, 6375 RSC.
- H. Tsutsui, M. Moriyama, D. Nakayama, R. Ishii and R. Akashi, Macromolecules, 2006, 39, 7 CrossRef.
- X. Hu and L. A. Lyon, ACS Macro Lett., 2015, 4, 302–307 CrossRef CAS.
- S. N. Habisreutinger, T. Leijtens, G. E. Eperon, S. D. Stranks, R. J. Nicholas and H. J. Snaith, Nano Lett., 2014, 14, 5561–5568 CrossRef CAS PubMed.
- T. Gatti, S. Casaluci, M. Prato, M. Salerno, F. Di Stasio, A. Ansaldo, E. Menna, A. Di Carlo and F. Bonaccorso, Adv. Funct. Mater., 2016, 26, 7443–7453 CrossRef CAS.
- K. Aitola, K. Domanski, J.-P. Correa-Baena, K. Sveinbjörnsson, M. Saliba, A. Abate, M. Grätzel, E. Kauppinen, E. M. J. Johansson, W. Tress, A. Hagfeldt and G. Boschloo, Adv. Mater., 2017, 29, 1606398 CrossRef PubMed.
- J.-Y. Wang, F.-C. Hsu, J.-Y. Huang, L. Wang and Y.-F. Chen, ACS Appl. Mater. Interfaces, 2015, 7, 27676–27684 CAS.
- J. Xiao, J. Shi, H. Liu, Y. Xu, S. Lv, Y. Luo, D. Li, Q. Meng and Y. Li, Adv. Energy Mater., 2015, 5, 1401943 CrossRef.
- J. Yang, B. D. Siempelkamp, D. Liu and T. L. Kelly, ACS Nano, 2015, 9, 1955–1963 CrossRef CAS PubMed.
- C.-Y. Chang, K.-T. Lee, W.-K. Huang, H.-Y. Siao and Y.-C. Chang, Chem. Mater., 2015, 27, 5122–5130 CrossRef CAS.
- Z. Liu, B. Sun, T. Shi, Z. Tang and G. Liao, J. Mater. Chem. A, 2016, 4, 10700–10709 CAS.
- M. Z. Mokhtar, M. Chen, E. Whittaker, B. Hamilton, N. Aristidou, S. Ramadan, A. Gholinia, S. A. Haque, P. O'Brien and B. R. Saunders, Phys. Chem. Chem. Phys., 2017, 19, 7204–7214 RSC.
- N. J. Jeon, J. H. Noh, Y. C. Kim, W. S. Yang, S. Ryu and S. I. Seok, Nat. Mater., 2014, 13, 897–903 CrossRef CAS PubMed.
- J. H. Heo and S. H. Im, Phys. Status Solidi RRL, 2014, 8, 816–821 CrossRef CAS.
- M. M. Lee, J. Teuscher, T. Miyasaka, T. N. Murakami and H. J. Snaith, Science, 2012, 338, 643–647 CrossRef CAS PubMed.
- S. Schmidt, H. Motschmann, T. Hellweg and R. von Klitzing, Polymer, 2008, 49, 749–756 CrossRef CAS.
- S. Wellert, D. Kesal, S. Schön, R. von Klitzing and K. Gawlitza, Langmuir, 2015, 31, 2202–2210 CrossRef CAS PubMed.
- M. Xiao, F. Huang, W. Huang, Y. Dkhissi, Y. Zhu, J. Etheridge, A. Gray-Weale, U. Bach, Y.-B. Cheng and L. Spiccia, Angew. Chem., Int. Ed., 2014, 126, 10056–10061 CrossRef.
- Y. Lee, J. K. Kim, C.-H. Chiu, Y.-K. Lan and C.-I. Huang, Polymer, 2009, 50, 4944–4949 CrossRef CAS.
- M. Fang, Z. Tang, H. Lu and S. Nutt, J. Mater. Chem., 2012, 22, 109–114 RSC.
- D. Bi, G. Boschloo and A. Hagfeldt, Nano, 2014, 09, 1440001 CrossRef.
- J. H. Heo, S. H. Im, J. H. Noh, T. N. Mandal, C.-S. Lim, J. A. Chang, Y. H. Lee, H.-J. Kim, A. Sarkar, K. NazeeruddinMd, M. Gratzel and S. I. Seok, Nat. Photonics, 2013, 7, 486–491 CrossRef CAS.
- S. H. Hwang, J. Roh, J. Lee, J. Ryu, J. Yun and J. Jang, J. Mater. Chem. A, 2014, 2, 16429–16433 CAS.
- W.-T. Wang, S. K. Das and Y. Tai, ACS Appl. Mater. Interfaces, 2017, 9, 10743–10751 CAS.
- X.-H. Zhang, J.-J. Ye, L.-Z. Zhu, H.-Y. Zheng, X.-P. Liu, X. Pan and S.-Y. Dai, ACS Appl. Mater. Interfaces, 2016, 8, 35440–35446 CAS.
- G. E. Eperon, V. M. Burlakov, A. Goriely and H. J. Snaith, ACS Nano, 2014, 8, 591–598 CrossRef CAS PubMed.
- H.-W. Kang, J.-W. Lee, D.-Y. Son and N.-G. Park, RSC Adv., 2015, 5, 47334–47340 RSC.
- Y. Tu, J. Wu, M. Zheng, J. Huo, P. Zhou, Z. Lan, J. Lin and M. Huang, Nanoscale, 2015, 7, 20539–20546 RSC.
- J. H. Heo, H. J. Han, M. Lee, M. Song, D. H. Kim and S. H. Im, Energy Environ. Sci., 2015, 8, 2922–2927 CAS.
- J. H. Heo, H. J. Han, D. Kim, T. K. Ahn and S. H. Im, Energy Environ. Sci., 2015, 8, 1602–1608 CAS.
- S. C. Hong, G. Lee, K. Ha, J. Yoon, N. Ahn, W. Cho, M. Park and M. Choi, ACS Appl. Mater. Interfaces, 2017, 9, 7879–7884 CAS.
- I. J. Park, M. A. Park, D. H. Kim, G. D. Park, B. J. Kim, H. J. Son, M. J. Ko, D.-K. Lee, T. Park, H. Shin, N.-G. Park, H. S. Jung and J. Y. Kim, J. Phys. Chem. C, 2015, 119, 27285–27290 CAS.
- W. Hu, T. Liu, X. Yin, H. Liu, X. Zhao, S. Luo, Y. Guo, Z. Yao, J. Wang, N. Wang, H. Lin and Z. Guo, J. Mater. Chem. A, 2017, 5, 1434–1441 CAS.
- M. Salado, J. Idigoras, L. Calio, S. Kazim, M. K. Nazeeruddin, J. A. Anta and S. Ahmad, ACS Appl. Mater. Interfaces, 2016, 8, 34414–34421 CAS.
- C. Eames, J. M. Frost, P. R. F. Barnes, B. C. O'Regan, A. Walsh and M. S. Islam, Nat. Commun., 2015, 6, 7497 CrossRef CAS PubMed.
- P. Calado, A. M. Telford, D. Bryant, X. Li, J. Nelson, B. C. O'Regan and P. R. F. Barnes, Nat. Commun., 2016, 7, 13831 CrossRef CAS PubMed.
- M. Ahmadi, Y.-C. Hsiao, T. Wu, Q. Liu, W. Qin and B. Hu, Adv. Energy Mater., 2017, 7, 1601575 CrossRef.
- J.-M. Cha, J.-W. Lee, D.-Y. Son, H.-S. Kim, I.-H. Jang and N.-G. Park, Nanoscale, 2016, 8, 6341–6351 RSC.
- N. Arora, S. Orlandi, M. I. Dar, S. Aghazada, G. Jacopin, M. Cavazzini, E. Mosconi, P. Gratia, F. De Angelis, G. Pozzi, M. Graetzel and M. K. Nazeeruddin, ACS Energy Lett., 2016, 1, 107–112 CrossRef CAS.
- D. E. Motaung, G. F. Malgas, C. J. Arendse and S. E. Mavundla, Mater. Chem. Phys., 2012, 135, 401–410 CrossRef CAS.
- M. Xiaoyan, Q. L. Jun, R. S. Brock, M. J. Kenneth, Y. Ping and H. Xin-Hua, Phys. Med. Biol., 2003, 48, 4165 CrossRef.
- P. Du, P. Jing, D. Li, Y. Cao, Z. Liu and Z. Sun, Small, 2015, 11, 2454–2462 CrossRef CAS PubMed.
- S. Nootchanat, A. Pangdam, R. Ishikawa, K. Wongravee, K. Shinbo, K. Kato, F. Kaneko, S. Ekgasit and A. Baba, Nanoscale, 2017, 9, 4963–4971 RSC.
- L.-H. Wang and R. S. Porter, J. Appl. Polym. Sci., 1983, 28, 1439–1445 CrossRef CAS.
- I. Hwang, I. Jeong, J. Lee, M. J. Ko and K. Yong, ACS Appl. Mater. Interfaces, 2015, 7, 17330–17336 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7nr02650a |
| This journal is © The Royal Society of Chemistry 2017 |