
Nanoparticle-encapsulated baicalein markedly modulates pro-inflammatory response in gingival epithelial cells†
 a
Lijian
Jin
*a
a
Lijian
Jin
*a
Abstract
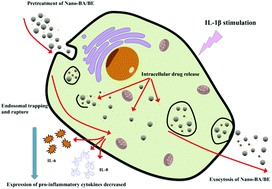
Severe gum disease (periodontitis), which is one of the major global oral diseases, results from microbe-host dysbiosis and dysregulated immuno-inflammatory responses. It seriously affects oral health and general wellbeing with significant socio-economic implications. It has been well documented that natural flavonoids such as baicalin (BA) and baicalein (BE) possess potent anti-inflammatory effects. However, their intrinsic poor solubility and low bioavailability severely limit their biomedical applications. In the present study, BA and BE were encapsulated in our synthesized and amine-modified mesoporous silica nanoparticles (MSNs) (Nano-BA and Nano-BE, respectively), and their loading efficiencies and releasing profiles were investigated. Their cytotoxicity was examined on primary human gingival epithelial cells (hGECs), and the cellular uptake of Nano-BA or Nano-BE was visualized via a transmission electron microscope. Their anti-inflammatory effects were evaluated in IL-1β-treated hGECs using the cytokine array and enzyme-linked immunosorbent assay. The present study shows that the amine-modified MSNs could encapsulate BA and BE, and nano-encapsulation greatly enhances the drug delivery rate and prolongs the release of BA and BE up to 216 h. Moreover, both Nano-BA and Nano-BE could be internalized by hGECs and retained intracellularly in nanoparticle-free media for at least 24 h. Note that Nano-BE pre-treatment effectively down-regulates the IL-1β-induced expression of IL-6 and IL-8 in hGECs. In conclusion, nanoparticle-encapsulated BE exhibits notable anti-inflammatory effects through effective release and cellular internalization approaches. This study may facilitate the development of novel drug delivery systems for improving oral care.
- This article is part of the themed collection: 2017 Nanoscale HOT Article Collection
 Please wait while we load your content...
Please wait while we load your content...

