
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSDS-assisted protein transport through solid-state nanopores†
Laura
Restrepo-Pérez‡
 a,
Shalini
John‡
b,
Aleksei
Aksimentiev
*b,
Chirlmin
Joo
*a and
Cees
Dekker
*a
a,
Shalini
John‡
b,
Aleksei
Aksimentiev
*b,
Chirlmin
Joo
*a and
Cees
Dekker
*a
aDepartment of Bionanoscience, Kavli Institute of Nanoscience, Delft University of Technology, van der Maasweg 9, 2629 HZ Delft, The Netherlands. E-mail: c.dekker@tudelft.nl; c.joo@tudelft.nl
bDepartment of Physics, University of Illinois at Urbana; Champaign, Urbana, Illinois 61801, USA. E-mail: aksiment@illinois.edu
First published on 12th July 2017
Abstract
Using nanopores for single-molecule sequencing of proteins – similar to nanopore-based sequencing of DNA – faces multiple challenges, including unfolding of the complex tertiary structure of the proteins and enforcing their unidirectional translocation through nanopores. Here, we combine molecular dynamics (MD) simulations with single-molecule experiments to investigate the utility of SDS (Sodium Dodecyl Sulfate) to unfold proteins for solid-state nanopore translocation, while simultaneously endowing them with a stronger electrical charge. Our simulations and experiments prove that SDS-treated proteins show a considerable loss of the protein structure during the nanopore translocation. Moreover, SDS-treated proteins translocate through the nanopore in the direction prescribed by the electrophoretic force due to the negative charge impaired by SDS. In summary, our results suggest that SDS causes protein unfolding while facilitating protein translocation in the direction of the electrophoretic force; both characteristics being advantageous for future protein sequencing applications using solid-state nanopores.
Introduction
The proteome contains essential information for the understanding of biological processes and diseases. Despite its importance, only a handful of methods are available for proteome analysis. Mass spectrometry is currently the gold standard for protein sequencing. However, analysis of complex biological samples remains a challenge for the mass spectrometry method, in particular, as many proteins are present in low-copy numbers.1 Techniques operating at the single-molecule level could provide ultimate sensitivity, enabling detection of proteins of low abundance.Nanopores have emerged as a platform for fast and label-free detection of single molecules.2–4 In a nanopore measurement, a thin membrane containing a nanopore is placed between two compartments filled with electrolytes. A voltage bias across the membrane produces a current of ions that flow from one compartment to the other. The presence of a biomolecule in a nanopore is sensed from transient blockades of the ionic current that the biomolecule produces. In the case of linear nucleic acids such as DNA or RNA, the ionic current signal carries information about the nucleotide content of the molecules.5–10
Nanopores have also been used to detect proteins. Biological and solid-state nanopores were utilized to detect the presence of proteins bound to DNA,11–14 characterize proteins captured by the nanopore,15–19 and distinguish different states of proteins.16,20–22 A motor protein was used to produce unidirectional protein translocation.23 First attempts to distinguish the type of individual amino acids in a nanopore were made both experimentally24 and theoretically,25 including detection of post-translational modifications.26
Nanopore-based protein sequencing presents a number of challenges. Proteins, contrary to nucleic acids, do not adopt a random coil conformation but fold into complex tertiary structures. The local charge of a polypeptide chain is non-uniform, alternating between neutral and positive and negative values. The direction of the electrophoretic transport through the nanopore is influenced by both the charge of the protein and the electroosmotic flow in the nanopore.27 As a result, a unidirectional and single-file transport of protein through a nanopore, which is a prerequisite for nanopore-based sequencing, is not ensured.
Detergent treatment of proteins may help achieve better control over the process of protein translocation. Polyacrylamide gel electrophoresis (PAGE) in the presence of sodium dodecyl sulfate (SDS) is a common laboratory technique used in molecular biology to separate proteins according to their size. SDS denatures the proteins and imparts a negative charge to the linearized peptide chains. When SDS-treated proteins are placed on a gel and an electric field is applied, proteins travel to the positive electrode due to the negative charge imparted by the detergent. These two properties, linearization and uniform charge distribution, are promising to promote linear translocation of proteins through a nanopore. In a previous study, Li and colleagues made a survey of different denaturants that facilitate the translocation of unfolded proteins through nanopores.28 However, no detailed study has been reported on the use of SDS for nanopore translocation of proteins. More recently, Kennedy et al. used SDS to translocate proteins through a sub-nanometer pore, however, no information on the detergent–protein interaction has been provided.29
Here we employ all-atom molecular dynamics (MD) simulations and nanopore experiments to determine how SDS influences the protein transport through nanopores. Our integrated approach elucidates the microscopic conformation and the transport modality of the SDS–protein assemblies. We show that the transport of protein–SDS complexes is determined by electrophoresis, unlike that of native proteins, which is governed by the electroosmotic flow. This is consistent with a prevailing model that SDS imparts uniform negative charges on proteins. SDS-unfolded proteins exhibit a lower current blockade than native proteins do and the blockade duration increases with the molecular weight of the protein. We also explore the use of SDS at different concentrations and show that it can be used both above and below its Critical Micelle Concentration (CMC).
Fig. 1 illustrates the concept of protein translocation through a nanopore. We focused our investigation on three different proteins: titin I27 domain (13 kDa, a well-characterized protein often used in protein unfolding studies), titin I27 dimer (26 kDa), and β-amylase (200 kDa, a globular protein with a high molecular weight). We performed the majority of our measurements and simulations using silicon nitride (SiN) nanopores 6 or 10 nm in diameter and an electrolyte solution containing 0.4 M NaCl. Higher electrolyte concentrations or potassium-based buffers caused SDS precipitation in experiments.
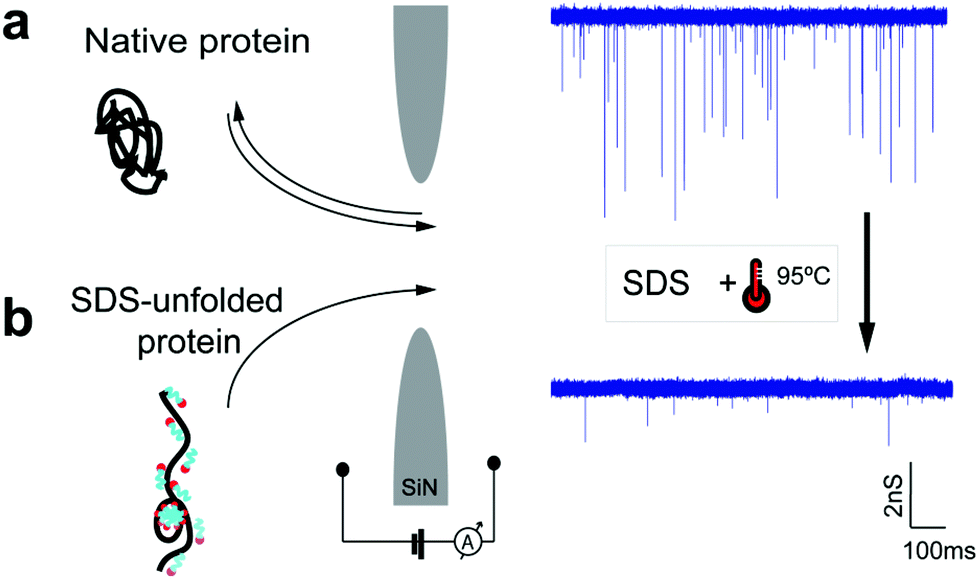 | ||
| Fig. 1 Schematic illustration and representative current traces of the translocation of native proteins (a) and SDS-unfolded proteins (b) through a solid-state nanopore. The traces correspond to the translocation of β-amylase through a 10 nm nanopore. | ||
Molecular dynamics simulations
We explored the effects of SDS treatment on proteins and its consequences for nanopore translocation. First, we obtained microscopic models of SDS/protein complexes by simulating spontaneous self-assembly of unfolded proteins with SDS molecules in the electrolyte solution (0.4 M NaCl). In a typical simulation (Fig. 2a and ESI Movie 1†), SDS molecules were observed to aggregate around an unfolded protein at boiling temperature (373 K) forming a complex that contains approximately one SDS molecule per two amino acids of the protein, in agreement with previous experimental findings.30 The complex remained stable upon subsequent cooling to room temperature. Fig. 2b shows examples of final room temperature SDS/protein complexes obtained from such self-assembly simulations. The structures of such SDS/protein assemblies appear to be in accord with a decorated micelle model30 in which the protein wraps around multiple micelle-like agglomeration of SDS molecules. | ||
| Fig. 2 Microscopic models of SDS/protein complexes. (a) MD simulation of a titin/SDS complex self-assembly. The sequence of snapshots illustrates the microscopic state of the system during a 300 ns explicit solvent all-atom MD simulation. The protein backbone is shown in blue, SDS molecules in cyan and red, and water and 0.4 M NaCl are not shown. (b) Typical microscopic conformations of the SDS/protein assemblies obtained at the end of the self-assembly simulations. The SDS molecules are shown using the molecular bond representation: carbon, sulfur and oxygen atoms are shown in cyan, yellow and red, respectively; hydrogen atoms are not shown. Each protein molecule is shown as a trace of the protein backbone. | ||
To simulate nanopore translocation (Fig. 3), atomic models of different substrates (folded proteins, SDS micelles, and SDS–protein complexes) were combined with atomic models of solid-state nanopores; water and ions were added to produce a rectangular volume of NaCl solution. An external electric field was applied normal to the membrane to produce a transmembrane bias of desired magnitude. Subject to the transmembrane bias, biomolecules move through a nanopore altering the nanopore ionic current. ESI Table S1† provides a summary of nanopore translocation simulations.
 | ||
| Fig. 3 MD simulation of SDS/protein nanopore translocation. (a–d) Simulated ionic current blockades produced by translocation of various molecular species through a 6 nm diameter nanopore (System 1). The left column illustrates typical microscopic conformations observed during nanopore translocation simulations; the black arrow indicates the direction of the positive transmembrane bias. Protein conformation is depicted as a trace of the protein backbone; SDS molecules are shown as molecular bonds; water and ions are not shown for clarity. Right columns shows the ionic current traces recorded from the translocation simulations and the open pore current level. Because of the periodic boundary conditions employed in our MD simulations, individual ionic current traces feature multiple translocation events. Ionic current traces from independent simulations are delineated by the “//” mark. Black circles and horizontal black bars indicate the average blockade current of individual blockade events and the duration of each event, respectively. With the exception of native titin, the blockade events were defined by the reduction of the nanopore current below 75% of the open pore value; the blockade event in the native titin simulation was characterized using the titin's center of mass coordinates. (e) Sequence of snapshots illustrating the nanopore translocation of a β-amylase/SDS complex. (f) Same as in panels (a–d) but for an SDS micelle. (g) Average translocation time of the SDS/protein assemblies. The color of the bars indicates the transmembrane voltage. (h) The average conductance blockade amplitudes. To enable direct comparison with experiment, the conductance blockades computed from MD simulations were scaled by the ratio of the experimental and simulation bulk conductivity of 0.4 NaCl (3.6/4.8). Open bars indicate conductance blockades obtained from the molecular assemblies placed at the center of a 10 nm diameter nanopore (System 2) at a 500 mV bias; the molecular configurations are defined in ESI Fig. S3.† | ||
Our simulations of a folded protein (titin) translocation (Fig. 3a) demonstrate that a protein's encounter with a nanopore is hardly determined by the electrophoretic force exerted due to the transmembrane bias. In five independent simulations of the folded titin system (60 ns each; ±500 mV bias), folded titin rarely translocated through a 6 nm wide nanopore (ESI Fig. S1 and Movie 2†). We ascribe this behaviour to the low net charge of native titin at pH 7.5. In the only simulation where a protein translocation event was observed, the protein was seen moving through the nanopore in the direction set by the electro-osmotic flow.
In contrast, the translocation of SDS–protein assemblies was clearly governed by electrophoresis (Fig. 3b–d). A complex of SDS molecules and either monomeric or dimeric titin permeated through a 6.5 nm diameter nanopore multiple times, producing sharp spike-like blockades. The SDS–titin assembly maintained its integrity. ESI Movies 3 and 4† illustrate the typical simulation trajectories. During the translocation, the SDS–titin assemblies noticeably deformed in the center of the nanopore, particularly in the case of the titin dimer. The protein–SDS assembly relaxed back to a compact globular state after each translocation. Repeating the simulations starting from a differently orientated SDS–titin assembly (Fig. 3b and c) or at a lower transmembrane bias (ESI Fig. S2†) produced similar outcomes.
Pronounced deformations of the SDS–protein assemblies were observed during the simulations of the translocation of large SDS-unfolded β-amylase complexes through a 6.5 nm diameter nanopore (Fig. 3d and e). The translocation of the SDS–β-amylase assembly began when two or more peptide chains detached from the assembly, entering the nanopore in a “multi-chained” conformation. The peptide chains stretched through the nanopore, pulling the rest of the protein in a train-like fashion, reminiscent of DNA translocation. Fig. 3e and ESI Movie 5† illustrate one of the permeation trajectories. Despite such major conformational changes, the number of SDS molecules associated with the protein remained constant during the nanopore translocation. Note that the physical size of β-amylase in its native folded conformation (∼10 nm) is greater than the 6.5 nm diameter of the simulated nanopore.
SDS micelles alone also translocated through the nanopore in the direction prescribed by the electrophoretic force. Each permeation event produced a pronounced reduction of the ionic current (Fig. 3f). The shape of the ionic current blockades—a downward spike—was consistent with that produced by a spherical particle passing through a larger pore.31 During the translocation process, the micelle was observed to deform slightly, recovering its spherical shape after each passage. ESI Movie 6† illustrates a typical simulation trajectory. At all transmembrane biases tested, the SDS micelle maintained its integrity with no SDS molecules leaving the micelle.
Fig. 3g plots the average duration of the translocation events for different molecular assemblies and under several transmembrane bias conditions. At a given transmembrane bias, SDS micelles moved through the nanopore considerably faster than SDS–protein assemblies. Predictably, the translocation of an SDS–titin monomer complex occurred faster than that of an SDS–titin dimer complex. Both complexes moved through the nanopore considerably faster than the large SDS–β-amylase complex, despite a stronger bias used in the simulation of the latter system. These trends suggest that the translocation time increases with the molecular weight of the protein.
The translocation of different SDS–protein assemblies and SDS micelles through a 6 nm diameter nanopore produced conductance blockades of similar amplitudes (Fig. 3h). This result is expected since all molecular assemblies had a similar cross sectional area in the narrowest part of the pore when permeating through a 6 nm diameter nanopore. By contrast, considerably different blockade amplitudes were observed when the same molecular assemblies were placed at the center of a larger, 10 nm diameter nanopore (Fig. 3h and ESI Fig. S3†). This observation indicates that the amplitude of nanopore blockades produced by a compact object, such as a folded protein or an SDS micelle, depends on the nanopore size.31
Encouraged by our observation of SDS-treated β-amylase translocation through a nanopore that was smaller in cross-section than the folded β-amylase protein, we investigated if a titin–SDS complex could be electrophoretically driven through a 3 nm-diameter nanopore (System 3, see Materials and methods). The steric confinement of such a narrow nanopore was expected to produce single-file translocation of the SDS-treated protein. However, no fragment of the titin protein was observed to enter the 3 nm nanopore within multiple 80 ns simulations performed at 125 and 250 mV biases despite being placed in close proximity to the nanopore entrance, see ESI Movie 7.† Increasing the transmembrane bias to 1 V resulted in stripping of the SDS molecules away from the titin–SDS assembly but no protein translocation through the nanopore. A similar outcome was observed when the temperature of the system was increased to 373 K.
Summing up, our MD simulations show that SDS treatment provides proteins an overall negative charge, thereby enabling the electrophoretic transport through nanopores. Subject to the electric field and steric forces from the nanopore, the SDS-treated proteins deform and permeate through nanopores that are smaller in diameter than the unperturbed dimensions of the folded protein. The depth of ionic current blockades produced by the permeation of SDS–protein assemblies depends on both the diameter of the nanopore and the conformation of the protein. Realizing single-file translocation of a protein/SDS complex may require pre-stretching of the complex prior to reaching the nanopore entrance.
Single-molecule experiments
We experimentally measured the conductance blockades of native proteins and SDS-unfolded proteins (Fig. 1). We performed all our measurements using SiN nanopores with a diameter of 10 nm at 100 mV in an electrolyte solution containing 0.4 M NaCl, buffered to pH 7.5 with 10 mM Tris-HCl and 1 mM EDTA. SDS-denatured proteins were heated up to 95 °C for 5 min with 1 mM DTT. DTT was used to disrupt and prevent the formation of disulphide bonds. This procedure guaranteed protein denaturation facilitating SDS interaction. The denatured proteins were cooled down before measurement. Note that a denaturant is necessary to guarantee irreversible protein unfolding, which cannot be guaranteed by temperature on its own. Our experiments with native titin revealed that folded proteins translocate preferentially in the direction of the electro-osmotic flow, consistent with the MD simulations (Fig. 3a) and also reported by others.27 SDS–protein complexes, however, move in the direction set by the electrophoretic force, directly verifying the simulations.To elucidate the mechanism of the altered translocation direction caused by SDS, we analyzed the event rate of titin for translocations at several different SDS concentrations, Fig. 4a. In the regime of low SDS concentrations, below the CMC, we observed a high event rate in the direction of the electro-osmotic flow (Fig. 4b). At SDS concentrations above the CMC, however, the event rate associated with the electrophoretic force was dominant. This result is in agreement with the direction of translocation observed in our simulations, which were carried out at SDS concentrations above the CMC. Using dynamic light scattering, we estimated the CMC under our experimental conditions to be between 0.01% and 0.05% (ESI Note S1 and Fig. S4†).
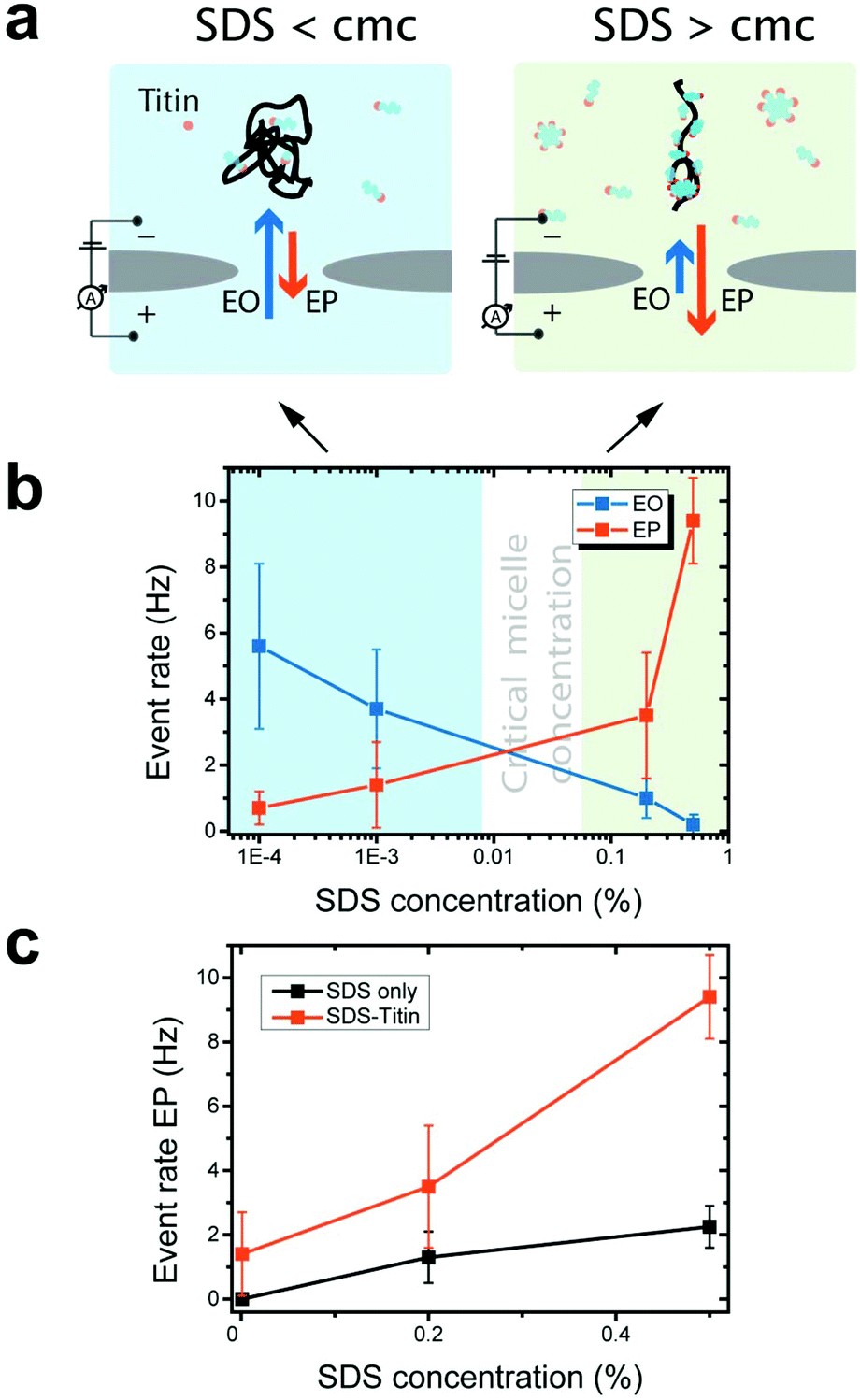 | ||
| Fig. 4 (a) Schematic of the dominant mechanisms of protein translocation at SDS concentrations below and above the CMC. (b) Event rates of the translocation of titin–SDS complexes through a 10 nm solid-state nanopore at different SDS concentrations. At SDS concentrations below the CMC (blue) electro-osmosis is the dominant transport mechanism of titin–SDS through solid-state nanopores. At SDS concentrations above the CMC (green) electrophoresis dominates. (c) Plot of the event rate vs. SDS concentration for SDS micelles alone and SDS-unfolded proteins. | ||
Our observation that electrophoresis dominates at high SDS concentrations is consistent with previous studies on protein unfolding. Above the CMC, SDS forms micelles and cooperatively binds to the polypeptide chain.32–34 Cooperative binding of SDS above the CMC imparts a strong negative charge to the denatured protein, thus increasing the electrophoretic force acting on it. Below the CMC, SDS monomers individually interact with the polypeptide via hydrophobic and electrostatic interactions.
When measurements were performed above the CMC, the translocation of SDS micelles was observed. We performed control experiments in which we characterized these translocations at different SDS concentrations in the absence of any protein (Fig. 4c). SDS micelles moved towards the positive electrode, as expected from their negative charge. Micelles produced well-defined conductance blockades of 3.5 ± 0.5 nS with low event rates (only ∼2 events per s at 0.5% SDS, equivalent to 17 mM) and unusually long dwell times (∼1.2 ms) (Fig. 5). From these characteristics, we speculate that the observed events are from those micelles that interact strongly with the SiN surface in the pore lumen, whereas micelles that do not interact with the pore surface pass through the nanopore much too quickly to be detected by the ionic current measurement. The low event rate (Fig. 4c) indicates that the SDS micelles alone contribute only to approximately 25% of the event rates presented in Fig. 4b.
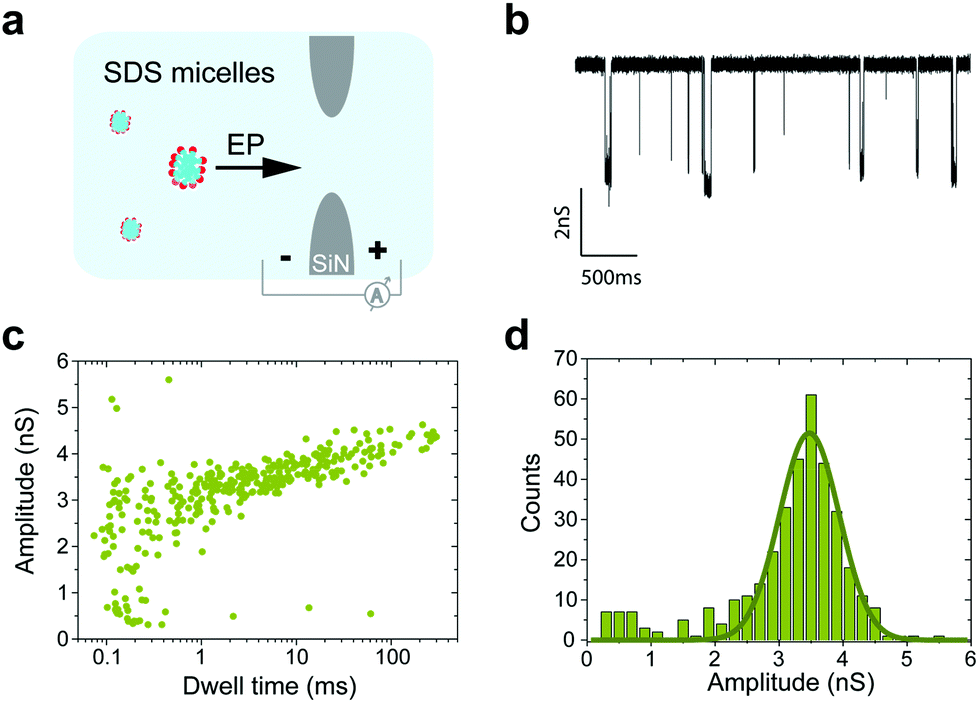 | ||
| Fig. 5 Translocation of SDS micelles through a 10 nm solid-state nanopore. (a) Schematic representation of the nanopore control experiment. (b) Example of a current trace measured showing the translocation of SDS micelles. (c) Dwell time vs. conductance blockade scatter plot for SDS-micelles and SDS-treated protein. (d) Conductance blockade histogram of the translocation of micelles. | ||
Fig. 6 presents a key result of our study. It shows that we can distinguish folded and SDS-induced unfolded proteins in nanopore translocation experiments. Fig. 6a shows examples of translocations of β-amylase proteins, in which it is clear that SDS-treated proteins exhibit a much lower conductance blockade level than native proteins. This is evidently observed in a scatter plot of conductance blockade vs. dwell time for native and SDS-unfolded β-amylase (Fig. 6b). The translocation of native proteins was measured in the direction prescribed by the electro-osmotic flow, whereas SDS-treated proteins were found to translocate in the direction of the electrophoretic force. These measurements were performed at low SDS concentrations (0.005%) below the CMC to unambiguously ensure that the observed blockades are due to SDS–protein complexes, and not due to SDS micelles. We verified that SDS disrupts the structure of β-amylase even at SDS concentrations below the CMC using tryptophan fluorescence measurements (ESI Note S2 and Fig. S5†).
 | ||
| Fig. 6 (a) Current traces produced by the translocation of native β-amylase and SDS-unfolded β-amylase through a 10 nm pore at 10 kHz bandwidth. Typical events are shown in the right panels. (b) Scatter plot of the conductance blockade vs. dwell time of native β-amylase and SDS-treated β-amylase. Native protein translocations are measured in the direction of the electro-osmotic flow, SDS-treated proteins are measured in the electrophoretic direction. (c) Histograms of conductance blockades of native protein (left), SDS-treated protein below the CMC (middle), and SDS-treated protein above the CMC (right). The conductance blockade of the native protein has a value of 3.7 ± 1.7 nS. The blockade for the SDS-treated protein below the CMC is 0.53 ± 0.29 nS. Above the CMC, two peaks are observed. The first has a value of 0.40 ± 0.27 nS, and the second one, presumably due to SDS micelles, has a value of 4.3 ± 0.4 nS. | ||
Our data suggest that SDS treatment causes significant protein unfolding, as also observed in our simulations. As shown in the scatter plot (Fig. 6b) and the histograms presented in Fig. 6c (left and middle), a pronounced decrease in the amplitude of the blockade was observed when β-amylase was treated with SDS, from 3.7 ± 1.7 nS (mean ± standard deviation) for folded proteins to 0.53 ± 0.29 nS for SDS-unfolded proteins. This significant decrease in the conductance blockade is consistent with the protein adopting an elongated structure of a smaller cross section than its native folded conformation. Note that for the folded β-amylase the conductance values are lower than expected, presumably due to the well-known bandwidth limitations when measuring protein translocations.18
We also carried out measurement of SDS-treated β-amylase at an SDS concentration above the CMC. As Fig. 6c (right panel) shows, a low conductance blockade peak (at ∼0.4 nS) persisted, while an additional blockade peak appeared at 4.26 ± 0.37 nS. The presence of the first peak suggests that a population of SDS-associated proteins remains, which translocates in an elongated fashion without binding to micellar SDS. The additional blockade can be attributed to SDS micelles.
In our measurements with titin, no appreciable difference was observed between the native and the SDS-denatured protein, as both gave ∼0.4 nS blockades (ESI Fig. S6†). This is explained by the small size of the protein, which even in the native conformation produces blockades of ∼0.4 nS, a value that unfortunately coincides with the low value expected for the SDS-unfolded protein. Although the conductance blockade of titin did not change significantly upon treatment with SDS, the change in the direction of translocation (Fig. 4) clearly indicates the interaction of SDS with the protein.
The conductance blockades for SDS-unfolded β-amylase and SDS-unfolded titin proteins are comparable (0.40 ± 0.17 nS for titin and 0.49 ± 0.18 nS for β-amylase), suggesting that SDS-denatured proteins translocate through a nanopore with a similar cross-section regardless of the protein molecular weight. For the SDS-treated β-amylase, most of our simulations show a deeper conductance blockade (∼9 nS) compared to our experiments. However, note that, in most of our simulations, the protein–SDS complex adopts a “decorated micelle” structure (see Fig. 2b) and, hence, multiple polypeptide chains simultaneously translocate through the nanopore when driven by a transmembrane bias. Because translocation of such decorated micelle structures is very fast (at the sub-microsecond time scale), they would likely escape experimental detection because of the bandwidth limitations.18 By contrast, events measured in the experiments suggest that the protein adopts an elongated conformation, given the low conductance blockade measured. Indeed, when a single polypeptide chain was simulated in a 10 nm pore (ESI Fig. S7†), a much lower conductance blockade value of ∼2 nS was observed, regardless of whether SDS molecules were bound to the peptide chain or not. The simulated and experimentally measured ionic current blockades are in much better agreement in the case of pure SDS micelle systems, suggesting that experiment and simulation characterize similar molecular configurations of biomolecules within the nanopore. Thus, the low conductance blockades and the relatively slow translocation kinetics observed in the experiments are consistent with the notion that the translocation of protein/SDS complexes is affected by interactions with the nanopore surface.
To summarize, our experimental results suggest that SDS treatment of proteins has drastic effects on the process of protein translocation through solid-state nanopores: the treatment reverses the translocation direction and can reduce the conductance blockade. At high SDS concentrations, proteins preferentially translocate by electrophoretic forces as shown both experimentally and computationally. SDS causes protein unfolding, which can present the polypeptide chain in an elongated conformation to the nanopore volume. For example, the measured conductance blockade of SDS-unfolded β-amylase is clearly lower than the blockade produced by that protein in its folded conformation. The low conductance blockade level is consistent with the conductance blockade observed in our MD simulations of a single peptide chain threaded through the nanopore. This correspondence suggests that single-chain translocations are being experimentally detected in the case of SDS-treated proteins. Further studies are necessary to verify this hypothesis.
Conclusions
Through nanopore measurements combined with MD simulations, we have shown that upon SDS treatment, proteins can translocate through nanopores in an unfolded fashion. Additionally, our simulations show that the dwell time of the SDS-treated proteins in a nanopore increases with the protein molecular weight. These results suggest that a nanoscaled single-molecule version of SDS-PAGE, in which proteins are differentiated according to their translocation time, could be created using solid-state nanopores. Our study also shows that proteins move through the nanopore by electrophoresis, overcoming the electroosmotic flow that runs in the reverse direction. This can be attributed to the enhanced negative charge imparted to the protein by the SDS sulfate groups.We also explored a wide range of SDS concentrations to show that measurements are feasible both above and below the CMC. Characterization of the micelles produced by SDS above the CMC is also presented.
Altogether, our results suggest that SDS causes protein unfolding while facilitating protein translocation in the direction of the electrophoretic force, both characteristics could be advantageous for future protein sequencing applications using solid-state nanopores.
Materials and methods
Nanopore fabrication
20 nm-thick freestanding silicon nitride membranes were fabricated using trans-chip illumination lithography as previously described by Janssen et al.35 TEM is used to drill nanopores on the membranes and then the chips are manually PDMS painted to reduce the membrane capacitance and improve the signal-to-noise ratio.36Nanopore measurements
Before each measurement, the nanopores are cleaned using oxygen plasma and mounted in a PEEK flow cell. Buffer containing 0.4 M NaCl, 10 mM Tris pH 7.5 and 1 mM ETDA is used for all measurements. SDS is added at different concentrations, as indicated. Measurements are recorded at 100 kHz using an Axopatch 200B (Molecular Devices, LLC) and digitized at 500 kHz using a National instruments Digidata 1322A DAQ. The recorded signals were further low-pass filtered at 10 kHz and analyzed using the Transalyzer Matlab package.37Protein substrates
Titin I27 V13P containing an N-terminal His6-tag and a C-terminal SsrA tag was purified by affinity chromatography as follows. A 1-L culture of E. coli strand BL21ai containing the plasmid pRSETa was incubated at 37 °C in LB medium supplemented with ampicillin. At an OD600 of ∼0.8, arabinose (0.2%) was added and the culture was grown for an additional 4 h at 37 °C before harvesting the cells and freezing them at −80 °C. Pellets were re-suspended in lysis buffer (50 mM of sodium phosphate pH 8.0, 300 mM of NaCl, 50 mM of imidazole) and lysed by sonication. The lysate was centrifuged (17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm, 30 min) and titin was purified from the supernatant using Ni2+-NTA affinity resin and eluted with imidazole (200 mM). The sample was dialyzed overnight at 4 °C against PBS pH 7.4. β-Amylase from sweet potato was purchased from Sigma-Aldrich.
000 rpm, 30 min) and titin was purified from the supernatant using Ni2+-NTA affinity resin and eluted with imidazole (200 mM). The sample was dialyzed overnight at 4 °C against PBS pH 7.4. β-Amylase from sweet potato was purchased from Sigma-Aldrich.
General MD methods
All MD simulations were performed using the NAMD2 software package,38 periodic boundary conditions, a 2 fs integration time step and a multiple time-stepping integration scheme.39 Proteins, SDS molecules, water and ions were described using the CHARMM36 force field.40 A custom CHARMM-compatible force field was used for SiO241 along with custom NBFIX corrections to non-bonded interactions of sodium ions and the sulphate groups of SDS or acetate groups of the proteins.42,43 SETTLE44 and RATTLE45 algorithms were applied to the covalent bonds involving hydrogen atoms in water and protein, respectively. The van der Waals forces were cut off smoothly starting at 10 Å and cut off completely at 12 Å. Long range electrostatic interactions were evaluated every third time step using the particle mesh Ewald method46 over a 0.11 nm-spaced grid. The temperature was controlled using a Langevin thermostat acting on the membrane's atoms with a damping constant of 0.5 ps−1. In the NPT (i.e. constant number of particles, pressure, and temperature) simulations, Nosé–Hoover Langevin piston47 maintained the pressure at 1 atm by adjusting the systems’ dimensions along the direction normal to the membrane (z axis in our setup). In all production simulations of the nanopore systems, atoms of the inorganic membrane were harmonically restrained to their coordinates attained at the end of the annealing simulation with the force constant of 20 kcal mol−1 Å−2.
Atomic-scale models of SDS–protein assemblies
Computational models of SDS–protein assemblies were prepared by combining an unfolded conformation of a protein with randomly distributed SDS molecules in a 1:1 amino acid/SDS molecule ratio. The systems were submerged in 0.4 M NaCl solution. Following 10000 steps of energy minimization and 1.5 ns of equilibration in an NPT ensemble, the systems were simulated at 373 K over the course of 200 ns. Following that, the systems were cooled down to 300 K in eight simulation steps, decreasing the temperature of the system by 10 K between each step. Following that, the SDS–protein systems were simulated for another 50 ns at 300 K. ESI Movie 1† illustrates a typical self-assembly trajectory. SDS molecules that were not in contact with the protein at the end of the cooling phase were removed from the systems. The remaining SDS–protein assembly was used for the nanopore translocation simulations.
Atomic-scale models of nanopore systems
The atomic-scale models of solid-state nanopores were obtained by simulated annealing of amorphous silica subject to a shape-defining grid-based potential.48 The initial models of the nanopores were made by removing atoms from a slab of a crystalline SiO2 membrane. The systems were then simulated using the BKS49 force field in a vacuum at 7000, 5000, 2000, and 300 K for 160, 160, 400, and 400 ps, respectively, and in the presence of a grid-based potential50 that confined the atoms to a predefined volume. The nanopores produced using this method are electrically neutral but contain negatively charged oxygen atoms at the nanopore surface, which makes the nanopore ionic current slightly cation selective.51 Three nanopore models were built, differing by the dimensions of the SiO2 membrane (15 × 15 nm2 cross-section/6 nm thickness, System 1; 15 × 15 nm2 cross-section/10 nm thickness, System 2 and 10 × 10 nm2 cross section/5 nm thickness, System 3). Each membrane contained a single hourglass-shaped nanopore with the constriction/entrance diameters of 6/7.5 (System 1), 10/13 (System 2) or 3/4.5 (System 3) nm, respectively. The nanopore models produced by the annealing procedures were merged with the atomic-scale models of the biomolecules and solvated with pre-equilibrated volumes of water. Sodium and chloride ions were added to produce a 0.4 M solution; the precise number of ions was chosen to make the entire system electrically neutral.Following the assembly, the systems were minimized using a conjugate gradient method and equilibrated in the NPT ensemble for 2 ns each at 1 atm pressure and 295 K temperature. During the equilibration, the protein and/or SDS atoms were restrained to their initial coordinates with a force constant of 10 kcal mol−1 Å−2 for 1 ns; then the atoms were restrained with a force constant of 5 kcal mol−1 Å−2 for another 1 ns. All subsequent simulations were performed in the presence of a grid-based potential that prevented a direct contact interaction between protein and SDS atoms with the nanopore surface,50 thereby eliminating non-specific binding; the grid potential did not apply to water molecules or ions. All simulations of the nanopore systems under applied electric field conditions were carried out in the NVT ensemble (i.e. constant number of particles, volume, and temperature). The center of mass of the biomolecules was restrained to remain at the symmetry axis of the nanopore using steered molecular dynamics (SMD) features of NAMD2. Such restraints allowed the SDS protein assembly to undergo considerable conformational transformations and move through the nanopore while maintaining its center of mass aligned with the geometrical center of the nanopore. A transmembrane potential V was induced by applying a constant electric field E = −ΔV/lz normal to the membrane, where lz is the length of the simulation system normal to the membrane. ESI Table S1† provides a complete list of simulations performed. Open pore systems were built and simulated using the same procedure as described above. Atomic coordinates were recorded every 9.6 ps.
Acknowledgements
The authors thank Malwina Szczepaniak, James Wilson, and Maxim Belkin for insightful discussions. We also would like to acknowledge Pawel Tulinski for his help during titin purification. Titin protein plasmids were a generous gift from Victor Muñoz and Jörg Schönfelder. This work was supported by the National Human Genome Research Institute of the National Institute of Health under Award Number R01-HG007406. The C. D. lab was further supported by the ERC Advanced Grant SynDiv (no. 669598). C. J. was funded by the Foundation for Fundamental Research on Matter (12PR3029). L. R., C. J. and C. D. were funded by The Netherlands Organization of Scientific Research (NWO/OCW) as part of the Frontiers of the Nanoscience Program. The supercomputer time provided through XSEDE Allocation Grant MCA05S028 and the Blue Waters petascale supercomputer system (UIUC).Notes and references
- P. Mitchell, Nat. Biotechnol., 2002, 21, 233–237 CrossRef PubMed.
- B. N. Miles, A. P. Ivanov, K. a. Wilson, F. Doğan, D. Japrung and J. B. Edel, Chem. Soc. Rev., 2013, 42, 15–28 RSC.
- C. Dekker, Nat. Nanotechnol., 2007, 2, 209–215 CrossRef CAS PubMed.
- M. Wanunu, Phys. Life Rev., 2012, 9, 125–158 CrossRef PubMed.
- J. J. Kasianowicz, E. Brandin, D. Branton and D. W. Deamer, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 13770–13773 CrossRef CAS.
- A. Meller, L. Nivon, E. Brandin, J. Golovchenko and D. Branton, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 1079–1084 CrossRef CAS.
- M. Akeson, D. Branton, J. J. Kasianowicz, E. Brandin and D. W. Deamer, Biophys. J., 1999, 77, 3227–3233 CrossRef CAS PubMed.
- G. M. Cherf, K. R. Lieberman, H. Rashid, C. E. Lam, K. Karplus and M. Akeson, Nat. Biotechnol., 2012, 30, 344–348 CrossRef CAS PubMed.
- E. A. Manrao, I. M. Derrington, A. H. Laszlo, K. W. Langford, M. K. Hopper, N. Gillgren, M. Pavlenok, M. Niederweis and J. H. Gundlach, Nat. Biotechnol., 2012, 30, 349–353 CrossRef CAS PubMed.
- A. H. Laszlo, I. M. Derrington, B. C. Ross, H. Brinkerhoff, A. Adey, I. C. Nova, J. M. Craig, K. W. Langford, J. M. Samson, R. Daza, K. Doering, J. Shendure and J. H. Gundlach, Nat. Biotechnol., 2014, 32, 829–833 CrossRef CAS PubMed.
- B. Hornblower, A. Coombs, R. D. Whitaker, A. Kolomeisky, S. J. Picone, A. Meller and M. Akeson, Nat. Methods, 2007, 4, 315–317 CAS.
- Q. Zhao, G. Sigalov, V. Dimitrov, B. Dorvel, U. Mirsaidov, S. Sligar, A. Aksimentiev and G. Timp, Nano Lett., 2007, 7, 1680–1685 CrossRef CAS PubMed.
- S. W. Kowalczyk, A. R. Hall and C. Dekker, Nano Lett., 2010, 10, 324–328 CrossRef CAS PubMed.
- C. Plesa, J. W. Ruitenberg, M. J. Witteveen and C. Dekker, Nano Lett., 2015, 15, 3153–3158 CrossRef CAS PubMed.
- M. M. Mohammad, S. Prakash, A. Matouschek and L. Movileanu, J. Am. Chem. Soc., 2008, 130, 4081–4088 CrossRef CAS PubMed.
- K. J. Freedman, S. R. Haq, J. B. Edel, P. Jemth and M. J. Kim, Sci. Rep., 2013, 3, 1638 CrossRef PubMed.
- E. C. Yusko, J. M. Johnson, S. Majd, P. Prangkio, R. C. Rollings, J. Li, J. Yang and M. Mayer, Nat. Nanotechnol., 2011, 6, 253–260 CrossRef CAS PubMed.
- C. Plesa, S. W. Kowalczyk, R. Zinsmeester, A. Y. Grosberg, Y. Rabin and C. Dekker, Nano Lett., 2013, 13, 658–663 CrossRef CAS PubMed.
- E. C. Yusko, B. R. Bruhn, O. M. Eggenberger, J. Houghtaling, R. C. Rollings, N. C. Walsh, S. Nandivada, M. Pindrus, A. R. Hall, D. Sept, J. Li, D. S. Kalonia and M. Mayer, Nat. Nanotechnol., 2016, 12, 360–367 CrossRef PubMed.
- D. S. Talaga and J. Li, J. Am. Chem. Soc., 2009, 131, 9287–9297 CrossRef CAS PubMed.
- D. Rodriguez-Larrea and H. Bayley, Nat. Nanotechnol., 2013, 8, 288–295 CrossRef CAS PubMed.
- L. Movileanu, S. Howorka, O. Braha and H. Bayley, Nat. Biotechnol., 2000, 18, 1091–1095 CrossRef CAS PubMed.
- J. Nivala, D. B. Marks and M. Akeson, Nat. Biotechnol., 2013, 31, 247–250 CrossRef CAS PubMed.
- Y. Zhao, B. Ashcroft, P. Zhang, H. Liu, S. Sen, W. Song, J. Im, B. Gyarfas, S. Manna, S. Biswas, C. Borges and S. Lindsay, Nat. Nanotechnol., 2014, 9, 466–473 CrossRef CAS PubMed.
- J. Wilson, L. Sloman, Z. He and A. Aksimentiev, Adv. Funct. Mater., 2016, 26, 4830–4838 CrossRef CAS PubMed.
- C. B. Rosen, D. Rodriguez-Larrea and H. Bayley, Nat. Biotechnol., 2014, 32, 179–181 CrossRef CAS PubMed.
- M. Firnkes, D. Pedone, J. Knezevic, M. Döblinger and U. Rant, Nano Lett., 2010, 10, 2162–2167 CrossRef CAS PubMed.
- J. Li, D. Fologea, R. Rollings and B. Ledden, Protein Pept. Lett., 2014, 21, 256–265 CrossRef CAS PubMed.
- E. Kennedy, Z. Dong, C. Tennant and G. Timp, Nat. Nanotechnol., 2016, 11, 968–976 CrossRef CAS PubMed.
- D. Otzen, Biochim. Biophys. Acta, Proteins Proteomics, 2011, 1814, 562–591 CrossRef CAS PubMed.
- L. Luo, S. R. German, W.-J. Lan, D. A. Holden, T. L. Mega and H. S. White, Annu. Rev. Anal. Chem., 2014, 7, 513–535 CrossRef CAS PubMed.
- A. Lee, S. K. Y. Tang, C. R. MacE and G. M. Whitesides, Langmuir, 2011, 27, 11560–11574 CrossRef CAS PubMed.
- M. M. Nielsen, K. K. Andersen, P. Westh and D. E. Otzen, Biophys. J., 2007, 92, 3674–3685 CrossRef CAS PubMed.
- D. E. Otzen, Biophys. J., 2002, 83, 2219–2230 CrossRef CAS PubMed.
- X. J. a. Janssen, M. P. Jonsson, C. Plesa, G. V. Soni, C. Dekker and N. H. Dekker, Nanotechnology, 2012, 23, 475302 CrossRef PubMed.
- V. Tabard-cossa, D. Trivedi, M. Wiggin, N. N. Jetha and A. Marziali, Nanotechnology, 2007, 18, 305505 Search PubMed.
- C. Plesa and C. Dekker, Nanotechnology, 2015, 26, 1–7 CrossRef PubMed.
- J. C. Phillips, R. Braun, W. Wang, J. Gumbart, E. Tajkhorshid, E. Villa, C. Chipot, R. D. Skeel, L. Kalé and K. Schulten, J. Comput. Chem., 2005, 26, 1781–1802 CrossRef CAS PubMed.
- P. F. Batcho, D. A. Case and T. Schlick, J. Chem. Phys., 2001, 115, 4003–4018 CrossRef CAS.
- A. D. MacKerell, D. Bashford, M. Bellott, R. L. Dunbrack, J. D. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph-McCarthy, L. Kuchnir, K. Kuczera, F. T. K. Lau, C. Mattos, S. Michnick, T. Ngo, D. T. Nguyen, B. Prodhom, W. E. Reiher, B. Roux, M. Schlenkrich, J. C. Smith, R. Stote, J. Straub, M. Watanabe, J. Wiórkiewicz-Kuczera, D. Yin and M. Karplus, J. Phys. Chem. B, 1998, 102, 3586–3616 CrossRef CAS PubMed.
- E. R. Cruz-chu, A. Aksimentiev and K. Schulten, Society, 2006, 110, 21497–21508 CAS.
- J. Yoo and A. Aksimentiev, J. Phys. Chem. Lett., 2012, 3, 45–50 CrossRef CAS.
- J. Yoo and A. Aksimentiev, J. Chem. Theory Comput., 2016, 12, 430–443 CrossRef CAS PubMed.
- S. Miyamoto and P. A. Kollman, J. Comput. Chem., 1992, 13, 952–962 CrossRef CAS.
- H. C. Andersen, J. Comput. Phys., 1983, 52, 24–34 CrossRef CAS.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089 CrossRef CAS.
- G. J. Martyna, D. J. Tobias and M. L. Klein, J. Chem. Phys., 1994, 101, 4177 CrossRef CAS.
- S. Banerjee, J. Wilson, J. Shim, M. Shankla, E. A. Corbin, A. Aksimentiev and R. Bashir, Adv. Funct. Mater., 2015, 25, 936–946 CrossRef CAS PubMed.
- B. W. H. Van Beest, G. J. Kramer and R. A. Van Santen, Phys. Rev. Lett., 1990, 64, 1955–1958 CrossRef CAS PubMed.
- D. B. Wells, V. Abramkina and A. Aksimentiev, J. Chem. Phys., 2007, 127, 125101 CrossRef PubMed.
- M. Shankla and A. Aksimentiev, J. Phys. Chem. B, 2017, 121, 3724–3733 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7nr02450a |
| ‡ These authors contributed equally to this study. |
| This journal is © The Royal Society of Chemistry 2017 |