
In situ crystal growth of gold nanocrystals on upconversion nanoparticles for synergistic chemo-photothermal therapy†
 *a
*a
Abstract
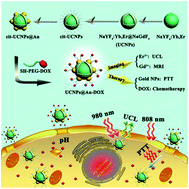
A multifunctional cancer therapy nanocomposite was proposed and synthesized by linking the pH-responsive SH-PEG-DOX prodrug onto gold nanocrystals that were grown in situ on the surface of upconversion nanoparticles (UCNPs). In the structure of the SH-PEG-DOX prodrug, a hydrazone bond was utilized for subsequent pH-responsive drug release in the intracellular acidic microenvironment of cancer cells. This innovative assembly method is facile and mild, and can be used to obtain nanocomposites of UCNPs and gold, which show excellent photostability and biocompatibility. The final UCNPs@Au-DOX nanocomposites offer efficient treatment effects in vitro under irradiation with an 808 nm laser due to the synergistic effect of chemotherapy and photothermal therapy. In addition, the UCNPs@Au-DOX nanocomposites show excellent intracellular locating ability via upconversion luminescence (UCL) imaging with Er3+ ions and magnetic resonance imaging (MRI) with Gd3+ ions, indicating that they have potential as a visual tracking agent in cancer treatment. Therefore, the presented bioimaging-guided multifunctional synergistic therapy nanocomposites are promising tools for imaging-guided cancer therapy.
 Please wait while we load your content...
Please wait while we load your content...

