
Selective protein transport through ultra-thin suspended reduced graphene oxide nanopores†

Abstract
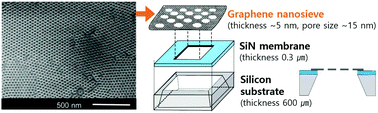
The nanoporous free-standing graphene membrane is of great interest in high performance separation technology. In particular, the separation of biological molecules with similar sizes is one of the key challenges in the purification of biomaterials. Here, we report a reliable, cost-effective, and facile method for the fabrication of a graphene-based nanosieve and its application in the separation of similar-size proteins. A suspended reduced graphene oxide (rGO) nanosieve with ultra-thin, large-area, well-ordered, and dense 15 nm-sized pores was fabricated using block copolymer (BCP) lithography. The fabricated 5 nm-ultrathin nanosieve with an area of 200 μm × 200 μm (an ultra-high aspect ratio of ∼40 000) endured pressure up to 1 atm, and effectively separated hemoglobin (Hb) from a mixture of hemoglobin and immunoglobulin G (IgG), the common proteins in human blood, in a highly selective and rapid manner. The use of the suspended rGO nanosieve is expected to provide a simple and manufacturable platform for practical biomolecule separation offering high selectivity and a large throughput.
 Please wait while we load your content...
Please wait while we load your content...

