
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Insulated molecular wires: inhibiting orthogonal contacts in metal complex based molecular junctions†
Oday A.
Al-Owaedi‡
ab,
Sören
Bock‡
c,
David C.
Milan‡
 d,
Marie-Christine
Oerthel
e,
Michael S.
Inkpen
f,
Dmitry S.
Yufit
e,
Alexandre N.
Sobolev
cg,
Nicholas J.
Long
f,
Tim
Albrecht
f,
Simon J.
Higgins
d,
Martin R.
Bryce
e,
Richard J.
Nichols
*d,
Colin J.
Lambert
*a and
Paul J.
Low
*c
d,
Marie-Christine
Oerthel
e,
Michael S.
Inkpen
f,
Dmitry S.
Yufit
e,
Alexandre N.
Sobolev
cg,
Nicholas J.
Long
f,
Tim
Albrecht
f,
Simon J.
Higgins
d,
Martin R.
Bryce
e,
Richard J.
Nichols
*d,
Colin J.
Lambert
*a and
Paul J.
Low
*c
aDepartment of Physics, University of Lancaster, Lancaster, LA1 4YB, UK. E-mail: paul.low@uwa.edu.au
bDepartment of Laser Physics, Women Faculty of Science, Babylon University, Hilla, Iraq
cSchool of Molecular Sciences, University of Western Australia, 35 Stirling Highway, Perth 6009, Australia
dDepartment of Chemistry, University of Liverpool, Crown St, Liverpool, L69 7ZD, UK
eDepartment of Chemistry, Durham University, South Rd, Durham, DH1 3LE, UK
fDepartment of Chemistry, Imperial College London, London SW7 2AZ, UK
gCentre for Microscopy Characterization and Analysis, University of Western Australia, 35 Stirling Highway, Perth 6009, Australia
First published on 26th June 2017
Abstract
Metal complexes are receiving increased attention as molecular wires in fundamental studies of the transport properties of metal|molecule|metal junctions. In this context we report the single-molecule conductance of a systematic series of d8 square-planar platinum(II) trans-bis(alkynyl) complexes with terminal trimethylsilylethynyl (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CSiMe3) contacting groups, e.g. trans-Pt{CCC6H4CCSiMe3}2(PR3)2 (R = Ph or Et), using a combination of scanning tunneling microscopy (STM) experiments in solution and theoretical calculations using density functional theory and non-equilibrium Green's function formalism. The measured conductance values of the complexes (ca. 3–5 × 10−5G0) are commensurate with similarly structured all-organic oligo(phenylene ethynylene) and oligo(yne) compounds. Based on conductance and break-off distance data, we demonstrate that a PPh3 supporting ligand in the platinum complexes can provide an alternative contact point for the STM tip in the molecular junctions, orthogonal to the terminal CCSiMe3 group. The attachment of hexyloxy side chains to the diethynylbenzene ligands, e.g. trans-Pt{CCC6H2(Ohex)2CCSiMe3}2(PPh3)2 (Ohex = OC6H13), hinders contact of the STM tip to the PPh3 groups and effectively insulates the molecule, allowing the conductance through the full length of the backbone to be reliably measured. The use of trialkylphosphine (PEt3), rather than triarylphosphine (PPh3), ancillary ligands at platinum also eliminates these orthogonal contacts. These results have significant implications for the future design of organometallic complexes for studies in molecular junctions.
CSiMe3) contacting groups, e.g. trans-Pt{CCC6H4CCSiMe3}2(PR3)2 (R = Ph or Et), using a combination of scanning tunneling microscopy (STM) experiments in solution and theoretical calculations using density functional theory and non-equilibrium Green's function formalism. The measured conductance values of the complexes (ca. 3–5 × 10−5G0) are commensurate with similarly structured all-organic oligo(phenylene ethynylene) and oligo(yne) compounds. Based on conductance and break-off distance data, we demonstrate that a PPh3 supporting ligand in the platinum complexes can provide an alternative contact point for the STM tip in the molecular junctions, orthogonal to the terminal CCSiMe3 group. The attachment of hexyloxy side chains to the diethynylbenzene ligands, e.g. trans-Pt{CCC6H2(Ohex)2CCSiMe3}2(PPh3)2 (Ohex = OC6H13), hinders contact of the STM tip to the PPh3 groups and effectively insulates the molecule, allowing the conductance through the full length of the backbone to be reliably measured. The use of trialkylphosphine (PEt3), rather than triarylphosphine (PPh3), ancillary ligands at platinum also eliminates these orthogonal contacts. These results have significant implications for the future design of organometallic complexes for studies in molecular junctions.
Introduction
The development of methods that allow the formation and experimental determination of the electrical response of single-molecule metal|molecule|metal junctions has driven rapid advances in molecular electronics.1–6 Studies of simple systems such as α,ω-alkane dithiols contacted between two gold electrodes have shed light on issues such as non-resonant charge transport and the importance of molecular conformation within the junction,7–9 and inspired innovations in designs of wire-like molecules. Moving beyond the identification of the influence of the medium on electrical properties of the junction10–14 and introduction of redox-active molecules to molecular junctions has led to systems capable of modulating charge transport in response to an external electrochemical or chemical gate.15–20Whilst the majority of studies have been directed towards organic molecules within molecular junctions, metal complexes are now also attracting attention.21–24 Metal complexes offer the potential to tune energies of key molecular orbitals through choice of metal and supporting ligands, whilst the possibilities to control the metal complex redox state at modest potentials leads to exciting applications in electrostatically and electrochemically gated junctions.25,26 Of the various metal complexes that have been studied in molecular junctions, including multi-metallic strings,27 clusters,13,28,29 porphyrins,30,31 and ferrocene derivatives,32,33 compounds and complexes of the group 8 metals within bipyridyl-,34–36 bis(terpyridyl)-37 or trans-bis(alkynyl)-based38,39,40–46 structures feature prominently. In contrast, analogous wire-like trans-bis(alkynyl) platinum complexes have been little explored in molecular junctions.47–49 This is likely due to the perceptions of low conductivity arising from the smaller contribution of Pt(5d) orbitals to the frontier orbitals of square planar trans-Pt(CCR)2L2 (L = phosphine) complexes compared with the more extensive delocalisation in the HOMOs of octahedral complexes trans-M(CCR)2L4 (M = Fe, Ru, Os) along the π-d-π-conjugated molecular backbone.45,50–53 However, as discussed by McGrady in the context of pseudo-1D metal string complexes, efficient through-molecule conductance can be achieved through frontier orbitals which are distributed near, and energetically aligned with, the electrodes and need not be evenly distributed along the entire molecular backbone.54,55 Recently, the molecular conductances of Pt(II) complexes trans-Pt{CCC6H2(Ohex)2CCC6H4SMe}2(PPh3)2 (1.8 × 10−5G0) and trans-Pt{CCC6H2(Ohex)2CCC5H4N}2(PPh3)2 (9.8 × 10−6G0) have been measured, and found to be similar to those of analogous ruthenium complexes trans-Ru{CCC6H2(Ohex)2CCC6H4SMe}2(dppe)2 (1.8 × 10−5G0) and trans-Ru{CCC6H2(Ohex)2CCC5H4N}2(dppe)2 (4.5 × 10−6G0),56 (Ohex = OC6H13) making trans-bis(alkynyl) Pt(II) complexes an attractive target for further study.
Through numerous studies of molecular junctions, the nature of the molecule-electrode contact has proven important,57 and recognition of the role that anchor groups play in the electrical performance of the junction has led to the development of a wide range of contacting groups and electrode materials.58–63 However, even well-known contacting groups such as thiolates bind to a range of surface sites, including terraces or near to adatoms as well as idealised pristine atomically flat terraces, giving rise to a range of molecular conductance signatures from a given molecular backbone.64 Consequently, interest has increasingly focussed on molecular junctions in which the molecule is contacted to the electrode by strong electrode-carbon covalent bonds.65,66 Other strategies to minimise the range of conductance signatures observed include the use of bulky anchor groups such as trimethylsilylethynyl, which give rise only to junctions of appreciable conductance in a restricted range of configurations, simplifying conductance profiles and allowing more precise assessment of low conductance contacts.14,67
However, even when well-defined contacts are placed within a molecule, the contact of the electrodes to different parts of the compound, particularly in the case of long, conjugated molecules, can result in complex electrical behavior. Examples include the potentiometric-like response of thiol-terminated oligoenes, where electrical contact can be made at either the thiol or oligoene π-system,68 and related observations in oligoyne-based69 and other molecular wires70,71 in which the π-system of the molecular backbone interacts with the electrodes at shorter electrode separations leading to enhanced conductance (short circuits).
Observations of electrical contact directly to the molecular backbone, and conceptual analogies to conventional wires, inspire the design of ‘insulated’ wire-like compounds.72,73 For example, the Gladysz group has reported a series of bimetallic, polyyndiyl-bridged platinum complexes in which the wire-like polyyndiyl core is wrapped in helical alkyl chains (Fig. 1a).74–79 A related concept involving the introduction of highly fluorous trialkylphosphines as ancillary ligands to bimetallic platinum polyynydiyl complexes to shield the wire-like carbon-chain through aggregation of the fluorine- rich aliphatic segments has also been proposed (Fig. 1b).80 Rotaxane-like encapsulation has also proven to be a popular concept in the design of insulated wire-like structures, with prominent recent examples from the Anderson and Tykwinski,81,82 Gladysz83,84 and Terao85–89 groups being reported recently (Fig. 1c). In each of these cases, the conceptual design principle involves shielding the π-conjugated backbone of the molecule from adventitious contact within a junction.90
 | ||
| Fig. 1 Representative sketches of different insulating strategies for metal-complex based molecular wires: (a) helical alkyl chains arranged around a conjugated chain (bold line); (b) fluorous alkyl chains self-assembled around a conjugated chain; (c) encapsulation of a conjugated chain in a rotaxane-like structure; (d) protection of the ancillary ligands on the metal complex by alkyloxy side-chains; (e) single molecule conductance can be determined by placing the molecule within a junction represented here by an STM tip and a conducting substrate. After initial contact with the molecule, the current flowing from the tip to the substrate through the molecule is determined whilst the STM tip is withdrawn. As the tip is withdrawn the molecule may rearrange within the junction until eventually the tip|molecule|substrate contact is broken.1 | ||
The motivation for the present study was twofold: (i) to explore the electrical properties and behavior of new trans-bis(alkynyl) platinum complexes within molecular junctions, and (ii) to address issues concerning the design of insulated molecular wires. We demonstrate that when contacted through the trimethylsilylethynyl group installed at the remote ends of the molecular backbone, the trans- bis(alkynyl) platinum complexes examined here display single molecule conductances of similar magnitude to organic oligo(phenylene ethynylene)- and oligo(yne)-based molecular wires. In addition, hexyloxy side chains, initially introduced to promote solubility (Fig. 1d), are shown to play an important role in preventing adventitious contacts (‘short circuits’) by hindering formation of molecular junctions contacted through ancillary triphenylphosphine (PPh3) ligands.
Results and discussion
The compounds 1a,b–4a,b (Chart 1) were chosen to allow further exploration of the behavior of trans-bis(alkynyl) Pt(II) complexes and the influence of supporting ligands in single-molecule junctions. The trimethylsilylethynyl group (CCSiMe3) has been shown to be a useful anchor in the construction of self-assembled monolayers (SAMs) of unsaturated hydrocarbons on gold, with the electron-withdrawing ethynyl moiety playing an essential role in stabilizing the gold–molecule interaction.91–94 As a contacting group in molecular junctions, the CCSiMe3 moiety is known to favor only a single, albeit low conductance, contact.14,95–97 This is due to the fact that the CCSiMe3 group makes effective electrical contact to defect sites on the gold electrode surfaces in a restricted range of geometries, limited in part due to the steric bulk of the ancillary methyl groups.67 Thus, in even π-rich oligophenylene ethynylene based molecules, the large footprint and limited range of conducting configurations that are available to CCSiMe3 contacts gives rise to conductance histograms featuring only a single conductance peak without complications from alternative conductive molecular orientations or contacts within the junction14,14,67,96 As will be seen in the discussion that follows, the relatively weak nature of the CCSiMe3–gold interaction (estimated at ca. 0.4–0.7 eV)14,67 permits competitive, if adventitious, interactions between the STM tip and other regions of the molecule.
 | ||
| Chart 1 The compounds 1a,b–4a,b used in this work, and reference compound 5. | ||
The complexes were synthesized from CuI-catalyzed ligand exchange reactions of cis-PtCl2(PPh3)2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 98 or a mixture of cis- and trans-PtCl2(PEt3)299 with the appropriate alkyne in diethyl-, triethyl- or diisopropyl-amine solvent.100 The compounds were characterized by the usual suite of 1H, 13C and 31P NMR spectroscopies, IR spectroscopy, mass spectrometry, and elemental analysis. Complexes 2a and 2b were also characterized by single crystal X-ray diffraction studies, the results of which are summarized in the ESI.† Conductance data were acquired using the STM I(s) technique (Fig. 1e and Fig. S1–S8†),101 with measurements made in mesitylene solution.14 These experimental measurements were also supported by calculations of model junctions carried out using a combination of DFT and non-equilibrium Green's function formalism (Fig. 2).
98 or a mixture of cis- and trans-PtCl2(PEt3)299 with the appropriate alkyne in diethyl-, triethyl- or diisopropyl-amine solvent.100 The compounds were characterized by the usual suite of 1H, 13C and 31P NMR spectroscopies, IR spectroscopy, mass spectrometry, and elemental analysis. Complexes 2a and 2b were also characterized by single crystal X-ray diffraction studies, the results of which are summarized in the ESI.† Conductance data were acquired using the STM I(s) technique (Fig. 1e and Fig. S1–S8†),101 with measurements made in mesitylene solution.14 These experimental measurements were also supported by calculations of model junctions carried out using a combination of DFT and non-equilibrium Green's function formalism (Fig. 2).
 | ||
| Fig. 2 The relaxed geometries from DFT model molecular junctions and plots showing selected comparisons of calculated conductance as a function of the Fermi energy for all molecular junctions. Black dashed lines show the chosen Fermi energy (EF = −0.4 eV). | ||
Initial STM I(s) studies of the compounds 1a ((3.1 ± 0.9) × 10−5G0 or 2.4 ± 0.7 nS) and 1b ((3.2 ± 0.8) × 10−5G0 or 2.5 ± 0.6 nS) bearing solubilizing hexyloxy groups revealed essentially identical single-molecule conductance values, and hence little influence of the supporting phosphine ligands (1a, PPh3; 1b, PEt3) on the conductance properties (Table 1, Fig. 3).47 These values compare with that of the analogous organic compound Me3SiCCC6H4CCC6H4CCC6H4CCSiMe3 (5, (2.75 ± 0.55) × 10−5G0 or 2.13 ± 0.43 nS, Chart 1) of comparable molecular length (1a 2.12 nm; 1b 2.05 nm; 5 2.45 nm), also measured by the I(s) method.96 Break-off distances obtained from the I(s) data from 1a and 1b are in reasonable agreement with these estimates of molecular length (Table 1).
 | ||
| Fig. 3 I(s) conductance histograms of 1a and 1b constructed from 500 traces. | ||
| Exp. G/G0a |
Th. G/G0b |
Z*c (nm) |
Z (nm) | Si⋯Si distancee (nm) | Anchor group | ||
|---|---|---|---|---|---|---|---|
| a The experimental conductance determined from I(s) measurements. b The calculated conductance values G/G0 at EF − EDFTF = −0.4 eV from model junctions. c Experimental break-off distance from I(s) measurments. d The calculated electrode separation in a relaxed junction, Z = dAu–Au − 0.25 nm, where 0.25 nm is the calculated center-to-center distance of the apex atoms of the two opposing gold electrodes when conductance = G0 in the absence of a molecule. e Si⋯Si distance from DFT optimised gas-phase geometries. f See Fig. S11. g Crystallographically determined distance 2.37 nm. | |||||||
| 1a |
trans-Pt{CCC6H2(Ohex)2CCSiMe3}2(PPh3)2 |
3.1 ± 0.9 × 10−5 | 3.1 × 10−5 | 1.84 ± 0.1 | 2.11 | 2.39 | CCSiMe3 |
| 1b |
trans-Pt{CCC6H2(Ohex)2CCSiMe3}2(PEt3)2 |
3.2 ± 0.8 × 10−5 | 3.2 × 10−5 | 2.05 ± 0.2 | 2.21 | 2.39 | CCSiMe3 |
| 2a |
trans-Pt{CCC6H4CCSiMe3}2(PPh3)2 |
7.9 ± 1.1 × 10−5 | 8.0 × 10−5 | 1.70 ± 0.1 | 1.73 | 2.40g | PPh3 |
| 2b |
trans-Pt{CCC6H4CCSiMe3}2(PEt3)2 |
3.2 ± 1.3 × 10−5 | 3.5 × 10−5 | 2.1 ± 0.15 | 2.31 | 2.40g | CCSiMe3 |
| 3a |
trans-Pt{CCCCSiMe3}2(PPh3)2 |
5.2 ± 1.6 × 10−5 | 5.7 × 10−5 | 1.38 ± 0.1 | 1.37 | 1.53 | PPh3 |
| 3b |
trans-Pt{CCCCSiMe3}2(PEt3)2 |
4.9 ± 1.0 × 10−5 | 4.5 × 10−5 | 1.70 ± 0.17 | 1.72 | 1.53 | CCSiMe3 |
| 4a |
trans-Pt{CCC6H4But}2(PPh3)2 |
4.1 ± 0.6 × 10−5 | 1.98 × 10−5 | 1.46 ± 0.21 | 1.05/1.40f nm | PPh3 | |
| 4b |
trans-Pt{CCC6H4But}2(PEt3)2 |
No peak | 2.46 × 10−8 | — | 2.23 | — | |
The model junction constructed from 1a contacted via the trimethylsilylethynyl moieties (Fig. 2) gave excellent agreement with the experimental results at EF − EDFTF = −0.4 eV (Fig. 2). The tail of the molecular LUMO states aligns with this Fermi level (Fig. 2), indicating a charge transport mechanism involving tunnelling though the LUMO. A Mülliken Population Analysis of the frontier orbitals of 1a and 1b indicates that the energetically low-lying Pt orbitals make only a small contribution to the HOMO (1a, ε −4.53 eV, 7% Pt; 1b, ε −4.62 eV, 8% Pt) and LUMO (1a, ε −0.94 eV, 8% Pt; 1b, ε −0.95 eV, 8% Pt); instead these orbitals are largely diethynylbenzene in character (Fig. 4). Whilst the LUMO of 1b is rather well separated from another combination of the ligand π* orbitals which forms LUMO+1 (ε −0.72 eV), for 1a the LUMO+1 lies only some 0.02 eV higher in energy (at this level of theory) and is delocalised over the Ph3P–Pt–PPh3 fragment (Pt 14%, PPh3, 80%) (Fig. 4), and contributions from the PPh3 ligands also dominate the next 14 unoccupied orbitals.
 | ||
| Fig. 4 Plots of selected frontier orbitals of 1a (a, LUMO+1; b, LUMO; c, HOMO) and 1b (d, LUMO+1; e, LUMO; f, HOMO). In this and all other plots, iso-surfaces are shown at ±0.03 (e per bohr3)1/2. | ||
Schull and colleagues have previously reported studies of self-assembled films of the platinum complexes trans-Pt(CCC6H4SAc)2L2 (L = PCy3, PBu3, PPh3, P(OEt)3, P(OPh)3) within crossed-wire molecular junctions.47 Despite the modest (0.08 eV) variation in the HOMO–LUMO gap as a function of the supporting ligand, L, the I–V characteristics of these film-based junctions were also identical within experimental error. On the basis of these conductance results from both crossed-wire thin-films of Schull and those from I(s) single-molecule based junctions of 1a and 1b described here, it could initially be concluded that the supporting ligands L play no significant role in the electrical characteristics of the junctions formed from trans-Pt(CCR)2L2 complexes.
However, a more complex picture emerged when the study was extended to the complexes 2a,b (Chart 1) of similar structure and comparable molecular length (Table 1). Solubilizing alkyloxy groups, such as those present in 1a and 1b, play little role in tuning the conductance behavior of all-organic oligo(phenylene ethynylene)-based molecular wires,102 yet the conductance histogram of triphenylphosphine-supported 2a revealed a much less pronounced peak, at significantly higher conductance ((7.9 ± 1.1) × 10−5G0 or 6.1 ± 0.85 nS) and a shorter break-off distance (1.70 ± 0.1 nm), than found for 1a and 1b (Table 1 and Fig. 5). However, the experimentally determined conductance of the triethylphosphine complex 2b forms a more well-defined peak in the histogram ((3.2 ± 1.3) × 10−5G0 or 2.5 ± 1.0 nS) which is entirely in line with the values expected from 1a and 1b, and with better agreement between the experimental break-off distance (2.1 ± 0.15 nm) and the estimated Si⋯Si distance (2.40 nm) (Table 1 and Fig. 5). Computationally derived model junctions constructed for 2a (3.1 × 10−5G0 or 2.4 nS) (Table S1 and Fig. S9† (Model C)) and 2b (3.5 × 10−5G0 or 2.7 nS) (Table 1 and Fig. 2) with contact through the trimethylsilylethynyl moieties gave computed conductance values close to those expected, causing further consideration of the experimental result for 2a.
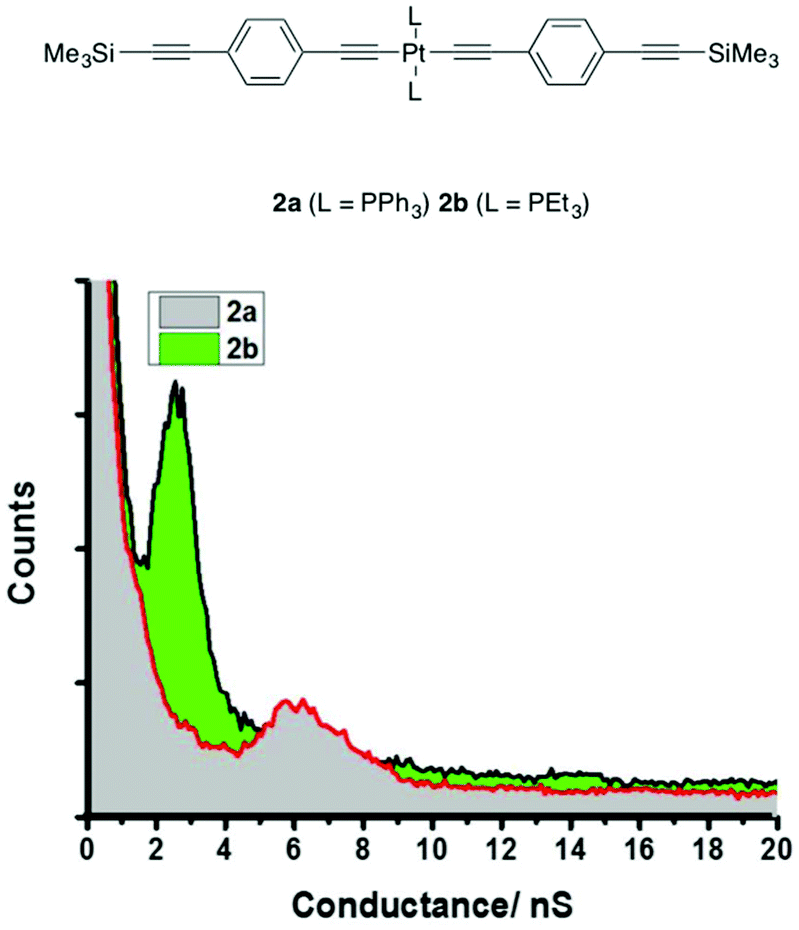 | ||
| Fig. 5 I(s) conductance histograms of 2a and 2b constructed from 500 traces. | ||
As noted above, the trimethylsilylethynyl molecule-gold contact has been studied in a number of contexts,91–96 and is well described in terms of physisorption of the methyl groups, augmented by a degree of charge transfer when residing near to surface defects.14,67 It is only these latter situations that give rise to CCSiMe3-contacted junctions of sufficient conductance to be measured. However, the potential for molecules to bind in a number of distinct configurations within a molecular junction, each offering a distinct contact and hence conductance value, has been recognized. As phenyl rings are known to bind at defect sites to give rise to molecular junctions,64,103 and the PPh3 ligands make a significant contribution to the low-lying unoccupied orbitals of 1a, a second model junction was explored in which a molecule of 2a is contacted by the STM tip at one of the ostensibly ancillary PPh3 ligands (Fig. 2 and Table 1). The calculated conductance of this alternative contact geometry (8.0 × 10−5G0) is in excellent agreement with the experimental value ((7.9 ± 1.1) × 10−5G0 or 6.1 ± 0.85 nS). Moreover, a calculated break-off distance (1.73 nm) in agreement with experiment (1.70 ± 0.1 nm) was obtained (Table 1). A further model in which the junction is formed by contact across both trans-disposed PPh3 ligand sets (Table S1 and Fig. S9,† Model B) gave a significantly higher calculated conductance (3.44 × 10−4G0 or 26.6 nS) and a much shorter break-off distance (1.05 nm) than the experimental values (see also discussion below).
It is therefore quite probable that the peak observed in the conductance histogram of 2a (Fig. 5) is derived from adventitious contact of the STM tip to one of the bulky PPh3 ligands rather than to the ‘designated’ CCSiMe3 contact. The flexible hexyloxy chains, initially introduced to improve the solubility of compounds 1a and 1b, therefore also appear to play a role in preventing the approach of the STM tip to the PPh3 ligand, effectively ‘insulating’ the PPh3 ligands from contacting to the electrodes. Similarly, the absence of any significant electron density on the alkyl chains of the PEt3 ligands in the frontier orbitals of 1b and 2b ensures more conductive molecular junctions are formed by contacts between the electrodes and the trimethylsilylethynyl groups. Thus, for the identically-contacted compounds 1a, 1b, and 2b the junction conductances are identical (Table 1), whilst the higher values measured for 2a can be attributed to the binding of the molecule in the junction through one of the PPh3 ligands (Fig. 2). Interestingly, molecular junctions formed with the bis(platinum) octatetrayndiyl complex [Pt(SAc){P(p-tol)3}2]2(μ-CCCCCCCC) (p-tol = 4-MeC6H4-) by the mechanically controlled break junction (MCBJ) method were observed to be rather unstable, with broadening of peaks in the dI/dV plots ascribed to various instabilities in the junction including structural fluctuations.49 Given the steric encumbrance around the thiolate group in this complex, adventitious contact through the tolyl moieties of the supporting phosphine ligands may now be suggested as a contributing factor to these junction instabilities and peak broadening.
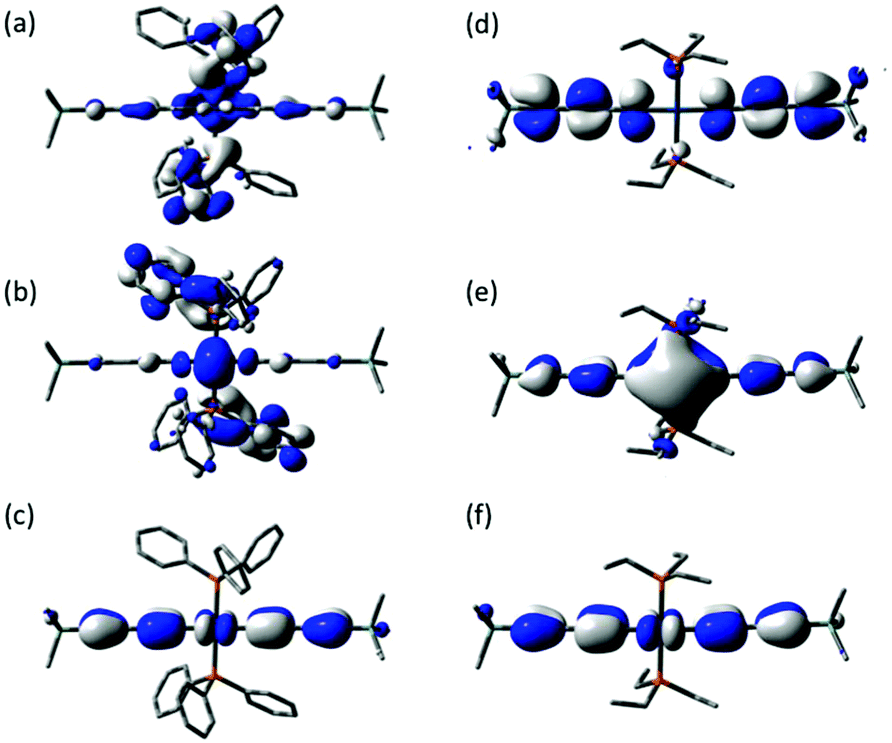
The frontier orbitals of 2a and 2b were analyzed (Fig. 6) in part to explore if such junction geometries might be predicted from the molecular electronic structure, which, in turn, might aid future molecular design concepts. Whilst the HOMOs are also significantly diethynylbenzene in character, with a small contribution from the Pt atom, (2a HOMO Pt 8%; 2b HOMO 11%), the LUMO of 2a is now found to be largely PPh3 in character (76%) and somewhat separated from the other frontier orbitals (ΔE HOMO–LUMO 3.92 eV; ΔE LUMO–(LUMO+1) 0.09 eV). The LUMO and LUMO+1 of 2b are both delocalized over the diethynylbenzene and Pt centre. Although the conductance channel cannot be predicted from simple inspection of MOs, the presence of the low-lying phosphine π* orbitals is consistent with the results from the experimental and model junctions.
 | ||
| Fig. 6 Plots of selected frontier orbitals of 2a (a, LUMO+1; b, LUMO; c, HOMO) and 2b (d, LUMO+1; e, LUMO; f, HOMO). | ||
To explore this concept further, the electronic structures of the bis(diynyl) complexes trans-Pt(CCCCSiMe3)2(PPh3)2 (3a) and trans-Pt(CCCCSiMe3)2(PEt3)2 (3b) were also compared and the compounds examined in both experimental and computational molecular junctions. The HOMO of 3a (−5.04 eV) and 3b (−5.17 eV) are of similar energy and almost identically composed (Fig. 7). However, again the exchange of PPh3 for PEt3 has a significant influence on the energy and composition of the unoccupied frontier molecular orbitals. In the case of 3a the PPh3 ligands contribute almost exclusively to the LUMO (−1.09 eV) and the next 12 unoccupied orbitals. The LUMO+13 of 3a (+0.16 eV), whilst featuring only a small contribution from the Pt center (2%), is the lowest lying orbital to be largely comprised of contributions from atoms within the diynyl ligands. In contrast, the LUMO of 3b (−0.97 eV), which is well removed from the other frontier orbitals, is delocalized over the 11-atom Si–C4–Pt–C4–Si chain.
 | ||
| Fig. 7 Plots of selected frontier orbitals of 3a (a, LUMO+1; b, LUMO; c, HOMO) and 3b (d, LUMO+1; e, LUMO; f, HOMO). | ||
The single-molecule conductance measurements initially belie the influence of these differences in electronic structure on the characteristics of the junction, with both 3a ((5.2 ± 1.6) × 10−5G0 or 4.0 ± 1.2 nS) and 3b ((4.9 ± 1.0) × 10−5G0 or 3.8 ± 0.8 nS) giving rise to conductance histograms with peaks at similar conductance values (Fig. 8). However, the experimentally determined break-off distance is somewhat shorter for the PPh3 ligated complex 3a (1.38 ± 0.10 nm) than PEt3 substituted 3b (1.70 ± 0.17 nm), which compares with the Si⋯Si distance of 1.53 nm in each case (Table 1). From the model junction with two trimethylsilylethynyl contacts (Fig. S9,† Model C), a calculated conductance value of 4.23 × 10−5G0 or 3.28 nS was obtained from 3a (Fig. S10†), which although in fairly good agreement with the experimental value, gave an estimated break-off distance of 1.72 nm, considerably longer than the experimental value.
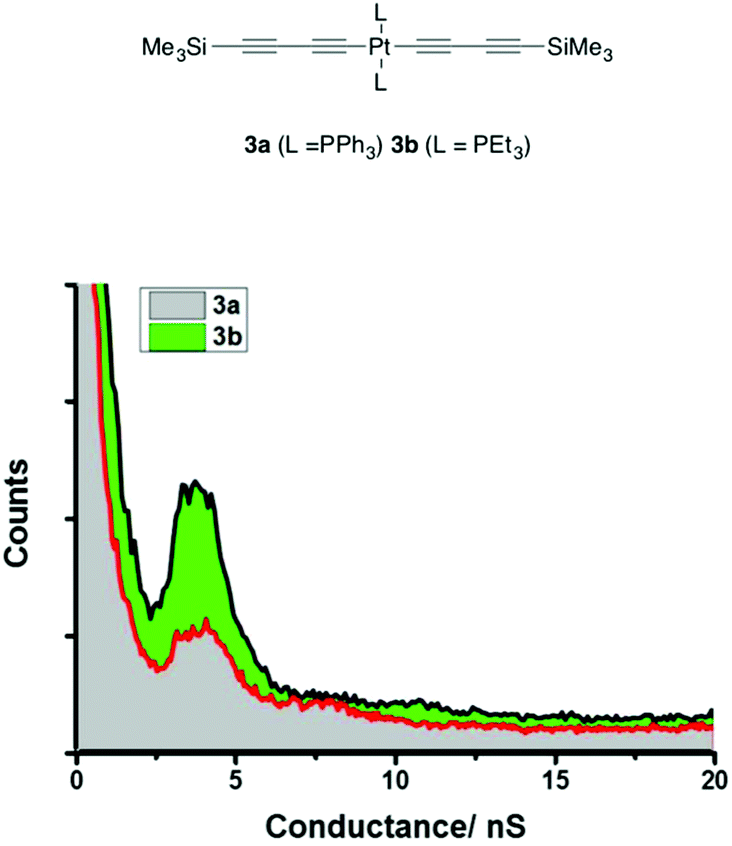 | ||
| Fig. 8 I(s) conductance histograms of 3a and 3b constructed from 500 traces. | ||
As with 2a, a model junction of 3a with two triphenylphosphine contacts (Fig. S9,† Model B) gave a calculated conductance value of 5.66 × 10−4G0 or 7.22 nS (Fig. S10 and Table S1†) and an estimated break-off distance of 1.03 nm, in poor agreement with experiment (G = 5.2 ± 1.6 × 10−5G0 or 4.0 ± 1.2 nS; Z* = 1.38 ± 0.1 nm). However, a model junction in which the STM tip is allowed to contact one of the PPh3 ligands in 3a (Fig. 2 and Fig. S9,† Model A) gave better overall agreement with a calculated conductance of 5.70 × 10−5G0 or 4.41 nS and break-off distance of 1.37 nm. The model junction constructed from trimethylsilylethynyl-contacted 3b (Fig. 2) gave good agreement between the experimental ((4.9 ± 1.0) × 10−5G0 or 3.8 ± 0.8 nS; break-off distance 1.70 ± 0.17 nm) and calculated (4.50 × 10−5G0 or 3.49 nS; break-off distance 1.72 nm).
To conclusively demonstrate the significance of the PPh3 contacts in these Pt-complex based molecular junctions, the model compounds trans-Pt(CCC6H4But)2(PPh3)2 (4a) and trans-Pt(CCC6H4But)2(PEt3)2 (4b) were studied (Fig. 9 and Table 1). The alkynyl ligands were chosen to model the electronic effects of the alkynyl ligands in 1a,b–3a,b, with the tert-butyl substituent (tBu) introduced to prevent adventitious junction formation to that phenyl ring.103 The I(s) studies of 4a revealed a peak in the conductance histogram ((4.1 ± 0.6) × 10−5G0 or 3.17 ± 0.46 nS). The experimental break-off distance (1.46 ± 0.21 nm) compares poorly with the estimated separation of the quaternary carbons of the tBu groups of 1.81 nm, but is much more consistent with the dimensions across the aryl rings of the phosphine ligands (ca. 1.4 nm). Three computational model junctions were constructed from 4a, contacted through either a tert-butyl group and one PPh3 ligand (Fig. S9,† Model A), both PPh3 ligands (Fig. 2 and Fig. S9,† Model B) or both the tert-butyl moieties (Fig. S9,† Model C) were constructed (Table S1†). Only the bis(PPh3)-contacted junction gave conductance value and break-off distance consistent with the experimental data (Fig. S9–S11†). In contrast, I(s) studies of 4b bearing the trialkyl phosphine PEt3 revealed no traces containing the current plateaus characteristic of single-molecule junction formation (Fig. S8†).
 | ||
| Fig. 9 I(s) conductance histograms of 4a and 4b constructed from 500 traces. | ||
The binding energies of 3a, chosen as a representative example of the compounds in the series, in a phosphine-contacted model junction were calculated over a range of electrode separations to provide further support for the hypothesis of competing contacts to different regions of the molecule (Fig. S12 and Table S2†). The trimethylsilylethynyl-gold binding energy has been estimated to fall between −0.40 and −0.74 eV over a range of gold- surface features.67 As the phosphine-contacted junction is evolved to simulate the pulling of the junction, the binding energy naturally decreases from −3.46 eV to −0.12 eV (Table S2†). It is therefore likely that I(s) junctions formed as the STM tip approaches and withdraws from the surface involve at least one PPh3-based contact. Although bis(PPh3)-contacted junctions (Fig. S9,† Model B), are energetically favorable at the shortest electrode separations (Table S2†), the formation of these junctions are likely geometrically limited by the closest tip-substrate approach distance in the I(s) measurements (>1 nm) and the dimensions of the prolate-shaped molecules (Fig. S11†).
Finally, the conductance behavior of these platinum complexes in comparison with closely related organic compounds measured under similar conditions is deserving of comment. The electrical properties and conductance behavior of oligo(phenylene ethynylene)102,104–108 and oligo(yne) derivatives14,69,109,110 have been explored extensively. The oligo(phenylene ethynylene) compound Me3SiCCC6H4CCC6H4CCC6H4CCSiMe3 (5) has an estimated Si⋯Si distance of 2.69 nm, and gives rise to a conductance value of (2.75 ± 0.56) × 10−5G0 or 2.13 ± 0.43 nS.96 The trimethylsilylethynyl-contacted platinum complexes 1a, bearing the insulating hexyloxy side chains, 1b and 2b offer comparable molecular lengths (2.39–2.40 nm) and similar conductances (Table 1). Clearly, the platinum center cannot be considered an insulating fragment per se, but in the present series is comparable in effect to the central phenylene ring in Me3SiCCC6H4CCC6H4CCC6H4CCSiMe3 (5).
For the trimethylsilylethynyl-contacted bis(diynyl) complex 3b ((4.9 ± 1.0) × 10−5G0 or 3.8 ± 0.8 nS, Si⋯Si distance 1.53 nm) the observed conductance of the molecular junction may be compared with those reported earlier for the organic oligoynes Me3SiCCCCSiMe3 (2.01 × 10−5G0 or 1.56 nS, Si⋯Si distance 0.76 nm), Me3SiCCCCCCSiMe3 (1.63 × 10−5G0 or 1.26 nS, Si⋯Si distance 1.06 nm), Me3SiCCCCCCCCSiMe3 (1.42 × 10−5G0 or 1.10 nS, Si⋯Si distance 1.33 nm) and Me3SiCCCCCCCCCCSiMe3 (0.90 × 10−5G0 or 0.70 nS, Si⋯Si distance 1.60 nm) in the same solvent.14 The relative values of conductance obtained from 3b and the octatetrayne indicate that introduction of the trans-Pt(PEt3)2 fragment within the oligoyne chain gives rise to molecules of greater conductance than those prepared by homo-coupling through a carbon–carbon sigma bond, or insertion of another alkyne –CC– moiety. We can, therefore, conclude that although the platinum center makes little contribution to the LUMO, there is no detrimental effect on the conductance of these molecules with LUMO-based conduction channels. Similar concepts have been proposed to account for the high conductance of Cr3-metal strings despite the absence of extensively delocalized frontier orbitals.54,55
Conclusion
The d8 square-planar platinum complexes 1a ((3.1 ± 0.9) × 10−5G0), 1b ((3.2 ± 0.8) × 10−5G0), 2b ((3.2 ± 1.3) × 10−5G0) and 3b ((4.9 ± 1.0) × 10−5G0) form molecular junctions in I(s) experiments with conductance commensurate with similarly structured all-organic oligo(phenylene ethynylene) and oligoyne compounds. The PPh3 supporting ligands in the series 2a–4a provide an alternative contact point in the molecular junctions, although the introduction of solubilising hexyloxy side chains to the diethynylbenzene ligands in 1a hinders this contact and effectively insulates these alternative contact points. Thus, despite the wide-spread use of PPh3 and other aryl-phosphines as ancillary ligands in organometallic chemistry, these moieties can clearly have unintended consequences for single-molecule conductance measurements arising from adventitious contacts and formation of un-anticipated molecular junctions. The hexyloxy groups in 1a serve to effectively insulate these alternate contacts. Otherwise, the use of trialkylphosphines, such as PEt3 here, is effective in both maintaining sufficient compound solubility and preventing adventitious junction formation through the ancillary ligands. It is also important to note the simplicity with which trans-bis(alkynyl)platinum(II) complexes can be synthesized. These results provide a considerable body of information concerning the design of organometallic complexes for use in molecular electronics.Acknowledgements
C. J. L. and O. A. A. acknowledge financial support from the Ministry of Higher Education and Scientific Research of Iraq. C. J. L. and M. R. B. acknowledges funding from the EU through the FP7 ITN MOLESCO (project number 212942). M. R. B. thanks the EPSRC for funding (grant EP/K039423/1). P. J. L. holds an ARC Future Fellowship (FT120100073) and gratefully acknowledges funding for this work from the ARC (DP140100855, LE150100148). S. B. holds an International Postgraduate Research Scholarship and gratefully acknowledges support from the University of Western Australia. R. J. N. and S. J. H. thank EPSRC for funding (grant EP/H035184/1 and EP/K007785/1). M. S. I., N. J. L. and T. A. acknowledge the Leverhulme Trust (RPG 2012-754) for funding. M. S. I. is supported by a Marie Skłodowska Curie Global Fellowship (MOLCLICK: 657247) within the Horizon 2020 Programme.Notes and references
- R. J. Nichols, W. Haiss, S. J. Higgins, E. Leary, S. Martín and D. Bethell, Phys. Chem. Chem. Phys., 2010, 12, 2801–2815 RSC.
- R. M. Metzger, Chem. Rev., 2015, 115, 5056–5115 CrossRef CAS PubMed.
- S. G. Lemay, S. Kang, K. Mathwig and P. S. Singh, Acc. Chem. Res., 2013, 46, 369–377 CrossRef CAS PubMed.
- C. Li, A. Mishchenko and T. Wandlowski, in Unimolecular and Supramolecular Electronics II, Springer, Berlin, Heidelberg, 2011, vol. 313, pp. 121–188 Search PubMed.
- D. Xiang, X. Wang, C. Jia, T. Lee and X. Guo, Chem. Rev., 2016, 116, 4318–4440 CrossRef CAS PubMed.
- L. Sun, Y. A. Diaz-Fernandez, T. A. Gschneidtner, F. Westerlund, S. Lara-Avila and K. Moth-Poulsen, Chem. Soc. Rev., 2014, 43, 7378–7411 RSC.
- W. Sheng, Z. Y. Li, Z. Y. Ning, Z. H. Zhang, Z. Q. Yang and H. Guo, J. Chem. Phys., 2009, 131, 244712 CrossRef CAS PubMed.
- W. Haiss, H. Van Zalinge, D. Bethell, J. Ulstrup, D. J. Schiffrin and R. J. Nichols, Faraday Discuss., 2006, 131, 253–264 RSC.
- C. Li, I. Pobelov, T. Wandlowski, A. Bagrets, A. Arnold and F. Evers, J. Am. Chem. Soc., 2008, 130, 318–326 CrossRef CAS PubMed.
- V. Fatemi, M. Kamenetska, J. B. Neaton and L. Venkataraman, Nano Lett., 2011, 11, 1988–1992 CrossRef CAS PubMed.
- B. Capozzi, J. Xia, O. Adak, E. J. Dell, Z. F. Liu, J. C. Taylor, J. B. Neaton, L. M. Campos and L. Venkataraman, Nat. Nanotechnol., 2015, 10, 522–527 CrossRef CAS PubMed.
- M. Kotiuga, P. Darancet, C. R. Arroyo, L. Venkataraman and J. B. Neaton, Nano Lett., 2015, 15, 4498–4503 CrossRef CAS PubMed.
- B. Choi, B. Capozzi, S. Ahn, A. Turkiewicz, G. Lovat, C. Nuckolls, M. L. Steigerwald, L. Venkataraman and X. Roy, Chem. Sci., 2016, 7, 2701–2705 RSC.
- D. C. Milan, O. A. Al-Owaedi, M. C. Oerthel, S. Marqués-González, R. J. Brooke, M. R. Bryce, P. Cea, J. Ferrer, S. J. Higgins, C. J. Lambert, P. J. Low, D. Z. Manrique, S. Martín, R. J. Nichols, W. Schwarzacher and V. M. García-Suárez, J. Phys. Chem. C, 2016, 120, 15666–15674 CAS.
- N. J. Kay, S. J. Higgins, J. O. Jeppesen, E. Leary, J. Lycoops, J. Ulstrup and R. J. Nichols, J. Am. Chem. Soc., 2012, 134, 16817–16826 CrossRef CAS PubMed.
- H. M. Osorio, S. Catarelli, P. Cea, J. B. G. Gluyas, F. Hartl, S. J. Higgins, E. Leary, P. J. Low, S. Martín, R. J. Nichols, J. Tory, J. Ulstrup, A. Vezzoli, D. C. Milan and Q. Zeng, J. Am. Chem. Soc., 2015, 137, 14319–14328 CrossRef CAS PubMed.
- A. Vezzoli, I. Grace, C. Brooke, K. Wang, C. J. Lambert, B. Xu, R. J. Nichols and S. J. Higgins, Nanoscale, 2015, 7, 18949–18955 RSC.
- C. Huang, A. V. Rudnev, W. Hong and T. Wandlowski, Chem. Soc. Rev., 2015, 44, 889–901 RSC.
- M. Baghernejad, X. Zhao, K. Baruël Ørnsø, M. Füeg, P. Moreno-García, A. V. Rudnev, V. Kaliginedi, S. Vesztergom, C. Huang, W. Hong, P. Broekmann, T. Wandlowski, K. S. Thygesen and M. R. Bryce, J. Am. Chem. Soc., 2014, 136, 17922–17925 CrossRef CAS PubMed.
- Z. Li, H. Li, S. Chen, T. Froehlich, C. Yi, C. Schönenberger, M. Calame, S. Decurtins, S. X. Liu and E. Borguet, J. Am. Chem. Soc., 2014, 136, 8867–8870 CrossRef CAS PubMed.
- P. J. Low, Dalton Trans., 2005, 2821–2824 RSC.
- S. Rigaut, Dalton Trans., 2013, 42, 15859–15863 RSC.
- T. Ren, Organometallics, 2005, 24, 4854–4870 CrossRef CAS.
- J. Ponce, C. R. Arroyo, S. Tatay, R. Frisenda, P. Gaviña, D. Aravena, E. Ruiz, H. S. J. van der Zant and E. Coronado, J. Am. Chem. Soc., 2014, 136, 8314–8322 CrossRef CAS PubMed.
- E. A. Osorio, K. Moth-Poulsen, H. S. J. van der Zant, J. Paaske, P. Hedegård, K. Flensberg, J. Bendix and T. Bjørnholm, Nano Lett., 2010, 10, 105–110 CrossRef CAS PubMed.
- F. Schwarz, G. Kastlunger, F. Lissel, C. Egler-Lucas, S. N. Semenov, K. Venkatesan, H. Berke, R. Stadler and E. Lörtscher, Nat. Nanotechnol., 2016, 11, 170–176 CrossRef CAS PubMed.
- S. A. Hua, M. C. Cheng, C. H. Chen and S. M. Peng, Chem. Ber., 2015, 2015, 2510–2523 CAS.
- X. Roy, C. L. Schenck, S. Ahn, R. A. Lalancette, L. Venkataraman, C. Nuckolls and M. L. Steigerwald, Angew. Chem., Int. Ed., 2012, 51, 12473–12476 CrossRef CAS PubMed.
- E. Leary, H. Van Zalinge, S. J. Higgins, R. J. Nichols, F. Fabrizi De Biani, P. Leoni, L. Marchetti and P. Zanello, Phys. Chem. Chem. Phys., 2009, 11, 5198–5202 RSC.
- T. Tanaka and A. Osuka, Chem. Soc. Rev., 2015, 44, 943–969 RSC.
- M. Jurow, A. E. Schuckman, J. D. Batteas and C. M. Drain, Coord. Chem. Rev., 2010, 254, 2297–2310 CrossRef CAS PubMed.
- S. A. Getty, C. Engtrakul, L. Wang, R. Liu, S. H. Ke, H. U. Baranger, W. Yang, M. S. Fuhrer and L. R. Sita, Phys. Rev. B: Condens. Matter, 2005, 71, 241401 CrossRef.
- Y. Y. Sun, Z. L. Peng, R. Hou, J.-H. Liang, J. F. Zheng, X. Y. Zhou, X. S. Zhou, S. Jin, Z. J. Niu and B.-W. Mao, Phys. Chem. Chem. Phys., 2014, 16, 2260–2267 RSC.
- T. Albrecht, A. Guckian, A. M. Kuznetsov, J. G. Vos and J. Ulstrup, J. Am. Chem. Soc., 2006, 128, 17132–17138 CrossRef CAS PubMed.
- T. Albrecht, K. Moth-Poulsen, J. B. Christensen, A. Guckian, T. Bjørnholm, J. G. Vos and J. Ulstrup, Faraday Discuss., 2006, 131, 265–279 RSC.
- T. Albrecht, A. Guckian, J. Ulstrup and J. G. Vos, Nano Lett., 2005, 5, 1451–1455 CrossRef CAS PubMed.
- R. Sakamoto, S. Katagiri, H. Maeda and H. Nishihara, Coord. Chem. Rev., 2013, 257, 1493–1506 CrossRef CAS.
- B. Kim, J. M. Beebe, C. Olivier, S. Rigaut, D. Touchard, J. G. Kushmerick, X. Y. Zhu and C. D. Frisbie, J. Phys. Chem. C, 2007, 111, 7521–7526 CAS.
- L. Luo, A. Benameur, P. Brignou, S. H. Choi, S. Rigaut and C. D. Frisbie, J. Phys. Chem. C, 2011, 115, 19955–19961 CAS.
- A. Mulas, Y. M. Hervault, L. Norel, S. Rigaut and C. Lagrost, ChemElectroChem, 2015, 2, 1799–1805 CrossRef CAS.
- A. Mulas, Y. M. Hervault, X. He, E. Di Piazza, L. Norel, S. Rigaut and C. Lagrost, Langmuir, 2015, 31, 7138–7147 CrossRef CAS PubMed.
- F. Meng, Y. M. Hervault, Q. Shao, B. Hu, L. Norel, S. Rigaut and X. Chen, Nat. Commun., 2014, 5, 3023 Search PubMed.
- F. Meng, Y. M. Hervault, L. Norel, K. Costuas, C. Van Dyck, V. Geskin, J. Cornil, H. H. Hng, S. Rigaut and X. Chen, Chem. Sci., 2012, 3, 3113–3118 RSC.
- F. Lissel, F. Schwarz, O. Blacque, H. Riel, E. Lörtscher, K. Venkatesan and H. Berke, J. Am. Chem. Soc., 2014, 136, 14560–14569 CrossRef CAS PubMed.
- F. Schwarz, G. Kastlunger, F. Lissel, H. Riel, K. Venkatesan, H. Berke, R. Stadler and E. Lörtscher, Nano Lett., 2014, 14, 5932–5940 CrossRef CAS PubMed.
- K. Liu, X. Wang and F. Wang, ACS Nano, 2008, 2, 2315–2323 CrossRef CAS PubMed.
- T. L. Schull, J. G. Kushmerick, C. H. Patterson, C. George, M. H. Moore, S. K. Pollack and R. Shashidhar, J. Am. Chem. Soc., 2003, 125, 3202–3203 CrossRef CAS PubMed.
- M. Mayor, C. Von Hänisch, H. B. Weber, J. Reichert and D. Beckmann, Angew. Chem., Int. Ed., 2002, 41, 1183–1186 CrossRef CAS PubMed.
- S. Ballmann, W. Hieringer, D. Secker, Q. Zheng, J. A. Gladysz, A. Görling and H. B. Weber, ChemPhysChem, 2010, 11, 2256–2260 CrossRef CAS PubMed.
- G. Frapper and M. Kertesz, Inorg. Chem., 1993, 32, 732–740 CrossRef CAS.
- M. Parthey, K. B. Vincent, M. Renz, P. A. Schauer, D. S. Yufit, J. A. K. Howard, M. Kaupp and P. J. Low, Inorg. Chem., 2014, 53, 1544–1554 CrossRef CAS PubMed.
- S. Marqués-González, M. Parthey, D. S. Yufit, J. A. K. Howard, M. Kaupp and P. J. Low, Organometallics, 2014, 33, 4947–4963 CrossRef.
- P. J. West, M. P. Cifuentes, T. Schwich, M. D. Randles, J. P. Morrall, E. Kulasekera, S. Petrie, R. Stranger and M. G. Humphrey, Inorg. Chem., 2012, 51, 10495–10502 CrossRef CAS PubMed.
- V. P. Georgiev and J. E. McGrady, J. Am. Chem. Soc., 2011, 133, 12590–12599 CrossRef CAS PubMed.
- V. P. Georgiev, P. J. Mohan, D. DeBrincat and J. E. McGrady, Coord. Chem. Rev., 2013, 257, 290–298 CrossRef CAS.
- O. A. Al-Owaedi, D. C. Milan, M. C. Oerthel, S. Bock, D. S. Yufit, J. A. K. Howard, S. J. Higgins, R. J. Nichols, C. J. Lambert, M. R. Bryce and P. J. Low, Organometallics, 2016, 35, 2944–2954 CrossRef CAS.
- M. G. Reuter, T. Seideman and M. A. Ratner, J. Chem. Phys., 2011, 134, 154708 CrossRef PubMed.
- R. Frisenda, S. Tarkuç, E. Galán, M. L. Perrin, R. Eelkema, F. C. Grozema and H. S. J. van der Zant, Beilstein J. Nanotechnol., 2015, 6, 1558–1567 CrossRef CAS PubMed.
- J. Hihath and N. Tao, Semicond. Sci. Technol., 2014, 29, 054007 CrossRef.
- C. Jia and X. Guo, Chem. Soc. Rev., 2013, 42, 5642–5660 RSC.
- S. Y. Quek, M. Kamenetska, M. L. Steigerwald, H. J. Choi, S. G. Louie, M. S. Hybertsen, J. B. Neaton and L. Venkataraman, Nat. Nanotechnol., 2009, 4, 230–234 CrossRef CAS PubMed.
- T. A. Su, J. R. Widawsky, H. Li, R. S. Klausen, J. L. Leighton, M. L. Steigerwald, L. Venkataraman and C. Nuckolls, J. Am. Chem. Soc., 2013, 135, 18331–18334 CrossRef CAS PubMed.
- V. Kaliginedi, A. V. Rudnev, P. Moreno-García, M. Baghernejad, C. Huang, W. Hong and T. Wandlowski, Phys. Chem. Chem. Phys., 2014, 16, 23529–23539 RSC.
- W. Haiss, S. Martín, E. Leary, H. Van Zalinge, S. J. Higgins, L. Bouffier and R. J. Nichols, J. Phys. Chem. C, 2009, 113, 5823–5833 CAS.
- Z. L. Cheng, R. Skouta, H. Vazquez, J. R. Widawsky, S. Schneebeli, W. Chen, M. S. Hybertsen, R. Breslow and L. Venkataraman, Nat. Nanotechnol., 2011, 6, 353–357 CrossRef CAS PubMed.
- Y. Fu, S. Chen, A. Kuzume, A. Rudnev, C. Huang, V. Kaliginedi, M. Baghernejad, W. Hong, T. Wandlowski, S. Decurtins and S. X. Liu, Nat. Commun., 2015, 6, 6403 CrossRef CAS PubMed.
- R. R. Ferradás, S. Marqués-González, H. M. Osorio, J. Ferrer, P. Cea, D. C. Milan, A. Vezzoli, S. J. Higgins, R. J. Nichols, P. J. Low, V. M. García-Suárez and S. Martín, RSC Adv., 2016, 6, 75111–75121 RSC.
- J. S. Meisner, M. Kamenetska, M. Krikorian, M. L. Steigerwald, L. Venkataraman and C. Nuckolls, Nano Lett., 2011, 11, 1575–1579 CrossRef CAS PubMed.
- P. Moreno-García, M. Gulcur, D. Z. Manrique, T. Pope, W. Hong, V. Kaliginedi, C. Huang, A. S. Batsanov, M. R. Bryce, C. Lambert and T. Wandlowski, J. Am. Chem. Soc., 2013, 135, 12228–12240 CrossRef PubMed.
- L. Xiang, T. Hines, J. L. Palma, X. Lu, V. Mujica, M. A. Ratner, G. Zhou and N. Tao, J. Am. Chem. Soc., 2016, 138, 679–687 CrossRef CAS PubMed.
- S. T. Schneebeli, M. Kamenetska, Z. Cheng, R. Skouta, R. A. Friesner, L. Venkataraman and R. Breslow, J. Am. Chem. Soc., 2011, 133, 2136–2139 CrossRef CAS PubMed.
- C. Pan, C. Zhao, M. Takeuchi and K. Sugiyasu, Chem. – Asian J., 2015, 10, 1820–1835 CrossRef CAS PubMed.
- H. Masai, J. Terao and Y. Tsuji, Tetrahedron Lett., 2014, 55, 4035–4043 CrossRef CAS.
- J. Stahl, J. C. Bohling, E. B. Bauer, T. B. Peters, W. Mohr, J. M. Martín-Alvarez, F. Hampel and J. A. Gladysz, Angew. Chem., Int. Ed., 2002, 41, 1871–1876 CrossRef CAS.
- J. Stahl, W. Mohr, L. de Quadras, T. B. Peters, J. C. Bohling, J. M. Martín-Alvarez, G. R. Owen, F. Hampel and J. A. Gladysz, J. Am. Chem. Soc., 2007, 129, 8282–8295 CrossRef CAS PubMed.
- L. de Quadras, E. B. Bauer, J. Stahl, F. Zhuravlev, F. Hampel and J. A. Gladysz, New J. Chem., 2007, 31, 1594–1604 RSC.
- G. R. Owen, S. Gauthier, N. Weisbach, F. Hampel, N. Bhuvanesh and J. A. Gladysz, Dalton Trans., 2010, 39, 5260–5271 RSC.
- L. de Quadras, E. B. Bauer, W. Mohr, J. C. Bohling, T. B. Peters, J. M. Martín-Alvarez, F. Hampel and J. A. Gladysz, J. Am. Chem. Soc., 2007, 129, 8296–8309 CrossRef CAS PubMed.
- G. R. Owen, J. Stahl, F. Hampel and J. A. Gladysz, Organometallics, 2004, 23, 5889–5892 CrossRef CAS.
- M. C. Clough, T. Fiedler, N. Bhuvanesh and J. A. Gladysz, J. Organomet. Chem., 2016, 812, 34–42 CrossRef CAS.
- L. D. Movsisyan, M. Franz, F. Hampel, A. L. Thompson, R. R. Tykwinski and H. L. Anderson, J. Am. Chem. Soc., 2016, 138, 1366–1376 CrossRef CAS PubMed.
- L. D. Movsisyan, D. V. Kondratuk, M. Franz, A. L. Thompson, R. R. Tykwinski and H. L. Anderson, Org. Lett., 2012, 14, 3424–3426 CrossRef CAS PubMed.
- H. Sahnoune, Z. Baranová, N. Bhuvanesh, J. A. Gladysz and J.-F. Halet, Organometallics, 2013, 32, 6360–6367 CrossRef CAS.
- N. Weisbach, Z. Baranová, S. Gauthier, J. H. Reibenspies and J. A. Gladysz, Chem. Commun., 2012, 48, 7562–7564 RSC.
- J. Terao, T. Hosomi, H. Masai, W. Matsuda, S. Seki, T. Fujihara and Y. Tsuji, Chem. Lett., 2014, 43, 1289–1291 CrossRef CAS.
- J. Terao, K. Homma, Y. Konoshima, R. Imoto, H. Masai, W. Matsuda, S. Seki, T. Fujihara and Y. Tsuji, Chem. Commun., 2014, 50, 658–660 RSC.
- J. Terao, H. Masai, T. Fujihara and Y. Tsuji, Chem. Lett., 2012, 41, 652–653 CrossRef CAS.
- T. Hosomi, H. Masai, W. Matsuda, S. Seki, T. Fujihara, Y. Tsuji and J. Terao, Chem. Lett., 2016, 45, 931–933 CrossRef CAS.
- H. Masai, J. Terao, S. Seki, S. Nakashima, M. Kiguchi, K. Okoshi, T. Fujihara and Y. Tsuji, J. Am. Chem. Soc., 2014, 136, 1742–1745 CrossRef CAS PubMed.
- M. Kiguchi, S. Nakashima, T. Tada, S. Watanabe, S. Tsuda, Y. Tsuji and J. Terao, Small, 2012, 8, 726–730 CrossRef CAS PubMed.
- N. Katsonis, A. Marchenko, S. Taillemite, D. Fichou, G. Chouraqui, C. Aubert and M. Malacria, Chem. – Eur. J., 2003, 9, 2574–2581 CrossRef CAS PubMed.
- N. Katsonis, A. Marchenko, D. Fichou and N. Barrett, Surf. Sci., 2008, 602, 9–16 CrossRef CAS.
- A. Nion, N. Katsonis, A. Marchenko, C. Aubert and D. Fichou, New J. Chem., 2013, 37, 2261–2265 RSC.
- A. Marchenko, N. Katsonis, D. Fichou, C. Aubert and M. Malacria, J. Am. Chem. Soc., 2002, 124, 9998–9999 CrossRef CAS PubMed.
- G. Pera, S. Martín, L. M. Ballesteros, A. J. Hope, P. J. Low, R. J. Nichols and P. Cea, Chem. – Eur. J., 2010, 16, 13398–13405 CrossRef CAS PubMed.
- S. Marqués-González, D. S. Yufit, J. A. K. Howard, S. Martín, H. M. Osorio, V. M. García-Suárez, R. J. Nichols, S. J. Higgins, P. Cea and P. J. Low, Dalton Trans., 2013, 42, 338–341 RSC.
- D. Millar, L. Venkataraman and L. H. Doerrer, J. Phys. Chem. C, 2007, 111, 17635–17639 CAS.
- J. C. Bailar and H. Itatani, Inorg. Chem., 1965, 4, 1618–1620 CrossRef CAS.
- G. W. Parshall, Inorg. Synth., 1970, 12, 26–33 CAS.
- K. Sonogashira, T. Yatake, Y. Tohda, S. Takahashi and N. Hagihara, J. Chem. Soc., Chem. Commun., 1977, 291–292 RSC.
- W. Haiss, H. Van Zalinge, S. J. Higgins, D. Bethell, H. Höbenreich, D. J. Schiffrin and R. J. Nichols, J. Am. Chem. Soc., 2003, 125, 15294–15295 CrossRef CAS PubMed.
- R. Huber, M. T. González, S. Wu, M. Langer, S. Grunder, V. Horhoiu, M. Mayor, M. R. Bryce, C. Wang, R. Jitchati, C. Schönenberger and M. Calame, J. Am. Chem. Soc., 2008, 130, 1080–1084 CrossRef CAS PubMed.
- S. Martín, I. Grace, M. R. Bryce, C. Wang, R. Jitchati, A. S. Batsanov, S. J. Higgins, C. J. Lambert and R. J. Nichols, J. Am. Chem. Soc., 2010, 132, 9157–9164 CrossRef PubMed.
- J. M. Tour, Acc. Chem. Res., 2000, 33, 791–804 CrossRef CAS PubMed.
- X. Xiao, L. A. Nagahara, A. M. Rawlett and N. Tao, J. Am. Chem. Soc., 2005, 127, 9235–9240 CrossRef CAS PubMed.
- L. T. Cai, H. Skulason, J. G. Kushmerick, S. K. Pollack, J. Naciri, R. Shashidhar, D. L. Allara, T. E. Mallouk and T. S. Mayer, J. Phys. Chem. B, 2004, 108, 2827–2832 CrossRef CAS.
- Q. Lu, K. Liu, H. Zhang, Z. Du, X. Wang and F. Wang, ACS Nano, 2009, 3, 3861–3868 CrossRef CAS PubMed.
- V. Kaliginedi, P. Moreno-García, H. Valkenier, W. Hong, V. M. García-Suárez, P. Buiter, J. L. H. Otten, J. C. Hummelen, C. J. Lambert and T. Wandlowski, J. Am. Chem. Soc., 2012, 134, 5262–5275 CrossRef CAS PubMed.
- C. Wang, A. S. Batsanov, M. R. Bryce, S. Martín, R. J. Nichols, S. J. Higgins, V. M. García-Suárez and C. J. Lambert, J. Am. Chem. Soc., 2009, 131, 15647–15654 CrossRef CAS PubMed.
- S. Ballmann, W. Hieringer, R. Härtle, P. B. Coto, M. R. Bryce, A. Görling, M. Thoss and H. B. Weber, Phys. Status Solidi B, 2013, 250, 2452–2457 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic procedures and characterisation data. Details of computational analysis, descriptions of alternate junction geometries, plots showing calculated conductance as a function of Fermi energy for these different models and tabulated results. Plots of NMR spectra. Data from the single molecule studies is available from the University of Liverpool data catalogue (DOI: 10.17638/datacat.liverpool.ac.uk/212). See DOI: 10.1039/c7nr01829k |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2017 |