
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNanoparticle discrimination based on wavelength and lifetime-multiplexed cathodoluminescence microscopy
Mathijs W. H.
Garming
a,
I. Gerward C.
Weppelman
a,
Pascal
de Boer
b,
Felipe Perona
Martínez
c,
Romana
Schirhagl
 c,
Jacob P.
Hoogenboom
*a and
Robert J.
Moerland
*a
c,
Jacob P.
Hoogenboom
*a and
Robert J.
Moerland
*a
aDelft University of Technology, Lorentzweg 1, NL-2628CJ Delft, The Netherlands. E-mail: j.p.hoogenboom@tudelft.nl; r.j.moerland@tudelft.nl
bUniversity Medical Center Groningen, Department of Cell Biology, Antonius Deusinglaan 1, NL-9713AW Groningen, The Netherlands
cUniversity Medical Center Groningen, Department of Biomedical Engineering, Antonius Deusinglaan 1, NL-9713AW Groningen, The Netherlands
First published on 18th August 2017
Abstract
Nanomaterials can be identified in high-resolution electron microscopy images using spectrally-selective cathodoluminescence. Capabilities for multiplex detection can however be limited, e.g., due to spectral overlap or availability of filters. Also, the available photon flux may be limited due to degradation under electron irradiation. Here, we demonstrate single-pass cathodoluminescence-lifetime based discrimination of different nanoparticles, using a pulsed electron beam. We also show that cathodoluminescence lifetime is a robust parameter even when the nanoparticle cathodoluminescence intensity decays over an order of magnitude. We create lifetime maps, where the lifetime of the cathodoluminescence emission is correlated with the emission intensity and secondary-electron images. The consistency of lifetime-based discrimination is verified by also correlating the emission wavelength and the lifetime of nanoparticles. Our results show how cathodoluminescence lifetime provides an additional channel of information in electron microscopy.
Introduction
High-resolution identification of materials in electron microscopy images is a challenge throughout many areas of science. The detection of visible photons emitted resulting from electron irradiation, so-called cathodoluminescence (CL),1 provides a straightforward analytical alternative to the electron signals. Typically, spectrally selective CL detection is used to identify materials with different composition, especially in materials and geosciences.2–8 Recently, CL microscopy is gaining interest also in the biological, optical, and nano-sciences as electron beam excitation provides a natural solution to circumvent the diffraction limit of photon-excited fluorescence microscopy.9–15 In addition, compared to correlative fluorescence and electron microscopy (CLEM),16 detection of CL may alleviate the need to register images obtained on different microscopes, i.e., light- versus electron-based.17 A major challenge in CL microscopy, especially in the biological sciences, is the rapid degradation of CL signals, particularly from organic molecules, under electron irradiation.18 Cathodoluminescent nanoparticles (CNPs), for example phosphors composed of metal oxides13,19–21 and fluorescent nanodiamonds (FNDs),22 are an attractive alternative for labeling in biological sciences with size and thus resolution that can go down to a few nanometers.23–25 Multicolor experiments can be realized by employing various types of emitters, such as rare earth ions13 and FNDs with engineered defects to modify the emission spectrum.22 However, as in fluorescence microscopy, the analytical power of spectral discrimination is inherently limited due to overlap of emission bands and availability of filters.In fluorescence microscopy, lifetime imaging26,27 provides additional or otherwise unobtainable data, on, for example, the local (optical) environment28–30 or state of fluophores.31 Moreover, lifetime imaging can be used to differentiate between various emitters.26 With strong CL signals, the fluorescence lifetime can be obtained by fitting exponential curves to decay curves after excitation with continuous beams.32,33 For low signals, single photon counting can still accurately resolve lifetimes by determining, e.g., antibunching curves34 or photon arrival time histograms.35 The latter option can be used to minimize electron-induced damage to the sample by exciting the sample with short electron pulses, while also maintaining the native resolution of the Scanning EM (SEM).36,37
Here, we demonstrate the ability to discriminate between various CNPs in the time domain. We show that we can measure the emission intensity of CNPs while exciting them with a pulsed electron beam, which is generated by a commercial beam blanker.37 For each pixel in the intensity map we measure the intrinsic lifetime associated with the type of emitter at that particular location. To confirm our discrimination based on the time-domain data, we measure spectrally-selective CL intensity maps for comparison.
Results & discussion
We have modified a standard electron microscope to include a high-NA optical objective, which is part of an inverted optical microscope,14,38,39 and have added a pulsed electron-beam mode suitable for time-resolved imaging (see Fig. 1). With this setup, we obtained electron pulses with lengths down to 80–90 ps. We employ a Time-Correlated Single-Photon Counting (TCSPC) scheme (PicoQuant PicoHarp 300), which is often used in fluorescence microscopy to determine the fluorescence lifetime of fluophores. In that case, the TCSPC clock signal is typically issued by a pulsed laser. Here we drive the blanker with a pulse generator and use the same signal to synchronize with the TCSPC hardware.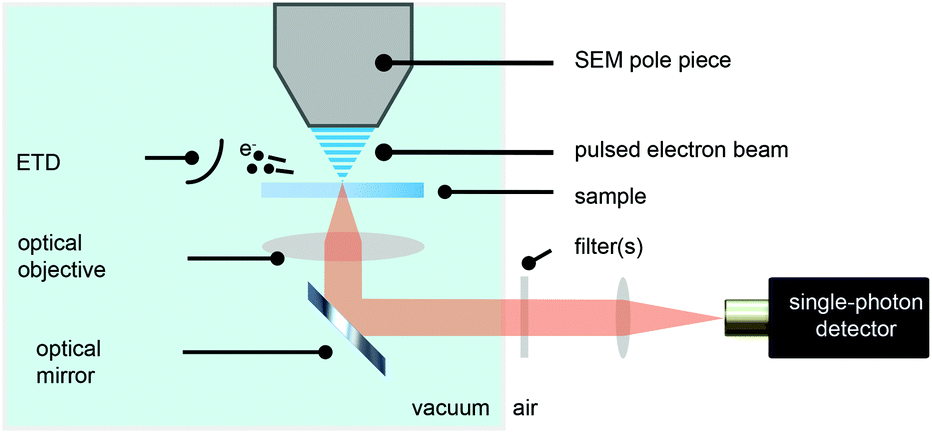 | ||
| Fig. 1 Schematic depiction of an integrated optical and electron microscope. A pulsed electron beam strikes the sample with a fixed frequency. An Everhart–Thornley detector (ETD) collects secondary electrons used for imaging with the SEM. Cathodoluminescence, induced by the energetic electrons, is collected with a 0.95 NA optical objective and imaged onto a single-photon counting detector. | ||
Samples with dispersed CNPs are obtained by spincoating from solution. In order to establish the distribution of lifetimes of each type of nanoparticle, we first prepare two samples, where each sample contains only one type of particle. Here, we use cerium-doped Lu3Al5O12 (LuAG:Ce) particles (Boston Applied Technology, Inc.) that exhibit a transition from the 5d level to the 4f ground state40 of Ce3+, and FNDs (Adamas) that exhibit a transition from the 2A state to the 2E state,41 associated with the display of CL by primarily the neutral NV0 center.42 Typical photon arrival time histograms are shown in Fig. 2a. We approximate the measured arrival times with a single exponential, which yields a typical decay time of the CNP. We collect the decay time of 28 FNDs and 46 LuAG:Ce CNPs and determine the distribution of lifetimes (Fig. 2b). The difference in lifetime distribution between the two particles is clearly visible; LuAG:Ce nanoparticles have a typical lifetime of about 50 ns, with a width of 14 ns (2σ). The FNDs on the other hand have a substantially shorter lifetime of typically 30 ns, with a width of 10 ns (2σ). Finally, in Fig. 2c, the measured emission spectra of both types of CNPs are shown.
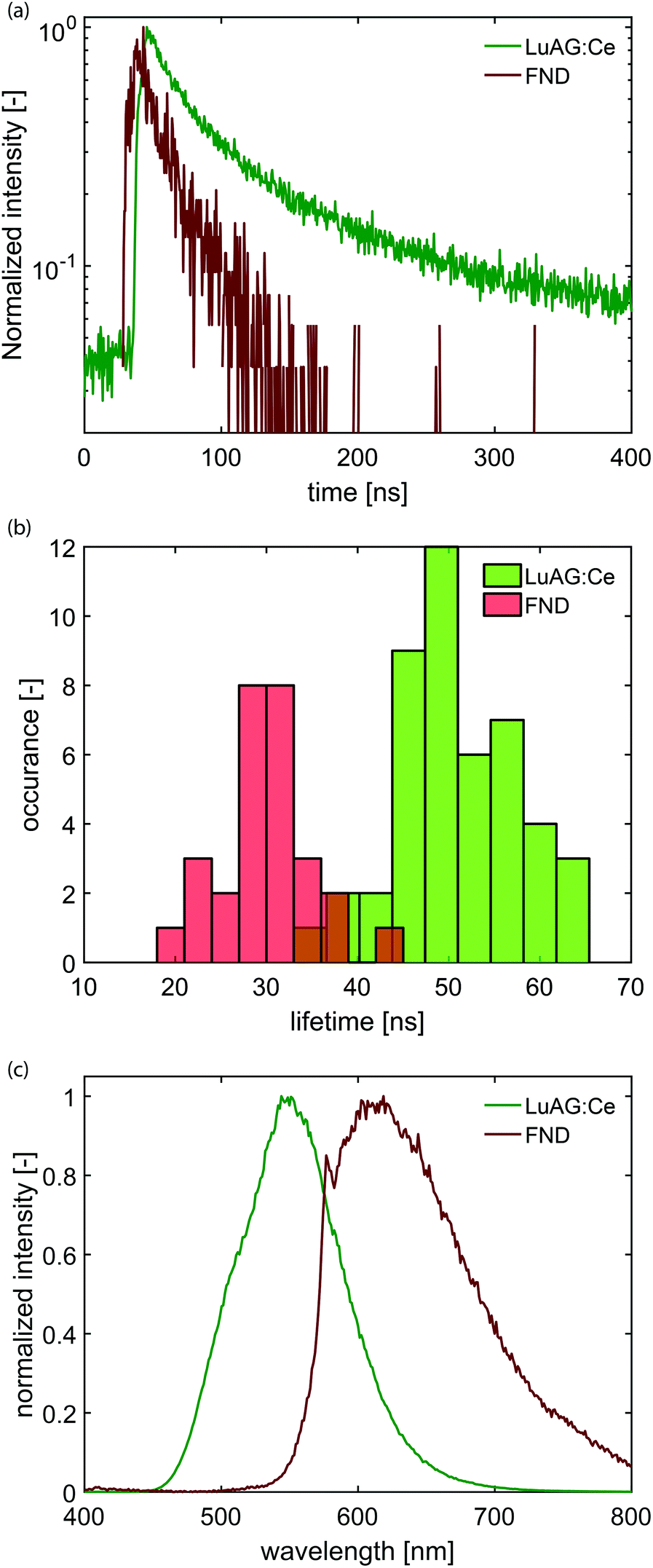 | ||
| Fig. 2 Emission properties of the two types of particles used here. (a) Typical photon arrival time histograms for Lu3Al5O12:Ce3+ (LuAG:Ce) particles (green) and fluorescent nanodiamonds (FNDs, red). (b) Histogram of lifetimes of various nanoparticles, as obtained by time-correlated single-photon counting. The average lifetime of FNDs is 30 ns with a width of 10 ns (2σ). The average lifetime of the LuAG:Ce particles is about 50 ns with a width of 14 ns (2σ). (c) Emission spectrum of LuAG:Ce (green) and FNDs (red). | ||
Stability of lifetime under e-beam irradiation
Next, we investigated the stability of the lifetime measurements over the irradiation time. In contrast to intensity, which changes over time during the irradiation with charged particles, lifetime is a relatively stable quantity. This is highlighted in Fig. 3a–d. Fig. 3a displays the measured lifetime of three FNDs over a time span of 10 minutes. The photon arrival histograms were determined every 5 seconds. The number of photons in every histogram is displayed in Fig. 3b over the same time span. Similar measurements for the LuAG:Ce particles are shown in Fig. 3c and d. Despite the fact that the CL intensity varies (likely due to bleaching and drift of the electron beam), the measured lifetime remains extremely robust. As a particularly extreme example, the lifetime of the FND displayed with a blue curve in Fig. 3a varies between a maximum of 27 ns and a minimum of 20 ns and has a mean of 25.0 ns ± 12% (3σ). This happens while the intensity drops by a factor of 29. Whereas bleaching of FNDs has been reported in literature,43 the exact mechanism is still subject of research. Potentially the creation or annihilation of surface charge traps,44 dynamics of impurities or charge carriers in the vicinity of the vacancy centers,45–48 or quenching of nitrogen-vacancy centers near the surface by a thin carbon contamination layer grown by e-beam-induced deposition play a role. This is subject to further investigation.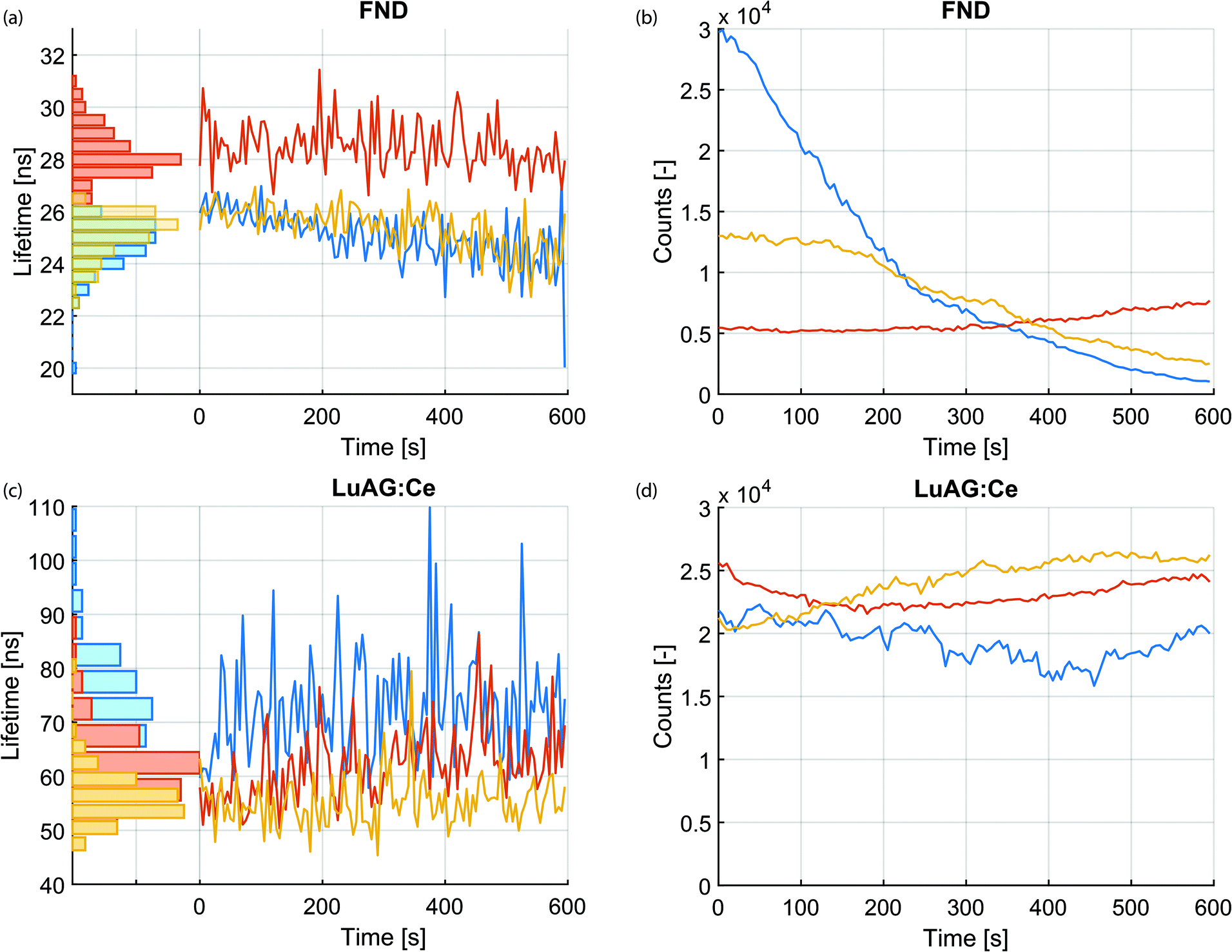 | ||
| Fig. 3 Stability of lifetime and intensity versus irradiation with the electron beam, for three particles of each type (LuAG:Ce and FND). Lifetime versus e-beam irradiation duration, determined every 5 seconds, of three FNDs and LuAG:Ce particles is shown in panel (a) and (c), respectively. Panels (b) and (d) show the corresponding CL intensity during the measurement. Despite (severe) differences in intensity, the lifetime of the nanoparticles is a robust parameter during the measurement. | ||
Cathodoluminescence lifetime maps
Subsequently, a sample is prepared that contains both types of CNPs, with approximately a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of the number of particles of each type. A secondary-electron (SE) image of a 6 × 6 μm region on the sample is displayed in Fig. 4a. We scan the same area again, but now with a pulsed electron beam at a step size of 100 nm and a dwell time of 0.5 seconds. For every pixel, the photon arrival histogram is determined, which is then fitted with a single exponential decay. Thus, we simultaneously measure the intensity and lifetime of the emitters on the sample. The intensity of both type of particles can differ greatly, which on a linear scale could render the less intense particles invisible. Therefore, we show the CL intensity map in Fig. 4b on a logarithmic scale. Note that this leads to an apparently larger size of the particle. The correlation between the CL intensity and the SE image can be seen in Fig. 4c, where the intensity is shown in a false color scale for identification purposes only.
:1 ratio of the number of particles of each type. A secondary-electron (SE) image of a 6 × 6 μm region on the sample is displayed in Fig. 4a. We scan the same area again, but now with a pulsed electron beam at a step size of 100 nm and a dwell time of 0.5 seconds. For every pixel, the photon arrival histogram is determined, which is then fitted with a single exponential decay. Thus, we simultaneously measure the intensity and lifetime of the emitters on the sample. The intensity of both type of particles can differ greatly, which on a linear scale could render the less intense particles invisible. Therefore, we show the CL intensity map in Fig. 4b on a logarithmic scale. Note that this leads to an apparently larger size of the particle. The correlation between the CL intensity and the SE image can be seen in Fig. 4c, where the intensity is shown in a false color scale for identification purposes only.
 | ||
| Fig. 4 (a) Secondary electron (SE) image a typical area of a sample containing a mixture of LuAG:Ce and FND nanoparticles. The nanoparticles are deposited onto a glass cover slide with a thin indium-tin-oxide coating which provides the optical transparency and electric conductance required. In (b), the cathodoluminescence (CL) is displayed on a logarithmic scale. In (c), a false-color image where the SE image and the (logarithmic) CL intensity image are overlaid is shown, showing the clear correlation between the two. | ||
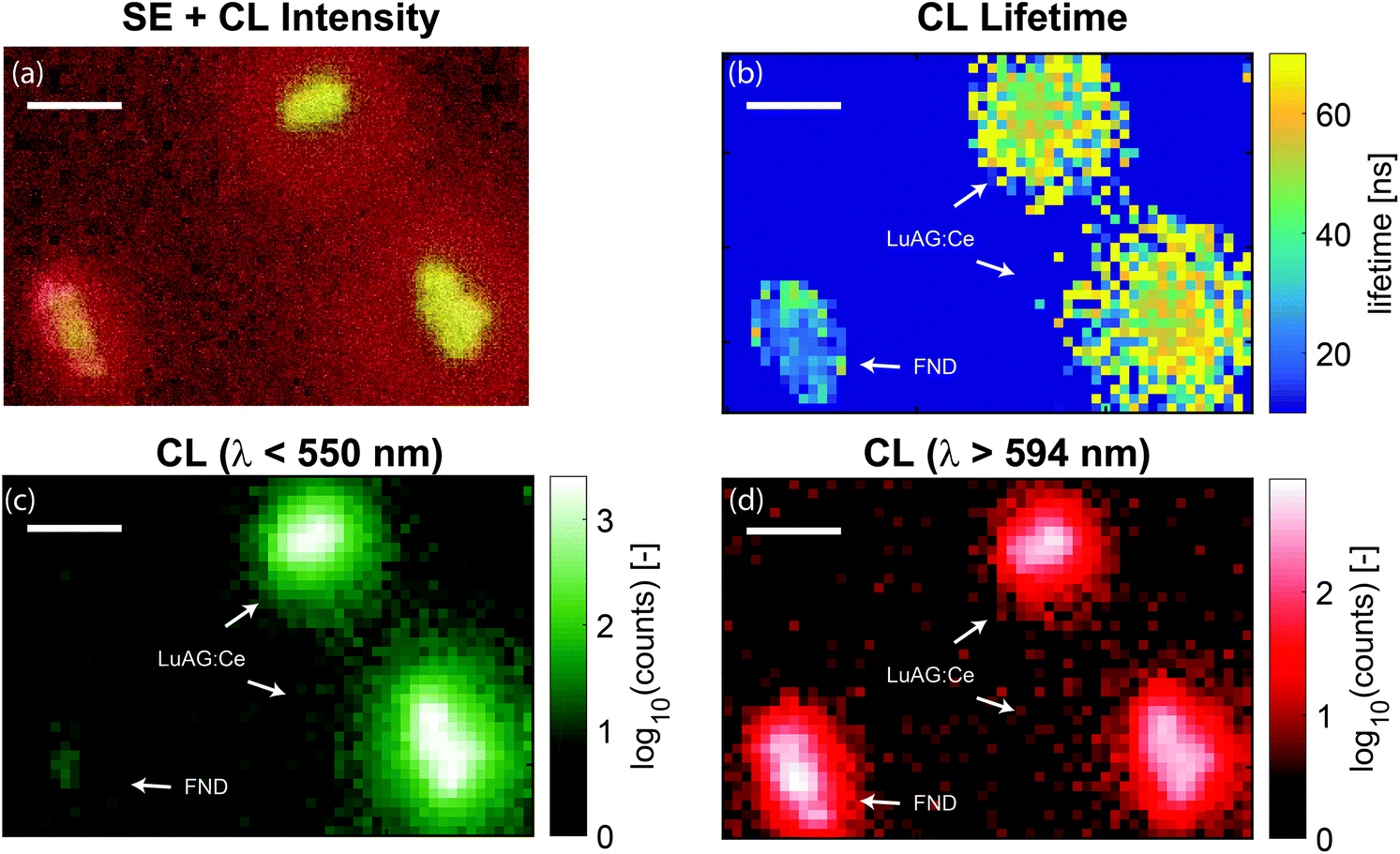
The lifetime map that we obtain from the fitting procedure is shown in Fig. 5a. Here, the lifetime map is thresholded with the intensity, such that areas without CNPs do not show as noise in the lifetime maps. Clearly, there are two dominant lifetimes in Fig. 5a, corresponding to the distribution of lifetimes as measured before and displayed in Fig. 2b. Particles with a lifetime of around 20–30 ns correspond to FNDs, and particles with a lifetime of about 50–70 ns correspond to LuAG:Ce particles. For comparison, the scans were repeated, inserting optical filters to discriminate between the two particle types: a short pass filter with an edge wavelength of 550 nm in order to only see LuAG:Ce particles, and a long pass filter in order to see the emission by the FNDs. Due to the broad spectrum and high CL intensity, the LuAG:Ce particles are suppressed in intensity but visible as well. The results for the short pass filter are shown in Fig. 5b, whereas the results for the long pass filter are shown in Fig. 5c. As an example, the low-lifetime FNDs highlighted by a white square in Fig. 5a clearly only show up in the longer-wavelength emission channel in Fig. 5c, corresponding to the bulk of the emission spectrum of FNDs. Thus, the spectral data confirm the validity of our CL lifetime-based discrimination.
 | ||
| Fig. 5 Lifetime and spectral maps of LuAG and FND nanoparticles. In (a), the measured lifetime as a function of position is shown, with typical times around 40–60 ns for LuAG:Ce particles and around 20–30 ns for FNDs. The squares highlight examples of the type of particle as determined by the lifetime and the histograms of Fig. 2b. Here, no wavelength selection was applied. In (b), the CL intensity for light with wavelengths less than 550 nm is shown, corresponding to the emission of LuAG:Ce particles. In (c), the CL intensity for light with wavelength longer than 594 nm is shown, corresponding to a tail in the emission by LuAG:Ce and the bulk of the FND emission. The FNDs are absent in panel (b) and clearly show up in panel (c). The broad spectrum of LuAG:Ce means that this type of particle shows up in both measurements. However, for wavelengths longer than 597 nm the intensity is greatly reduced. In panel (a)–(c), the scale bar is 500 nm. | ||
For higher resolution CL lifetime imaging, the step size of the electron beam can be reduced. In Fig. 6 we show three different nanoparticles scanned with a step size of 50 nm. As the current with pulsed electron beam operation is reduced, image acquisition times are longer compared to continuous operation and thus choice of step size is a balance between resolution required for particle identification and field of view size. The results shown in Fig. 6 confirm the above conclusion that we can discriminate the particles based on lifetime, where here we are limited by the particle size.
 | ||
| Fig. 6 High-resolution lifetime and spectral maps of LuAG:Ce and FND nanoparticles. (a) False-color image of SE overlaid with the (logarithmic) CL intensity. (b) Measured lifetime as a function of position. The arrows indicate the two types of particles as determined by the lifetime and the histograms of Fig. 2b. No wavelength selection was applied. (c) CL intensity for light with wavelengths less than 550 nm is shown (LuAG:Ce particles). (d) CL intensity for light with wavelength longer than 594 nm is shown (bulk of the FND emission). The FNDs are nearly absent in panel (c) and clearly show up in panel (d). In panel (a)–(d), the scale bar is 500 nm. | ||
CL lifetime in stained biological sections
Application of CL lifetime imaging for nanostructural characterization may range from geosciences and materials sciences, phosphor particle detector development, to visualization of labels in biological sciences. As an application example, we here demonstrate that CL lifetime imaging can be conducted on biological sections after fixation, staining, and sectioning protocols. Fig. 7a shows a back-scattered electron (BSE) image of a 300 nm section of macrophages incubated with the FNDs. The image shows good ultrastructural preservation and fine intracellular details are well resolved. The FNDs are however not visible in the BSE images. In Fig. 7b and c we show a zoom of an area where CL was detected, presumably from uptaken FNDs. The CL lifetime map in Fig. 7e indeed unequivocally identifies the FNDs as being the source of the CL emission logarithmically plotted in Fig. 7d (cf.Fig. 2b). The weaker CL signal for one of the three FNDs may be due to an FND buried deeper within the 300 nm section. Indeed, while the other two particles stood out in the SE image as well (data not shown), as expected for surface-exposed FNDs,25 no SE signal above background tissue signal was recorded at this location. The BSE-CL overlay allows for high-resolution localization of the CNPs in the macrophage ultrastructural environment and lifetime data is useful to identify these as FNDs. | ||
| Fig. 7 CL lifetime measurements of FNDs in stained sections after uptake in macrophages. (a) Backscattered electron image of part a section showing macrophage ultrastructure (b) zoom-in of the dashed area in (a), which is a region where CL was detected.(c) Logarithmic CL intensity map from area (b) overlaid on the BSE image. (d) Logarithmic CL intensity, and (e) simultaneously acquired CL lifetime map for the area in (b), clearly showing CL lifetime values typical for FNDs. | ||
Conclusions
We have shown that lifetime-based imaging and particle discrimination is a stable modality in electron microscopy, useful for analytical and correlative imaging purposes. Based on lifetime, we demonstrated discrimination of particle type in a mixture of two CNPs dispersed over a surface, and CL lifetime imaging of CNPs in tissue sections. The lifetime of the CNPs measurements show very good robustness over time, with only mild variations even when the intensity itself diminishes significantly.For applications, we believe that correlative SE and CL lifetime imaging is suitable for high-resolution characterization and imaging throughout the nanosciences. Particular areas of interest may include materials sciences and geosciences in general, and characterization of the homogeneity of phosphors for use in particle detectors or of nanoparticle assemblies in particular. In addition, the pulsed electron excitation paired with the possibility to obtain accurate lifetime estimates from histograms with low photon counts35 may be particularly beneficial for systems suffering from electron-beam degradation, such as labels in biological tissue sections. Moreover, instead of or combined with multi-color CL imaging, the lifetime of the nanoparticles may be used to increase the number of labels discernible in EM; e.g., green-fluorescing nanodiamonds and LuAG:Ce have very similar optical emission spectra but have different lifetimes,49 which is sufficient to distinguish the particles. Time-resolved CL imaging can also be beneficial in combination with other characterization techniques such as energy-dispersive X-ray spectroscopy,50e.g., to distinguish materials with similar elemental composition such as different (carbon-based) FNDs and native biological tissue or polymeric materials. Finally, CL lifetime microscopy may be used to recover more information about the local environment of the nanoparticle, such as structures which modify the optical local density of states,37,51 or the actual (surface) structure or doping concentration of the nanoparticle itself.52–54
Methods
Particle samples
Macrophage samples
000g) and subsequent microfiltration. The protein was purified by selective precipitation with ammonium sulfate (at 45% saturation). After a dialysis against 50 mM formic acid the protein was lyophilized and the protein powder was stored at room temperature. Before coating the polymer was diluted in MilliQ water to a concentration of 7.36 mg ml−1. The protein was passed through a 0.22 μm filter to remove aggregates.
The diamond particles were coated with the C4K12 protein polymers by mixing the stock solution of nanodiamonds (100 μg mL−1 in water), with the protein solution followed by incubating for 30 minutes. The final particles contain roughly 4 C4K12 molecules per 100 nm2 of nanodiamond surface. All solutions were prepared under sterile conditions. J774A.1 macrophages (LGC Standards, Germany) were cultured in Dulbecco's Modified Eagle Medium (DMEM), supplemented with 10% FBS, 1% penicillin/streptomycin. Cells were incubated with 1 μg ml−1 coated FND in cell culture medium for 5 hours at 37 °C and 5% CO2. After removal of culture medium with FNDs, cells were fixed with 4% paraformaldehyde/0.1% glutaraldehyde in 0.1 M cacodylate buffer, pH 7.4 for 30 minutes at room temperature. After washing with 0.1 M cacodylate buffer, cells were incubated with 1% osmiumtetroxide/1.5% potassiumferrocyanide in 0.1 M cacodylate buffer for 30 minutes at 4 °C, followed by washing with water. Next, the cells were dehydrated through an increasing graded ethanol series and left overnight in 1:1 ethanol and Epon (Serva) mixture at room temperature, which was replaced by pure Epon (4 times) and finally polymerised overnight at 58 °C. The cover glass of the imaging dish was removed using hydrogen fluoride. Areas containing cells were selected using a stereo microscope and sawn from the Epon block. Subsequently, 300 nm sections were cut with an ultramicrotome (Leica EM UC7) using a glass knife and put on an ITO-coated cover glass.
As BSE detection provides better contrast on biological tissue than SE detection, and our time-resolved setup is not equipped with a BSE detector, the biological structure of the cell was imaged with an FEI Verios 460 SEM. By referring to the SE images obtained during the time-resolved measurement, the region of interest is retrieved, and SE and BSE images are simultaneously obtained with a 3 kV, 0.8 nA continuous electron beam.
Conflicts of interest
Our integrated microscope and cathodoluminescence detection set-up served as a prototype for a product by Delmic BV. JPH is co-founder and shareholder in Delmic.Acknowledgements
This research was supported by the Netherlands Organization for Fundamental Research (NWO) and the Stichting voor Fundamenteel Onderzoek der Materie (FOM, Foundation for Fundamental Research on Matter) grant number 10PR2826. RS acknowledges support from NWO-FOM via grant number FOM-G-36, PdB from Stichting Technische Wetenschappen (STW) – Perspectief voor de Topsectoren – Microscopy Valley grant number 12718, project leader Ben Giepmans. FPM is supported by A CONICYT scholarship from the Chilean government. Part of the work has been performed in the UMCG Microscopy and Imaging Center (UMIC), sponsored by ZonMW grant 91111.006 and NWO 175-010-2009-023.References
- E. F. Bond, D. Beresford and G. H. Haggis, J. Microsc., 1974, 100, 271–282 CrossRef CAS PubMed.
- B. G. Yacobi and D. B. Holt, J. Appl. Phys., 1986, 59, R1–R24 CrossRef CAS.
- D. Poelman, D. Wauters, J. Versluys and R. L. V. Meirhaeghe, J. Appl. Phys., 2001, 90, 248–251 CrossRef CAS.
- K. Milliken, S.-J. Choh, P. Papazis and J. Schieber, Sediment. Geol., 2007, 198, 221–232 CrossRef.
- B. E. Sørensen and R. B. Larsen, Contrib. Mineral. Petrol., 2008, 157, 147 CrossRef.
- M. Kostova, M. Batentschuk, F. Goetz-Neunhoeffer, S. Gruber, A. Winnacker, P. Greil and C. Zollfrank, Mater. Chem. Phys., 2010, 123, 166–171 CrossRef CAS.
- J. Bruckbauer, P. R. Edwards, T. Wang and R. W. Martin, Appl. Phys. Lett., 2011, 98, 141908 CrossRef.
- M. F. Hamers and M. R. Drury, Meteorit. Planet. Sci., 2011, 46, 1814–1831 CrossRef CAS.
- P. V. Hough, W. R. McKinney, M. C. Ledbeter, R. E. Pollack and H. W. Moos, Proc. Natl. Acad. Sci. U. S. A., 1976, 73, 317–321 CrossRef CAS.
- J. Silver, R. Withnall, T. G. Ireland, G. R. Fern and S. Zhang, Nanotechnology, 2008, 19, 095302 CrossRef CAS PubMed.
- M. Kuttge, E. J. R. Vesseur, A. F. Koenderink, H. J. Lezec, H. A. Atwater, F. J. García de Abajo and A. Polman, Phys. Rev. B: Condens. Matter, 2009, 79, 113405 CrossRef.
- B. Dierre, X. Yuan and T. Sekiguchi, Sci. Technol. Adv. Mater., 2010, 11, 043001 CrossRef PubMed.
- H. Niioka, T. Furukawa, M. Ichimiya, M. Ashida, T. Araki and M. Hashimoto, Appl. Phys. Express, 2011, 4, 112402 CrossRef.
- A. C. Narváez, I. G. C. Weppelman, R. J. Moerland, N. Liv, A. C. Zonnevylle, P. Kruit and J. P. Hoogenboom, Opt. Express, 2013, 21, 29968–29978 CrossRef PubMed.
- C. I. Osorio, T. Coenen, B. J. M. Brenny, A. Polman and A. F. Koenderink, ACS Photonics, 2016, 3, 147–154 CrossRef CAS.
- P. de Boer, J. P. Hoogenboom and B. N. G. Giepmans, Nat. Methods, 2015, 12, 503–513 CrossRef CAS PubMed.
- D. R. Glenn, H. Zhang, N. Kasthuri, R. Schalek, P. K. Lo, A. S. Trifonov, H. Park, J. W. Lichtman and R. L. Walsworth, Sci. Rep., 2012, 2, 865 CrossRef CAS PubMed.
- J.-i. Niitsuma, H. Oikawa, E. Kimura, T. Ushiki and T. Sekiguchi, J. Electron Microsc., 2005, 54, 325 CrossRef CAS PubMed.
- K. Kömpe, H. Borchert, J. Storz, A. Lobo, S. Adam, T. Möller and M. Haase, Angew. Chem., Int. Ed., 2003, 42, 5513–5516 CrossRef PubMed.
- Y. Nawa, W. Inami, A. Chiba, A. Ono, A. Miyakawa, Y. Kawata, S. Lin and S. Terakawa, Opt. Express, 2012, 20, 5629–5635 Search PubMed.
- T. Furukawa, H. Niioka, M. Ichimiya, T. Nagata, M. Ashida, T. Araki and M. Hashimoto, Opt. Express, 2013, 21, 25655–25663 CrossRef PubMed.
- Y. Nawa, W. Inami, S. Lin, Y. Kawata, S. Terakawa, C.-Y. Fang and H.-C. Chang, ChemPhysChem, 2014, 15, 721–726 CrossRef CAS PubMed.
- I. E. G. Morrison, A. Samilian, P. Coppo, T. G. Ireland, G. R. Fern, J. Silver, R. Withnall and P. J. O'Toole, J. Chem. Biol., 2015, 8, 169–177 CrossRef.
- S. Nagarajan, C. Pioche-Durieu, L. H. G. Tizei, C.-Y. Fang, J.-R. Bertrand, E. Le Cam, H.-C. Chang, F. Treussart and M. Kociak, Nanoscale, 2016, 8, 11588–11594 RSC.
- S. R. Hemelaar, P. de Boer, M. Chipaux, W. Zuidema, T. Hamoh, F. P. Martinez, A. Nagl, J. P. Hoogenboom, B. N. G. Giepmans and R. Schirhagl, Sci. Rep., 2017, 7, 720 CrossRef CAS PubMed.
- J. R. Lakowicz and K. W. Berndt, Rev. Sci. Instrum., 1991, 62, 1727–1734 CrossRef CAS.
- X. F. Wang, A. Periasamy, B. Herman and D. M. Coleman, Crit. Rev. Anal. Chem., 1992, 23, 369–395 CrossRef CAS.
- K. H. Drexhage, J. Lumin., 1970, 1–2, 693–701 CrossRef.
- R. A. L. Vallée, N. Tomczak, L. Kuipers, G. J. Vancso and N. F. van Hulst, Phys. Rev. Lett., 2003, 91, 038301 CrossRef PubMed.
- R. J. Moerland and J. P. Hoogenboom, Optica, 2016, 3, 112–117 CAS.
- P. R. Selvin, Nat. Struct. Biol., 2000, 7, 730–734 CrossRef CAS PubMed.
- D. Poelman and P. F. Smet, Physica B, 2014, 439, 35–40 CrossRef CAS.
- D. den Engelsen, P. G. Harris, T. G. Ireland, G. R. Fern and J. Silver, Ultramicroscopy, 2015, 157, 27–34 CrossRef PubMed.
- S. Meuret, L. H. G. Tizei, T. Auzelle, R. Songmuang, B. Daudin, B. Gayral and M. Kociak, ACS Photonics, 2016, 3, 1157–1163 CrossRef CAS.
- M. Köllner and J. Wolfrum, Chem. Phys. Lett., 1992, 200, 199–204 CrossRef.
- D. S. Yang, O. F. Mohammed and A. H. Zewail, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 14993–14998 CrossRef CAS PubMed.
- R. J. Moerland, I. G. C. Weppelman, M. W. H. Garming, P. Kruit and J. P. Hoogenboom, Opt. Express, 2016, 24, 24760–24772 CrossRef PubMed.
- A. C. Zonnevylle, R. F. Van Tol, N. Liv, A. C. Narváez, A. P. Effting, P. Kruit and J. P. Hoogenboom, J. Microsc., 2013, 252, 58–70 CrossRef CAS PubMed.
- A. C. Narváez, I. G. C. Weppelman, R. J. Moerland, J. P. Hoogenboom and P. Kruit, Appl. Phys. Lett., 2014, 104, 251121 CrossRef.
- H.-L. Li, X.-J. Liu and L.-P. Huang, Opt. Mater., 2007, 29, 1138–1142 CrossRef CAS.
- S. Felton, A. M. Edmonds, M. E. Newton, P. M. Martineau, D. Fisher and D. J. Twitchen, Phys. Rev. B: Condens. Matter, 2008, 77, 081201 CrossRef.
- A. Zaitsev, Optical Properties of Diamond, Springer-Verlag, Berlin, Heidelberg, 2001 Search PubMed.
- C. Bradac, T. Gaebel, N. Naidoo, M. J. Sellars, J. Twamley, L. J. Brown, A. S. Barnard, T. Plakhotnik, A. V. Zvyagin and J. R. Rabeau, Nat. Nanotechnol., 2010, 5, 345–349 CrossRef CAS PubMed.
- R. Schirhagl, K. Chang, M. Loretz and C. L. Degen, Annu. Rev. Phys. Chem., 2014, 65, 83–105 CrossRef CAS PubMed.
- K.-M. C. Fu, C. Santori, P. E. Barclay and R. G. Beausoleil, Appl. Phys. Lett., 2010, 96, 121907 CrossRef.
- L. Rondin, G. Dantelle, A. Slablab, F. Grosshans, F. Treussart, P. Bergonzo, S. Perruchas, T. Gacoin, M. Chaigneau, H.-C. Chang, V. Jacques and J.-F. Roch, Phys. Rev. B: Condens. Matter, 2010, 82, 115449 CrossRef.
- K. Beha, A. Batalov, N. B. Manson, R. Bratschitsch and A. Leitenstorfer, Phys. Rev. Lett., 2012, 109, 097404 CrossRef CAS PubMed.
- C. Zachreson, A. A. Martin, I. Aharonovich and M. Toth, ACS Appl. Mater. Interfaces, 2014, 6, 10367–10372 CAS.
- G. Laporte and D. Psaltis, Biomed. Opt. Express, 2016, 7, 34–44 CrossRef CAS PubMed.
- M. Scotuzzi, J. Kuipers, D. I. Wensveen, P. de Boer, C. W. Hagen, J. P. Hoogenboom and B. N. G. Giepmans, Sci. Rep., 2017, 7, 45970 CrossRef CAS PubMed.
- R. Carminati, A. Cazé, D. Cao, F. Peragut, V. Krachmalnicoff, R. Pierrat and Y. D. Wilde, Surf. Sci. Rep., 2015, 70, 1–41 CrossRef CAS.
- C. L. Melcher and J. S. Schweitzer, IEEE Trans. Nucl. Sci., 1990, 37, 161–164 CrossRef CAS.
- J. Tisler, G. Balasubramanian, B. Naydenov, R. Kolesov, B. Grotz, R. Reuter, J.-P. Boudou, P. A. Curmi, M. Sennour, A. Thorel, M. Börsch, K. Aulenbacher, R. Erdmann, P. R. Hemmer, F. Jelezko and J. Wrachtrup, ACS Nano, 2009, 3, 1959–1965 CrossRef CAS PubMed.
- Y. Lu, J. Zhao, R. Zhang, Y. Liu, D. Liu, E. M. Goldys, X. Yang, P. Xi, A. Sunna, J. Lu, Y. Shi, R. C. Leif, Y. Huo, J. Shen, J. A. Piper, J. P. Robinson and D. Jin, Nat. Photonics, 2014, 8, 32–36 CrossRef CAS.
- M. W. T. Werten, W. H. Wisselink, T. J. Jansen-van den Bosch, E. C. de Bruin and F. A. de Wolf, Protein Eng., Des. Sel., 2001, 14, 447 CrossRef CAS.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671–675 CrossRef CAS PubMed.
- P. Thévenaz, U. Ruttimann and M. Unser, IEEE Trans. Image Process., 1998, 7, 27–41 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2017 |