
Condition responsive nanoparticles for managing infection and inflammation in keratitis†
 a
and
Ch. Mohan
Rao
*a
a
and
Ch. Mohan
Rao
*a
Abstract
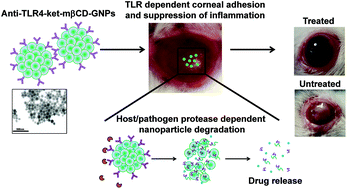
Keratitis is a major cause of avoidable visual impairment. About 30% of patients with fungal keratitis eventually become permanently blind in the developing world. Proteases, secreted by the pathogen and the host, damage the cornea before the infection is resolved. Treating keratitis is a challenge because both infection and inflammation need to be addressed. An additional challenge is to maintain a therapeutic dose at the corneal surface as blinking and tear film wash away the drugs, administered as eye drops. We have developed a nanoparticle-based drug delivery system that enhances the drug residence time by anchoring to the cornea, down-regulates inflammation and releases the antifungal drug: all in a condition-responsive manner. The expression of Toll-Like Receptors (TLR4) on the corneal epithelial cells increases in response to infection. We have conjugated anti-TLR4 antibodies on the surface of ketoconazole-encapsulated gelatin nanoparticles. The anti-TLR4 antibody not only facilitates binding of nanoparticles to the cornea, enhancing their residence time, but also reduces the levels of inflammatory cytokines. Host and fungal proteases degrade the gelatin nanoparticle, an alternative substrate for proteases, thereby reducing corneal damage and releasing the encapsulated drug, ketoconazole, proportional to the severity of infection. After testing the efficacy of the system with human corneal epithelial cells, we have extended our studies to a rat model of keratitis. The results show a significantly increased corneal retention, suppressed inflammation and resolution of infection in the infected eyes. We believe that this will be an excellent approach to manage keratitis as well as other topical ocular infections.
 Please wait while we load your content...
Please wait while we load your content...

