
Allosteric effects of gold nanoparticles on human serum albumin†
Qing
Shao
and
Carol K.
Hall
*
Department of Chemical and Biomolecular Engineering, North Carolina State University Raleigh, NC, 27695, USA. E-mail: hall@ncsu.edu
First published on 30th November 2016
Abstract
The ability of nanoparticles to alter protein structure and dynamics plays an important role in their medical and biological applications. We investigate allosteric effects of gold nanoparticles on human serum albumin protein using molecular simulations. The extent to which bound nanoparticles influence the structure and dynamics of residues distant from the binding site is analyzed. The root mean square deviation, root mean square fluctuation and variation in the secondary structure of individual residues on a human serum albumin protein are calculated for four protein–gold nanoparticle binding complexes. The complexes are identified in a brute-force search process using an implicit-solvent coarse-grained model for proteins and nanoparticles. They are then converted to atomic resolution and their structural and dynamic properties are investigated using explicit-solvent atomistic molecular dynamics simulations. The results show that even though the albumin protein remains in a folded structure, the presence of a gold nanoparticle can cause more than 50% of the residues to decrease their flexibility significantly, and approximately 10% of the residues to change their secondary structure. These affected residues are distributed on the whole protein, even on regions that are distant from the nanoparticle. We analyze the changes in structure and flexibility of amino acid residues on a variety of binding sites on albumin and confirm that nanoparticles could allosterically affect the ability of albumin to bind fatty acids, thyroxin and metals. Our simulations suggest that allosteric effects must be considered when designing and deploying nanoparticles in medical and biological applications that depend on protein–nanoparticle interactions.
Introduction
Nanoparticles (NPs) have been recognized as key to revolutionizing the healthcare,1,2 environment3–7 and daily product sectors.8–10 Their small size and large surface to volume ratios provide NPs with unique properties that bulk materials do not exhibit. As NPs directly interact with proteins in cells and blood, the success of NP-based medical and biotechnology applications depends on the nature of the NP–protein interactions.11,12 These interactions also determine whether NPs have negative effects on the biological functions of proteins. A thorough understanding of NP–protein interactions can help us to better design and deploy nanoparticles in medical and biotechnology applications that rely on these interactions.NPs can influence protein structure and flexibility. In designing NPs to interact with proteins, it is important to consider how NP size, surface chemistry, and shape impact the protein's ability to remain folded and its likelihood to bind to protein regions distant from the active site. Many investigations have been conducted with the aim of learning how to manipulate the ability of NPs to change protein structure and flexibility.13–18 For instance, Jonsson and his colleagues19 found that smaller nanoparticles (NPs) have weaker ability to denature proteins than larger ones. Others have found that coating nanomaterials with anti-biofouling agents such as polyethylene glycol and zwitterions helps keep proteins folded, likely by pushing them away from the NPs.20,21 Zuo et al.22 investigated the interactions between carbon nanotubes and WW domain proteins using molecular dynamics simulations. They found that the presence of a hydrophobic carbon nanotube prevents a WW domain protein from binding to its ligand by forming a protein–nanotube complex. Ding et al.23 investigated the interactions between ubiquitin and silver NPs using molecular simulations and experiments. Their results suggest that ubiquitin proteins bind to silver NPs through multiple pathways and that the presence of the NPs destabilizes helical structures on the protein. Several comprehensive reviews14,24–28 provide insight into various aspects of NP–protein interactions.
Here we focus on the allosteric effect of NPs on proteins, a topic that has not often been addressed in the literature. The term “allosteric effects” refers to the ability of ligands to change the flexibility and structure of protein regions distant from their binding site, possibly altering the protein's biological activity even when the protein does not unfold.29,30 The molecular-level mechanisms that underlie allosteric effects remain under investigation and several models have been proposed. These include the Monod, Wyman and Changeux (MWC) model,30 the Koshland, Nemethy, and Filmer (KNF) model,31 and the more recent ensemble model.32 Despite the differences in these models, they all suggest that NPs do exert allosteric effects on proteins.
The objective of this work is to investigate the extent to which a bound NP influences the structure and dynamics of residues distant from its binding site. We examine the structure and flexibility of a human serum albumin (HSA) protein bound to a gold NP with a diameter of 4.0 nm using computer simulations. HSA is chosen as our model protein because it is the most abundant protein in human blood serum. The large size of an HSA protein also makes it likely that a NP can change its flexibility and secondary structure without denaturing it. We selected gold NPs because they are widely used in medical applications33 and gold materials serve as models for investigations of protein–substrate interactions. The 4.0 nm NP in this work is well suited to our purposes because it is large enough to affect the protein yet small enough to allow atomistic simulations to be conducted in a reasonable time frame. We neglect the formation of Au–S bonds between HSA and the gold NP here because we are mainly concerned with the fundamentals of protein–NP interactions as opposed to the specifics of HSA–gold NP interactions. Therefore, only non-bonded interactions between the protein and the nanoparticle, such as van der Waals and electrostatic interactions, are considered. The effects of water molecules are included by adding explicit water solvent.
The interactions between nanomaterials and serum albumin proteins have been investigated extensively in experiment. Some experiments are focused on the potential of albumin proteins to improve the biocompatibility of nanomaterials. For instance, Peng et al.34 showed that a preformed albumin corona can be used to inhibit further adsorption of plasma proteins on polymeric NPs and hence prolongs the blood circulation time of the NPs. Duan et al.35 suggested that the adsorption of albumin proteins on graphene nanosheets weakens their ability to disrupt cell membranes, and therefore attenuates the toxicity of graphene nanomaterials. Some experiments reveal the influence of nanomaterials on serum albumins. Treuel et al.36 investigated the influence of the surface composition of nanoparticles on their interactions with bovine serum albumin proteins using circular dichroism spectroscopy. They found that citrate-stabilized gold NPs with a diameter of 20 nm partially destabilize the helix structure of albumin proteins. Lindman et al.37 studied the interactions between human serum albumin and polymeric nanoparticles of varying hydrophobicity and size using isothermal titration calorimetry. They correlated the amount of albumin proteins adsorbed with NP size and hydrophobicity. Boulos et al.38 used static fluorescence quenching titration and affinity capillary electrophoresis to investigate the adsorption process for bovine serum albumin proteins on gold NPs of various sizes, shapes and surface charges. They concluded that bovine albumin proteins bind to gold NPs regardless of surface charge. However, they also observed that the results from their two experimental methods are significantly different, highlighting the challenges of precisely examining protein–NP interactions via experiment. Tsai et al.39 investigated the adsorption and conformation of bovine serum albumin proteins on gold NPs using dynamic light scattering, asymmetric-flow field flow fractionation, fluorescence spectrometry, and attenuated total reflectance-Fourier transform infrared spectroscopy. They suggested that the α-helix content of bovine albumin proteins decreases as they are adsorbed on NPs. Their results also suggest that the conformation of adsorbed serum proteins remains unchanged in pHs ranging from 3.4 to 7.3. The experiments point to the importance of understanding nanomaterial–albumin interactions, especially at the molecular level.
Determining the binding structure of proteins on NPs experimentally remains a challenge although progress has been made. Calzolai et al.40 showed that it is possible to identify the protein–NP interaction site on the amino acid scale using a combination of NMR, chemical shift perturbation analysis, and dynamic light scattering. The level of detail achievable with these methods also makes it possible to determine the orientation of proteins on NPs. Wang et al.41 showed that a combination of synchrotron radiation, X-ray absorption spectroscopy, microbeam X-ray fluorescent spectroscopy, circular dichroism and molecular dynamics simulations is able to reveal the structure of bovine serum albumin on gold nanorods.
Identifying the binding location and orientation of proteins on substrates is also a challenge in simulations. Zhou et al.42 conducted Monte Carlo simulations to search for the orientation of proteins on solid surfaces using a coarse-grained implicit solvent model. Deighan and Pfaendtner43 exhaustively explored all binding structures of short peptides on self-assembled monolayers using parallel tempering metadynamics in the well-tempered ensemble. Here we apply a method similar to Zhou et al.'s approach,42 but instead of using Monte Carlo simulations, which does not guarantee that the configuration with the lowest interaction energy will be found, we apply a brute-force search process that checks every possible binding configuration and identifies the ones with the global or local lowest interaction energy.
Our work deploys a two-step strategy: a brute-force search is conducted to identify HSA–NP binding complexes which are at the global or local minimum interaction energy. This search process uses an implicit-solvent coarse-grained protein/NP model developed in our group. The resulting complexes are used as initial configurations in explicit-solvent atomistic molecular dynamics simulations to investigate the structure and flexibility of an HSA protein bound to a NP. We calculate three protein properties: root mean square deviations (RMSD) of Cα atoms to determine if the protein remains folded, root mean square fluctuation (RMSF) of Cα atoms to examine the protein flexibility, and secondary structure distribution of individual residues to determine if the protein adopts certain changes. The rest of the paper is organized as follows: it first gives details about the simulations and parameters used in the paper, then presents results and discussion, and finally gives a conclusion.
Simulation method
Brute-force process to search for the NP binding sites on HSA
A brute-force search process based on an implicit-solvent coarse-grained protein/NP model is employed to identify HSA–NP binding complexes with global or local minimum interaction energy. The HSA protein (PDB ID: 1OA6) is represented by a two-sphere-per-residue model. Each amino-acid residue is modeled as a sphere at the Cα position, and another at the sidechain center of mass. The proline and glycine are represented by a Cα sphere only. The NP is represented by a sphere located at its center of mass.The interactions between protein spheres and the NP sphere are described by a tabulated potential obtained from previously-determined potentials of mean force between the amino acid sidechain and backbone entities and the gold NP in explicit water. These potentials of mean force were obtained from a series of well-tempered metadynamics simulations, whose details were described in a previous paper.44 The parameters are listed in Tables S1 and S2.†
The brute-force search process is conducted as follows. An HSA protein in its native state conformation is converted to the two-sphere-per-residue model and placed at the center of a box whose x, y and z lengths are 20.0 nm larger than those of the HSA protein. This box is divided into a grid with bins of size 0.1 nm in the x, y and z directions. The NP sphere is placed at the center of each grid cell and the NP–HSA interaction energy is calculated using the tabulated potentials shown in Tables S1 and S2.† Grid cells that cause a geometric overlap between the NP and the HSA protein are excluded from the search. The HSA protein is kept rigid during the searching process. The search is executed by a program written in our group. If a grid cell produces a negative protein–NP interaction energy, the program records its coordinate, the value of the interaction energy, and the residues that interact with the NP. Fig. 1 shows a schematic of the simulation box for the search process.
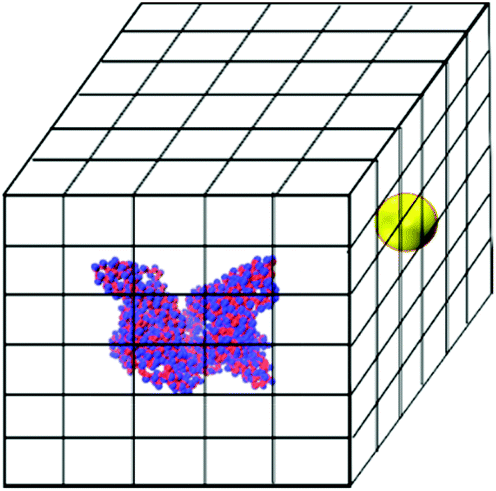 | ||
| Fig. 1 Schematic of the simulation box used for the brute-force search process. An HSA protein is put in the middle of the box. A gold NP is placed at the center of every grid cell and the protein–NP interaction energy is calculated. | ||
Atomistic molecular dynamics simulation
The NP–HSA complexes identified in the brute-force search process are converted to all-atom configurations and used to build the initial configurations for explicit solvent atomistic molecular dynamics (MD) simulations. The coarse-grained gold NP sphere is converted to an all-atom model at the same center of mass position. The all-atom HSA protein configuration is obtained by minimizing the RMSD between the positions of its Cα atom and the Cα spheres in the coarse-grained HSA protein model. The gold NP is shifted away from the protein slightly to avoid any close contact between the atoms on the protein and the gold NP.The atomistic MD simulation box is built as follows. An all-atom HSA–NP complex is placed at the center of a cubic box with sides of length 12.0 nm. A 2.5 nm-thick water layer is placed around this complex. This allows us to explicitly account for the effect of water on the HSA–NP complex but keeps the number of atoms in the simulation box to a manageable level so that the MD simulations can be run for 200 ns. Some water molecules are replaced by Na+ and Cl− to ensure that the net charge of the system is zero, and the NaCl concentration is 0.15 M. The final systems have more than 90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 atoms. Table S3† lists the number of water molecules and ions in the four simulation systems. Fig. 2 shows a snapshot of the initial configuration for atomistic MD simulations. The HSA protein and ions are described by the GROMOS54a7 force field.45 The gold atoms are described using the force field parameters developed by the Heinz group46 because this force field well describes the interfacial properties of gold materials. The water molecules are described using the SPC model47 as is recommended when using the GROMOS54a7 force field.
000 atoms. Table S3† lists the number of water molecules and ions in the four simulation systems. Fig. 2 shows a snapshot of the initial configuration for atomistic MD simulations. The HSA protein and ions are described by the GROMOS54a7 force field.45 The gold atoms are described using the force field parameters developed by the Heinz group46 because this force field well describes the interfacial properties of gold materials. The water molecules are described using the SPC model47 as is recommended when using the GROMOS54a7 force field.
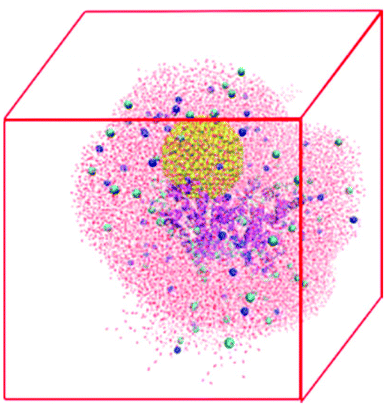 | ||
| Fig. 2 Snapshot of the initial configuration for the atomistic MD simulations of an HSA–NP complex in explicit solvent. The HSA protein is shown in cartoon model to emphasize its secondary structure. The gold NP is shown in the VDW model. The ions are shown in the VDW model and the water molecules are shown in a line model. | ||
Atomistic MD simulations are conducted to investigate the structure and flexibility of an HSA protein in the four complexes and in the bulk solution. A 200 ns canonical ensemble (NVT, T = 300 K) MD simulation with a 2 fs time step is conducted after an energy minimization for each system considered. The temperature is maintained at 300 K using the stochastic global thermostat.48 The positions of the gold atoms are fixed during the minimization and MD simulations. The non-bonded interaction energy is the sum of the short-range van der Waals interactions and long-range electrostatic interactions. The former are described by the Lennard-Jones 6–12 function with a 1.0 nm cut-off, and the latter are described by the Coulombic function and treated with particle mesh Ewald sum.49 The non-bonded interaction energy among the gold atoms is set to zero during the simulations. The bonds attached to the hydrogen atoms are constrained to their equilibrated lengths using the LINCS algorithm.50 The other bonded interactions are described as in the GROMOS54a7 force field. The data for the HSA–NP complexes are collected over the 100–200 ns simulation. A 100 ns MD simulation of an HSA in bulk solution (0.15 M NaCl) is conducted for reference. The atomistic MD simulations are conducted using Gromacs-5.0.4.51
Definition of ΔRMSFi
The RMSF difference parameter ΔRMSFi is defined to characterize the change in flexibility of residue i.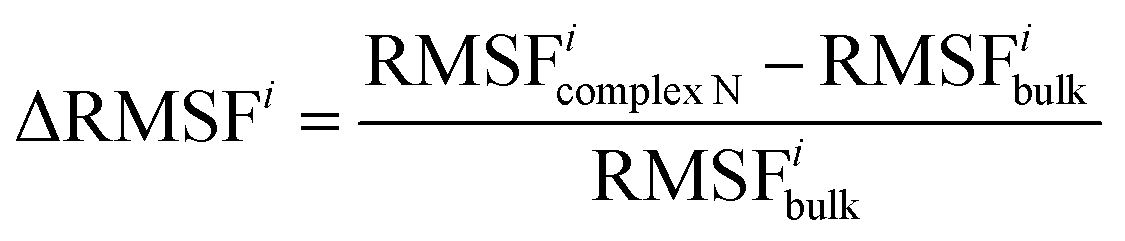 | (1) |
Definition of secondary structure score Sicoil, Sihelix, Sibend, and Siturn
We define four scores that measure the secondary structure for residue i: Sicoil, Sihelix, Sibend, and Siturn. They are calculated as Ncoil/Ntotal, Nhelix/Ntotal, Nbend/Ntotal, and Nturn/Ntotal, where Ncoil, Nhelix, Nbend, and Nturn are the numbers of frames in which residue i adopts the coil, helix, bend and turn structures in the trajectories and Ntotal is the total number of frames.Definition of ΔSeci
The parameter ΔSeci is defined to characterize the maximum change in secondary structure of residue i when the HSA protein binds to a gold NP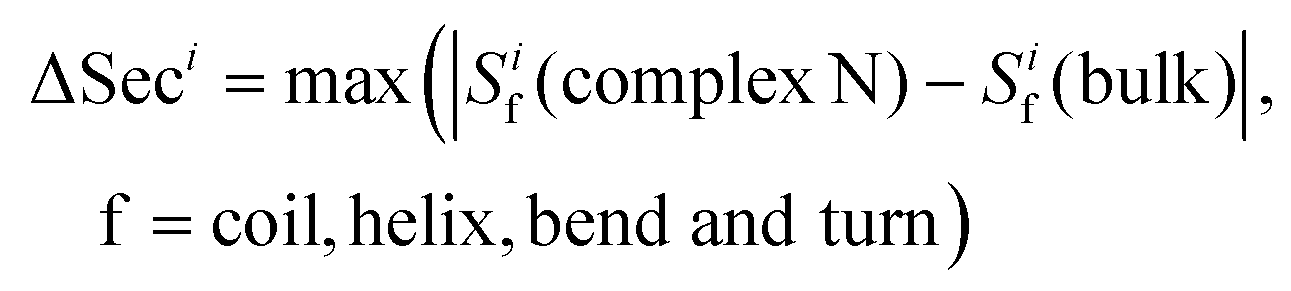 | (2) |
Results and discussion
Gold NP binds to multiple regions on HAS
The gold NP forms multiple binding configurations with the HSA protein as expected due to the chemical heterogeneity of the protein. Fig. 3 shows the atomistic configurations of the four NP–HSA binding structures that have the global and local lowest interaction energies. The protein–NP interaction energies of complex A (−1377 kJ mol−1) is lower than those of complexes B (−1061 kJ mol−1), C (−1054 kJ mol−1) and D (−1091 kJ mol−1). Complex A is the configuration that has the global minimum interaction energy, while the other three complexes are at local minimum interaction energies. Comparing the interaction energies of the four complexes shows that the probability for complex A to occur is higher than those for the other three complexes. Nevertheless, it is important to investigate complexes B, C, and D because they can still form even though the probability is low. Furthermore, switching from one complex to another may require overcoming a sizeable energy barrier, meaning all four complexes could be stable once formed. Thus, we investigate the influence of the gold NP on HSA structure and flexibility in all four complexes.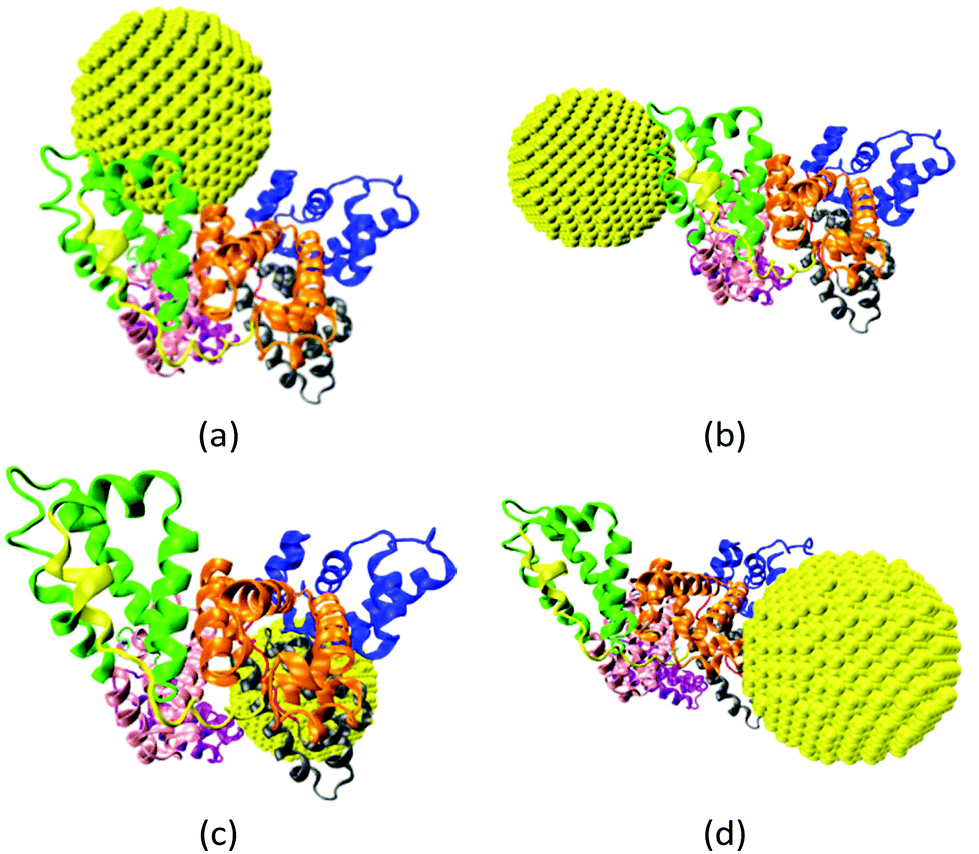 | ||
| Fig. 3 Four binding configurations of an HSA protein and a 4.0 nm gold NP. (a) Complex A, (b) complex B, (c) complex C and (d) complex D. The HSA protein is shown in the New Cartoon form and the gold atoms are shown in the VDW form. The subdomains of the HSA are shown in different colors (subdomain Ia: 5–105, Ia–Ib loop: 106–118, subdomain Ib: 119–195, subdomain IIa: 196–292, IIa–IIb loop: 293–313, subdomain IIb: 314–383, subdomain IIIa: 384–491, IIIa–IIb loop: 492–509, IIb subdomain: 510–582). The figures are generated using the VMD package.60 | ||
We analyze the number and spatial distribution of the residues in the “NP-interacting domain” in the four complexes. A residue is in the NP-interacting domain if the residue–NP interaction energy is negative in the complexes identified during the brute-force search process. The residues in the NP-interacting domain account for less than 5% of the total residues on an HSA. This indicates that the gold NP just binds to a small portion of the HSA. The NP-interacting domain is distributed over several HSA subdomains and the distribution varies among the complexes. For instance, in complex A the NP-interacting domain is distributed in four subdomains (Ia, IIa, IIb and IIIa), and in complex B it is distributed in a subdomain (IIIb) and a loop (IIa–IIb). The wide spatial distribution of the NP-interacting domain implies that the NP may influence properties of residues that are distributed widely on the HSA protein. All of this indicates that HSA protein exhibit different variations in structure and flexibility upon the NP binding.
The residues in the NP-interacting domain in the four complexes also exhibit a wide distribution of chemistries. The 20 amino acid residues are divided into six categories: hydrophobic (Ala, Gly, Ile, Leu, Met, Pro, and Val), hydrophilic (Cys, Ser, and Thr), aromatic (Phe, Tyr, and Trp), acidic (Asp and Glu), basic (Arg, His, and Lys), and amide (Asn and Gln). Table 1 lists the number of residues in the six categories. Residues in all six categories are found in the NP-interacting domain. Among the six categories, the hydrophobic, acidic and hydrophilic residues appear more than the others, accounting for 72.7% of the residues in the NP-interacting domain. The basic residues appear much less than the others; only complexes B and D have one and two basic residues in the NP-interacting domain. The distribution of residue chemistries in the NP-interacting domain depends not only on the interaction strength between individual amino-acid residues and the gold NP but also on the distribution of amino-acid residues on the HSA, which is the consequence of long-time evolution.
| Types of residues | ||||||
|---|---|---|---|---|---|---|
| Hydrophobic | Hydrophilic | Aromatic | Acidic | Basic | Amide | |
| A | 5 | 5 | 1 | 3 | 0 | 4 |
| B | 9 | 2 | 2 | 3 | 1 | 1 |
| C | 11 | 4 | 2 | 5 | 0 | 3 |
| D | 5 | 1 | 0 | 9 | 2 | 2 |
HSA protein remains folded when bound to gold NP
We first determine if the HSA protein in the four complexes remains folded during the simulations. A typical signal that a protein is unfolded or about to unfold is a significant increase in RMSD of the Cα atoms from the native state. Fig. 4 shows the RMSDs of Cα atoms of the HSA proteins in the four complexes (100–200 ns) and in the bulk solution (0–100 ns). The RMSD of the HSA protein in the bulk solution fluctuates from 0.35 to 0.5 nm, consistent with the RMSD fluctuation ranges reported by the Colina group52 and the Szleifer group53 in their simulations of an HSA protein in explicit solvent. The RMSDs for the HSA in the four complexes fluctuate in ranges similar to that of the HSA in bulk solution. The similarity of the RMSD ranges indicates that the HSA protein in the four complexes remains folded during the simulations.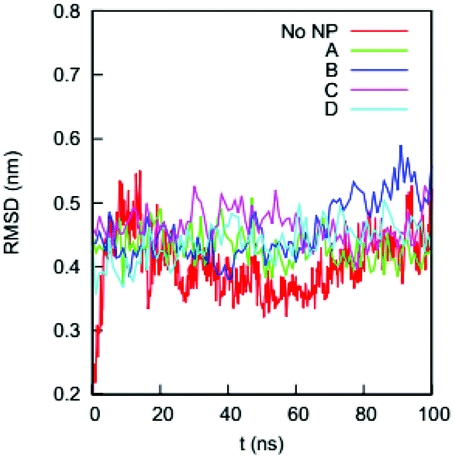 | ||
| Fig. 4 RMSDs of the Cα atoms on the HSA protein in the bulk solution and in the four complexes in explicit solvent. The crystal structure of the HSA (PDB ID: 1OA6) is used as the reference. | ||
We also determine if the binding of the gold NP causes the NP-interacting domain to unfold. (Raffaini et al.54 showed that the region close to the substrate likely unfolds first.) Fig. 5 shows the RMSDs of the Cα atoms of the residues in the NP-interacting domain on the HSA protein in the four complexes and the bulk solution. For complex A, the values for the RMSDs change from 0.2–0.3 nm (bulk) to 0.2 nm (complex); for complex B, they change from 0.2–0.4 nm (bulk) to 0.4 nm (complex); for complex C, they change from 0.3–0.8 nm (bulk) to 0.3 nm (complex); and for complex D, they change from 0.1–0.4 nm (bulk) to 0.4 nm (complex). The ranges of RMSD values for the complexes overlap with those in the bulk solution and do not increase significantly with time upon exposure to the NP binding. This excludes the possibility that the residues in the NP-interacting domain undergo an unfolding process.
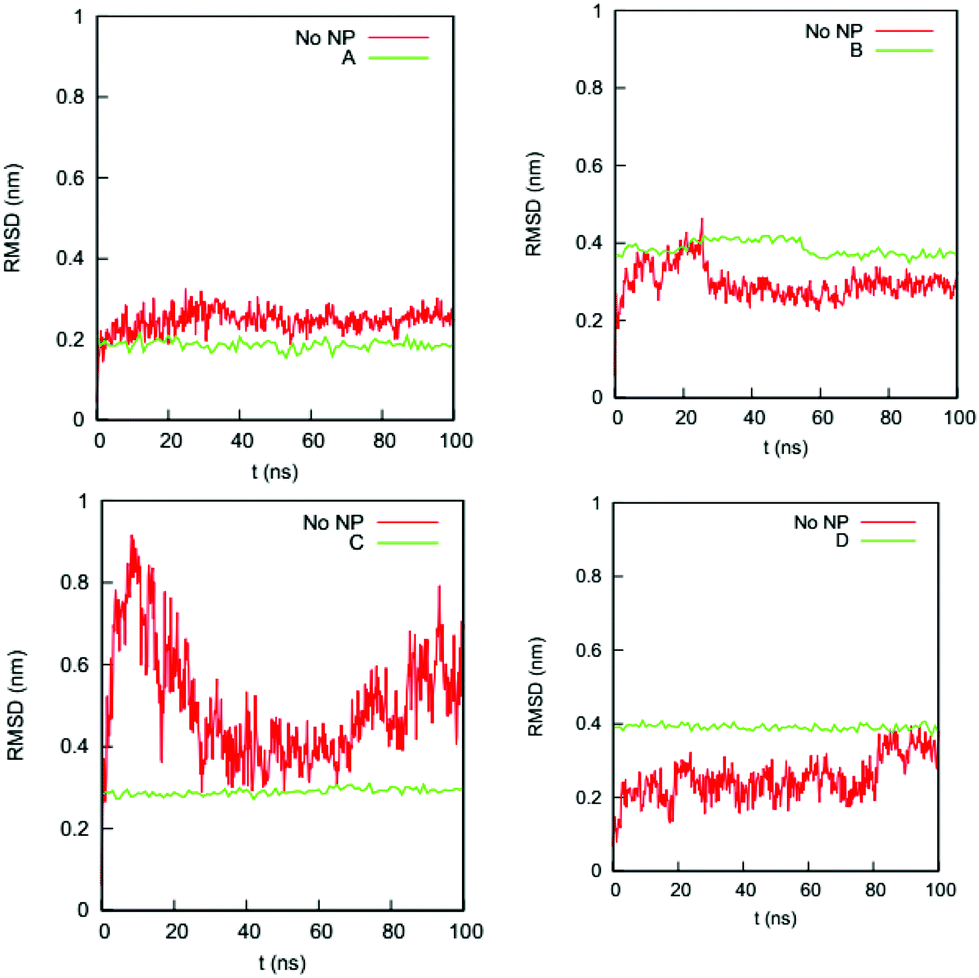 | ||
| Fig. 5 RMSDs of Cα atoms of the residues in the NP-interacting domain of the HSA protein in complexes (a) A, (b) B, (c) C, and (d) D and in the bulk solution (no NP). | ||
The narrow ranges of values for the Cα atom RMSD for the residues in the NP-interacting domain of the four complexes imply that the NP binding decreases the flexibility of these residues. As shown in Fig. 5, the fluctuations in the RMSD for the residues in the NP-interacting domain change from 0.1–0.5 nm to almost zero upon binding to the NP. This decrease in fluctuation could be caused by forces directly exerted by the gold NP on the protein or an adjustment in the hydration layer around the protein. The decrease in flexibility may constrain more than just the NP-interacting domain. It could spread through the HSA protein via the backbones and residue–residue non-bonded interactions. The spatial and chemical distributions of the NP-interacting domain may aid in this spread. To further probe how the gold NP constrains the HSA protein, we analyze RMSFs of individual residues in the next section.
Binding of gold NP constrains flexibility of many residues
The impact of the gold NP on the HSA's flexibility can be analyzed by comparing the RMSFs of individual Cα atoms in the complexes to those in the bulk solution. This is of interest because changes in protein flexibility can fundamentally influence a protein's biological function. Fig. 6 shows the RMSFs of individual Cα atoms on the HSA protein in the four complexes and the bulk solution. We find dissimilarities between the RMSF curves for the complexes and the bulk solution in residues across the whole amino acid sequence. The NP binding changes the flexibility of residues that are distributed not only on protein regions close to the NP-interacting domain but also on those distant from the domain. | ||
| Fig. 6 RMSFs of individual Cα atoms on the HSA protein in the four complexes and in the bulk solution (no NP). | ||
To further analyze the change in flexibility of individual residues on the HSA protein in the presence of a gold NP, we calculate the ΔRMSFi parameter defined in the simulation method section. Fig. S1† plots ΔRMSFivs. the distance (d) between the Cα atom and the center of mass (COM) of the gold NP for all the residues in the four complexes. The wide distribution of these dots shows that the change in flexibility occurs over the whole protein. As shown in Fig. S1,† we can even find residues with d > 7.0 nm that have ΔRMSFi < −0.5. The four complexes exhibit different changes in their flexibility. In complexes A and C, nearly all the residues have their flexibility decrease. In complexes B and D, although most of the residues have their flexibility decrease, some have their flexibility increase.
To quantitatively assess the change in flexibility and its relationship to the distance between the residue and the gold NP, we divide all the residues into six groups based on the values of ΔRMSFi and d. Since the HSA is a flexible protein, we chose ΔRMSFi = −0.5 and ΔRMSFi = 0.5 as the threshold values to determine if the flexibility of a residue decreases significantly, changes moderately, or increases significantly. We chose d = 5.0 nm as the threshold to determine if a residue is distant from the NP (d ≥ 5.0 nm) or not (d < 5.0 nm). Table 2 lists the numbers of residues in the six groups for the four complexes.
| 1a | 1b | 1c | 2a | 2b | 2c | |
|---|---|---|---|---|---|---|
| d (nm) | <5.0 | ≥5.0 | ||||
| ΔRMSFi | <−0.5 | [−0.5, 0.5] | >0.5 | <−0.5 | [−0.5, 0.5] | >0.5 |
| A | 204 | 201 | 0 | 83 | 90 | 0 |
| B | 34 | 179 | 0 | 16 | 349 | 0 |
| C | 266 | 87 | 0 | 93 | 131 | 0 |
| D | 11 | 355 | 8 | 25 | 203 | 6 |
Comparing the numbers of residues in the six groups among the four complexes shows that the binding position of the NP plays an important role in determining how significantly it changes the protein flexibility. For instance, the sums of the number of residues in groups 1a and 2a are 287 for complex A and 50 for complex B. Thus, in complex A, the NP binding decreases the RMSF of nearly 50% of the residues on the HSA protein by more than 50%, while in complex B, less than 10% of the residues on the HSA protein decrease their flexibility more than 50%.
Binding to the 4.0 nm gold NP can also increase the flexibility of some residues on an HSA protein. As shown in Table 2, 14 residues on complex D are in groups 1c and 2c, indicating that their flexibility increases by more than 50% upon NP binding. However, complex D is the only one that has a residue flexibility increase among the four complexes, and the number of such residues is smaller than the number of residues that decrease their flexibility. The increase in the flexibility of the residues shows that the NP binding can influence protein properties in unexpected ways. We will discuss the reasons for flexibility increases when we describe the mechanisms that govern the impact of NP binding on protein structure and flexibility.
The change in flexibility of so many residues in the four complexes supports our argument that NPs can induce toxicity without denaturing proteins. In the next section, we investigate how the binding of a gold NP influences the secondary structure of an HAS protein. We also discuss the mechanisms by which a NP can change the flexibility and secondary structure of a protein without denaturing it.
Binding of gold NP changes secondary structure of residues on HAS protein
Changing a protein's secondary structure can alter its biological function, generate new epitopes, or trigger further conformational changes. We determine the secondary structure of an HAS protein using the DSSP (Define Secondary Structure of Proteins) program.55 The residues are divided into coil, turn, bend, bridge, and helix categories based on their hydrogen bonding structures.We first analyze the numbers of residues in the five secondary structures on the HSA proteins in the complexes and in the bulk solution. The fraction of each type of secondary structure can be obtained from circular dichroism (CD) measurement, which is widely used to determine if proteins are folded. Fig. 7 shows the numbers of residues in the coil, helix, and bend + turn structures in the four complexes and the bulk solution. The number of residues in the bridge structure is neglected because it is very small. The HSA protein in bulk solution has around 400 residues in helix structure, more than 80 residues in coil structure, and around 40 residues in bend or turn structure. This secondary-structure distribution is consistent with the fact that the HSA protein is rich in helical structure. The secondary-structure distributions for the HSA proteins in the four complexes are very similar to that in the bulk solution. This similarity indicates that the HSA protein remains folded in the complex, consistent with our previous conclusion based on the RMSDs of Cα atoms. If we rely only on the number distributions in Fig. 7, binding to the gold NP does not appear to significantly change the secondary structure of the HSA protein. This would also likely be the conclusion from a CD measurement. However, the analysis of secondary structure of individual residues show that NP binding can change protein conformation. We use a parameter introduced in method to quantify the change in secondary structure per residue: the secondary structure change factor ΔSeci, which quantifies how the binding of the gold NP causes the change in secondary structure of residue i.
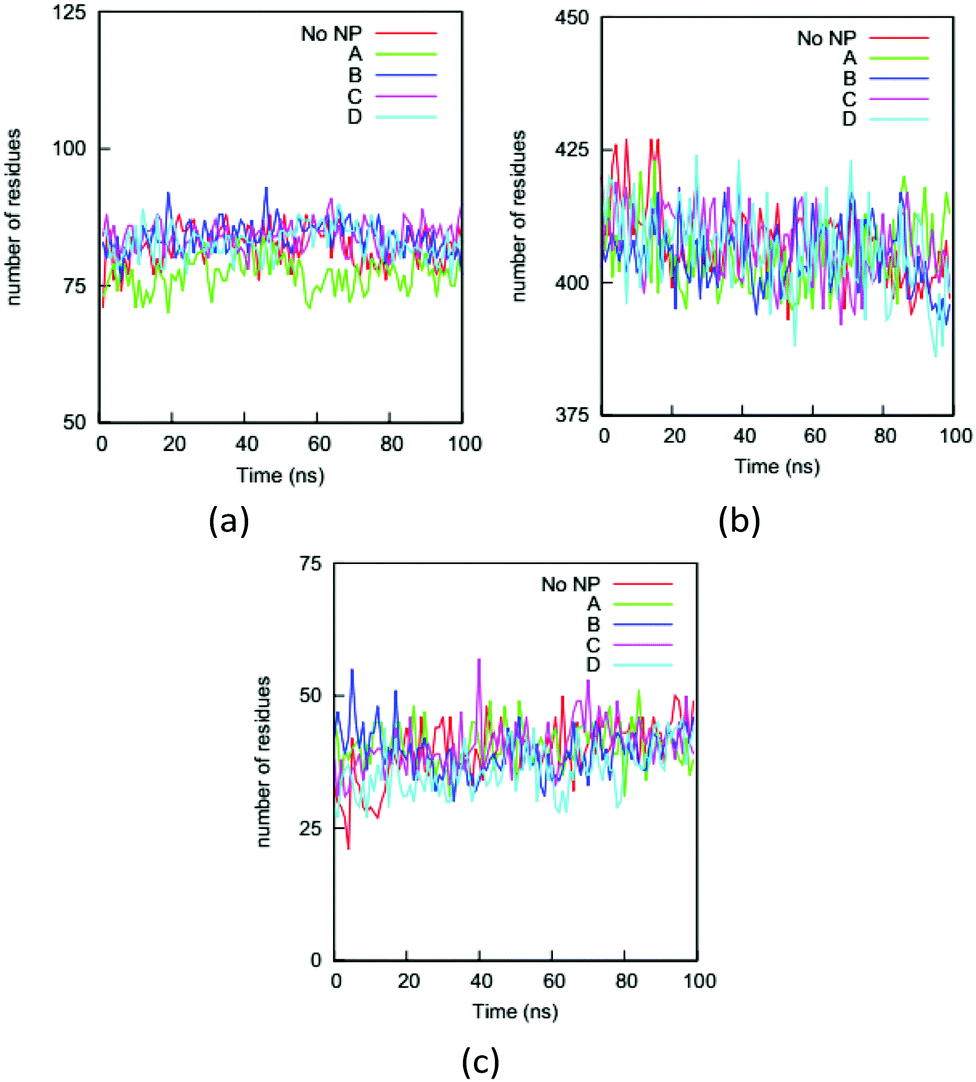 | ||
| Fig. 7 Numbers of residues on the HSA protein in the four complexes and the bulk solution (No NP) in (a) coil, (b) helix and (c) bend + turn during the simulations. As expected, the helix structure dominates the HSA protein. The similarity between the curves for the complexes and the bulk solution indicates that a CD measurement on the protein would not reflect the influence of the NP binding. | ||
The analysis of ΔSeci, indicates that many residues change their secondary structure upon NP binding, which the number distribution of secondary structure on the whole protein fails to reveal. Fig. S2† shows ΔSecivs. the distance d between the Cα atom on residue i and the COM of the gold NP for all the residues on the HSA protein in the four complexes. We divide the residues on the HSA protein into six groups based on the values of ΔSeci and d: residues that retain (ΔSeci < 0.2), have moderate change (0.2 ≤ ΔSeci ≤ 0.5) or significantly change their secondary structure (ΔSeci > 0.5), and residues close to (d ≤ 5.0 nm) or distant from NP (d ≥ 5.0 nm). Table 3 lists the numbers of residues in the six groups. Many residues belong to groups 1a and 2a. The total numbers of residues in groups 1a and 2a is 458 for complexes A and B, 468 for complex C, and 431 for complex D. These numbers indicate that 73–80% of the residues on an HSA retain their secondary structure when the protein binds to a 4.0 nm NP. They may contribute to stabilizing the protein structure.
| 1a | 1b | 1c | 2a | 2b | 2c | |
|---|---|---|---|---|---|---|
| d (nm) | <5.0 | ≥5.0 | ||||
| ΔSeci | <0.2 | [0.2, 0.5] | >0.5 | <0.2 | [0.2, 0.5] | >0.5 |
| A | 329 | 43 | 33 | 129 | 13 | 31 |
| B | 160 | 22 | 31 | 298 | 39 | 28 |
| C | 279 | 27 | 48 | 189 | 27 | 8 |
| D | 226 | 29 | 24 | 205 | 49 | 45 |
However, quite a few residues on the HSA protein change their secondary structure when the protein binds to the NP. As shown in Table 3, the sum of the residues in groups 1c and 2c are 64 for complex A, 59 for complex B, 56 for complex C and 69 for complex D. These numbers mean that around 10% of the residues on an HSA change their secondary structure significantly when it binds to a 4.0 nm NP. Changes in the secondary structures of even a few residues could significantly change biological function of proteins. These changes may alter the biological function of HSA, which suggests that the 4.0 nm NP could induce toxicity.
Fig. 8 shows the secondary structures of the 31 residues in complex A and the bulk solution that have ΔSeci > 0.5 and di > 5.0 nm over the simulation time. These residues are distant from the gold NP but all change their secondary structure significantly upon NP binding. Table S4† lists their residue IDs. Note that the 31 residues are distributed widely on the protein; here we number them continuously only for convenience in plotting the figure. Comparing the secondary structure types in complex A (Fig. 8a) and the bulk solution (Fig. 8b) illustrates the diversified and significant transfers that occur among secondary structures. For instance, residues labeled 3–4 and 16–17 change from the coil structure to the bend structure, while the residues labeled 5 to 15 change from the helix structure to the coil structure.
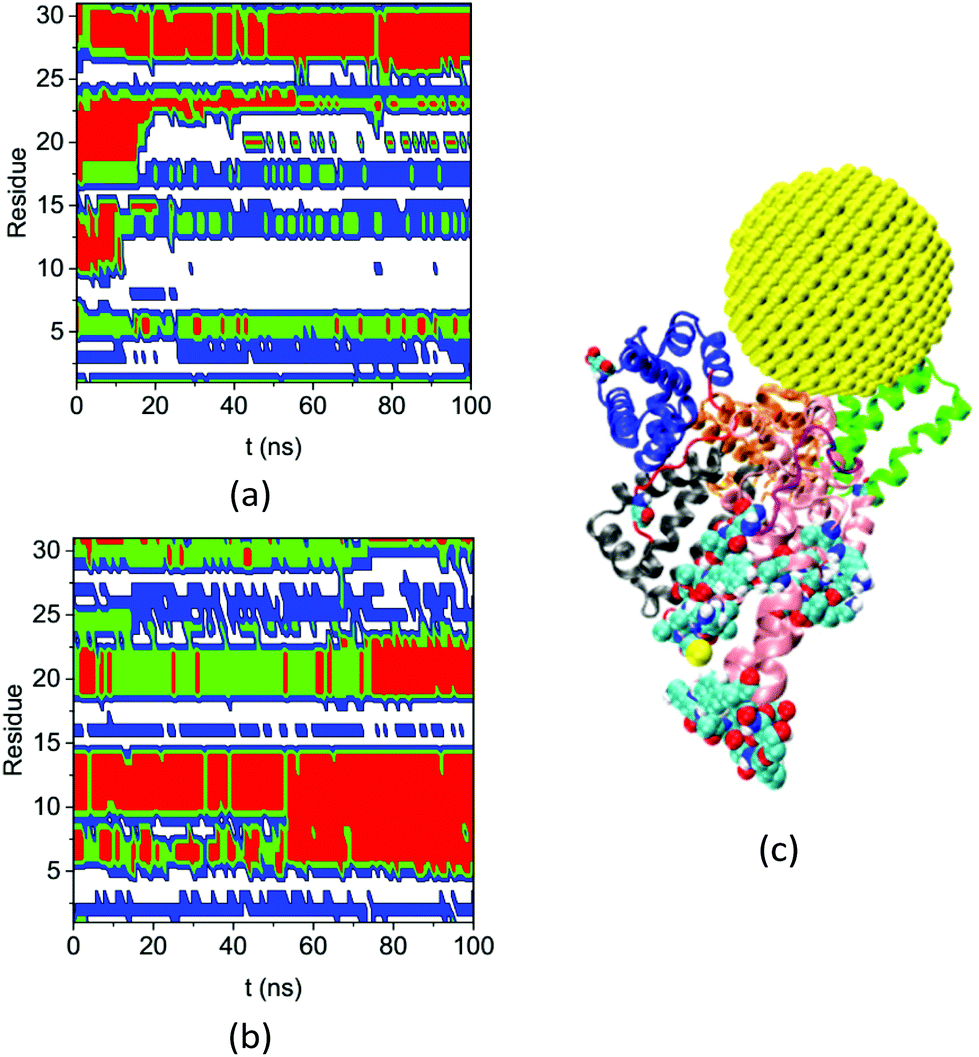 | ||
| Fig. 8 Secondary structure of residues that have ΔSeci > 0.5 and di > 5.0 nm on the HSA protein in (a) complex A and (b) bulk solution during the course of the simulation (coil: white, bend: blue: turn: green, and red: helix), (c) the configuration of complex A at 200 ns molecular dynamics (MD) simulation. The subdomains and loops are shown in secondary structure with the same color as in Fig. 3. The 31 residues are shown in the VDW model. | ||
The analyses of the RMSDs, RMSFs, and secondary structures presented above show that the binding of a NP changes the structure and flexibility of an HSA protein without denaturing it. The changes originate from contact between the NP and a small region on the protein, but propagate to a much wider protein region. For instance, as shown in our simulations, a 4.0 nm gold NP directly interacts with only ∼20 residues (<5%) on an HSA protein, but this binding causes approximately 50% of the residues to decrease their flexibility by a factor of 1/2 (Table 2), and around 10% of the residues to change their secondary structure significantly (Table 3). These residues are distributed on the whole HSA protein, not just on the region close to the NP binding site (Fig. S1 and S2†). If the ability of NPs to change protein structure and flexibility is a major source of their toxicity, our simulations imply that NPs could have a high risk of inducing toxicity even if they let proteins remain folded.
Allosteric effects depend on the complicated non-bonded and bonded interaction network within a protein. Every residue can be considered as a point on this network that receives and sends forces to or from other points. The behavior of individual residues is the synergetic consequence of these forces. Even though the binding of a NP affects the behavior of ∼20 residues directly, it changes the forces on these points, which then spread throughout the whole protein via the network, altering the behavior of other residues. The magnitude of the change in the forces on individual residues upon NP binding determines if they become more flexible than otherwise and consequently prefer to adopt a different secondary structure. The position of the NP binding site influences how it affects the network and consequentially the behavior of every residue. This explains why the HSA proteins in the four complexes exhibit different changes in flexibility and secondary structure and some residues even exhibit a flexibility increase upon NP binding. The allosteric effects of NPs on proteins should also depend on NP shape and size. Given the same surface composition, NP shape and size affect the number of amino acid residues that the NP interacts with and the interaction strength. For instance, in our previous work, we showed that the binding strength of amino acid residues on gold NPs changes as a function of nanoparticle diameter. The number of affected amino acids and the structure and flexibility of individual affected amino acids are one of the origins of the allosteric effects of the NP on proteins. Thus, we could expect that a change in NP size and shape would alter the NP's allosteric effect on proteins. We need to thoroughly understand the mechanistic details of the protein network to predict the consequences of the NP binding to the structure and flexibility of a protein.
Allosteric effects of small-molecule ligands on HSA proteins have been extensively studied.56 An HSA protein possesses multiple binding sites.57 Small ligands influence the binding affinities of each other even though their binding sites are far apart, strong evidence for their allosteric effect on HSA proteins.56 For instance, the binding of heme-Fe(III) can decrease the binding affinity of warfarin on Sudlow's site.58,59 We hypothesize that NPs should influence multiple binding sites on HSA proteins through an allosteric effect and that their presence changes the structure and flexibility of amino acid residues more strongly than the presence of small-molecule ligands. We will determine if the gold NP affects other binding sites on HSA protein in the next section.
Presence of gold NPs have the potential to allosterically regulate binding affinity of ligands on HSA protein
We investigate the allosteric effects of a 4.0 nm gold NP on biologically-relevant binding sites on HSA proteins by determining if any of the residues on these binding sites change their secondary structure or flexibility due to the presence of the gold NP. The considered binding sites are the nine fatty acid binding sites (FA1–FA9), the Heme binding site, the four thyroxin binding sites (Tr1–Tr4), the bacteria protein binding site (BacP) and several metal binding sites (Metal). The amino acid residues on these binding sites are listed in Table S5.† We assume that a binding site has a “high” possibility to be affected by the NP allosterically if an amino acid on the binding site has changes in secondary structure or flexibility upon NP binding.We find that 4.0 nm gold NP can allosterically affect multiple binding sites on HSA proteins. Table 4 lists the binding sites that could be changed through an allosteric regulation. In all four complexes, some binding sites on HSA protein have a high possibility to be affected allosterically upon gold NP binding. In complex A, the gold NP can allosterically affect the function of six binding sites (FA1, Heme, FA5, Tr2, Tr3 and Tr4); in complex B, the gold NP can affect the function of three binding sites (Heme, Tr3, Tr4 and Metal), in complex C, the gold NP can affect the function of nine binding sites (FA3, FA4, FA6, FA7, FA8, Tr1, Tr2, Tr3, Tr4 and BacP); and in complex D, the gold NP can affect the function of five binding sites (FA3, FA6, FA8, Tr3 and Tr4).
| Binding site | A | B | C | D |
|---|---|---|---|---|
| FA1 | High | |||
| Heme | High | High | ||
| FA2 | ||||
| FA3 | High | High | ||
| FA4 | High | |||
| FA5 | High | |||
| FA6 | High | High | ||
| FA7 | High | |||
| FA8 | High | High | ||
| FA9 | ||||
| Tr1 | High | |||
| Tr2 | High | High | ||
| Tr3/Tr4 | High | High | High | High |
| BacP | High | |||
| Metal | High | |||
The allosteric effect of a NP can be more profound than that of a small-molecule ligand. As shown in Table 4, a 4.0 nm gold NP can affect allosterically at least four binding sites on HSA proteins, while small-molecule ligands usually allosterically affect the function of only one binding site. The more profound allosteric effect of gold NPs is due to their stronger interactions with more amino acids on proteins, comparing to small-molecule ligands. NPs may provide a more effective way to modulate allosterically the function of some binding sites on proteins than small-molecule ligands.
Conclusions
Here we have shown that a nanoparticle can change the structure and flexibility of residues on protein region distant from its binding site. Computer simulations show that the binding of a gold nanoparticle with a diameter of 4.0 nm influences the structure and flexibility of a human serum albumin protein significantly without denaturing it. A brute-force search process is used to identify four gold NP–HSA complexes with the global and local lowest protein–nanoparticle interaction energies using a coarse-grained protein/nanoparticle model. We then calculate the RMSD and RMSF of the Cα atoms, and the secondary structures of the residues on the HSA protein in the four complexes and in the bulk solution using explicit solvent atomistic MD simulations. The proteins in the complexes exhibit RMSDs similar to that in the bulk solution, indicating that they remain folded when bound to the NP. Comparing RMSFs of the residues in the complexes and in the bulk solution shows, however, that many residues decrease their flexibility when the nanoparticle binds to the protein. The binding of the gold nanoparticle also induces 10% of the residues to change their secondary structure significantly. The mechanism by which the binding of the nanoparticle influences the behavior of so many residues is hypothesized to be an allosteric effect. The residues that change their flexibility and secondary structures are distributed on the whole protein instead of just on the protein region near the nanoparticle binding site. We analyze the changes in structure and flexibility of amino acid residues on a variety of binding sites on albumin and confirm that nanoparticles could allosterically affect the ability of albumin to bind fatty acid, thyroxin and metals.Current research into the molecular-level mechanisms underlying nanoparticle influence on protein structure and flexibility focuses mostly on regions nearby the nanoparticle binding site. Our simulations suggest that allosteric effects of nanoparticles are another must-consider source of their influence. The allosteric effects of nanoparticles allow them to distort the structure and flexibility of protein regions distant from the binding site. These changes could alter protein function even if the nanoparticles do not bind to the active sites directly. Our simulation results also show the importance of developing methods to detect structural and flexibility variation in local protein domains. The results show that changes in protein structure and flexibility can occur when the proteins remain folded. Thus, some widely-used biophysical measurements such as CD measurements which focus on signals from the whole protein may fail to discover these changes and thus give false information on the influence of NP bindings on protein conformation.
Acknowledgements
This work was supported by National Science Foundation (CBET-1236053) and the National Institutes of Health (EB006006). This work used the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation grant number ACI-1053575. This work was supported in part by the Research Triangle NSF-MRSEC on Programmable Soft Matter, DMR-1121107.References
- X. Yang, M. Yang, B. Pang, M. Vara and Y. Xia, Chem. Rev., 2015, 115, 10410–10488 CrossRef CAS PubMed.
- C. J. Cheng, G. T. Tietjen, J. K. Saucier-Sawyer and W. M. Saltzman, Nat. Rev. Drug Discovery, 2015, 14, 239–247 CrossRef CAS PubMed.
- M. Auffan, J. Rose, J. Y. Bottero, G. V. Lowry, J. P. Jolivet and M. R. Wiesner, Nat. Nanotechnol., 2009, 4, 634–641 CrossRef CAS PubMed.
- S. J. Klaine, P. J. J. Alvarez, G. E. Batley, T. F. Fernandes, R. D. Handy, D. Y. Lyon, S. Mahendra, M. J. McLaughlin and J. R. Lead, Environ. Toxicol. Chem., 2008, 27, 1825–1851 CrossRef CAS PubMed.
- G. V. Lowry, K. B. Gregory, S. C. Apte and J. R. Lead, Environ. Sci. Technol., 2012, 46, 6893–6899 CrossRef CAS PubMed.
- C. R. Thomas, S. George, A. M. Horst, Z. Ji, R. J. Miller, J. R. Peralta-Videa, T. Xia, S. Pokhrel, L. Mädler, J. L. Gardea-Torresdey, P. A. Holden, A. A. Keller, H. S. Lenihan, A. E. Nel and J. I. Zink, ACS Nano, 2011, 5, 13–20 CrossRef CAS PubMed.
- K. Simeonidis, S. Mourdikoudis, E. Kaprara, M. Mitrakas and L. Polavarapu, Environ. Sci.: Water Res. Technol., 2016, 2, 43–70 Search PubMed.
- L. Mu and R. L. Sprando, Pharm. Res., 2010, 27, 1746–1749 CrossRef CAS PubMed.
- T. V. Duncan, J. Colloid Interface Sci., 2011, 363, 1–24 CrossRef CAS PubMed.
- Y. Echegoyen and C. Nerín, Food Chem. Toxicol., 2013, 62, 16–22 CrossRef CAS PubMed.
- A. Nel, T. Xia, L. Mädler and N. Li, Science, 2006, 311, 622–627 CrossRef CAS PubMed.
- S. A. Love, M. A. Maurer-Jones, J. W. Thompson, Y.-S. Lin and C. L. Haynes, Annu. Rev. Anal. Chem., 2012, 5, 181–205 CrossRef CAS PubMed.
- S. H. D. P. Lacerda, J. J. Park, C. Meuse, D. Pristinski, M. L. Becker, A. Karim and J. F. Douglas, ACS Nano, 2010, 4, 365–379 CrossRef PubMed.
- M. P. Monopoli, D. Walczyk, A. Campbell, G. Elia, I. Lynch, F. Baldelli Bombelli and K. A. Dawson, J. Am. Chem. Soc., 2011, 133, 2525–2534 CrossRef CAS PubMed.
- J. Wang, U. B. Jensen, G. V. Jensen, S. Shipovskov, V. S. Balakrishnan, D. Otzen, J. S. Pedersen, F. Besenbacher and D. S. Sutherland, Nano Lett., 2011, 11, 4985–4991 CrossRef CAS PubMed.
- R. Huang, R. P. Carney, F. Stellacci and B. L. T. Lau, Nanoscale, 2013, 5, 6928–6935 RSC.
- M. J. Penna, M. Mijajlovic and M. J. Biggs, J. Am. Chem. Soc., 2014, 136, 5323–5331 CrossRef CAS PubMed.
- D. J. O'Connell, F. B. Bombelli, A. S. Pitek, M. P. Monopoli, D. J. Cahill and K. A. Dawson, Nanoscale, 2015, 7, 15268–15276 RSC.
- M. Lundqvist, I. Sethson and B.-H. Jonsson, Langmuir, 2004, 20, 10639–10647 CrossRef CAS PubMed.
- Z. Cao and S. Jiang, Nano Today, 2012, 7, 404–413 CrossRef CAS.
- B. Pelaz, P. del Pino, P. Maffre, R. Hartmann, M. Gallego, S. Rivera-Fernández, J. M. de la Fuente, G. U. Nienhaus and W. J. Parak, ACS Nano, 2015, 9, 6996–7008 CrossRef CAS PubMed.
- G. Zuo, Q. Huang, G. Wei, R. Zhou and H. Fang, ACS Nano, 2010, 4, 7508–7514 CrossRef CAS PubMed.
- F. Ding, S. Radic, R. Chen, P. Chen, N. K. Geitner, J. M. Brown and P. C. Ke, Nanoscale, 2013, 5, 9162–9169 RSC.
- J. J. Gray, Curr. Opin. Struct. Biol., 2004, 14, 110–115 CrossRef CAS PubMed.
- I. Lynch and K. A. Dawson, Nano Today, 2008, 3, 40–47 CrossRef CAS.
- A. E. Nel, L. Madler, D. Velegol, T. Xia, E. M. V. Hoek, P. Somasundaran, F. Klaessig, V. Castranova and M. Thompson, Nat. Mater., 2009, 8, 543–557 CrossRef CAS PubMed.
- M. Mahmoudi, I. Lynch, M. R. Ejtehadi, M. P. Monopoli, F. B. Bombelli and S. Laurent, Chem. Rev., 2011, 111, 5610–5637 CrossRef CAS PubMed.
- C. D. Walkey and W. C. W. Chan, Chem. Soc. Rev., 2012, 41, 2780–2799 RSC.
- F. Jacob and J. Monod, J. Mol. Biol., 1961, 3, 318–356 CrossRef CAS PubMed.
- J. Monod, J. Wyman and J.-P. Changeux, J. Mol. Biol., 1965, 12, 88–118 CrossRef CAS PubMed.
- D. E. Koshland, G. Némethy and D. Filmer, Biochemistry, 1966, 5, 365–385 CrossRef CAS PubMed.
- H. N. Motlagh, J. O. Wrabl, J. Li and V. J. Hilser, Nature, 2014, 508, 331–339 CrossRef CAS PubMed.
- E. C. Dreaden, A. M. Alkilany, X. Huang, C. J. Murphy and M. A. El-Sayed, Chem. Soc. Rev., 2012, 41, 2740–2779 RSC.
- Q. Peng, S. Zhang, Q. Yang, T. Zhang, X.-Q. Wei, L. Jiang, C.-L. Zhang, Q.-M. Chen, Z.-R. Zhang and Y.-F. Lin, Biomaterials, 2013, 34, 8521–8530 CrossRef CAS PubMed.
- G. Duan, S.-g. Kang, X. Tian, J. A. Garate, L. Zhao, C. Ge and R. Zhou, Nanoscale, 2015, 7, 15214–15224 RSC.
- L. Treuel, M. Malissek, J. S. Gebauer and R. Zellner, ChemPhysChem, 2010, 11, 3093–3099 CrossRef CAS PubMed.
- S. Lindman, I. Lynch, E. Thulin, H. Nilsson, K. A. Dawson and S. Linse, Nano Lett., 2007, 7, 914–920 CrossRef CAS PubMed.
- S. P. Boulos, T. A. Davis, J. A. Yang, S. E. Lohse, A. M. Alkilany, L. A. Holland and C. J. Murphy, Langmuir, 2013, 29, 14984–14996 CrossRef CAS PubMed.
- D.-H. Tsai, F. W. DelRio, A. M. Keene, K. M. Tyner, R. I. MacCuspie, T. J. Cho, M. R. Zachariah and V. A. Hackley, Langmuir, 2011, 27, 2464–2477 CrossRef CAS PubMed.
- L. Calzolai, F. Franchini, D. Gilliland and F. Rossi, Nano Lett., 2010, 10, 3101–3105 CrossRef CAS PubMed.
- L. Wang, J. Li, J. Pan, X. Jiang, Y. Ji, Y. Li, Y. Qu, Y. Zhao, X. Wu and C. Chen, J. Am. Chem. Soc., 2013, 135, 17359–17368 CrossRef CAS PubMed.
- J. Zhou, H.-K. Tsao, Y.-J. Sheng and S. Jiang, J. Chem. Phys., 2004, 121, 1050–1057 CrossRef CAS PubMed.
- M. Deighan and J. Pfaendtner, Langmuir, 2013, 29, 7999–8009 CrossRef CAS PubMed.
- Q. Shao and C. K. Hall, Langmuir, 2016, 32, 7888–7896 CrossRef PubMed.
- N. Schmid, A. Eichenberger, A. Choutko, S. Riniker, M. Winger, A. Mark and W. van Gunsteren, Eur. Biophys. J., 2011, 40, 843–856 CrossRef CAS PubMed.
- H. Heinz, R. A. Vaia, B. L. Farmer and R. R. Naik, J. Phys. Chem. C, 2008, 112, 17281–17290 CAS.
- H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren and J. Hermans, Intermolecular Forces Reidel, Dordrecht, 1981 Search PubMed.
- G. Bussi, D. Donadio and M. Parrinello, J. Chem. Phys., 2007, 126, 014101 CrossRef PubMed.
- U. Essmann, L. Perera, M. L. Berkowitz, T. Darden, H. Lee and L. G. Pedersen, J. Chem. Phys., 1995, 103, 8577–8593 CrossRef CAS.
- B. Hess, J. Chem. Theory Comput., 2008, 4, 116–122 CrossRef CAS PubMed.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1–2, 19–25 Search PubMed.
- M. M. Castellanos and C. M. Colina, J. Phys. Chem. B, 2013, 117, 11895–11905 CrossRef CAS PubMed.
- K. Baler, O. A. Martin, M. A. Carignano, G. A. Ameer, J. A. Vila and I. Szleifer, J. Phys. Chem. B, 2014, 118, 921–930 CrossRef CAS PubMed.
- G. Raffaini and F. Ganazzoli, Langmuir, 2010, 26, 5679–5689 CrossRef CAS PubMed.
- W. Kabsch and C. Sander, Biopolymers, 1983, 22, 2577–2637 CrossRef CAS PubMed.
- P. Ascenzi and M. Fasano, Biophys. Chem., 2010, 148, 16–22 CrossRef CAS PubMed.
- G. Fanali, A. di Masi, V. Trezza, M. Marino, M. Fasano and P. Ascenzi, Mol. Aspects Med., 2012, 33, 209–290 CrossRef CAS PubMed.
- J. Wyman Jr., in Advances in Protein Chemistry, ed. M. L. A. J. T. E. C. B. Anfinsen and M. R. Frederic, Academic Press, 1964, vol. 19, pp. 223–286 Search PubMed.
- E. Di Cera, in Methods in Enzymology, Academic Press, 1994, vol. 232, pp. 655–683 Search PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6nr07665c |
| This journal is © The Royal Society of Chemistry 2017 |