
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The importance of asking “how and why?” in natural product structure elucidation
Patrick D.
Brown
and
Andrew L.
Lawrence
 *
*
EaStCHEM School of Chemistry, University of Edinburgh, Joseph Black Building, David Brewster Road, Edinburgh, EH9 3FJ, UK. E-mail: a.lawrence@ed.ac.uk
First published on 29th August 2017
Abstract
Covering: up to 2017
This review highlights why careful consideration of the biosynthetic origin (the how) and the biological function (the why) of a natural product can be so useful during the determination of its structure. Recent examples of structural reassignments inspired by biosynthetic and functional insights will be presented. This review will demonstrate the importance of viewing the origin, structure and function of a natural product as intertwined threads of a single story, best viewed as a whole rather than as discrete topics.
 Patrick D. Brown | Patrick Brown (born in 1990) completed his Bachelor of Science (Hons 1st Class) at the Australian National University (ANU) in 2012 and was awarded the University Medal. He completed his PhD studies in the Lawrence research group in June 2016. His PhD research focused on the development of biomimetic approaches towards dimeric phenylethanoid natural products and the development of new domino reactions. |
 Andrew L. Lawrence | Andy Lawrence (born in 1984) studied chemistry at the University of Oxford and obtained his DPhil in 2010 working with Sir Jack E. Baldwin FRS and Robert M. Adlington. He then moved to Australia for postdoctoral studies with Michael S. Sherburn at the Australian National University (ANU) in Canberra. In 2012, Andy commenced his independent academic career at the ANU as an Australian Research Council (ARC) Research Fellow before being appointed as a Lecturer in organic chemistry at the University of Edinburgh in 2013. His research interests include biomimetic chemistry, natural product synthesis, and the development of new domino reactions. |
1 Introduction
Despite the power of modern spectroscopy, the structural elucidation of complex natural products is still a very difficult undertaking.1 This can be further compounded by a variety of issues: lack of sufficient material; difficulties in purification; a particularly challenging or unusual aspect of structural complexity; and human errors, which can occur all too easily when dealing with such complex data sets. Thankfully, other tools are available to chemists in their quest for certainty during structure elucidations. Total synthesis is a very robust method for the confirmation or reassignment of natural product structures, with excellent reviews concerning this topic already available.2 It is also clear that computational techniques for predicting and modeling spectroscopic data are emerging as very powerful tools, with relevant reviews again already available.3 This mini-review will highlight the often underappreciated power of simply considering the biosynthetic origin of a natural product when determining its structure.4 This involves no new equipment or expensive materials, it is not overly time-consuming, and can often provide added benefits to any associated synthetic efforts or functional investigations.5 The emerging, albeit currently limited, role of functional considerations during structure elucidations will also be highlighted.When faced with a natural product it is prudent to carefully consider how and why it exists. This will typically include some, or all, of the following steps:
(1) identification of likely biosynthetic building blocks.
(2) Proposal of feasible metabolic pathways.
(3) Direct comparison to biosynthetically related structures.
(4) Consideration of any known or proposed functional roles.
If nothing else, these relatively straightforward steps can serve as a reassuring check that any proposed structure, particularly if novel, makes logical sense. Obviously, this type of thought process does not provide hard evidence regarding a proposed structure, but it often leads to important insights and realizations. Then, if warranted, biomimetic synthetic studies or direct biosynthetic studies can be undertaken to probe the structure and origin of the natural product(s) further.
2 Case studies
This review is not intended to be exhaustive; instead, a limited number of examples will be used to illustrate some guiding principles. Firstly, the reassignment of the cytosporolides by George and Spence typifies how recognition of biosynthetically improbable structural features can lead to successful reassignments (Section 2.1). The process of deconstructing a natural product into its constituent metabolic building blocks is showcased in the reassignment of incargranine B (Section 2.2). Placement of a structure within its wider natural product family, thus revealing inconsistencies, is seen in Brill and Snyder's reassignment of the caraphenols (Section 2.3). The serendipitous reassignment of natural products during biomimetic syntheses is showcased in Calvert and Sperry's total synthesis of yuremamine (Section 2.4). Thomson's work on the cyclic prodigiosins is presented to illustrate how consideration of the function of a natural product can be used to successfully interrogate proposed structures (Section 2.5). Finally, reassignment of the tridachiahydropyrones by Moses and co-workers brings many of these principles together in a single case study (Section 2.6).2.1 Cytosporolides A–C
The cytosporolides (e.g., cytosporolide A, see Fig. 1) are caryophyllene-derived meroterpenoids isolated in 2010 by Che and colleagues from the fungus Cytospora sp.6 The carbon framework and oxygenation pattern of the caryophyllene-derived moiety was assigned using 1D and 2D NMR spectroscopy, assisted by comparison to the NMR data of known natural products 6-hydroxpunctaporonin B (2)7 and, the co-isolated, fuscoatrol A (3) (Fig. 2).8 On the basis of the downfield chemical shift of C(8) in cytosporolide A (1a) (87.5 ppm), compared to the analogous hydroxylated C(8) position of 6-hydroxypunctaporonin B (2) (74.2 ppm), Che and coworkers proposed that the cytosporolides contained an unusual 9-membered peroxylactone ring (Fig. 2).6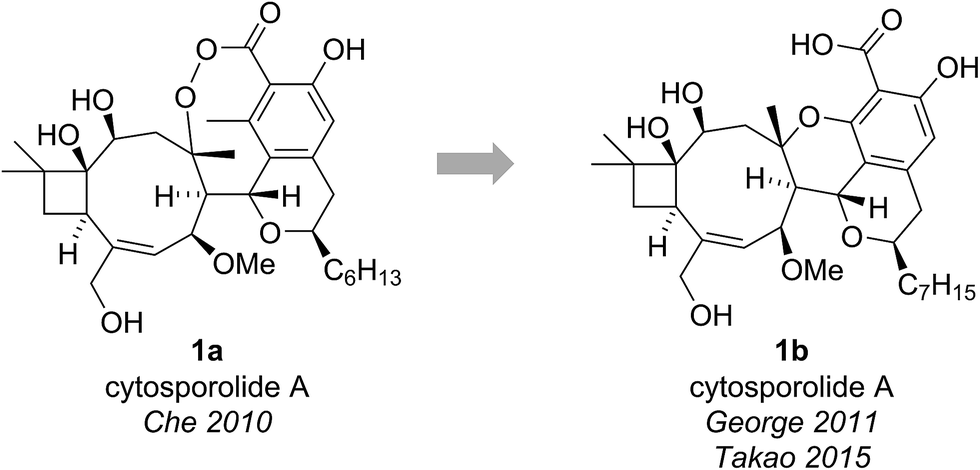 | ||
| Fig. 1 The originally proposed and revised structure of cytosporolide A. | ||
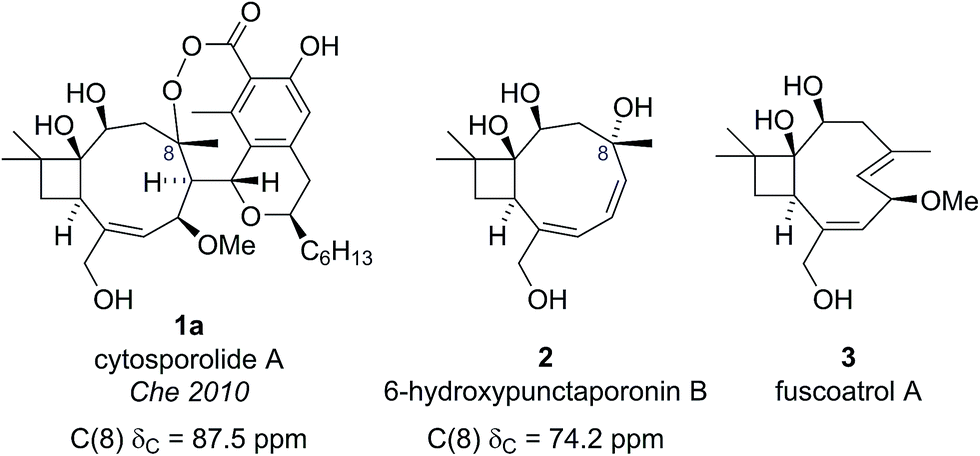 | ||
| Fig. 2 Che's proposed structure for cytosporolide A alongside known natural products used to assist the structural assignment. | ||
The presence of this unusual and highly strained structural feature led George and Spence to further investigate the reported characterization data.9 Comparison of the key C(8) chemical shift in cytosporolide A to the C(8) positions of the caryophyllene-derived natural products guajadial (4) (84.3 ppm)10 and psidial A (5) (88.0 ppm)11 suggested that a similar 6-membered aryl ether ring could account for the observed 13C NMR data (Scheme 1a). George speculated that a biosynthetic pathway analogous to that proposed by Lee for guajadial (4) and psidial A (5) (see Scheme 1a)12 could produce an alternative structure for cytosporolide A (1b), which might adequately account for the reported characterization data. Thus, a [4 + 2] cycloaddition between fuscoatrol A (3) and an ortho-quinone methide (6), derived from the known fungal metabolite CJ-12,373,13 would give structure 1b (Scheme 1b).
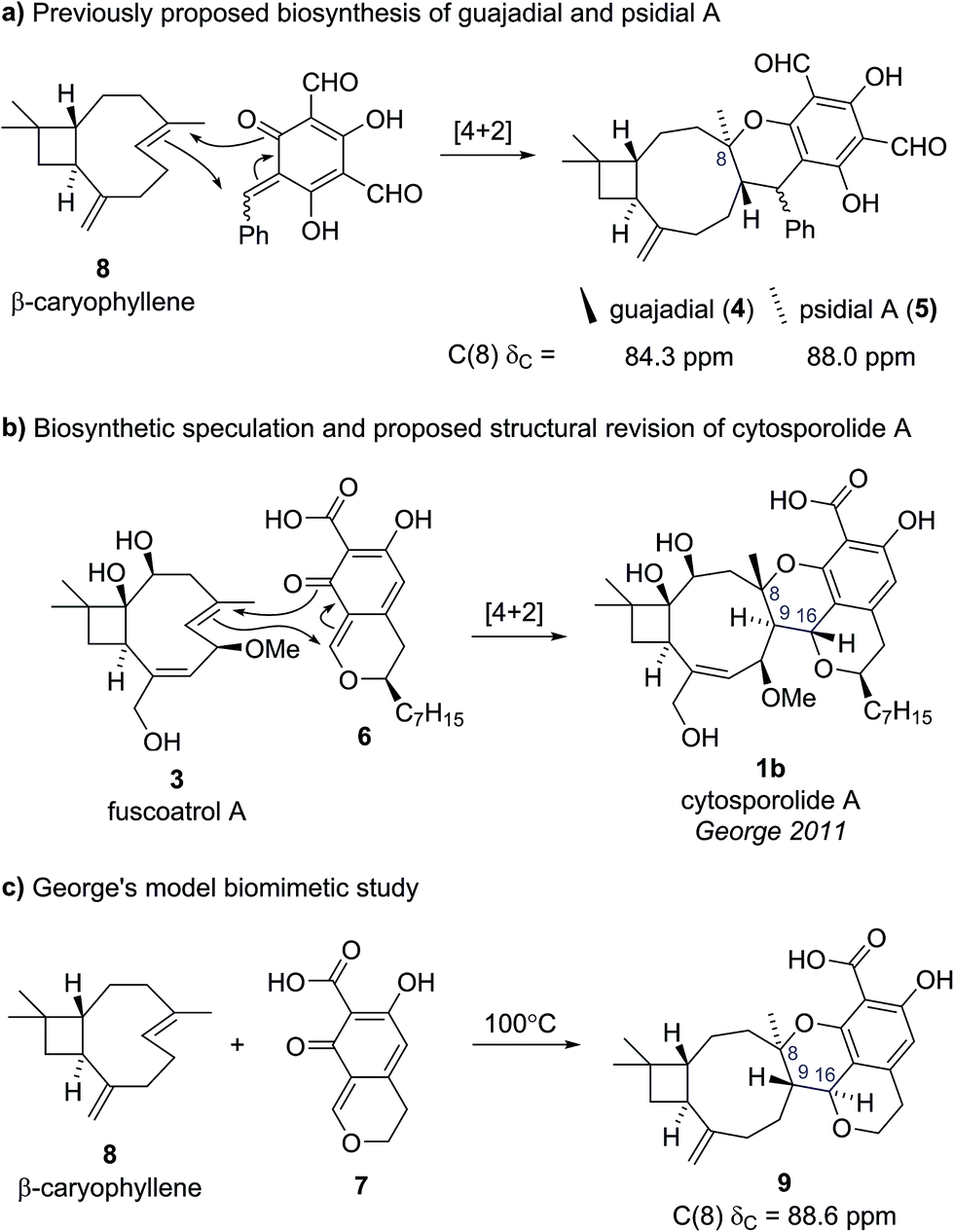 | ||
| Scheme 1 (a) Previously proposed biosynthesis of guajadial and psidial A. (b) George's biosynthetic proposal and proposed structural revision for cytosporolide A. (c) George's model biomimetic study. | ||
To test this biosynthetic hypothesis and structural revision, George and Spence conducted a biomimetic model study to produce the core framework of their proposed cytosporolide structure 1b.9 β-caryophyllene 8 was added to model ortho-quinone methide 7 and heating to 100 °C gave Diels–Alder adduct 9 in 53% yield as a single diastereomer (Scheme 1c). The structure of model compound 9 was confirmed by single crystal X-ray analysis and despite having opposite stereochemical configurations to the cytosporolides (e.g., 1b) at C(8), C(9) and C(16), the NMR and IR spectra of adduct 9 were found to be very similar to those reported for cytosporolides A–C. In particular, the key C(8) resonance in the model compound 9 occurred at 88.6 ppm, closely matching the cytosporolide A C(8) signal (88.0 ppm). George's proposed structural revision of cytosporolide A (1b) was subsequently confirmed by Takao and co-workers, in their 2015 total synthesis of cytosporolide A (1b),14 which followed a strategy analogous to George's biosynthetic hypothesis (Scheme 1b).9 Takao's total synthesis provides unequivocal validation of George's proposed structural reassignment and postulated biosynthesis.
2.2 Incargranine B
Incargranine B was isolated from Incarvillea mairei var. grandiflora in 2010 by Zhang and co-workers (Fig. 3).15 HR-ESI-MS revealed the molecular formula to be C36H50N2O12 and analysis of the 1D NMR data revealed the presence of two phenylethanoid units and two β-glucopyranose units. Extensive analysis of the 2D NMR data led Zhang and co-workers to propose a novel indolo[1.7]napthyridine alkaloid structure for incargranine B (10a).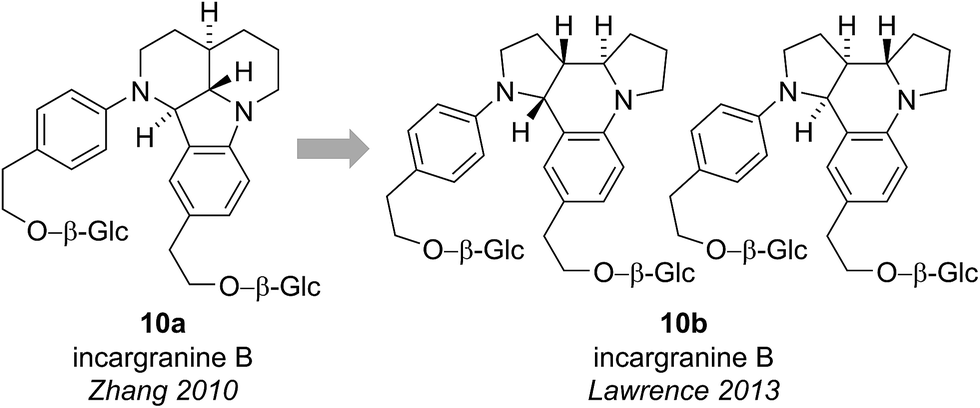 | ||
| Fig. 3 The originally proposed and revised structure of incargranine B. | ||
Incargranine B formulates as a dimer, containing two shikimate-derived C6C2 units, two ornithine-derived C4N units and two glucose units (Scheme 2a). Consideration of the biosynthetic origins of this previously unknown indolo[1.7]napthyridine framework 10a, however, led us to question the validity of this structural proposal. It should be stressed that our analysis of the available spectroscopic data did not lead us to question Zhang's proposal. Ostensibly, the proposed structure 10a did fit with the spectroscopic data reported for incargranine B,15 and it was only our biosynthetic considerations that revealed a problem may exist. Specifically, any potential biosynthesis of indolo[1.7]napthyridine 10a, involving ornithine-derived building blocks, would necessitate the cleavage and formation of an unusually high number of bonds. This biosynthetic puzzle led us to speculate that a more biosynthetically plausible dipyrroloquinoline structure 10b could account for the characterization data reported for incargranine B (Fig. 3).16
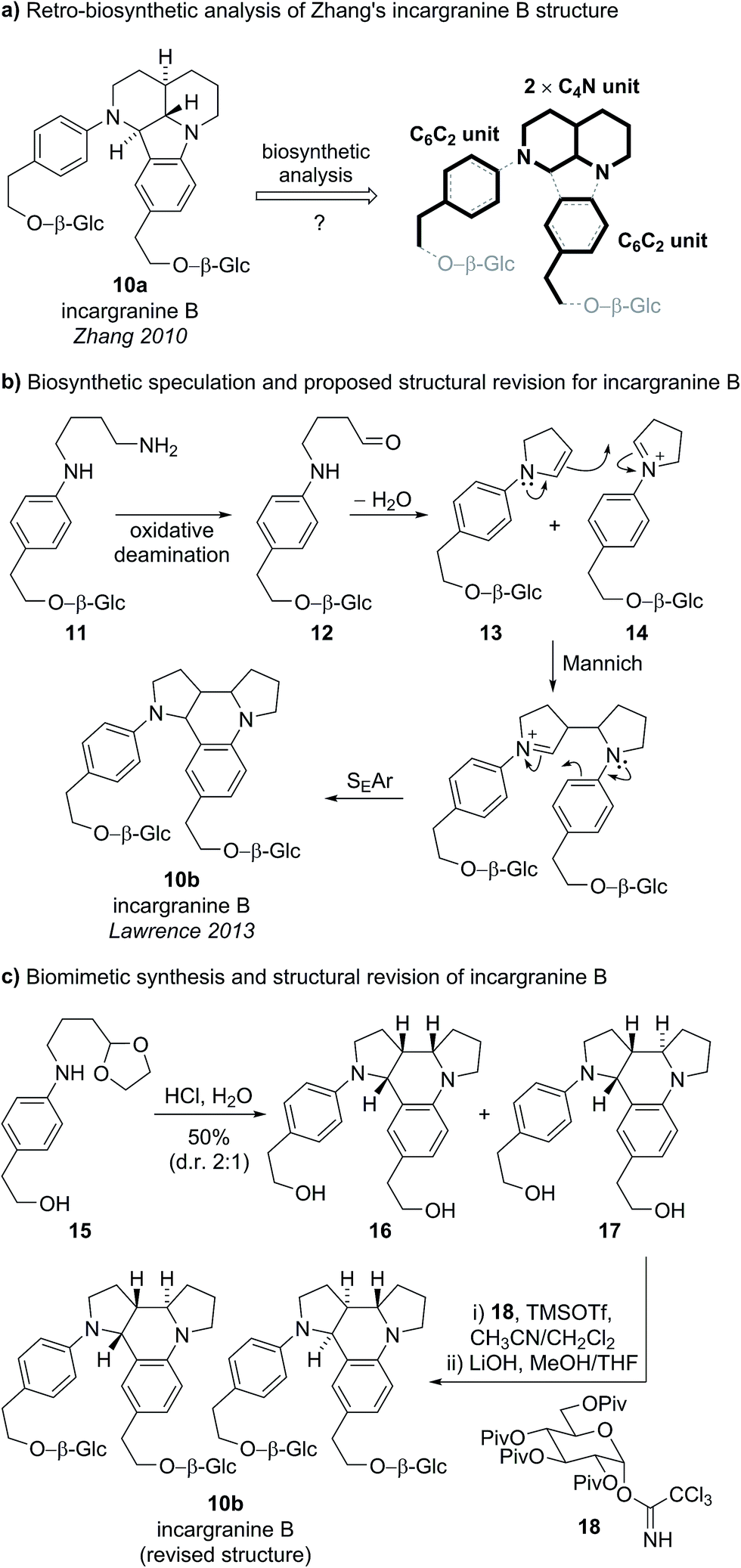 | ||
| Scheme 2 (a) Our attempted retro-biosynthetic analysis of Zhang's proposed structure for incargranine B. (b) Biosynthetic speculation and newly proposed structure for incargranine B. (c) Biomimetic synthesis and structural revision of incargranine B. | ||
We proposed phenylethanoid-diamine 11 represented a reasonable biosynthetic precursor to incargranine B (Scheme 2b). Oxidative deamination of diamine 11 could give an aldehyde 12, which, following intramolecular condensation, would give an N-aryl enamine 13. Enamine 13 could then dimerize with its corresponding iminium ion 14, via a domino Mannich/SEAr (electrophilic aromatic substitution) reaction sequence (i.e., a Povarov reaction), to give our proposed alternative structure for incargranine B (10b).17
To test both our proposed structural revision and new biosynthetic speculation, we undertook a short biomimetic synthesis of incargranine B (Scheme 2c).16 The acetal protected cyclisation precursor 15 (prepared in 3 steps) was exposed to 2 M aqueous HCl to deprotect the aldehyde and induce the condensation/Mannich/SEAr reaction sequence, giving a mixture of two dimeric products 16 and 17, in 50% isolated yield. Analysis of the 1H and 13C NMR spectra for the minor product, 17, revealed key similarities with the NMR data reported for incargranine B. Thus, dimer 17 was doubly glycosylated before global deprotection afforded the target structure 10b, as the expected 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of diastereomers (a result of glycosidation of a racemic aglycone). Data for this mixture of diastereomers, including optical rotation, matched extremely well with that reported for the natural product. Thus it is likely that natural incargranine B also exists as a mixture of diastereomers. Our proposed structural revision was, therefore, shown to be correct and the chemical feasibility of the biosynthetic hypothesis had been demonstrated.
:1 mixture of diastereomers (a result of glycosidation of a racemic aglycone). Data for this mixture of diastereomers, including optical rotation, matched extremely well with that reported for the natural product. Thus it is likely that natural incargranine B also exists as a mixture of diastereomers. Our proposed structural revision was, therefore, shown to be correct and the chemical feasibility of the biosynthetic hypothesis had been demonstrated.
2.3 Caraphenols B and C
Caraphenols B and C are resveratrol dimers, originally isolated from the roots of Caragana sinica by Hu and co-workers in 2001 (Fig. 4).18 On the basis of HRMS, UV-Vis and NMR data, Hu proposed substituted indane structures for caraphenol B (19a) and C (20a), differing in the hydroxylation pattern of the B and C-rings. Hu proposed relative stereochemistry featuring an all cis arrangement of the B, C and D-rings based on NOESY experiments.18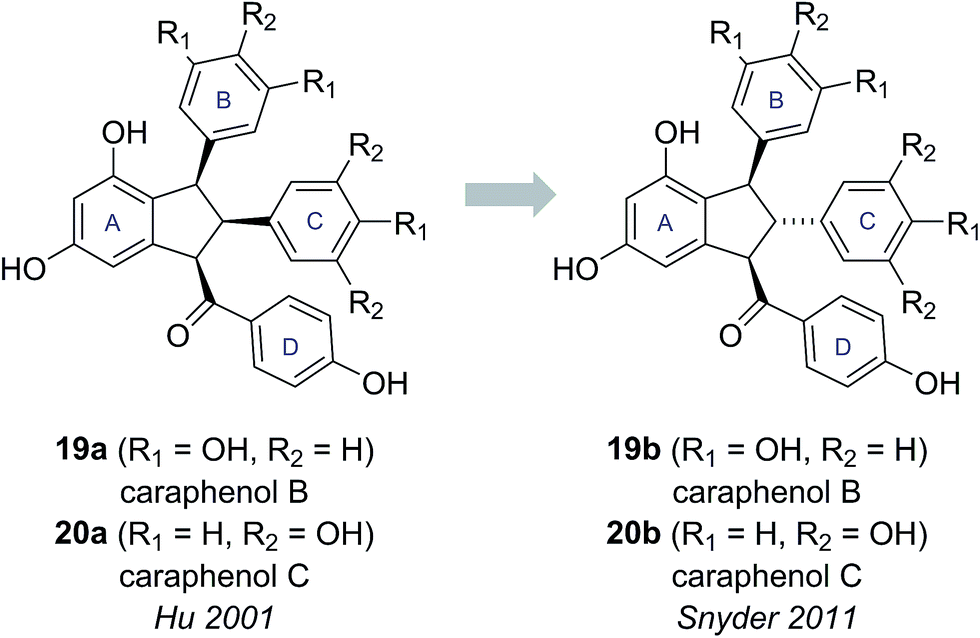 | ||
| Fig. 4 The originally proposed and revised structures of caraphenol B & C. | ||
This all cis arrangement, which is highly unusual within the wider family of resveratrol-derived indane natural products (cf. natural products 21–24, see Fig. 5),20–23 prompted Brill and Snyder to further investigate the structures of caraphenol B and C.19 They speculated that alternative structures with trans,trans stereochemistry (19b and 20b) could adequately account for all the spectroscopic data reported for caraphenol B and C (Fig. 4).19
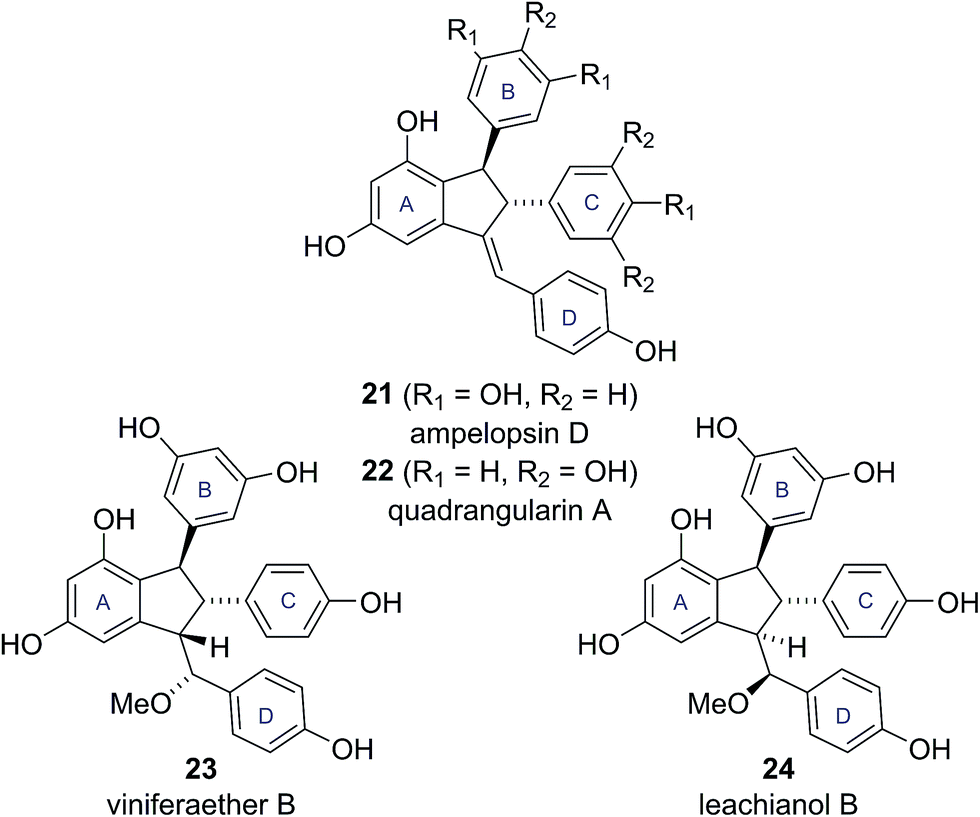 | ||
| Fig. 5 Known resveratrol-derived indane natural products. | ||
To test this hypothesis, Snyder and Brill set about preparing samples of both Hu's (19a, 20a) and their own (19b, 20b) proposed structures for caraphenols B and C. Hu's structure for caraphenol B (19a) was prepared in five steps from permethylated ampelopsin D (25) (Scheme 3a). X-ray analysis of a derivative of synthetic 19a confirmed the anticipated all cis stereochemistry. Analysis of the NMR data for synthetic 19a showed significant discrepancies to that reported for caraphenol B.19
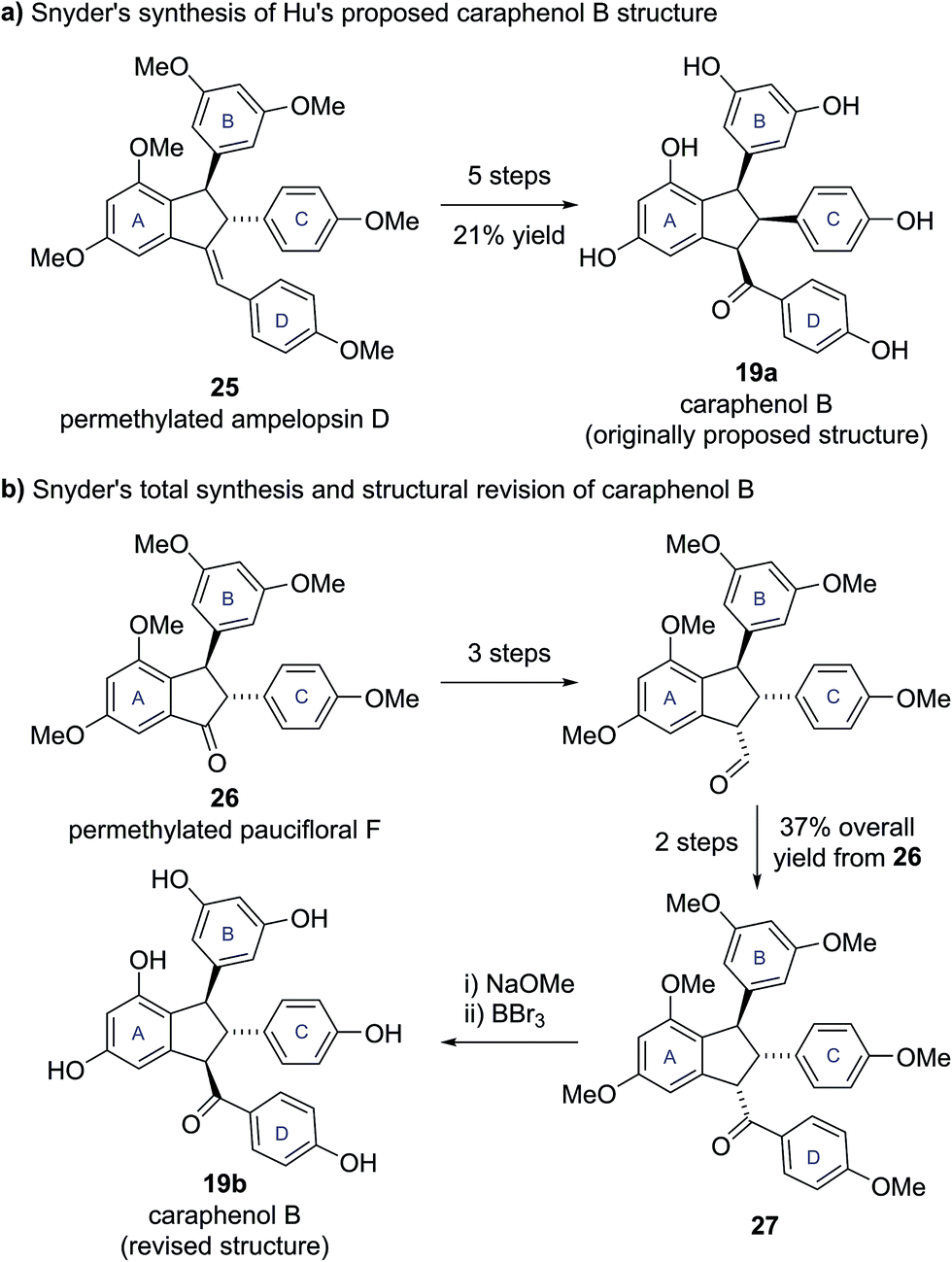 | ||
| Scheme 3 (a) Snyder's synthesis of Hu's proposed caraphenol B structure. (b) Snyder's total synthesis and structural revision of caraphenol B. | ||
Snyder's proposed trans,trans structure (19b) was accessed in seven steps from permethylated paucifloral F (26) (Scheme 3b), via formation of the trans,cis compound 27, which was epimerized to the desired trans,trans diastereomer prior to global deprotection to give caraphenol B (19b). Similar synthetic sequences allowed access to the original and newly proposed structures for caraphenol C (20a and 20b). In both cases the newly proposed structures (19b, 20b) matched the reported data for the natural products in all respects, allowing Snyder and Brill to confirm their proposed structural revisions.19 This revision of natural product stereochemistry highlights how conserved structural features within a family of biosynthetically related natural products (i.e., configuration of the core motif) can help identify potentially anomalous structures.
2.4 Yuremamine
Yuremamine was isolated in 2005 by Callaway and co-workers from the bark of Mimosa tenuiflora, a plant used in Brazil to prepare a psychoactive beverage. Callaway and co-workers proposed a pyrroloindole structure 28a, which has an intramolecular hydrogen bond. It was proposed that this hydrogen bond could protect yuremamine from monoamine oxidase metabolism, which could cause inhibition of the enzyme and facilitate the oral bioavailability of the N,N-dimethyltryptamine also present in the bark (Fig. 6).24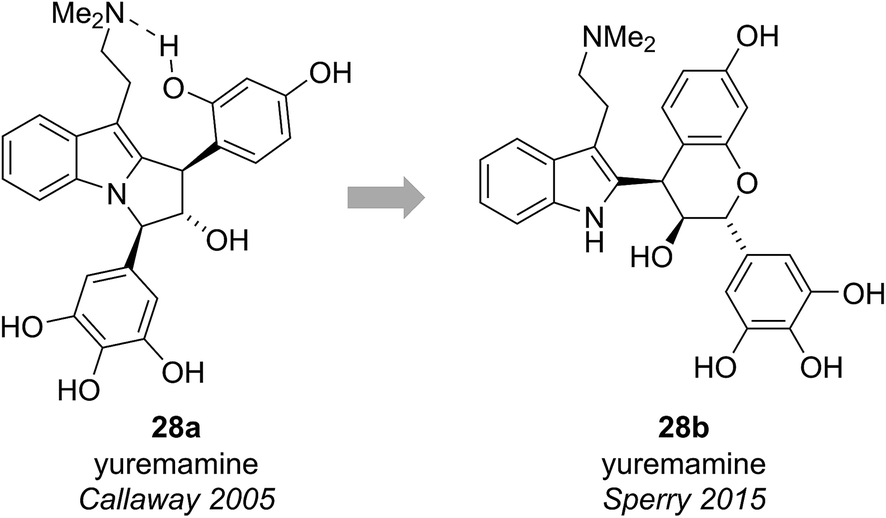 | ||
| Fig. 6 The originally proposed and revised structures of yuremamine. | ||
Calvert and Sperry proposed a biosynthetic pathway towards yuremamine (28a) featuring an initial diastereoselective coupling of the known natural product, leucorobinetinidin (29)26 with N,N-dimethyltryptamine to give the flavonoid indole 28b.25 Photochemically induced cleavage of the benzylic C–O bond could then give the para-quinone methide 30 which could be trapped via [1,6]-addition of the indole nitrogen to give yuremamine (28a) (Scheme 4).
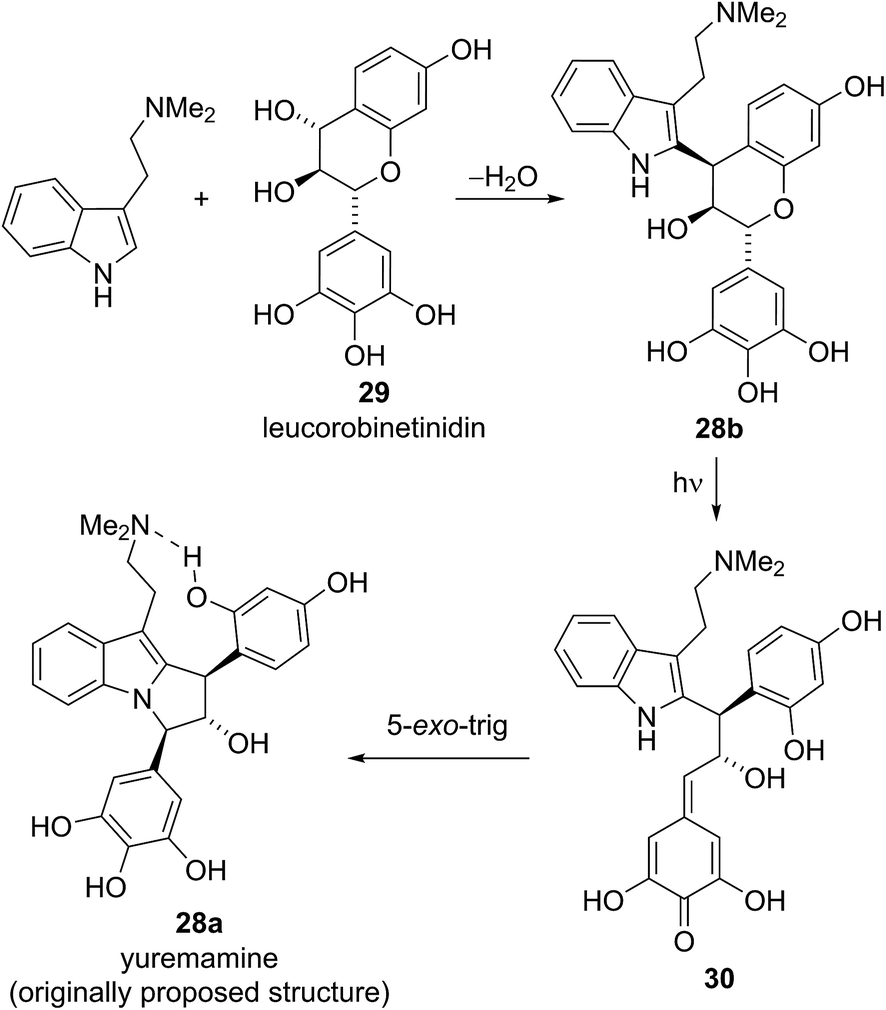 | ||
| Scheme 4 Sperry's proposed biosynthetic pathway for the originally proposed yuremamine structure. | ||
Based on this biosynthetic hypothesis, Calvert and Sperry undertook a short biomimetic synthesis, initially of the flavonoid indole 28b, which they hoped to subsequently rearrange to give the proposed structure of yuremamine (28a) (Scheme 5). They successfully accessed the key protected leucorobinetinidin analogue 31 in three steps from acetophenone 32 and aldehyde 33 in 32% yield. They were then able to diastereoselectively couple diol 31 with N,N-dimethyltryptamine in the presence of TMSOTf, followed by a global deprotection to give the postulated key biosynthetic intermediate 28b in a 56% yield over the two steps (Scheme 5).25 Intriguingly, the NMR data for this proposed biosynthetic intermediate 28b was found to be very similar to that reported for the natural product. On conversion to the TFA salt, which is how natural yuremamine was characterized, the spectroscopic data for this postulated intermediate 28b was found to match perfectly with that reported for the natural product.25 The possibility that the material had spontaneously rearranged to give 28a was ruled out by detailed analysis of 2D NMR data and the observation of an indole NH signal in the 1H-15N HSQC spectrum. Thus, Calvert and Sperry were able to conclusively reassign the natural product structure to that of their proposed biosynthetic intermediate 28b. This was further verified by Iwasawa and co-workers who synthesized the originally proposed structure of yuremamine 28a, plus all other diastereomers, and confirmed they all differ from the natural product.27 Calvert and Sperry's serendipitous discovery provides a striking example of the benefits of pursuing biomimetic strategies in total synthesis.
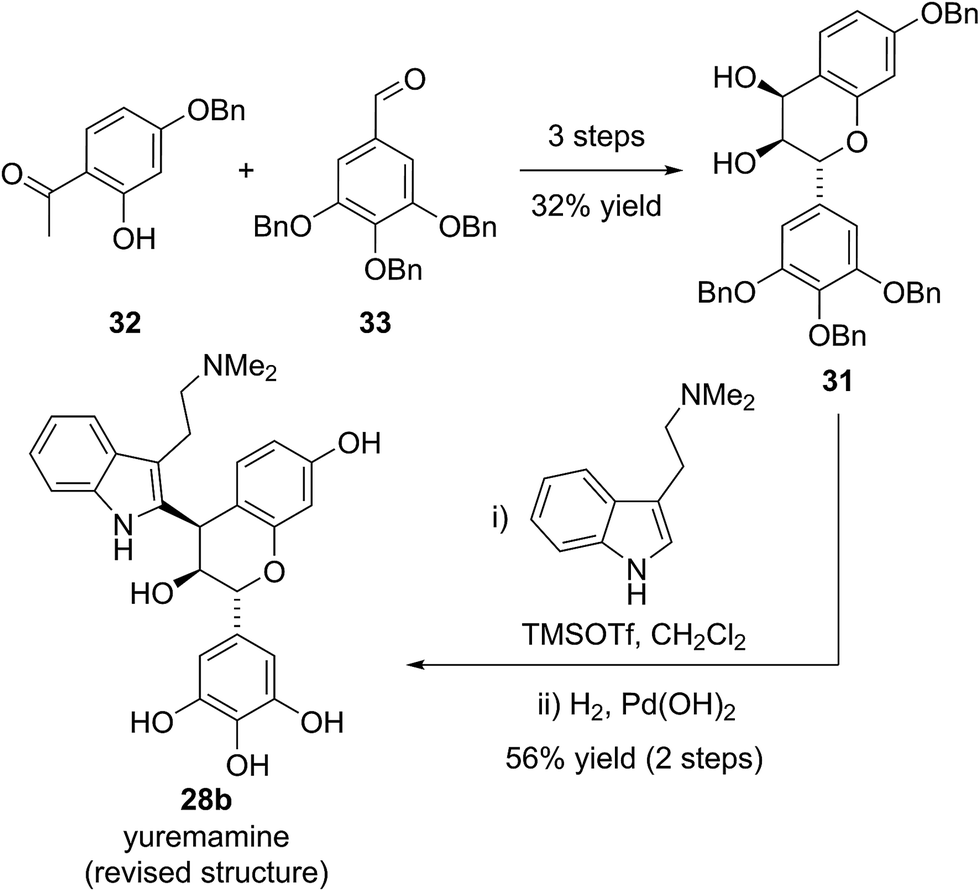 | ||
| Scheme 5 Sperry and Calvert's synthesis and reassignment of yuremamine. | ||
2.5 Streptorubin B/butylcycloheptylprodigiosin
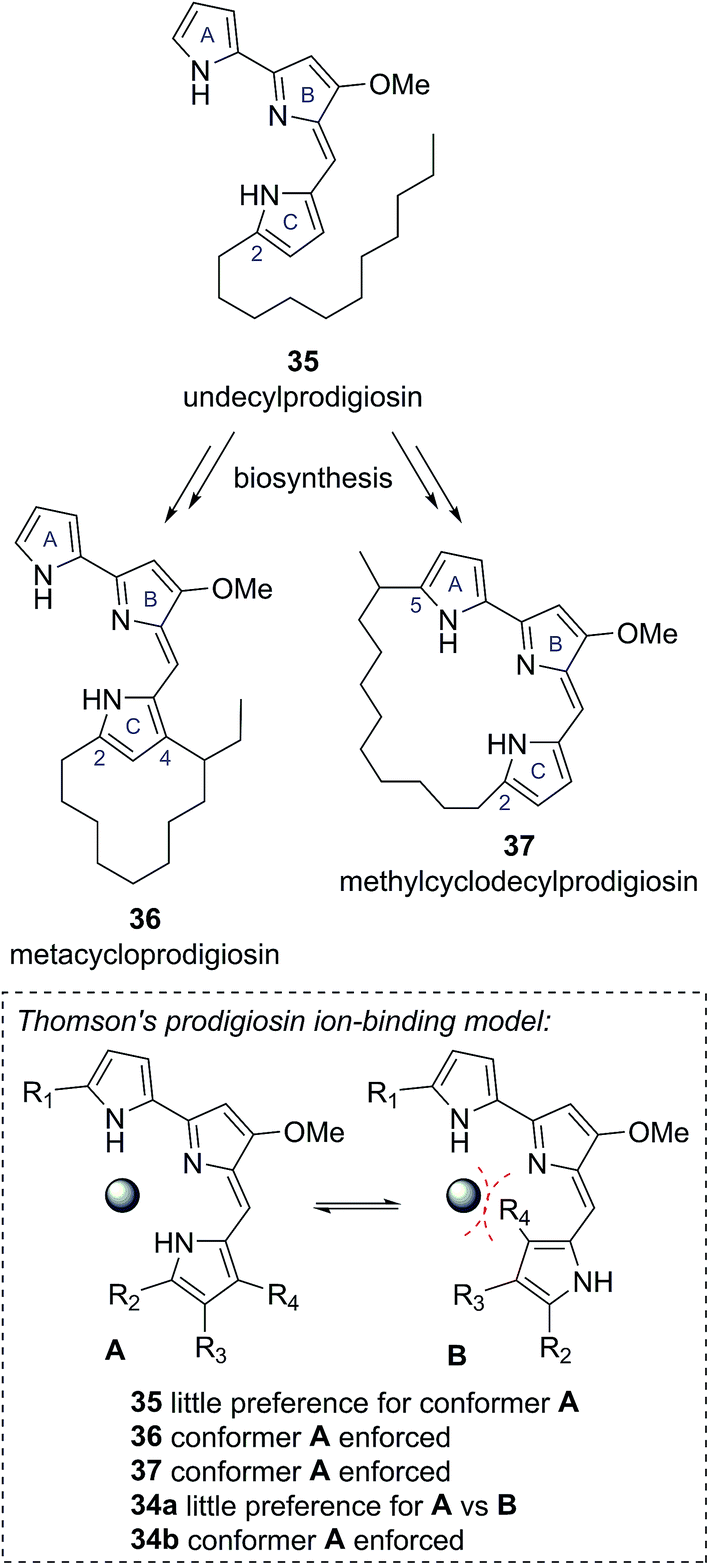
In 1975 Gerber reported the isolation of a cyclic prodigiosin natural product from the bacteria Streptomysces sp. Y-42 and S. rubrireticuli.28 An ortho-annulated structure 34a was proposed for this metabolite (Fig. 7), which was named butylcycloheptylprodigiosin.28 This structure was assigned based on analysis of MS, UV-Vis and limited NMR data; GC analysis of oxidative degradation products; and by comparison to the known prodigiosin natural products undecylprodigiosin (35) and metacycloprodigiosin (36) (Scheme 6). Gerber later concluded, however, that this originally assigned structure 34a was incorrect, identifying that the isolated material was the known natural product streptorubin B, which has a meta-annulated structure 34b (Fig. 7).29 Evidence, from other groups, however, was subsequently presented which supported butylcycloheptylprodigiosin (34a) as a distinct natural product from streptorubin B (34b). Floss reported the isolation of a pink pigment from Streptomyces coelicolor A3(2) in 1985 and assigned it as butylcycloheptylprodigiosin (34a).30 Furthermore, Fürstner and co-workers completed a total synthesis of butylcycloheptylprodigiosin (34a) in 2005 and, based on comparison to a sample of Floss' natural product (which was not pure), concluded it was a true natural product.31 However, in 2008 Challis, Reynolds and co-workers reported extensive NMR analyses that conclusively demonstrated that the carbocyclic derivative of undecylprodigiosin produced by Streptomyces coelicolor M511 (a derivative of the original A3(2) strain used by Floss) was streptorubin B, not butylcycloheptylprodigiosin.32a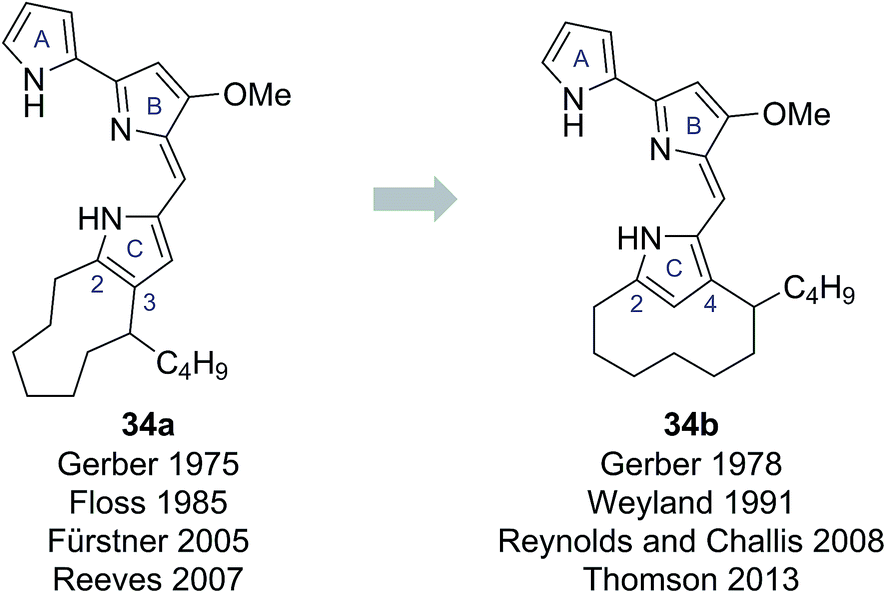 | ||
| Fig. 7 The proposed structures of butylcycloheptylprodigiosin and streptorubin B. | ||
 | ||
| Scheme 6 Other known cyclic prodigiosin natural products, which are biosynthetically derived from undecylprodigiosin, and Thomson's proposed ion-binding model. | ||
An ongoing interest in the synthesis and biosynthesis of cyclic prodigiosins, led Thomson and co-workers to also re-examine the proposed structure of butylcycloheptylprodigiosin (34a).32 Thomson noted that several carbocyclic prodigiosin derivatives are biosynthesized from a common precursor, undecylprodigiosin (35), via oxidative cyclization processes exclusively to C(4) of the C-ring pyrrole, or to C(5) of the A-ring pyrrole (e.g., metacycloprodigiosin (36) and methylcyclodecylprodigiosin (37), see Scheme 6).33 This highly conserved cyclisation pattern within the cyclic prodigiosin family led Thomson to speculate that cyclisation to these positions must provide an evolutionarily important function. They suggested that these cyclisation patterns provide a conformational bias between the A, B and C pyrrole rings, which allows for effective ion binding (Scheme 6). The unusual cyclisation to C(4) of the C-ring pyrrole in butylcycloheptylprodigiosin (34a) would not provide the same conformational bias, removing this presumed evolutionary function.32
Thomson and co-workers, re-synthesized the proposed butylcycloheptylprodigiosin structure 34a and compared this synthetic material to previously reported data for synthetic butylcycloheptylprodigiosin (34a) and the natural product isolated by Floss and co-workers.32 Although the NMR data for their synthetic material matched well with previously reported synthetic material, they found discrepancies to the data reported for the naturally occurring compound, which they were not able to resolve using NMR spectroscopy. As a result, they turned to a comparison of the EI mass spectra. From this they found that synthetic butylcycloheptylprodigiosin (34a) and synthetic streptorubin B (34b)34 showed characteristic differences in their EI mass spectra, with the data attributed to natural butylcycloheptylprodigiosin matching the mass spectrum of synthetic streptorubin B (34b) extremely well. On this basis, they concluded that butylcycloheptylprodigiosin (34a) was unlikely to represent a genuine natural product.32 Thomson's confirmation that butylcycloheptylprodigiosin (34a) is not a natural product provides a clear example of how consideration of the biological function of a compound can further highlight possible discrepancies in structural assignments.35 As the quantity and quality of available functional information increases we predict this will become an increasingly useful consideration for structure determinations.
2.6 Tridachiahydropyrones
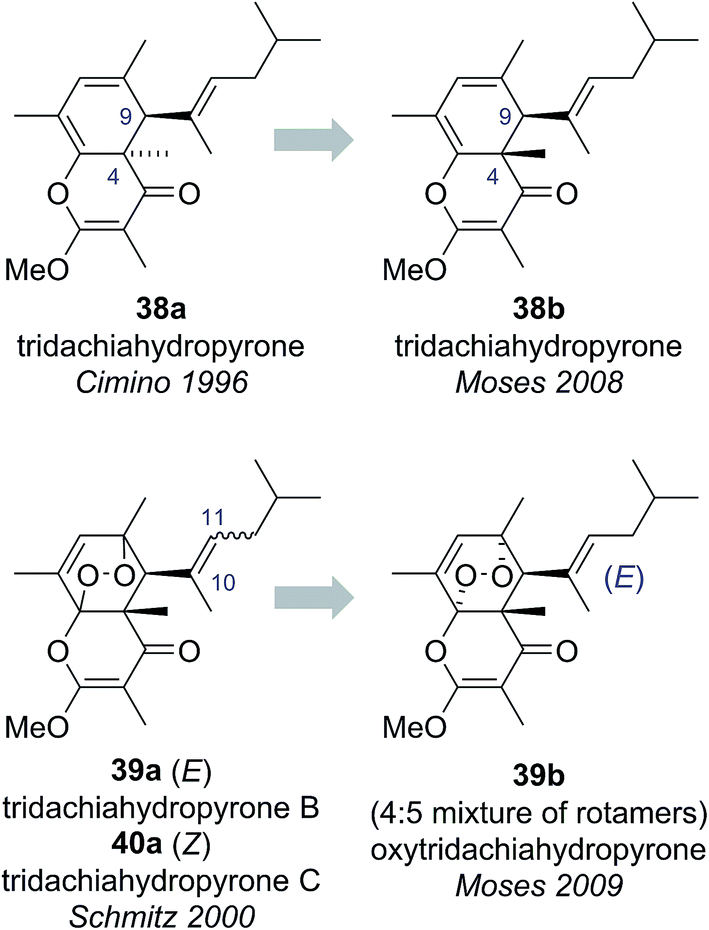
The tridachiahydropyrones are polyketide-derived pyrone natural products isolated from sacoglossan molluscs. Tridachiahydropyrone was isolated by Cimino and co-workers in 1996 from Tridachia crispata and assigned structure 38a on the basis of HRMS, 1D and 2D NMR data (Fig. 8).36 Tridachiahydropyrones B (39a) and C (40a) were isolated from another sacoglossan mollusc, Placobranchus ocellatus, by Schmitz and co-workers in 2000 as an inseparable 4:5 mixture (Fig. 8).37 Using HRMS, 1D and 2D NMR, IR and UV-Vis spectroscopy, Schmitz and co-workers were able to assign the connectivity of tridachiahydropyrones B (39a) and C (40a). Schmitz noted the close structural similarity to tridachiahydropyrone (38a) and suggested that the inseparable mixture of tridachiahydropyrones B (39a) and C (40a) was due to differing geometry around the C(10)–C(11) alkene; however, full assignment of the relative stereochemistry was not possible.37
 | ||
| Fig. 8 The originally proposed and revised structures of the tridachiahydropyrones. | ||
The sacoglossan molluscs have long been a source of great interest to chemists, providing a range of intriguing pyrone natural products. Previous biosynthetic and biomimetic studies by Faulkner, Ireland, Clardy and Solheim had demonstrated that many γ-pyrone natural products from Tridachiella and Tridachia molluscs are derived from a common biosynthetic pathway featuring a series of photochemically mediated rearrangements.38 Ireland and Scheuer further suggested that these strongly UV absorbing γ-pyrone natural products may serve an important biological function by acting as a sunscreen, protecting the molluscs from excessive UV exposure.39
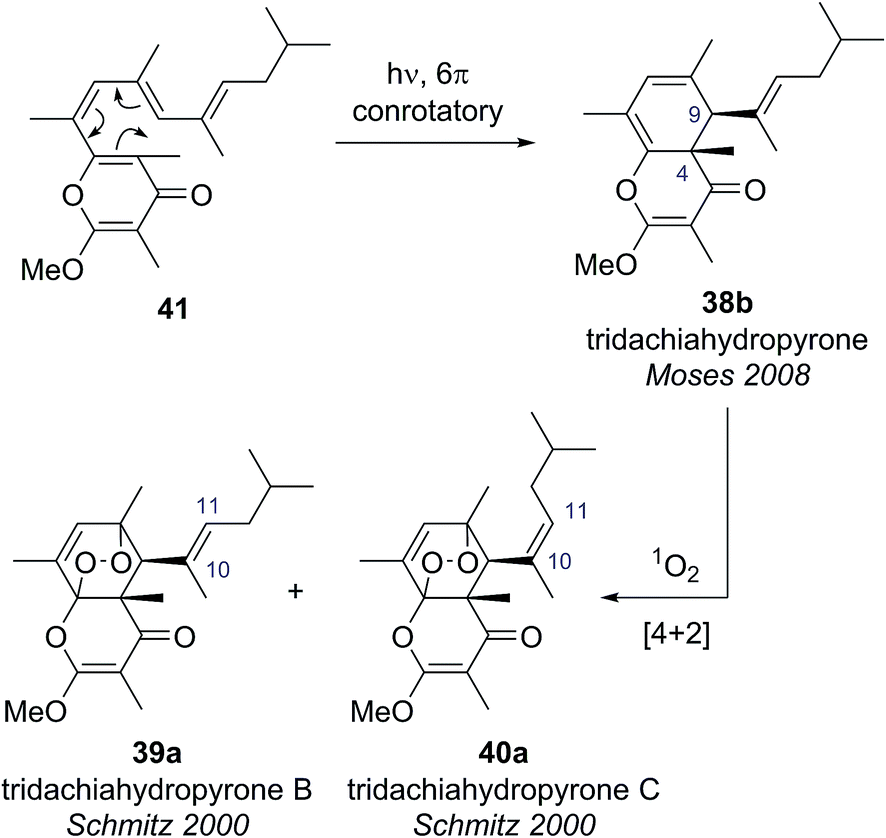
Building on this biosynthetic speculation, Moses and co-workers proposed that this family of γ-pyrone natural products could be derived from a common polyene biosynthetic intermediate 41 (Scheme 7).40 Moses proposed that tridachiahydropyrone could be derived biosynthetically from polyene 41 through a 6π electrocyclisation. Depending on whether this electrocyclisation occurred in nature under thermal or photochemical conditions, a tridachiahydropyrone structure with a trans (disrotatory cyclization, structure 38a) or a cis (conrotatory cyclization, structure 38b) relationship between the C(9) and C(4) substituents would result. A previous total synthesis of the proposed structure of tridachiahydropyrone (38a) by Perkins and co-workers had indicated that the trans structure was incorrect.41 Therefore, in light of the postulated biological function of these compounds, Moses proposed the cis structure (38b), originating from a photochemical 6π electrocyclization, was likely to represent the correct structure of tridachiahydropyrone.40a Moses also suggested that the closely related natural products tridachiahydropyrones B (39a) and C (40a) were likely to be derived from tridachiahydropyrone (38b) via a [4 + 2] cycloaddition of singlet oxygen.40b
 | ||
| Scheme 7 Moses' proposed biosynthetic pathway for the tridachiahydropyrones. | ||
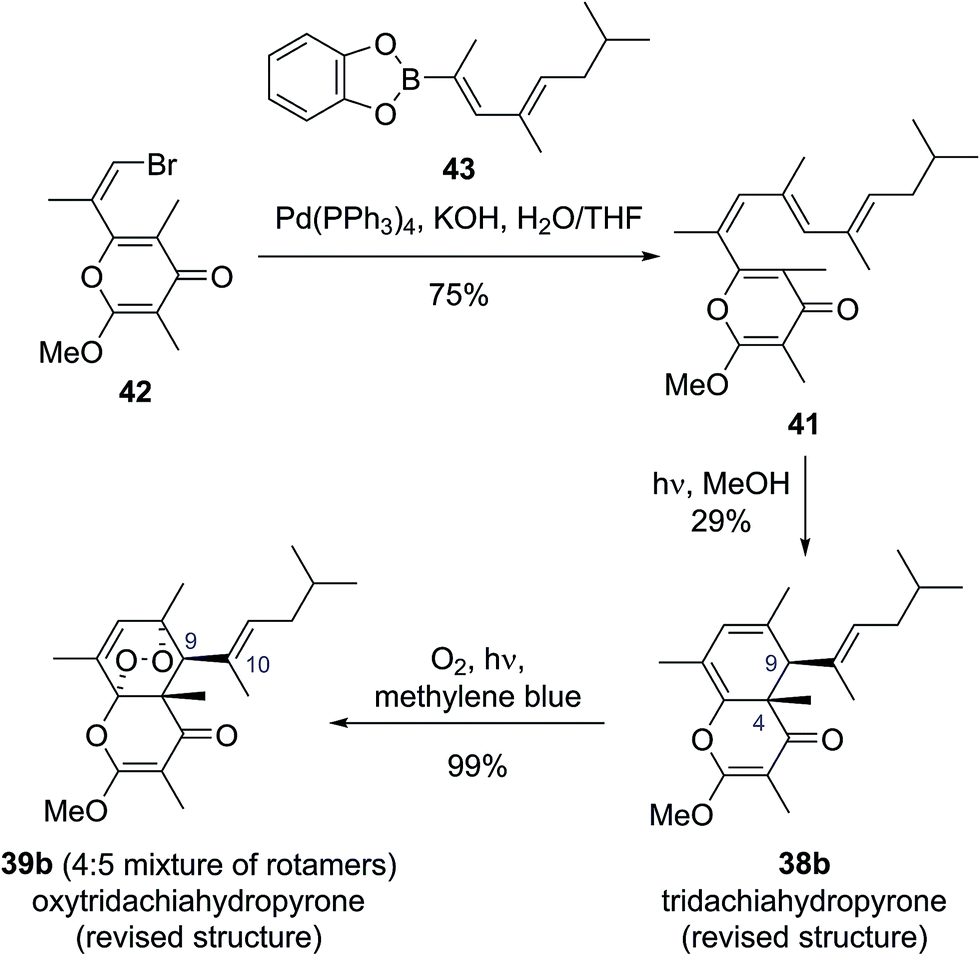
To test this biosynthetic hypothesis and proposed structural revision, Moses and co-workers undertook a biomimetic synthesis of the tridachiadhydropyrones (38b, 39a, 40a).40 The key polyene 41 was prepared in 75% yield by a Suzuki coupling of the pyrone bromide 42 and diene boronic ester 43 (Scheme 8). Polyene 41 was then subjected to both thermal and photochemical conditions to test their biosynthetic hypothesis. Heating polyene 41 to 150 °C produced no reaction; however, on exposure to sunlight for three days the desired tridachiahydropyrone 38b was formed in 29% yield, with all data matching that reported for the natural product (Scheme 8). NOESY and single crystal X-ray analysis of synthetic tridachiahydropyrone (38b) confirmed the anticipated cis stereochemistry, allowing Moses to confidently revise the structure of tridachiahydropyrone (38b) as well as lending support to the biosynthetic hypothesis.40a
 | ||
| Scheme 8 Total synthesis and structural reassignment of the tridachiahydropyrones. | ||
With samples of synthetic tridachiahydropyrone (38b) now available, Moses and co-workers were also able to test their biosynthetic hypothesis for tridachiahydropyrones B (39a) and C (40a). Exposure of tridachiahydropyrone (38b) to singlet oxygen gave quantitative conversion to a 4:5 mixture of two products, which matched the reported data for tridachiahydropyrones B and C. However, extensive NOE experiments on this mixture, supported by computational work on the possible product structures, indicated that only a single adduct is formed in the cycloaddition with singlet oxygen, with retention of the (E)-geometry at the C(10)–C(11) alkene (39b). The two products observed were, in fact, rotamers resulting from restricted rotation around the C(9)–C(10) bond. This observation was further confirmed through the use of variable temperature NMR experiments, allowing the reassignment of tridachiahydropyrones B and C as a single natural product 39b.40b
The success of this synthesis provides an excellent example of the broad range of benefits biosynthetic considerations can provide. Moses proposed a biosynthetic hypothesis, which linked three natural products together, and by taking into account the possible sunscreen function of these compounds suggested a structural revision of tridachiahydropyrone (38b). By investigating this hypothesis synthetically they were able to validate this proposed structural revision and serendipitously reassign the structures of tridachiahydropyrones B (39a) and C (40a) as two rotamers of oxytridachiahydropyrone (39b).40
3 Conclusions
Despite the constantly improving power of modern spectroscopic techniques, paying thoughtful consideration to the biosynthetic origin of a natural product will continue to serve an invaluable role in natural product structure elucidation. Additionally, as our understanding of natural product function increases it is likely that functional considerations will play an ever-greater role in structure determination.4 Conflicts of interest
There are no conflicts to declare.5 Acknowledgements
P. D. B. thanks the University of Edinburgh for the provision of a studentship. Dr Nicholas Green (University of Edinburgh), Dr Thomas Fallon (Massey University) and Dr Jonathan George (University of Adelaide) are thanked for proofreading this manuscript and making valuable suggestions. Prof. Guy Lloyd-Jones FRS (University of Edinburgh) and Prof. Alois Fürstner (Max-Planck-Institut für Kohlenforschung) are thanked for helpful discussions and advice.6 References
- (a) Progress in the Chemistry of Organic Natural Products, ed. A. D. Kinghorn, H. Falk and J. Kobayashi, Springer, New York, 2015, vol. 100 Search PubMed; (b) Handbook of Marine Natural Products, ed. E. Fattorusso, W. H. Gerwick and O. Taglialatela-Scafati, Springer, New York, 2012, vol. 1, pp. 387–674 Search PubMed; (c) K. Nakanishi in Comprehensive Natural Products Chemistry, ed. D. H. R. Barton, K. Nakanishi and O. Meth-Cohn, Elsevier, Amsterdam, 1999, vol. 1, pp. xxiii–xl Search PubMed; (d) C. Djerassi, Pure Appl. Chem., 1975, 41, 113–144 CrossRef CAS.
- (a) T. L. Suyama, W. H. Gerwick and K. L. McPhail, Bioorg. Med. Chem., 2011, 19, 6675–6701 CrossRef CAS PubMed; (b) M. E. Maier, Nat. Prod. Rep., 2009, 26, 1105–1124 RSC; (c) K. C. Nicolaou and S. A. Snyder, Angew. Chem. Int. Ed., 2005, 44, 1012–1044 ( Angew. Chem. , 2005 , 117 , 1036–1069 ) CrossRef CAS PubMed.
- For example, see: M. Elyashberg, A. J. Williams and K. Blinov, Nat. Prod. Rep., 2010, 27, 1296–1328 RSC.
- The use of genomics in natural product structure determinations can be viewed as an extension of this idea, but is beyond the scope of this mini-review. For more information, see: J. I. Tietz and D. A. Mitchell, Curr. Top. Med. Chem., 2016, 16, 1645–1694 CrossRef CAS PubMed.
- Biomimetic Organic Synthesis, ed. E. Poupon and B. Nay, Wiley–VCH, Weinheim, 2011, vol. 1–2 Search PubMed.
- Y. Li, S. Niu, B. Sun, S. Liu, X. Liu and Y. Che, Org. Lett., 2010, 12, 3144–13147 CrossRef CAS PubMed.
- S. T. Deyrup, D. C. Swenson, J. B. Gloer and D. T. Wicklow, J. Nat. Prod., 2006, 69, 608–611 CrossRef CAS PubMed.
- O. F. Smetanina, T. A. Kuznetsova, A. V. Gerasimenko, A. I. Kalinovsky, M. V. Pivkin, P. C. Dmitrenok and G. B. Elyakov, Russ. Chem. Bull., 2004, 53, 2643–2646 CrossRef CAS.
- J. T. J. Spence and J. H. George, Org. Lett., 2011, 13, 5318–5321 CrossRef CAS PubMed.
- X.-L. Yang, K.-L. Hsieh and J.-K. Liu, Org. Lett., 2007, 9, 5135–5138 CrossRef CAS PubMed.
- H.-Z. Fu, Y.-M. Luo, C.-J. Li, J.-Z. Yang and D.-M. Zhang, Org. Lett., 2010, 12, 656–659 CrossRef CAS PubMed.
- A. L. Lawrence, R. M. Adlington, J. E. Baldwin, V. Lee, J. A. Kershaw and A. L. Thompson, Org. Lett., 2010, 12, 1676–1679 CrossRef CAS PubMed.
- T. Inagaki, K. Kaneda, Y. Suzuki, H. Hirai, E. Nomura, T. Sakakibara, Y. Yamauchi, L. H. Huang, M. Norcia, L. M. Wondrack, J. A. Sutcliffe and N. Kojima, J. Antibiot., 1998, 51, 112–116 CrossRef CAS PubMed.
- K.-I. Takao, S. Noguchi, S. Sakamoto, M. Kimura, K. Yoshida and K.-I. Tadano, J. Am. Chem. Soc., 2015, 137, 15971–15977 CrossRef CAS PubMed.
- Y.-H. Shen, Y.-Q. Su, J.-M. Tian, S. Lin, H.-L. Li, J. Tang and W.-D. Zhang, Helv. Chim. Acta, 2010, 93, 2393–2396 CrossRef CAS.
- P. D. Brown, A. C. Willis, M. S. Sherburn and A. L. Lawrence, Angew. Chem., 2013, 125, 13515 ( Angew. Chem. Int. Ed. , 2013 , 52 , 13273–13275 ) CrossRef.
- There exists the possibility that this dimerization proceeds through a concerted cycloaddition mechanism, with dimers 16 and 17 the result of endo and exo processes, respectively. For further discussion, see: (a) D. A. Powell and R. A. Batey, Org. Lett., 2002, 4, 2913–2916 CrossRef CAS PubMed; (b) V. K. Kouznetsov, Tetrahedron, 2009, 65, 2721–2750 CrossRef CAS.
- (a) O. F. Smetanina, T. A. Kuznetsova, A. V. Gerasimenko, A. I. Kalinovsky, M. V. Pivkin, P. C. Dmitrenok and G. B. Elyakov, Russ. Chem. Bull., 2004, 53, 2643–2646 CrossRef CAS; (b) C. W. Choi, Y. H. Choi, M.-R. Cha, D. S. Yoo, Y. S. Kim, G. H. Yon, S. U. Choi, Y. H. Kim and S. Y. Ryu, Bull. Korean Chem. Soc., 2010, 31, 3448–3450 CrossRef CAS.
- S. A. Snyder and Z. G. Brill, Org. Lett., 2011, 13, 5524–5527 CrossRef CAS PubMed.
- Y. Takaya, K.-X. Yan, K. Terashima, J. Ito and M. Niwa, Tetrahedron, 2002, 58, 7259–7265 CrossRef CAS.
- S. A. Adesanya, R. Nia, M.-T. Martin, N. Boukamcha, A. Montagnac and M. Païs, J. Nat. Prod., 1999, 62, 1694–1695 CrossRef CAS.
- F. Fujii, Y.-H. He, K. Terashima, Y. Takaya and M. Niwa, Heterocycles, 2005, 65, 2461–2469 CrossRef CAS.
- M. Ohyama, T. Tanaka and M. Iinuma, Phytochemistry, 1995, 38, 733–740 CrossRef CAS.
- J. J. Vepsäläinen, S. Auriola, M. Tukiainen, N. Ropponen and J. C. Callaway, Planta Med., 2005, 71, 1053–1057 Search PubMed.
- M. B. Calvert and J. Sperry, Chem. Commun., 2015, 51, 6202–6205 RSC.
- Leucorobinetinidin (29) had previously been identified from an unspecified Mimosa extract, see: (a) A. Pizzi and A. Stephanou, J. Appl. Polym. Sci., 1993, 50, 2105–2113 CrossRef CAS; (b) A. A. Rahim, E. Rocca, J. Steinmetz, M. J. Kassim, M. S. Ibrahim and H. Osman, Food Chem., 2008, 107, 200–207 CrossRef CAS.
- T. Ohyama, M. Uchida, H. Kusama and N. Iwasawa, Chem.–Asian J., 2015, 10, 1850–1853 CrossRef CAS PubMed.
- (a) N. N. Gerber, J. Antibiot., 1975, 28, 194–199 CrossRef CAS PubMed; (b) N. N. Gerber and D. P. Stahly, Appl. Microbiol., 1975, 30, 807–810 CAS . Note that compound 34a is actually a cyclononyl, not a cycloheptyl, derivative.
- N. Gerber, A. G. McInnes, D. G. Smith, J. A. Walter, J. L. C. Wright and L. C. Vining, Can. J. Chem., 1978, 56, 1155–1163 CrossRef CAS , NMR studies conducted by Weyland and co-workers in 1991 support this reassignment; see: H. Laatsch, M. Kellner and H. Weyland, J. Antibiot., 1991, 44, 187–191 CrossRef PubMed.
- S. Tsao, B. Rudd, X. He, C. Chang and H. G. Floss, J. Antibiot., 1985, 38, 128–131 CrossRef CAS PubMed.
- (a) A. Fürstner, K. Radkowski and H. Peters, Angew. Chem., Int. Ed., 2005, 44, 2777–2781 CrossRef PubMed; (b) A. Fürstner, K. Radkowski, H. Peters, G. Seidel, C. Wirtz, R. Mynott and C. W. Lehmann, Chem. Eur. J., 2007, 13, 1929–1945 CrossRef PubMed, A second total synthesis was reported by Reeves in 2007, see: J. T. Reeves, Org. Lett., 2007, 9, 1879–1881 CrossRef CAS PubMed.
- (a) S. Mo, P. K. Sydor, C. Corre, M. M. Alhamadsheh, A. E. Stanley, S. W. Haynes, L. Song, K. A. Reynolds and G. L. Challis, Chem. Biol., 2008, 15, 137–148 CrossRef CAS PubMed; (b) B. T. Jones, D. X. Hu, B. M. Savoie and R. J. Thomson, J. Nat. Prod., 2013, 76, 1937–1945 CrossRef CAS PubMed.
- For a detailed study into the cyclization of undecylprodigiosin 35 into sterptorubin B (34b), see: D. M. Withall, S. W. Haynes and G. L. Challis, J. Am. Chem. Soc., 2015, 137, 7889–7897 CrossRef CAS PubMed.
- D. H. Hu, M. D. Clift, K. E. Lazarski and R. J. Thomson, J. Am. Chem. Soc., 2011, 133, 1799–1804 CrossRef CAS PubMed.
- For more information on the prodigiosin natural products, see: (a) D. X. Hu, D. M. Withall, G. L. Challis and R. J. Thomson, Chem. Rev., 2016, 116, 7818–7853 CrossRef CAS PubMed; (b) A. Fürstner, Angew. Chem., Int. Ed., 2003, 42, 3582–3603 CrossRef PubMed.
- M. Gavagnin, E. Mollo, G. Cimino and J. Ortea, Tetrahedron Lett., 1996, 37, 4259–4262 CrossRef CAS.
- X. Fu, E. P. Hong and F. J. Schmitz, Tetrahedron, 2000, 56, 8989–8993 CrossRef CAS.
- (a) C. Ireland, D. J. Faulkner, B. A. Solheim and J. Clardy, J. Am. Chem. Soc., 1978, 100, 1002–1003 CrossRef CAS; (b) C. Ireland, D. J. Faulkner and J. Clardy, J. Am. Chem. Soc., 1979, 101, 1275–1276 CrossRef CAS; (c) C. Ireland and D. J. Faulkner, Tetrahedron, 1981, 37, 233–240 CrossRef.
- C. Ireland and P. Scheuer, Science, 1979, 205, 922–923 CAS.
- (a) P. Sharma, N. Griffiths and J. E. Moses, Org. Lett., 2008, 10, 4025–4026 CrossRef CAS PubMed; (b) P. Sharma, B. Lygo, W. Lewis and J. E. Moses, J. Am. Chem. Soc., 2009, 131, 5966–5972 CrossRef CAS PubMed.
- (a) D. W. Jeffery, M. V. Perkins and J. M. White, Org. Lett., 2005, 7, 407–409 CrossRef CAS PubMed; (b) D. W. Jeffery, M. V. Perkins and J. M. White, Org. Lett., 2005, 7, 1581–1584 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |