
Pyranonaphthoquinones – isolation, biology and synthesis: an update
Briar J.
Naysmith
,
Paul A.
Hume
,
Jonathan
Sperry
and
Margaret A.
Brimble
*
School of Chemical Sciences, Maurice Wilkins Centre for Molecular Biodiscovery, University of Auckland, 23 Symonds Street, Auckland 1142, New Zealand
First published on 19th October 2016
Abstract
Covering: 2008 to 2015. A review on the isolation, biological activity and synthesis of pyranonaphthoquinone natural products from 2008–2015 is provided
This review discusses the isolation, biological activity and synthesis of pyranonaphthoquinone natural products, covering the years 2008–2015. The pyranonaphthoquinones are a group of metabolites sharing a common naphtho[2,3-c]pyran-5,10-dione ring system that have been isolated from a wide range of microorganisms, plants and insects. In addition to their synthetically challenging molecular structures, pyranonaphthoquinones exhibit a wide array of biological activity, including anti-bacterial, anti-fungal and anti-cancer properties. The therapeutic potential of these compounds has led to a dynamic interplay between total synthesis and biological evaluation.
 Briar J. Naysmith | Briar Naysmith was born in Blenheim, New Zealand. She completed her BSc(Hons) at the University of Otago in 2008, and her PhD at the University of Auckland under the supervision of Distinguished Professor Margaret Brimble in 2013. Her doctoral research was on the total synthesis of the griseusin family of natural products. Her postdoctoral research was on the synthesis of polymers for coating nanoparticles. She then accepted a position as lecturer of organic chemistry at the University of Auckland in 2014, before moving on in 2016 to begin studies towards becoming a patent attorney at AJ Park Intellectual Property. |
 Paul A. Hume | Paul Hume was born in New Zealand and completed his BSc(Hons) at the University of Auckland. He completed his PhD in 2014 under the supervision of Professor Margaret Brimble. His doctoral research concerned the total synthesis of the virgatolides, a family of spiroketal-containing natural products. He then accepted a postdoctoral position at the University of Nottingham with Professor David Amabilino, where his work focussed on the design, synthesis and characterisation of novel materials for use in organic solar energy devices. He returned to the University of Auckland in 2016, where he accepted a position as a lecturer. |
 Jonathan Sperry | Jonathan Sperry obtained his BSc (Hons) (2002) and PhD (2006, Professor Chris Moody) from the University of Exeter, UK. He spent 3.5 years as a postdoctoral researcher with Distinguished Professor Margaret Brimble at the University of Auckland, before taking up a lectureship at the same institution in 2009, where he is currently a Senior Lecturer and a Royal Society of New Zealand Rutherford Discovery Fellow. |
 Margaret A. Brimble | Margaret Brimble is a Distinguished Professor at the University of Auckland where her research program focuses on the synthesis of bioactive natural products and peptides. She has published >420 papers, 55 reviews, holds 30 patents, won the 2016 Marsden Medal, 2012 Rutherford Medal (NZ top science award), the 2010 RSC Natural Products Award, named an IUPAC Distinguished Women in Chemistry Award (2015), named L'Oreal-UNESCO Women in Science laureate for Asia-Pacific and conferred the Queen's Honour CNZM. She is President of IUPAC Organic and Biomolecular Division III, Chair of the Rutherford Foundation RSNZ, an Associate Editor for Organic and Biomolecular Chemistry and Past-President of the International Society of Heterocyclic Chemistry. |
1. Introduction
The pyranonaphthoquinones are a large group of over one hundred natural products primarily isolated from bacteria and fungi, many of which have been found to exhibit antibacterial, antifungal and anticancer properties.1 The basic structure of the pyranonaphthoquinones is the naphtho[2,3-c]pyran-5,10-dione ring system (Fig. 1, left). An additional structural feature often present in these natural products is a fused γ-lactone, whilst in others the lactone is ring-opened to the carboxylic acid side chain. Pyranonaphthoquinone natural products vary in the substitution of the pyran and aromatic rings, and can possess additional fused ring systems.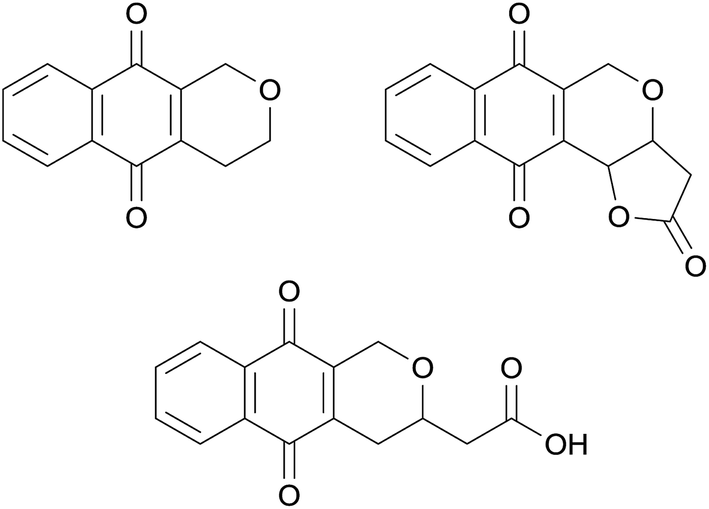 | ||
| Fig. 1 The basic structure of the pyranonaphthoquinone antibiotics. | ||
This review provides a timely addition to our two previous reviews discussing the isolation, biological activity and synthesis of pyranonaphthoquinones, published in 1999![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 1 and 2008,2 respectively. This review summarises all novel pyranonaphthoquinones have been isolated during the period 2008–2015, along with any new biological activity and synthetic studies towards this class of natural products. In keeping with our previous reviews, only pyranonaphthoquinones possessing the naphtho[2,3-c]pyran-5,10-dione ring systems outlined in Fig. 1 are discussed and in order to be as comprehensive as possible, material discussed in two recently published review articles3,4 is included where appropriate.
1 and 2008,2 respectively. This review summarises all novel pyranonaphthoquinones have been isolated during the period 2008–2015, along with any new biological activity and synthetic studies towards this class of natural products. In keeping with our previous reviews, only pyranonaphthoquinones possessing the naphtho[2,3-c]pyran-5,10-dione ring systems outlined in Fig. 1 are discussed and in order to be as comprehensive as possible, material discussed in two recently published review articles3,4 is included where appropriate.
2. Isolation and biological activity
In 2008, two novel pyranonaphthoquinones, ascomycones A (1) and B (2) together with a related metabolite, ascomycone C (3) were isolated from the ethyl acetate extract of the culture filtrate from an unidentified ascomycete (Fig. 2).5 The natural products contain a C3–C4 double bond, an unusual structural characteristic of this class of natural products. Ascomycone A (1) is the methyl ether of the lactol ascomycone B (2). The compounds were tested for growth inhibition of both bacteria and fungi, with ascomycone B (2) displaying greater activity against fungal strains than ascomycone A (1). Both natural products exhibited moderate activity against Gram-positive bacterial species, but were inactive against Gram-negative bacteria.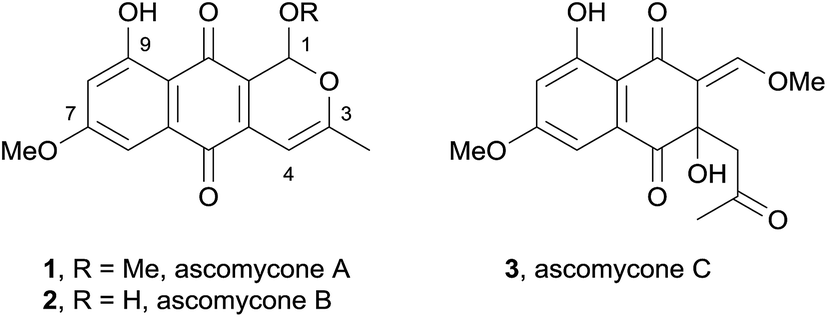 | ||
| Fig. 2 Ascomycones A–C. | ||
In the same year, the O-phenethylherbarin (4), a novel derivative of herbarin (5) was isolated by cytotoxicity-guided fractionation from the fungus IBWF79B-90A (Fig. 3).6 In contrast to herbarin (5) and several other derivatives, compound 4 exhibited no anti-microbial activity up to a concentration of 50 μg mL−1 against a range of bacterial species. The cytotoxicity of compound 4 was measured against four different cell lines, with IC50 values in the range of 2.5–15 μg mL−1.
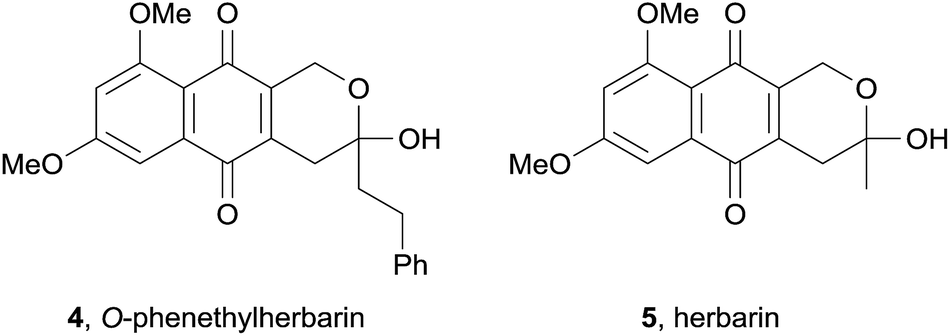 | ||
| Fig. 3 O-Phenethylherbarin and herbarin. | ||
In 2009, two new pyranonaphthoquinones, astropaquinones B (6) and C (7) and the related naphthoquinone astropaquinone A (8) were isolated from the ethyl acetate extract of the freshwater fungus Astrophaeriella papuana (Fig. 4).7 The astropaquinones are phenolic methyl ether derivatives of (−)-thysanone. These compounds were found to be active against Gibberella, Fusarium, Phyllosticta and two Alternaria fungal strains in addition to three Gram-positive bacterial strains. Astropaquinone B (6) also displayed activity against Escherichia coli, while astropaquinone C (7) was active against a Colletotrichum species.
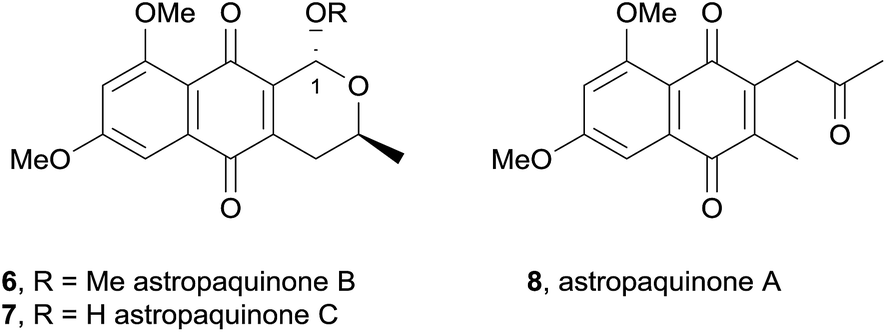 | ||
| Fig. 4 Astropaquinones A–C. | ||
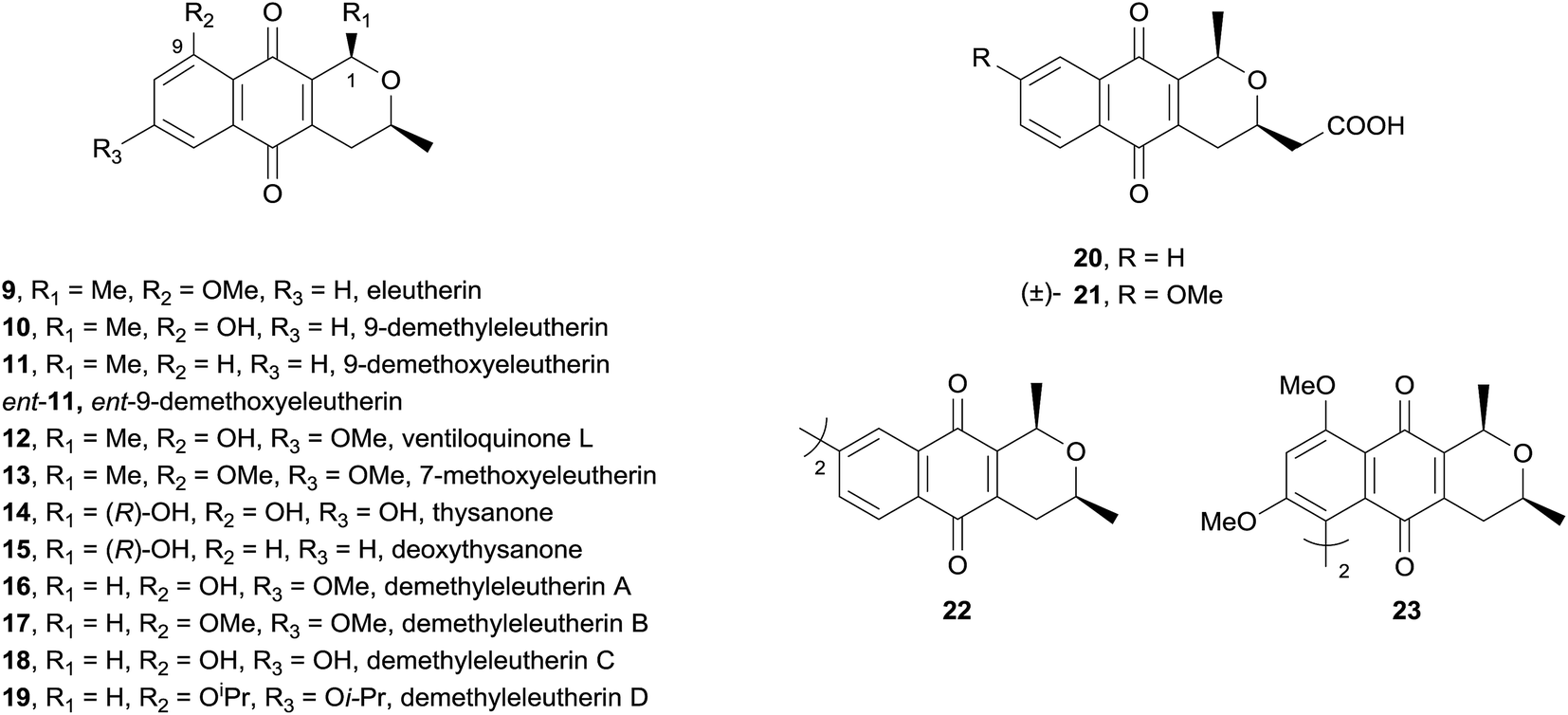
Also in 2009, a study examining the inhibitory activity of a number of synthetic and natural pyranonaphthoquinones against topoisomerase II was reported.8 Topoisomerases are good anti-tumour targets given they are highly active in cells which are rapidly proliferating. The compounds tested resulted from several synthetic projects conducted by the Brimble group (Fig. 5, also see Section 3).
 | ||
| Fig. 5 Pyranonaphthoquinones investigated for inhibition of topoisomerase II. | ||
All of the compounds tested were found to inhibit human topoisomerase IIα (hTopIIα) to varying degrees. Interestingly, the natural products ventiloquinone L (12) and thysanone (14) displayed only weak hTopIIα inhibition, suggesting that these antibiotics may have been under evolutionary selection pressure to not inhibit topoisomerases. As 9-demethoxyeleutherin (11) is less active than eleutherin (9), the oxygenated substituent at C9 appears important for inhibitory activity. The high inhibitory activity of C1-unsubstituted analogues 16–19 suggests that substitution at this position is detrimental to activity. The strong inhibition exhibited by 22 and 23 established for the first time that dimeric pyranonaphthoquinones possess topoisomerase inhibitory activity. The cytotoxicity of the compounds was also assessed by their ability to inhibit yeast growth. No correlation between cytotoxicity and hTopoIIα inhibition was observed. The most cytotoxic compounds were the nanaomycin A analogues 20 and 21, followed by ventiloquinone L (12) and derivatives thereof. Although this study provided some promising preliminary results, the strength of topoisomerase II inhibition correlates poorly with cytotoxicity, indicating that other molecular features in the pyranonaphthoquinone family must be considered to obtain highly specific topoisomerase II inhibitors.
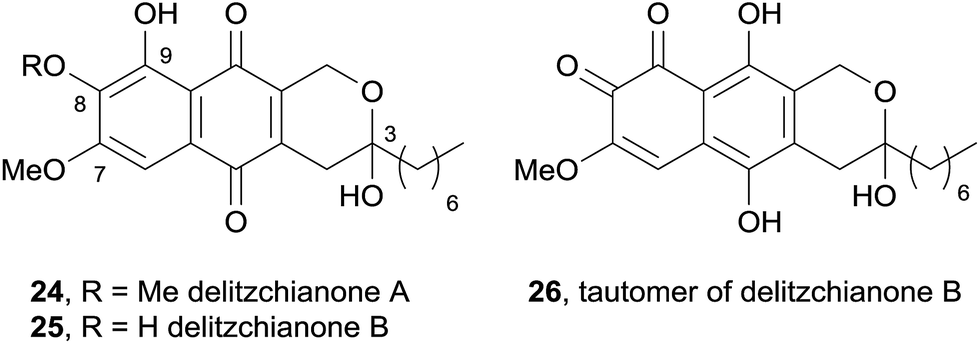
In 2011, two red pigments isolated from the endophytic fungus Delitzchia winteri were identified as pyranonaphthoquinones and named delitzchianones A (24) and B (25).9 These compounds contain unique structural features, including oxygenation at C3, C7, C8 and C9 sites, as well as an n-heptyl chain at C3 (Fig. 6). Delitzchianone B was isolated as a mixture of para- and ortho-naphthoquinone tautomers, 25 and 26 respectively. These compounds were tested in an assay against the malaria parasite Plasmodium falciparum, but were found to be inactive.
 | ||
| Fig. 6 Delitzchianones A and B. | ||
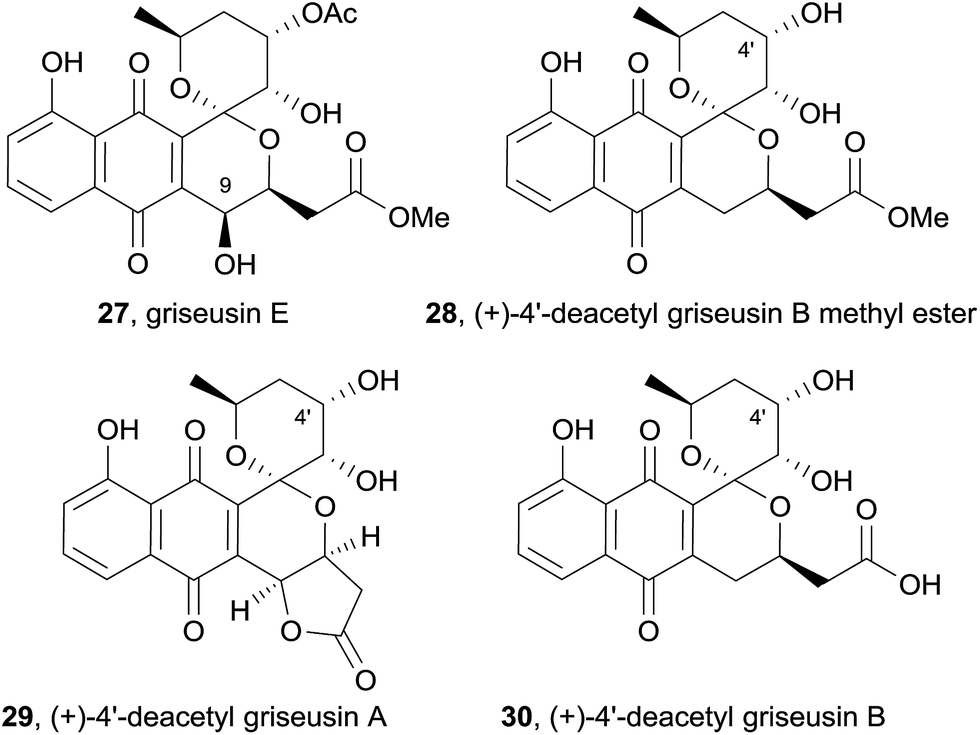
Five new natural products were isolated while screening for bioactive compounds produced by actinomycetes found in soil and seawater samples in Japan.10 Four of these compounds were identified as griseusin-subtype pyranonaphthoquinones using a combination of ESI mass spectrometry, UV spectroscopy, 1H NMR, 13C NMR and 2D NMR techniques. The first, named griseusin E (27), is a methyl ester derivative of griseusin B that also contains a novel 9-hydroxy substituent (Fig. 7). The relative stereochemistry of griseusin E (27) was determined using a combination of nOe correlations and coupling constants. Comparison of the CD spectra to other naturally occurring griseusins suggested that griseusin E (27) possesses the opposite absolute stereochemical configuration to griseusin A. Also isolated were the enantiomeric methyl ester derivative of known (−)-4′-deacetyl griseusin B (28) and (+)-enantiomers of 4′-deacetyl griseusin A (29) and B (30). These new pyranonaphthoquinones were analysed with respect to their ability to overcome cellular resistance to tumour necrosis factor-related apoptosis-inducing ligand (TRAIL). TRAIL is a potential anticancer agent due to its ability to selectively kill cancer cells; however, many cell-lines exhibit resistance. Some agents can work in combination with TRAIL in order to effect cancer cell death. When combined with TRAIL, all of the griseusins isolated resulted in decreased viability of cancer cells. (+)-4′-Deacetyl griseusin A (29) was the most effective in this regard.10
 | ||
| Fig. 7 Novel griseusins produced by actinomycetes. | ||
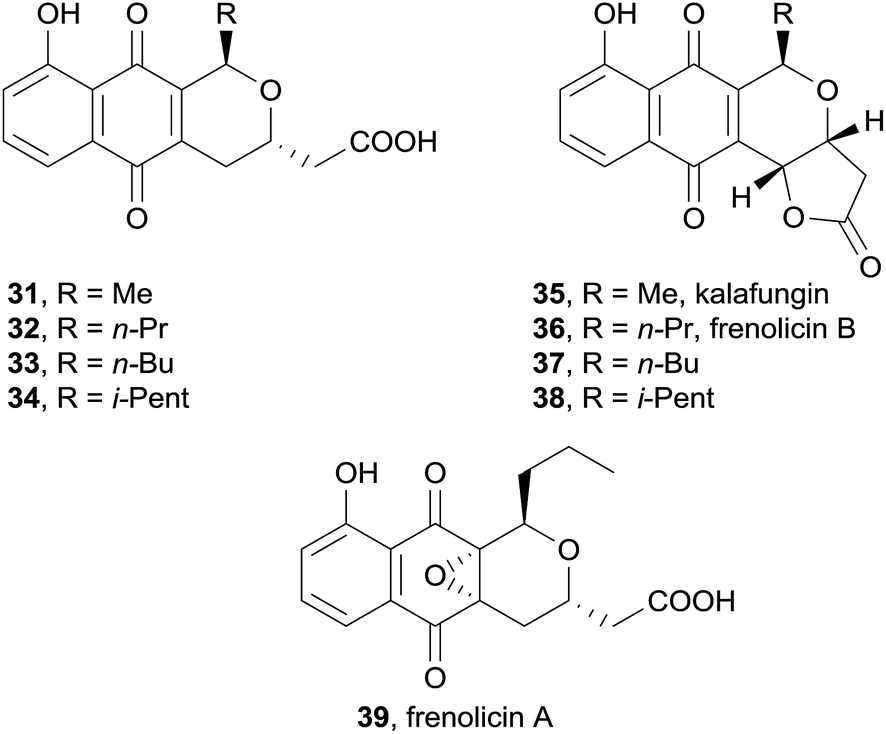
In 2011, Khosla and coworkers successfully biosynthetically engineered dihydrokalafungin (31), dihydrofrenolicin B (32) and frenolicin A (39) in the heterologous host bacteria Streptomyces coelicolor CH999 which had the actinorhodin biosynthetic gene cluster deleted (Fig. 8).11 By altering the gene pathway, novel analogues 33 and 34 were also produced. The free acids were converted to the corresponding lactones 35–38 synthetically. Both the natural product and the analogues inhibited the growth of Toxoplasma gondii in a manner that infers the substituent at C15 plays a major role. This work demonstrates that analogues of anti-fungal pyranonaphthoquinones can be obtained by reconstruction of a functional biosynthetic pathway, in what constitutes a new approach in anti-parasitic drug design.
 | ||
| Fig. 8 Biosynthetically engineered pyranonaphthoquinones. | ||
A report in 2011 showed that addition of known pyranonaphthoquinone actinorhodin (40, Fig. 9)12,13 to Streptomyces coelicolor activated the expression of SoxR genes in a SoxR dependent fashion.14 SoxR is a gene cluster that is upregulated in response to oxidants to protect the cell against oxidative damage.
 | ||
| Fig. 9 The dimeric pyranonaphthoquinone actinorhodin. | ||
Cipura paludosa plants from Brazil have been used traditionally for treatment of inflammation, infections, renal tract disorders and pain.15 Three known pyranonaphthoquinones: eleutherin (9, also see Fig. 5), isoeleutherin (42), and hongkonin (43) were isolated from methanol extracts of the bulbs of this plant, together with a novel pyranonaphthoquinone, identified as (R)-11-hydroxyeleutherin (41) using a combination of UV, mass and NMR analysis (Fig. 10). Eleutherin (9) and isoeleutherin (42) were then investigated for their anti-inflammatory effects by testing the reduction of carrageenan-induced oedema formation and plasma extravasation (leakage) in mice paws. Eleutherin (9) slightly reduced oedema formation but had no effect on plasma extravasation, while isoeleutherin (42) significantly decreased both oedema formation and plasma extravasation, displaying similar potency to the NSAID indomethacin. The compounds were also found to significantly restore mechanical hypernociception but had no effect on thermal nociception. In 2015, the same group published a follow-up study evaluating the activity of eleutherin and isoeleutherin from Cipura paludosa against glioma, breast, ovarian, kidney, lung, colon and leukaemia cell lines together with the non-tumour cell line HaCat.16 Pyranonaphthoquinones 9 and 42 were particularly active against glioma and breast cell lines, with eleutherin generally displaying higher activity than isoeleutherin.
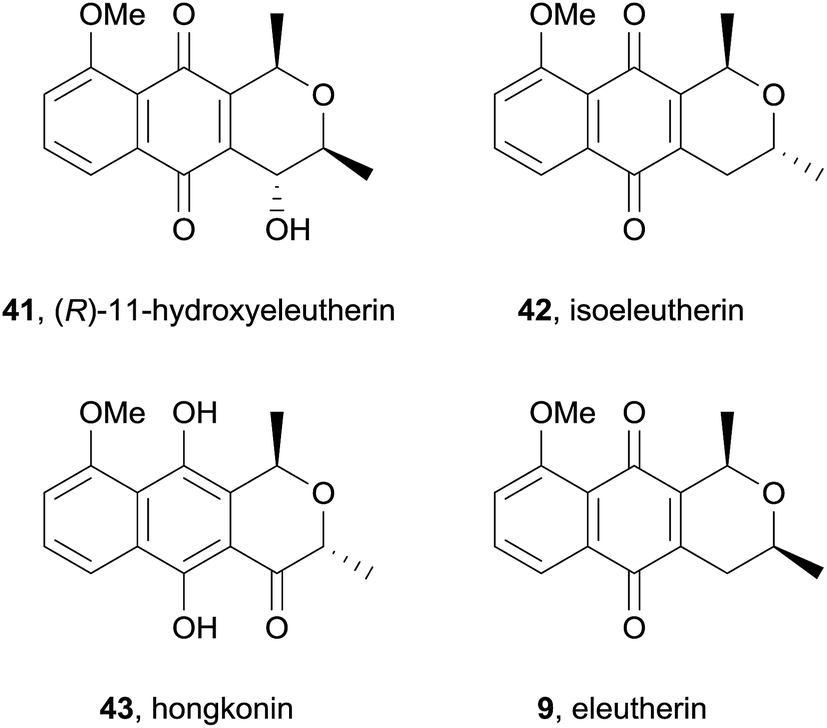 | ||
| Fig. 10 (R)-11-Hydroxyeleutherin and related metabolites isolated from Cipura paludosa. | ||
In 2012, the (−)-enantiomers of the previously reported astropaquinones B (6) and C (7) were isolated from the ethyl acetate extract of a Torula herbarum fungus from a sea hare collected in China (Fig. 11).17 Also isolated were the known pyranonaphthoquinones O-methylherbarin (44), herbarin (45), O-methylfusarubin (46), and dehydroherbarin (47), as well as a novel heptaketide named herbarone (48).
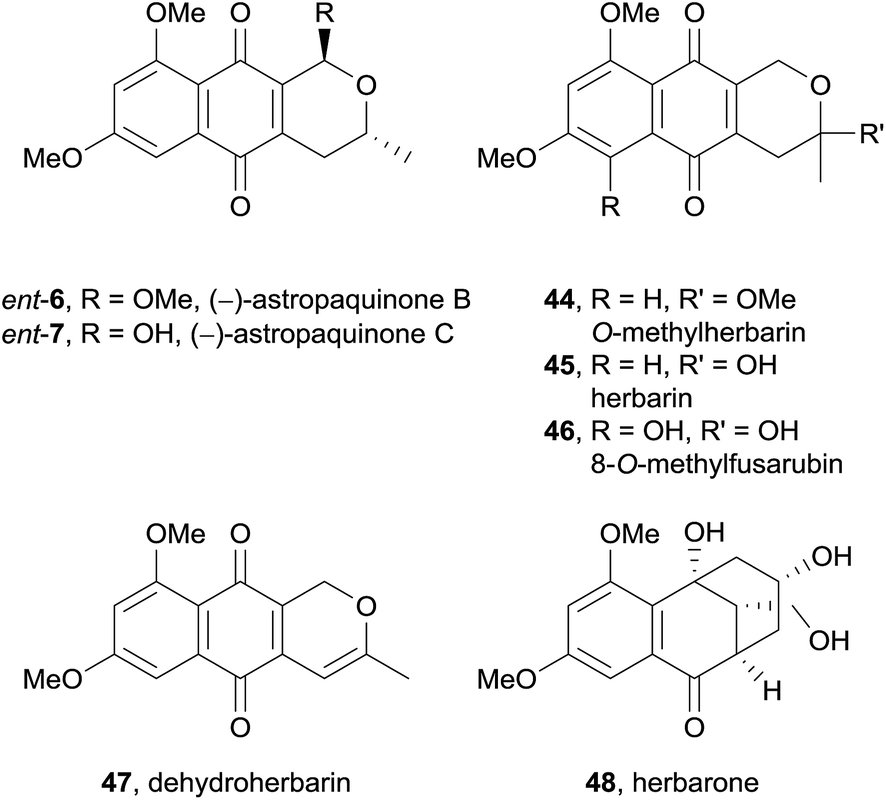 | ||
| Fig. 11 Isolation of herbarone and known pyranonaphthoquinones. | ||
Herbarone (48) was postulated to be biosynthetically related to partially reduced, ring-opened enol derivatives of the pyranonaphthoquinones via an intramolecular aldol reaction and reduction of the ketone at C-7 (Scheme 1).17 No biological testing was conducted on these compounds.
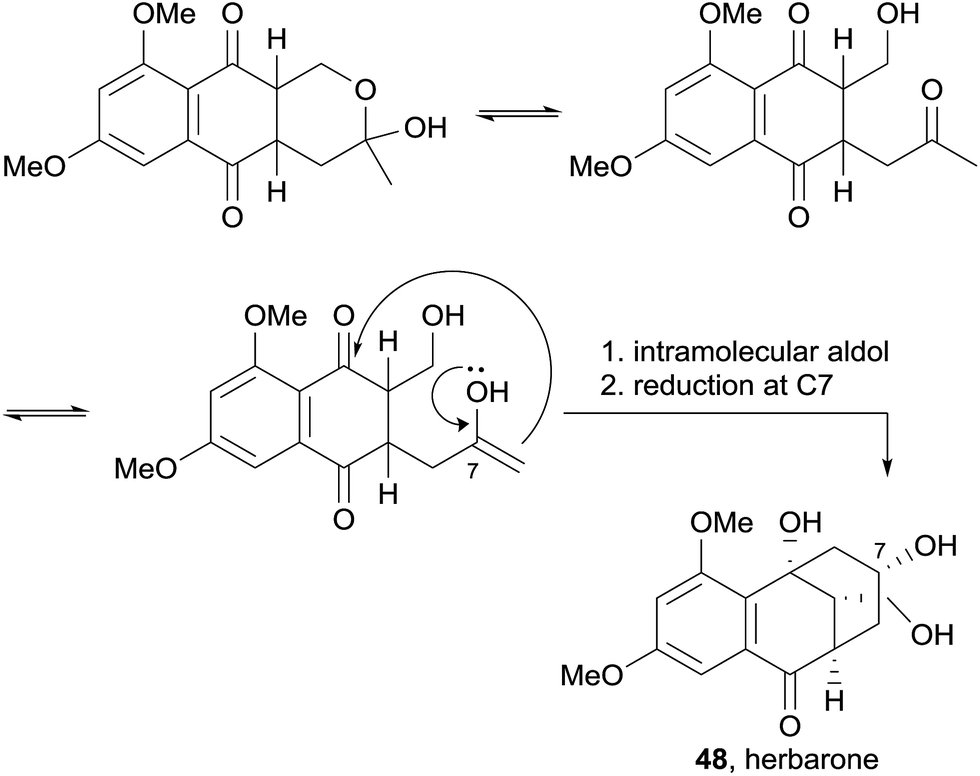 | ||
| Scheme 1 Proposed biosynthesis of herbarone. | ||
A Streptomyces coelicolor deletion mutant lacking the ActVA-ORF4 gene involved in the biosynthesis of actinorhodin (40) produced several pyranonaphthoquinone natural products.18 In particular, the actinorhodin monomer dihydrokalafungin (31) was isolated, together with 8-hydroxydihydrokalafungin (49, Fig. 12). The isolation of this compound and related compound 50, (from which 49 can be derived by keto–enol tautomerisation and subsequent oxidation) led the authors to conclude the importance of ActVA-ORF4 in the biosynthesis of actinorhodin (40), specifically with regard to dimerization of the monomeric units.
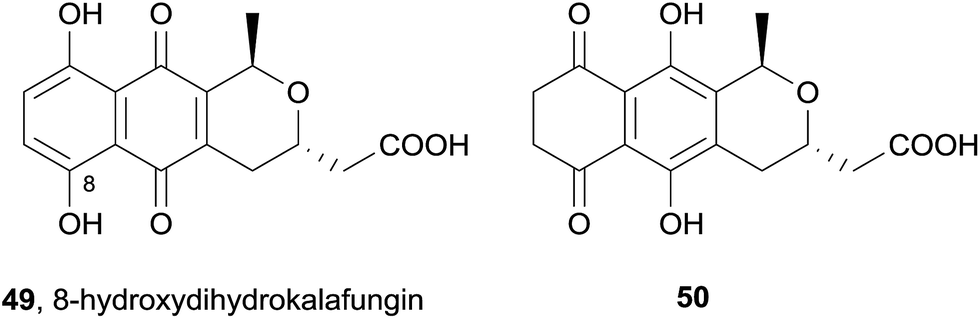 | ||
| Fig. 12 8-Hydroxydihydrokalafungin and derivative 50. | ||
Two known pyranonaphthoquinones, pentalongin (51) and psychorubrin (52), were isolated from the dichloromethane/methanol extracts of a sample of the East African medicinal plant Pentas longiflora (Fig. 13).19 These two compounds displayed antiplasmodial activity against both W2 chloroquine-resistant and D6 chloroquine-sensitive strains of P. falciparum (IC50 < 1 μg mL−1). Psychorubrin (52) was also isolated in 2012 from the Mitracarpus frigidus plant.20 The compound was found to be toxic to both leukemia (HL60 and Jurkat) and breast cancer (MCF-7) cell lines with IC50 values in the 1.1–5.6 μM range. Psychorubrin was shown to induce apoptosis via DNA fragmentation. Additionally, psychorubrin displayed activity against a protozoan parasite of the Leishmania genus, with IC50 values below 3 μM. Antibacterial activity against a variety of bacterial and yeast strains was moderate.
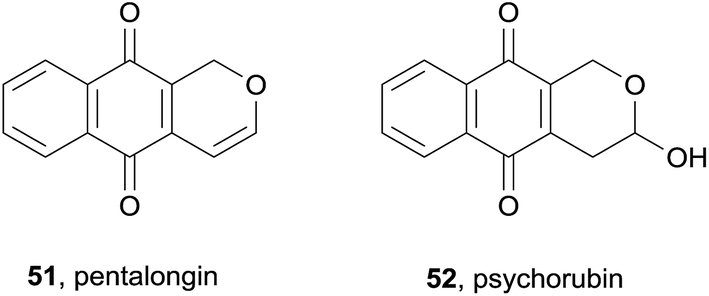 | ||
| Fig. 13 The pyranonaphthoquinones pentalongin and psychorubin. | ||
Also in 2012, a new griseusin derivative was isolated from Streptomyces sp. IFM 11307 collected from soil in Japan (Yoro Valley).21 The combined extracts of the culture broth and bacterial mycelium were purified by flash chromatography, followed by HPLC to afford the natural product as a yellow solid. The product reacted with Dragendorff's reagent, confirming that the product was an alkaloid, while the UV spectrum was indicative of a quinone chromophore. The structure and stereochemistry of the new product were further elucidated by 13C, DEPT and 2D NMR analyses. The griseusin-type natural product named yoropyrazone (53) is a structurally unprecedented naphthopyridazone bearing an amide side-chain (Fig. 14). The relative configuration of the stereocenters at C-17 and C-18 could not be assigned. In the presence of a TNF-related apoptosis-inducing ligand (TRAIL), yoropyrazone exhibits moderate TRAIL resistance-overcoming activity against human gastric AGS cells.
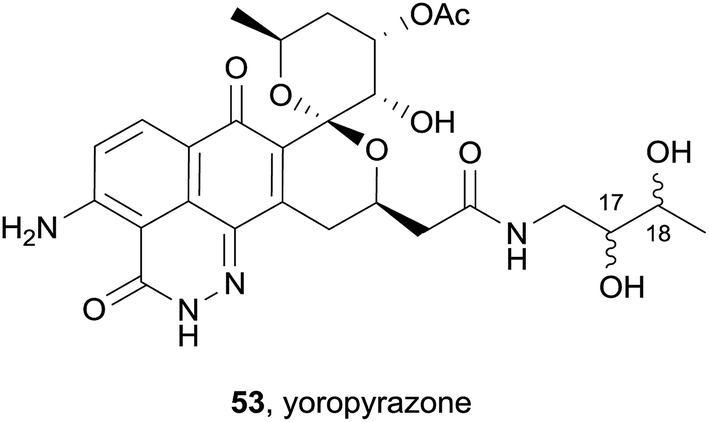 | ||
| Fig. 14 The griseusin-type pyranonaphthoquinone yoropyrazone. | ||
Two further griseusins were also isolated in the same year from the alkalophilic actinomycete Nocardiopsis sp. YIM DT266 from Chinese tin mine tailings (Fig. 15) and named griseusin F (54) and G (55).22 HRMS and extensive NMR analysis indicated that the compounds possess a griseusin A framework with a novel 2a-hydro-8a-(2-oxopropyl) substitution pattern. NOESY analysis of griseusin F (54) indicated a cis-relationship of the substituents with respect to the 2a,8a bond. The relative stereochemistry at C-9/10 was determined to be cis based on the observation of a nOe correlation between H-9 and H-10. The coupling constant of 9.2 Hz between H-3′ and H-4′ indicated a diaxial arrangement of these protons, an assignment further supported by an nOe correlation indicating a 1,3-diaxial interaction between H-3′ and H-5′ax. The relative configuration was then unequivocally established by single crystal X-ray crystallography. Finally, comparison of the computed CD spectrum with that observed enabled assignment of the absolute configuration of griseusin F (54). The molecular weight of griseusin G (55) was 2 Da lower than that of F (54), resulting from oxidation of the 4′-OH, as confirmed by the presence of the carbonyl resonance at δ 203.8 ppm in the 13C NMR spectrum. The CD curve was almost identical to that observed for griseusin F (54) and the absolute configuration of the remaining stereocenters were therefore assigned to be the same as grisuesin F (54). Griseusins F (54) and G (55) were tested against melanoma, breast, pancreatic, renal and colon cancer cell lines and were shown to exhibit high cytotoxicity, specifically against melanoma and breast cell lines – on a par with the well-known anticancer drug cisplatin. These natural products also exhibited antibacterial activity.22
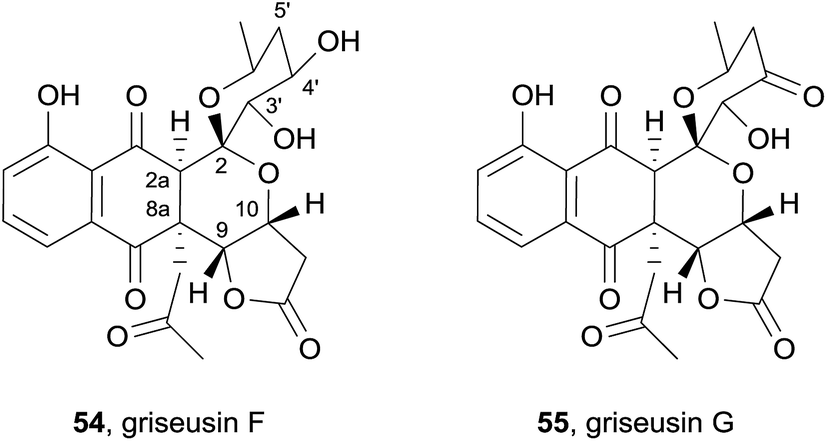 | ||
| Fig. 15 Griseusins F and G. | ||
In 2013, the ethyl acetate extract of a cultured Streptomyces species furnished a novel dimeric pyranonaphthoquinone, diepoxyactinorhodin (56, Fig. 16).23 The structure was elucidated by mass spectrometry and extensive spectroscopic analysis. This revealed the compound to be an actinorhordin (40) analogue in which the 4a/10a and 4a′/10a′ double bonds of 40 have undergone epoxidation. The stereochemical configurations at C4a/a′ and C10a/a′ were not determined. Pyranonaphthoquinone 56 displayed activity against the Gram-positive bacteria methicillin-resistant S. aureus (MRSA), methicillin-sensitive S. aureus (MSSA) and B. subtilis to a similar degree as actinorhodin, but was not active against Gram-negative bacteria or fungi.
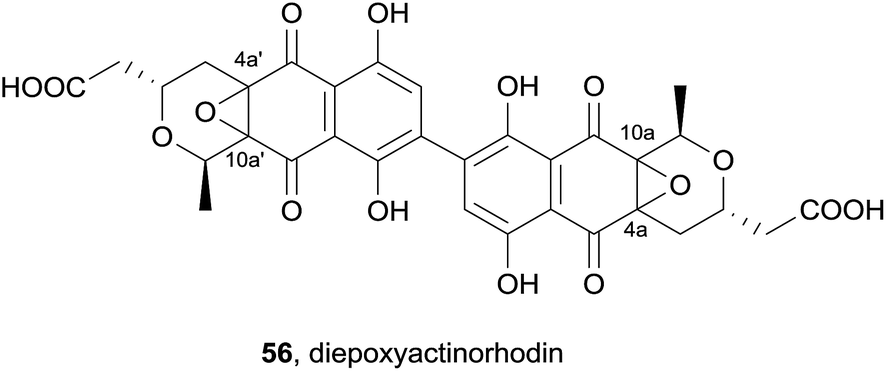 | ||
| Fig. 16 The dimeric pyranonaphthoquinone diepoxyactinorhodin. | ||
The previously known pyranonaphthoquinone fusarubin (57) was isolated from an ethyl acetate extract of the endophytic fungus Phomopsis sp. HCCB04730, alongside a novel dihydronaphthalenone and several other known naphthoquinones (Fig. 17).24 Fusarubin (57) displayed cytotoxicity against adenocarcinoma (A549), breast cancer (MDA-MB-231) and pancreatic cancer (PANC-1) cell lines, as well as good inhibitory activity against HIV-1.
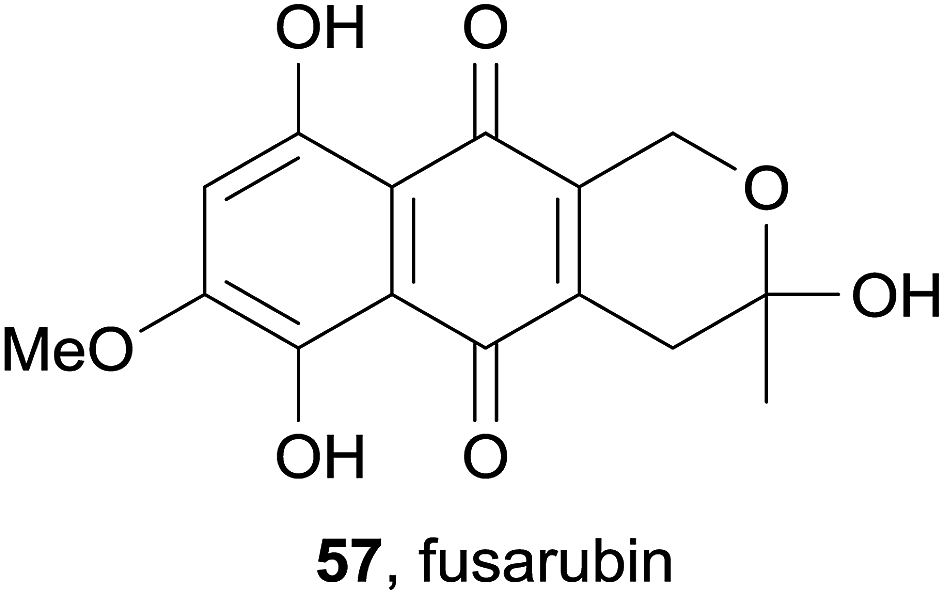 | ||
| Fig. 17 Fusarubin. | ||
Also in 2013, five new pyranonaphthoquinones were isolated from cultured Streptomyces sp. RM-4-15 (Fig. 18).25 The acetone extract of the mycelial cake and the methanol extract of the fermentation broth were shown to contain identical metabolites and were therefore combined prior to purification. Column chromatography and HPLC afforded the novel natural products, frenolicins C–F (58–61), as well as previously isolated compounds frenolicin B (36), UCF76-A and frenolicin. Frenolicin C (58) contains an acetylcysteine substituent at C4a and a hydroxyl group at C10a. Frenolicins D (59) and E (60) possess hydroxyl groups at C4a and C10a, while frenolicin F (61) contains the same C4a–C10a epoxy substitution as frenolicin, but has undergone reduction at C5. A second batch of Streptomyces sp. RM-4-15 was fermented in the presence of scandium chloride. The ethyl acetate extract provided frenolicin, frenolicin B (36), deoxyfrenolicin and the novel pyranonaphthoquinone frenolicin G (62). Frenolicin G (62) was determined to be a symmetric dimer, linked by a highly unusual thioether bridge at C4a, although the relative configuration at this position could not be determined. The natural products were assessed in an assay using lung carcinoma cells A549, however none of the new pyranonaphthoquinones displayed anticancer activity.
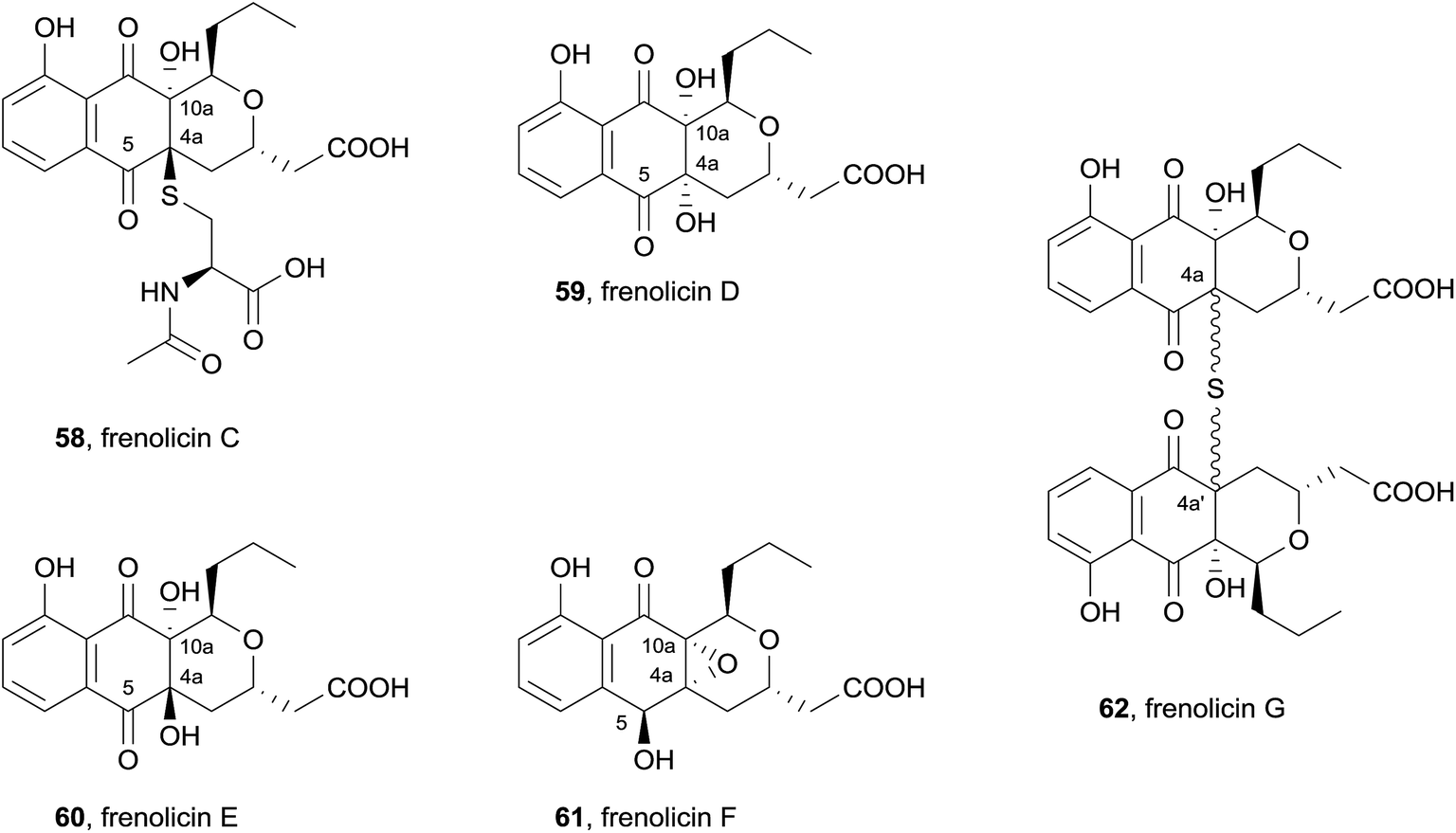 | ||
| Fig. 18 Novel frenolicins isolated from Streptomyces sp. RM-4-15. | ||
Indoleamine 2,3-dioxygenase-1 (IDO-1) is an enzyme involved in the breakdown of L-tryptophan that has been implicated in the pathology of diseases including Alzheimer's and cancer.26 Noting that the naturally occurring quinones annulins A–C (63–65) inhibit IDO-1, Brimble and coworkers evaluated a variety of synthetic pyranonaphthoquinones for inhibitory activity against IDO-1 (Fig. 19).
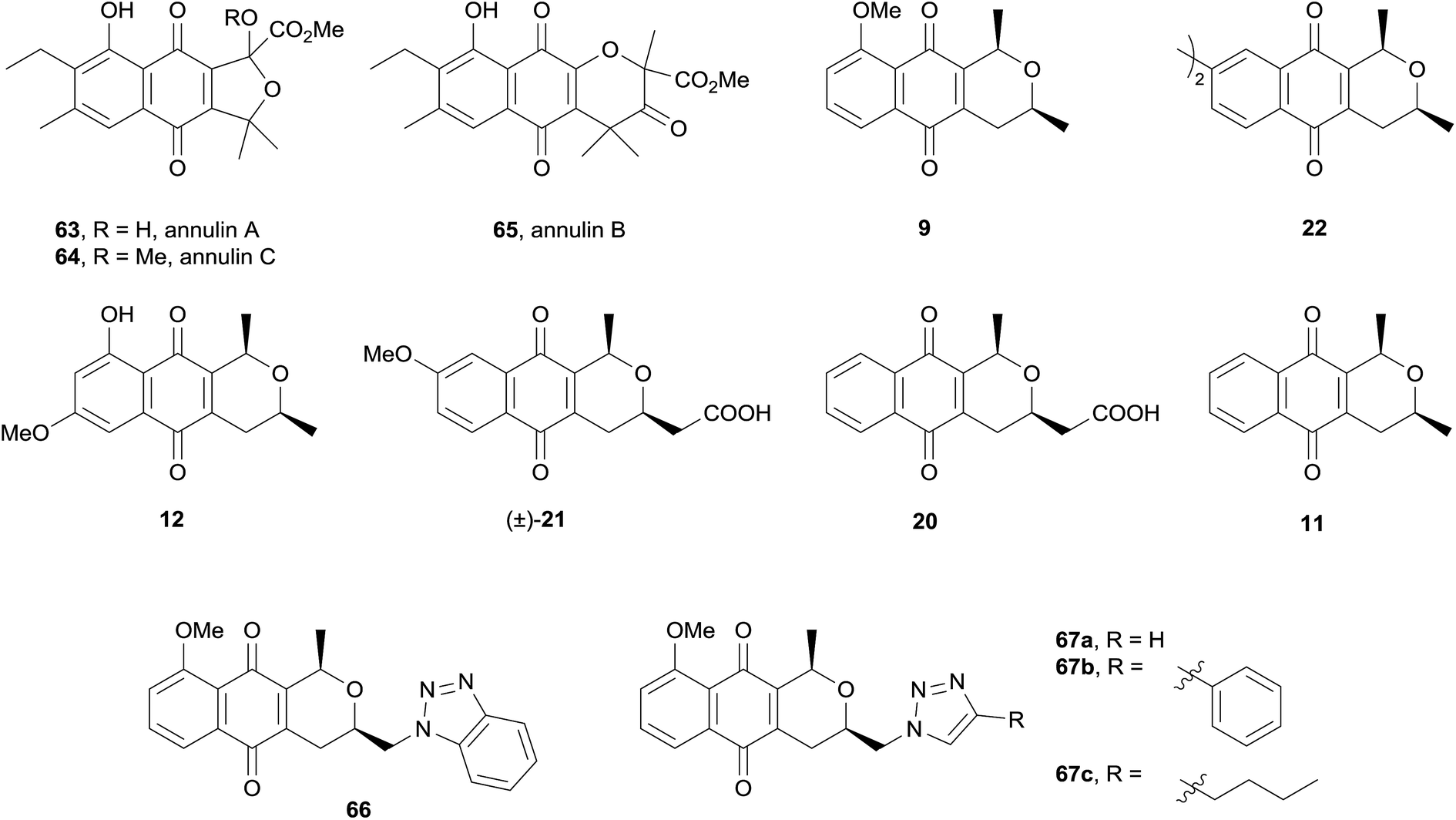 | ||
| Fig. 19 Annulins and synthetic pyranonaphthoquinones tested against IDO-1. | ||
All of the compounds tested displayed inhibition of IDO-1, some in the sub-micromolar region. The compounds were then evaluated against murine and human cancer cell lines. Only compound 21 showed over 50% inhibition of murine cancer cells. Compounds 21, 12, 22, 66 and 67a exhibited moderate activity against human cell lines. The final test was for IDO-1 inhibition in the cellular context, where once again only compound 21 displayed significant inhibitory activity.26
In 2014, two biosynthetic precursors to granaticin were isolated from Streptomyces sp. CPCC 200532.27 The structures of 6-deoxy-13-hydroxy-8,11-dione-dihydrogranaticin B (68b) and A (68a) were identified using mass spectrometry, 1H NMR, 13C NMR and 2D NMR analysis (Fig. 20). Compound 68a was proven to be an intermediate in granaticin biosynthesis, as it was successfully bioconverted into granaticin B (69b) by a mutant strain of S. vietnamensis GIMV4.000 that does not produce granaticin B. In general, compounds 68a and 68b were found to be less active against cancer cell lines than granaticin A (69a) and B (69b).
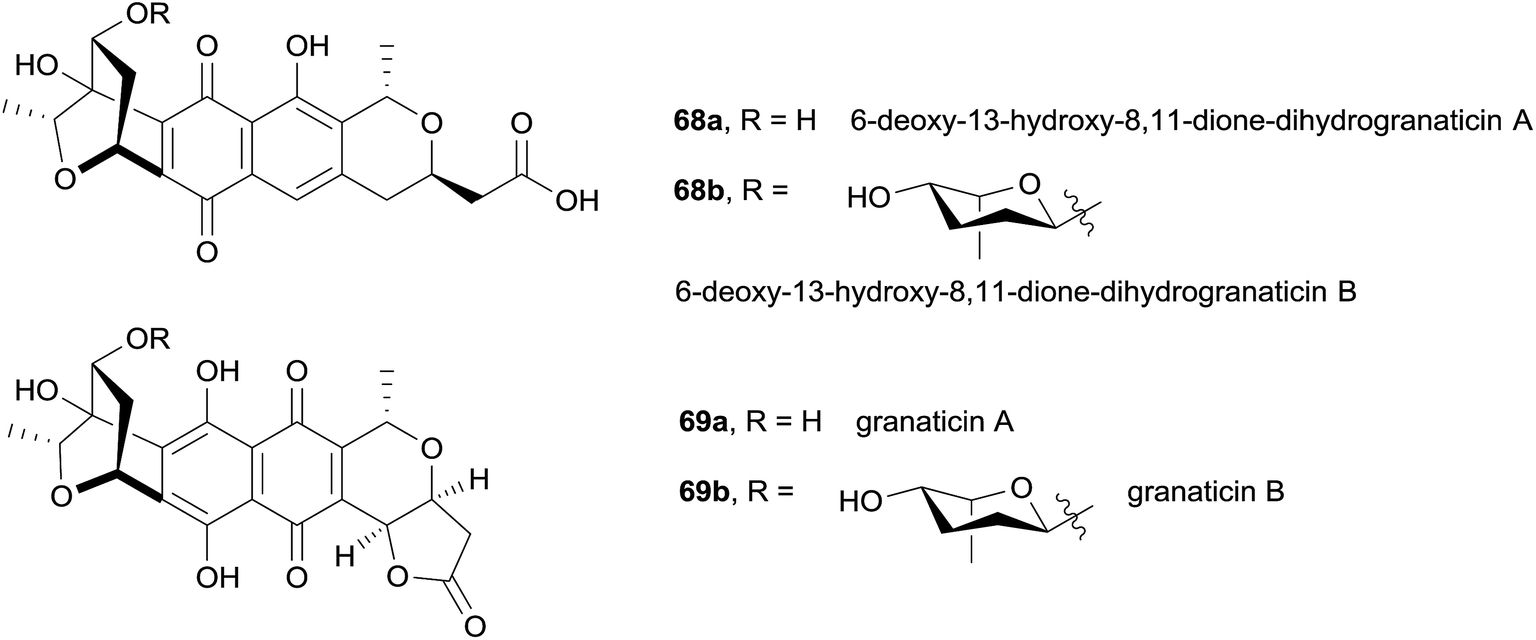 | ||
| Fig. 20 Biosynthetic intermediates of granaticin. | ||
In 2015, the dichloromethane extract of the fungus Biatriospora sp. CCF4378 yielded six known naphthoquinones, the two novel pyranonaphthoquinones pleorubins A (70) and B (71) and two furanonaphthoquinones pleorubrin C (72) and D (73) (Fig. 20).28 Pleorubrin A (70) is closely related to thysanone (14, see Fig. 5) and was previously synthesised by Brimble and coworkers during their synthesis of (+)-thysanone.29 Pleorubrin B (71) possesses (R)-stereochemistry at C3 and is an analogue of 70 which has undergone reduction at C5 and saturation of the C4a–C10a quinone double bond. Four of the isolated compounds: 6-deoxybostrycoidin, ascomycone B (2), 6-deoxyfusarubin and pleorubrin B (71), were tested against HeLa cells. 6-Deoxybostrycoidin and pleorubrin B (71), showed no effect up to 100 μg mL−1 whereas ascomycone B (2) and 6-deoxyfusarubin resulted in complete cell death after 24 hours incubation using a dose of 50 μg mL−1 (Fig. 21).
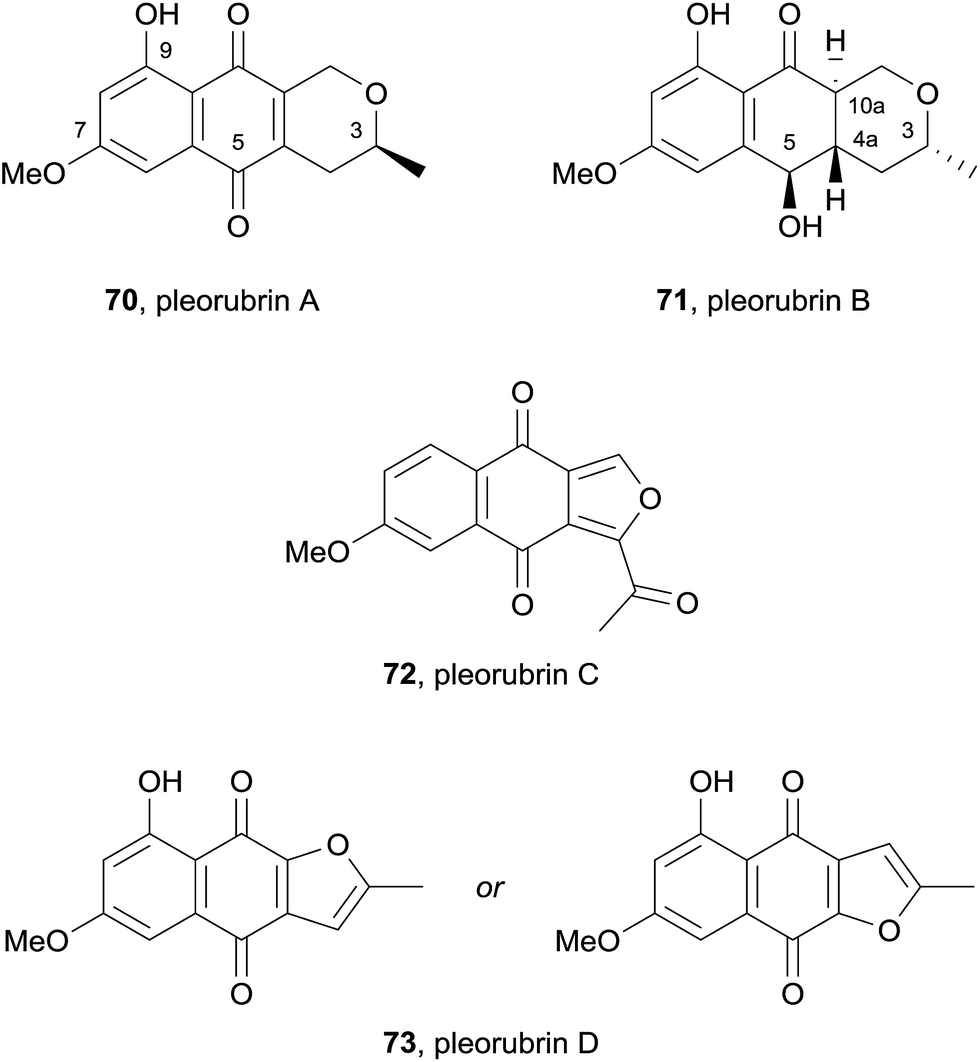 | ||
| Fig. 21 Pleorubrins A and B and related metabolites isolated from Biatriospora sp. CCF4378. | ||
Two new pyranonaphthoquinones 74a and 74b were isolated in the same year from the fermentation culture of the insect pathogenic fungus Conoideocrella luteorostrata Zimm. BCC 31648.30 Compound 74a had previously been synthesised,8 however this was the first time it had been isolated from a natural source (Fig. 22). Compound 74b was determined by analysis of the NMR and optical rotation data to be the 8-(4′-O-methyl-β-D-glucopyranoside) analogue of 74a. The compounds were tested for antimalarial activity against Plasmodium falciparum and cytotoxicity against KB, MCF-7, NCI-H187 and Vero cell lines. Compounds 74a and 74b both exhibited antimalarial activity with IC50 values of 3.31 and 3.65 μg mL−1 respectively, and low to moderate cytotoxicity.
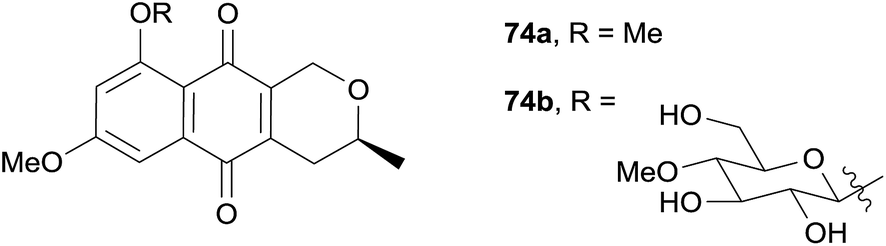 | ||
| Fig. 22 Pyranonaphthoquinones 74a and 74b. | ||
A new pyranonaphthoquinone was isolated from a Streptomyces sp. found in a karst cave in China.31 The UV-vis absorption spectrum of the new compound was similar to pyranonaphthoquinones containing a fused lactone such as kalafungin and was named xiakemyicin A (75, Fig. 23). Analysis of the 2D NMR spectra indicated substitution of the aromatic ring at C-6 and the substituent identified as a functionalised dihydropyran ring. The relative stereochemistry was determined by nOe analysis. The absolute configuration was not determined. Xiakemycin A (75) exhibited strong inhibitory activity against a variety of Gram-positive bacteria (MIC values of 4–16 μg mL−1) but weak activity against a number of Candida fungal species (MIC ≥ μg mL−1). Additionally, compound 75 exhibited reasonable cytotoxic activity against human lung cancer A549, breast cancer MCF-7, hepatoma HepG-2, cervical cancer HeLa, colon carcinoma HCT-116 cell p53 wt, neuroblastoma SH-SY5Y and human prostate cancer PC-3 cells with IC50 values of 2.77, 1.82, 1.54, 0.98, 0.59, 0.55 and 0.43 μM, respectively.
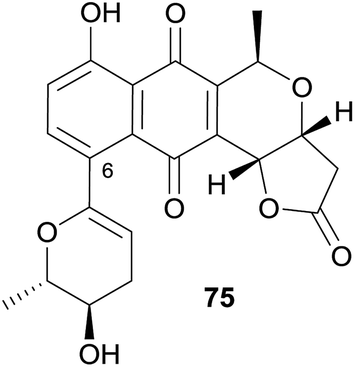 | ||
| Fig. 23 Xiakemycin A. | ||
Finally, a report in 2015 described the isolation of kalafungin (35) from medermycin-producing bacteria for the first time.32 The ethyl acetate extract of wild-type Streptomyces sp. AM-7161 was found to contain both medermycin (81) and kalafungin (35) in complementary quantities (increased production of 81 corresponded to decreased production of 35). A recombinant strain of S. coelicolor CH999 containing the pIK340 medermycin-producing gene cluster and a UV-mutant strain were also both shown to produce both medermycin and kalafungin, leading the authors to propose that the biosyntheses of these compounds are related. Based on previous discovery of (S)-DNPA (77, 4-dihydro-9-hydroxy-1-methyl-10-oxo-3H-naphtho[2,3-c]pyran-3-acetic acid) formed by a ketoreductase encoded by the medermycin gene cluster, the biosynthetic pathway in Scheme 2 was proposed. Reductive cyclisation of compound 76 gives chiral intermediate 77 which then undergoes further reduction to 6-deoxydihydrokalafungin (78), oxidation then provides dihydrokalafungin (31). Two pathways to medermycin (81) are then possible. In the first pathway, compound 31 is converted to kalafungin (35) via the quinone-methide 79. Subsequent C-glycosylation then provides medermycin (81). In the second pathway these two steps are reversed, proceeding through dihydromedermycin (82). Accumulation of kalafungin in Streptomyces sp. AM-7161 suggests that C-glycosylation is the rate-limiting step in medermycin biosynthesis, but does not conclusively determine which of the two pathways to medermycin dominates.
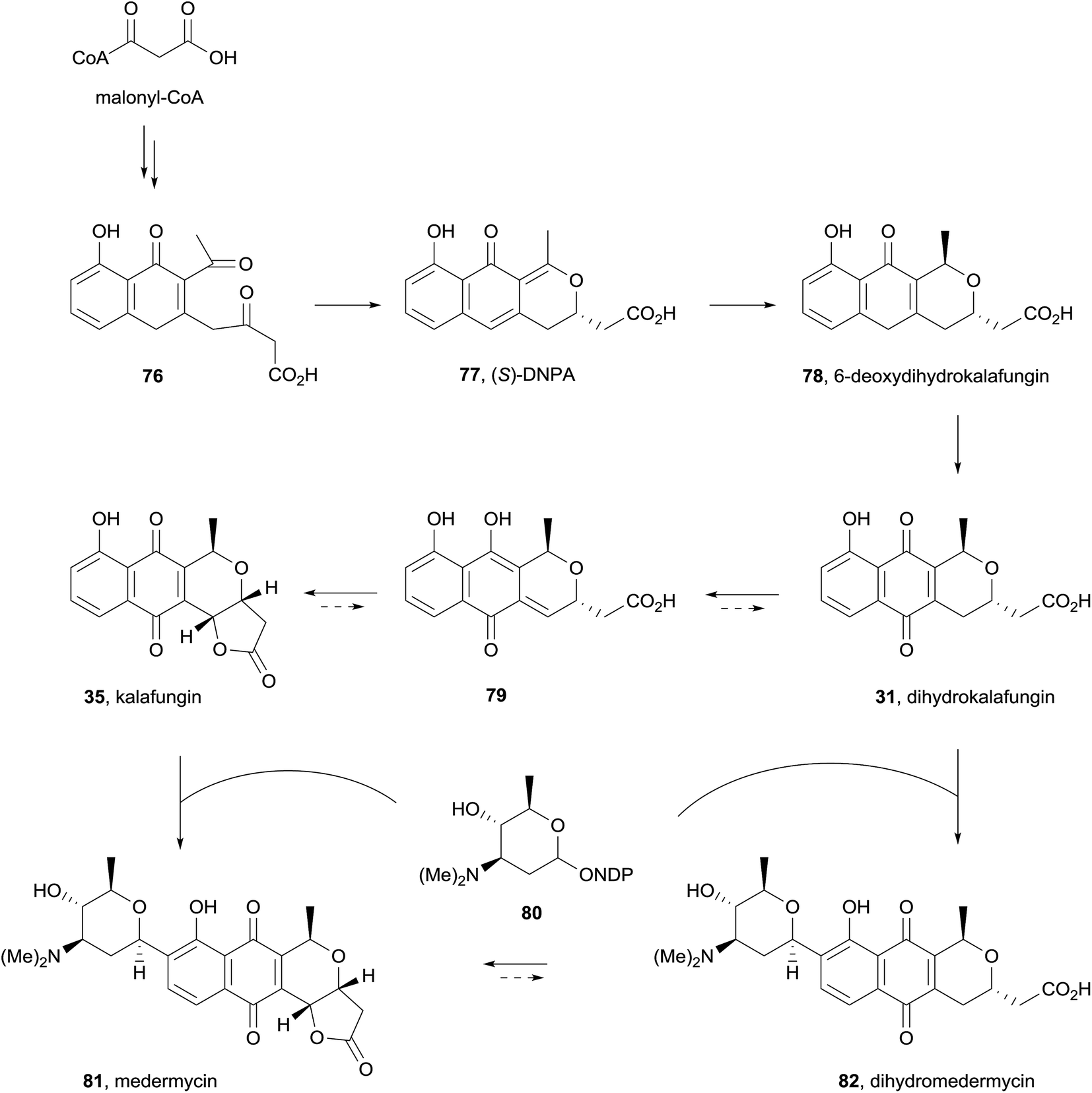 | ||
| Scheme 2 Proposed biosynthesis of medermycin. | ||
3. Synthetic strategies
3.1 Oxa-Pictet–Spengler reaction
Over the past decade or so, the oxa-Pictet–Spengler (OPS) reaction has increased in popularity as a means to access the pyranonaphthoquinone framework. In this reaction, a readily available 2-phenylethanol undergoes intramolecular electrophilic aromatic substitution after nucleophilic addition to an aldehyde or ketone (or synthetic equivalent). Indeed, the OPS reaction enables the rapid assembly of the pyranonaphthoquinone framework in enantiopure form, and it is of no surprise this approach has found utility in the synthesis of many pyranonaphthoquinones, including the more complex dimeric members. For example, Waghmode and Sawant used an OPS reaction to complete a formal synthesis of thysanone (Scheme 3).33 Ethyl-3-(2,5-dimethoxyphenyl)propionate (83) was converted to alcohol 84 in 5 steps by bromination of the aromatic ring and manipulation of the side-chain. Chirality was introduced during this sequence using L-proline-catalysed asymmetric α-aminooxylation. The key OPS reaction was conducted utilising methoxymethyl chloride and zinc chloride, affording isochromane 85 in high yield. Oxidative demethylation of isochromane 85 using CAN afforded the quinone 86, which has been previously employed in Diels–Alder reaction with a Danishefsky-type diene (87) to access (−)-thysanone (14).34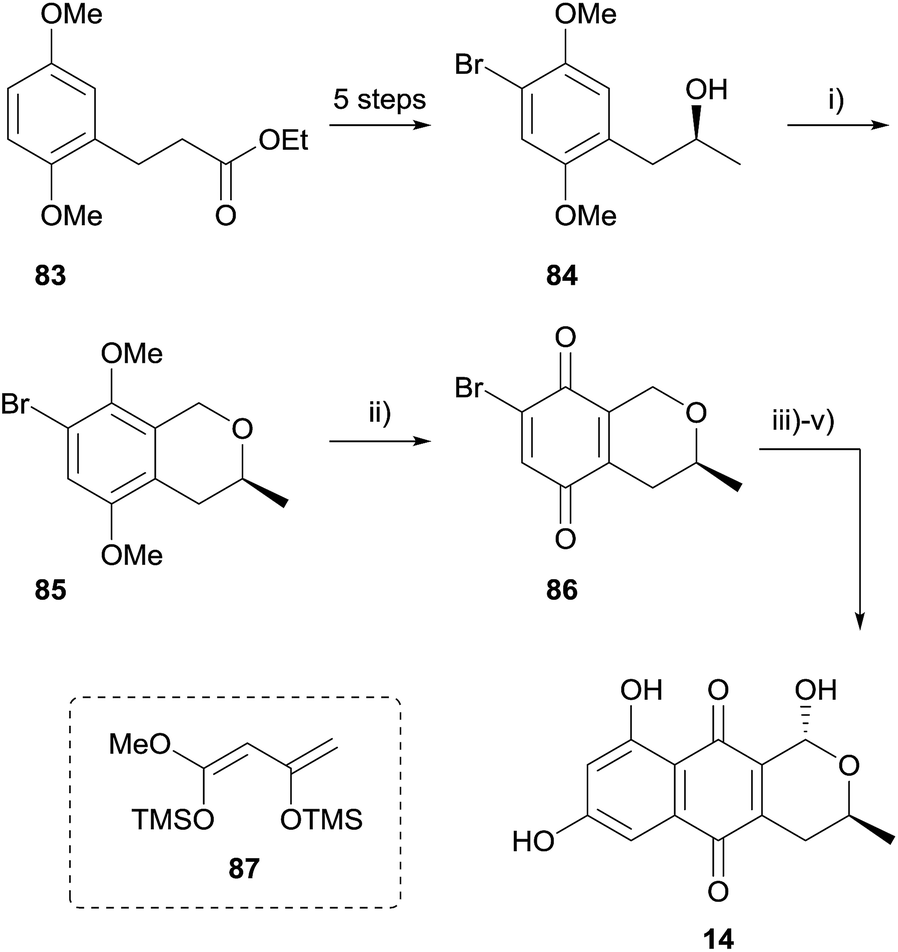 | ||
| Scheme 3 Reagents and conditions: (i) MeOCH2Cl, ZnCl2, Et2O, 0 °C to rt, 6 h, 80%; (ii) CAN, MeCN–H2O, 0 °C to rt, 25 min, 77%; (iii) 87, toluene, reflux, 73%; (iv) Br2, hν, CCl4, 30 min; (v) H2O, THF, rt, 1 h, 85% over 2 steps. | ||
A similar strategy was also employed by Waghmode and coworkers to synthesise deoxy analogues of a number of naturally-occurring pyranonaphthoquinones (Scheme 4).35 Diol 88 was protected as ethylidene acetal 89 which underwent a Lewis acid-mediated intramolecular OPS reaction to form isochromane 90. Manipulation of the side-chain and oxidative demethylation provided quinone 91, which was converted to (+)-deoxynanaomycin A methyl ester (92) following the Diels–Alder reaction conditions reported by Kometani and Yoshii.36 An attempt to synthesise nanaomycin A methyl ester by an analogous method employing substrate 93 was unsuccessful because the OPS reaction was low-yielding.
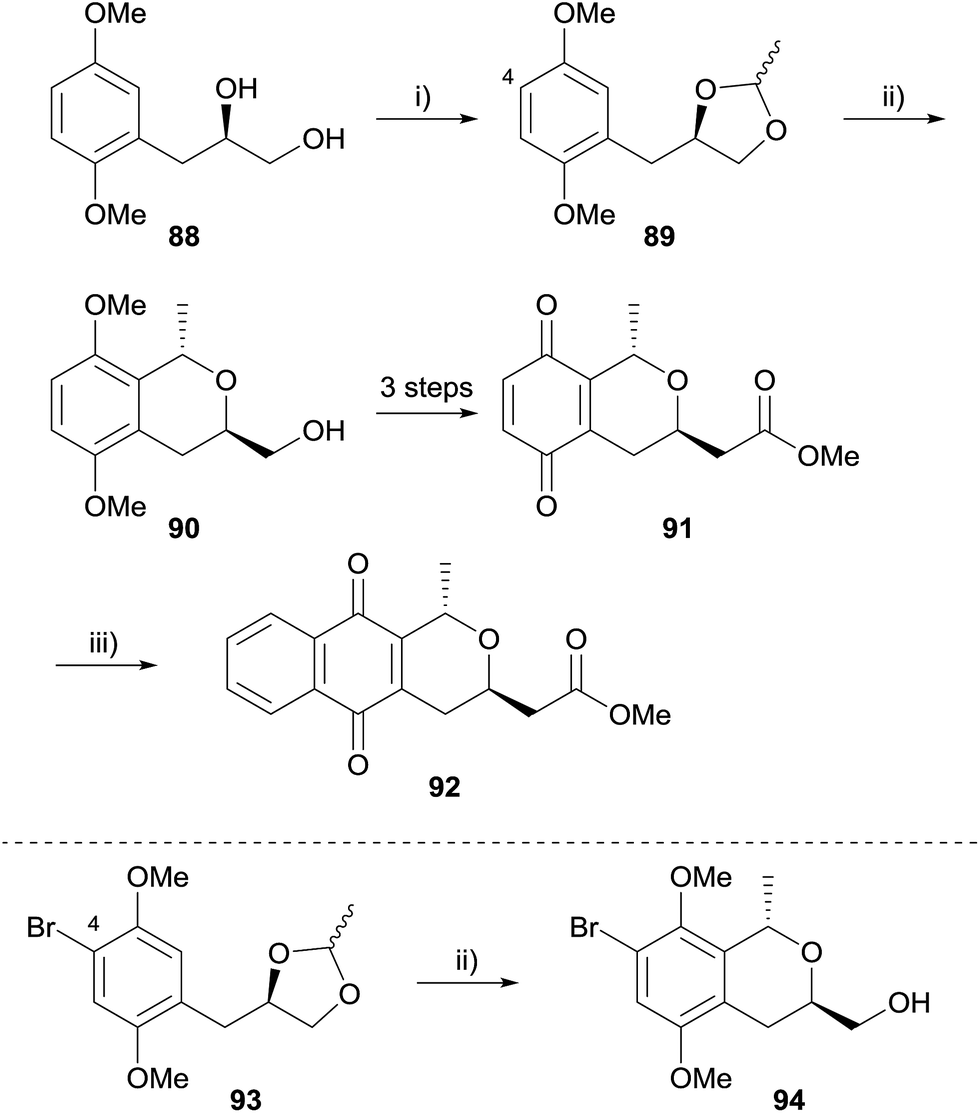 | ||
| Scheme 4 Reagents and conditions: (i) MeCH(OEt)2, PTSA, CH2Cl2, 0 °C to rt, 7 h, 98%; (ii) TiCl4, CH2Cl2, −30 °C, 3.5 h, 87% 90, 7% 94; (iii) 1-acetoxy-1,3-butadiene, toluene, rt, 48 h, then 1% aq. Na2CO3, EtOH, rt, 5 h, 78%. | ||
Similar attempts to employ an OPS reaction for the formation of (+)-deoxyeleutherin, (+)-deoxyalloeleutherin and (+)-deoxythysanone using C-4 brominated substrates were also unsuccessful. An alternative route starting from compound 95 was therefore designed (Scheme 5).35 OPS reaction of alcohol 95 with 1,1′-diethoxyethane gave a mixture of epimeric isochromanes 96 and 97, which were readily separable by column chromatography. Oxidative demethylation followed by Diels–Alder reaction with 1-acetoxy-1,3-butadiene according to literature procedures36 provided (+)-deoxyeleutherin (11) and (+)-deoxyalloeleutherin (98). In addition, OPS reaction of alcohol 95 with methoxymethyl chloride afforded isochromane 99, which was converted to (+)-deoxythysanone (15) in high yield following an analogous procedure.35
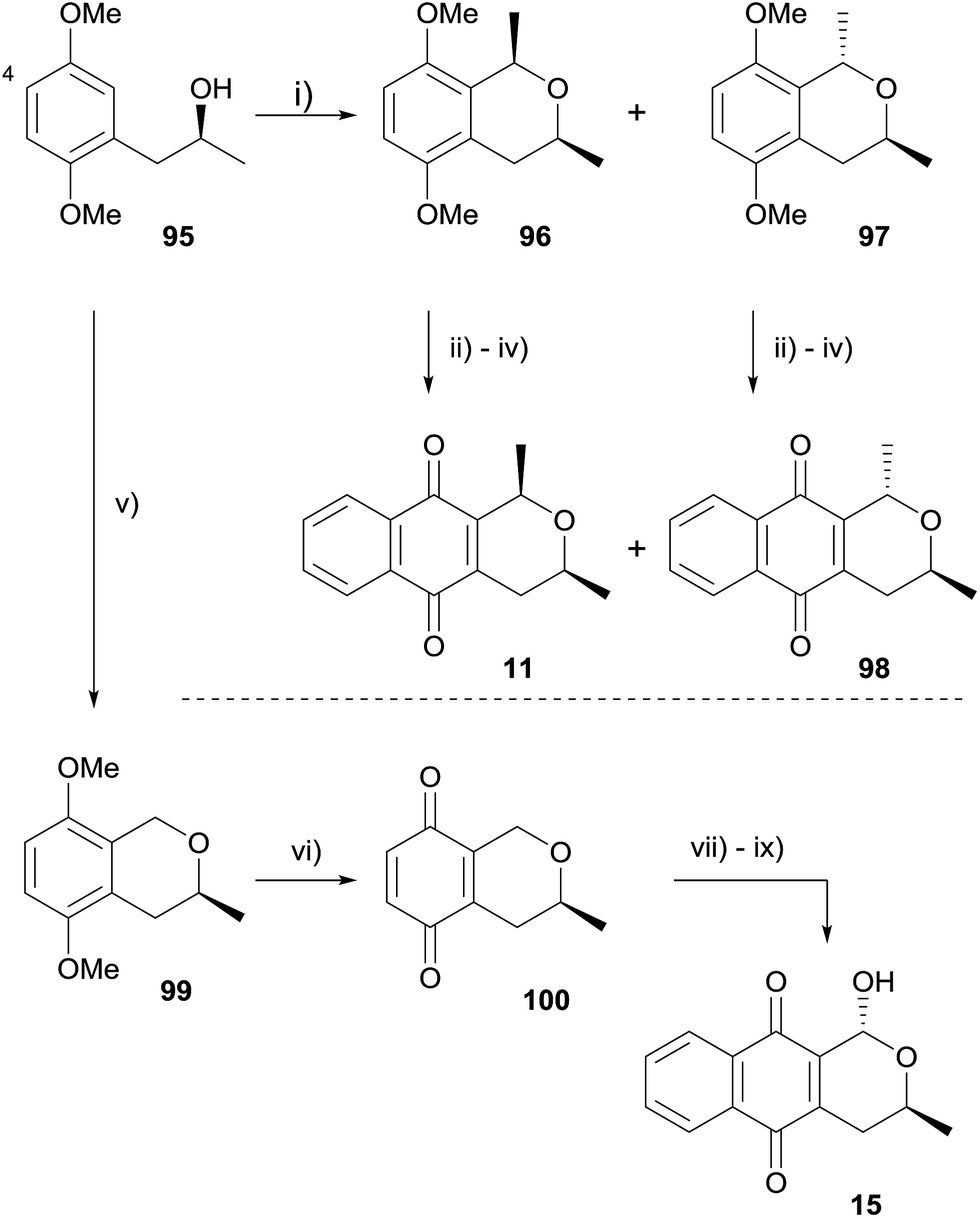 | ||
| Scheme 5 Reagents and conditions: (i) MeCH(OEt)2, BF3·OEt2, CH2Cl2, 0 °C to rt, 3.5 h, 21% 96, 65% 97; (ii) CAN, MeCN–H2O, 0 °C to rt, 0.5 h, 84% from 96, 78% from 97; (iii) 1-acetoxy-1,3-butadiene, toluene, rt, 72 h; (iv) 1% aq. Na2CO3, EtOH, rt, 5 h, 89% over two steps for 11, 91% over two steps for 98; (v) MeOCH2Cl, ZnCl2, Et2O, 0 °C to rt, 7 h, 83%; (vi) CAN, MeCN–H2O, 0 °C to rt, 25 min, 81%; (vii) 1-acetoxy-1,3-butadiene, toluene, rt, 48 h; (viii) 1% aq. Na2CO3, EtOH, rt, 5 h, 85% over 2 steps; (ix) Br2, CCl4, hν, 0.5 h then THF–H2O, rt, 1 h, 77% over 2 steps. | ||
In 2009, Eid and coworkers reported a concise synthesis of (+)-7-deoxyfrenolicin B (106a) and (+)-7-deoxykalafungin (106b) using an OPS reaction as the key step (Scheme 6).37 Heck coupling of bromide 101 and alkene 102 afforded alkene 103, which underwent Sharpless asymmetric dihydroxylation with concomitant lactonisation to give 104. Lewis-acid mediated OPS reaction of 104 with appropriate aldehydes gave naphthopyrans 105a and 105b in excellent yield and diastereoselectivity. Oxidative demethylation using CAN afforded the target compounds 106a and 106b. Computational analysis of the oxocarbenium ion intermediates involved in the OPS reaction indicated that the stereoselectivity of the reaction is due to an unfavourable geometry of the intermediate required for the formation of the diastereomer in which the substituents on C3a and C5 are cis.37
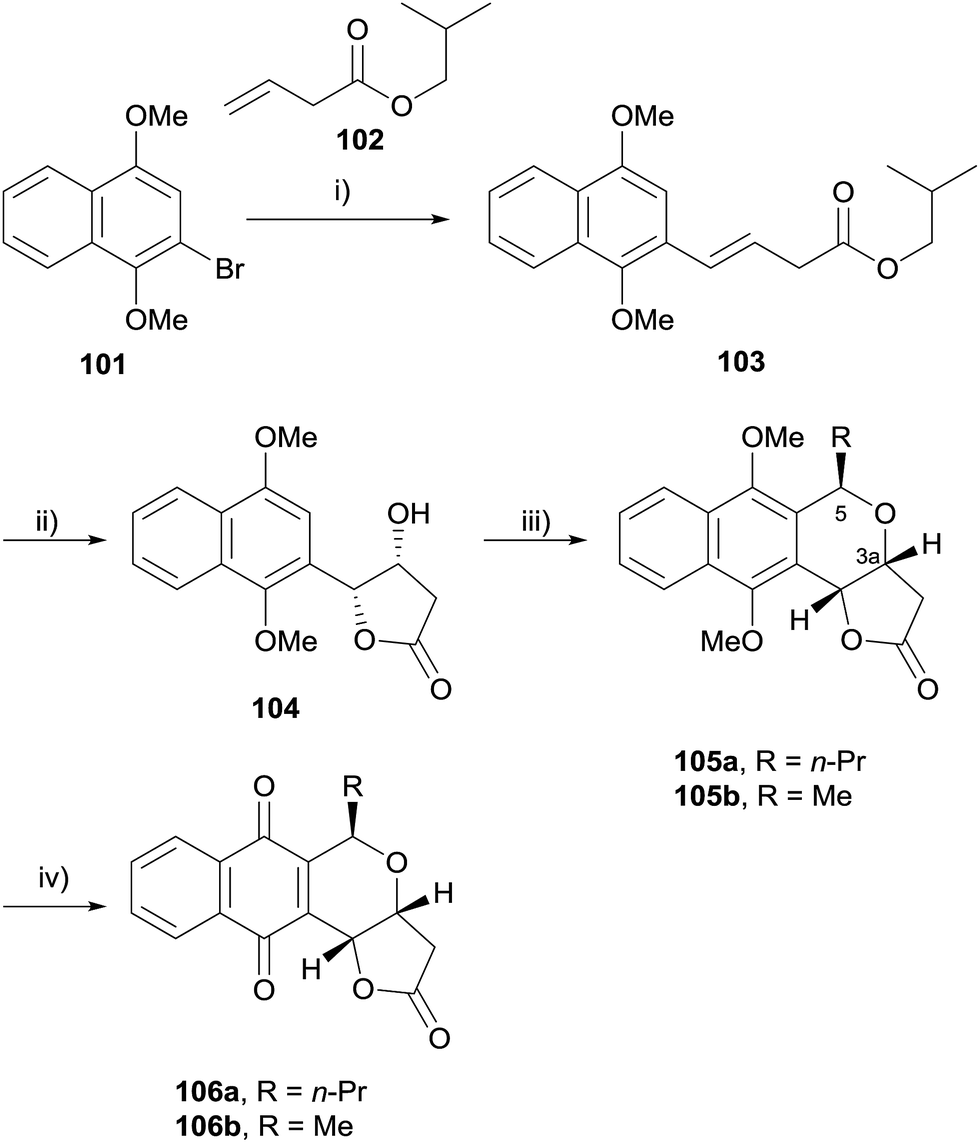 | ||
| Scheme 6 Reagents and conditions: (i) Pd(t-Bu3P)2, Cy2NMe, toluene, 100 °C, 80%; (ii) K3Fe(CN)6, K2CO3, (DHQD)2PHAL, K2OsO4·(H2O)2, MeSO2NH2, t-BuOH–H2O, 0 °C to rt, 18 h, 69%; (iii) BF3·OEt2, RCHO, CH2Cl2, 0 °C to rt, 88%, 88% de for 105a, R = Me 98%, 99+% de for 105b; (iv) CAN, MeCN, 0 °C, 68%, 99% de for 106a, R = Me 64%, 99% de for 106b. | ||
Also in 2009, it was shown that pyranonaphthoquinone antibiotics exhibit selective inhibitory activity against protein kinase B (also known as AKT), a serine/threonine kinase.38 Levels of activated AKT are often elevated in human cancer cells of the breast, colon, prostate and in acute myeloid leukaemia, which is thought to play a role in rapid and abnormal cellular proliferation. The known pyranonaphthoquinones lactoquinomycin (medermycin),39 kalafungin,40 frenolicin B,41 methoxyfrenolicin B and deoxyfrenolicin41 were shown to exhibit AKT inhibitory activity. Following this discovery, Salaski and coworkers synthesised a number of racemic and enantiopure analogues and conducted a structure–activity relationship (SAR) study. The analogues were rapidly assembled using an oxa-Pictet–Spengler-lactonisation strategy (Scheme 7).
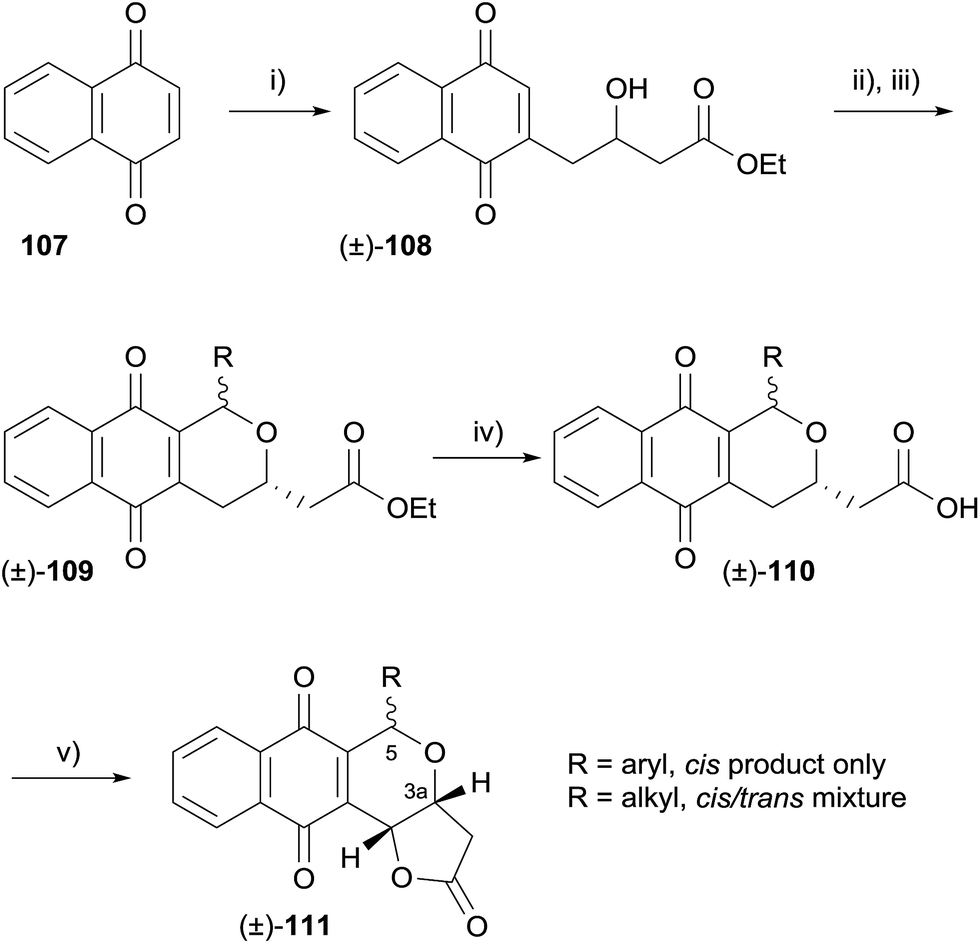 | ||
| Scheme 7 Reagents and conditions: (i) HO2CCH2CH(OH)CO2Et, (NH4)2S2O8, AgNO3, MeCN–H2O, 29%; (ii) H2, Pd/C, THF; (iii) RCHO, HCl (g), Et2O; (iv) 4 M HCl, THF; (v) MnO2, MeCN. | ||
Enantiopure analogues were synthesised via hydroxylactone 104, which was accessed by Sharpless asymmetric dihydroxylation (AD) of naphthalene 112 (Scheme 8).
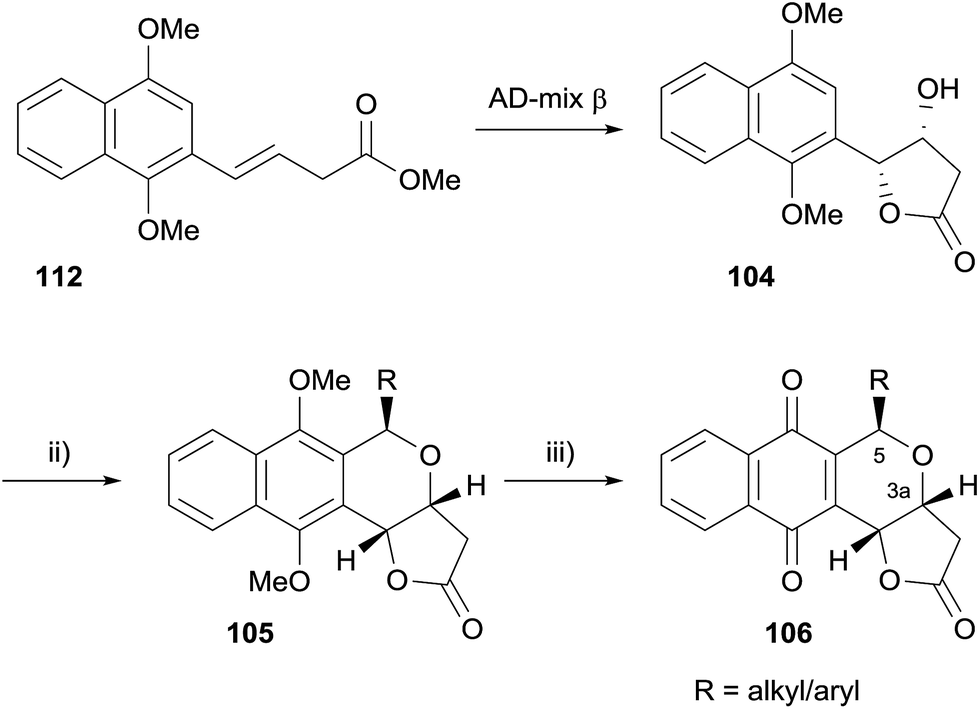 | ||
| Scheme 8 Reagents and conditions: (i) RCHO, BF3·OEt2, CH2Cl2; (ii) CAN, MeCN–H2O. | ||
The SAR study concluded that both the quinone and lactone moieties are essential for inhibition of AKT. Compounds with a free carboxylic acid capable of undergoing cyclisation to the lactone also possessed some activity. Although the absolute configuration did not affect activity, C3a–C5 trans isomers were more active than the corresponding cis isomers. Di-substitution at C5 was not tolerated. In the same publication, treatment of lactoquinomycin (113) with cysteine in water (Scheme 9A) resulted in the formation of monoadduct 115, a result that is in agreement with the thioalkylation of pyranonaphthoquinone 116 to form adducts 117a–c previously reported by Brimble and coworkers (Scheme 9B)42 and corroborating the bioreductive alkylation hypothesis proposed by Moore (Scheme 9C).43,44 Interestingly, this transformation did not require the use of a reductant such as sodium thiosulfate to generate the hydroquinone prior to formation of the quinone-methide, in contrast to the work of Brimble et al. Compound 115 displayed minimal inhibitory activity which was attributed to a decreased propensity for quinone-methide formation. Additionally, pyranonaphthoquinone 106 (R = 3-thienyl) was shown to bind to activated AKT, and appeared to selectively bind to AKT over other enzymes containing similar cysteine loops.38
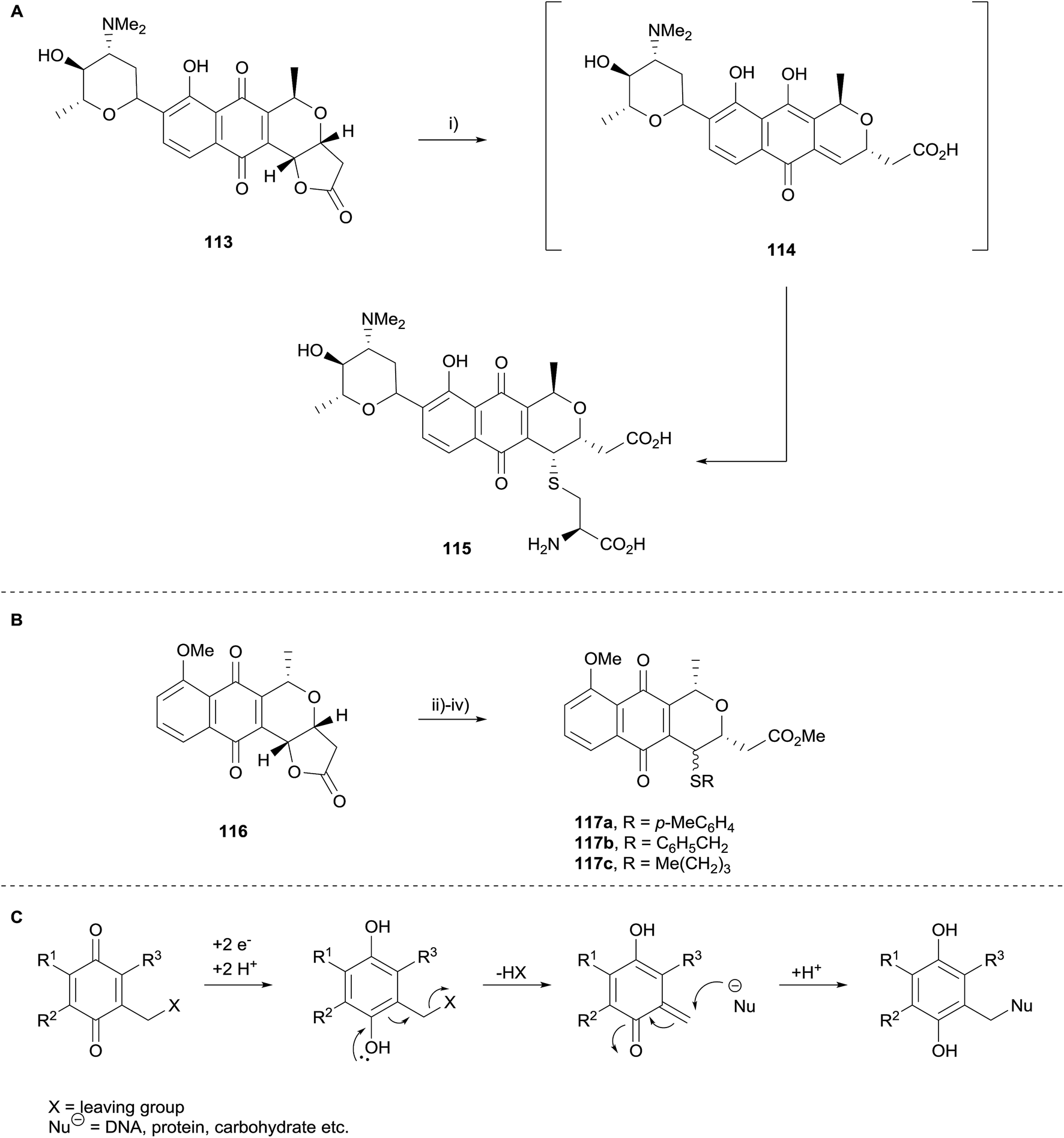 | ||
| Scheme 9 Reagents and conditions: (i) cysteine, H2O, 24 h, 75%; (ii) Na2S2O4, Trizma®, THF–MeOH, then RSH; (iii) aerial oxidation; (iv) CH2N2, Et2O, 63% 117a, 92% 117b, 52% 117c over three steps. | ||
Moore's hypothesis was also investigated in 2012, when Reynisson and coworkers carried out a computational investigation as to whether kalafungin40 could theoretically form DNA adducts (Scheme 9C).45 Guanine was used as a model for DNA due to its low oxidation potential and the deoxyribose moiety substituted with a methyl group for computational ease. A series of mono- and bis-alkylation possibilities were investigated. The calculations indicated that both mono- and bis-alkylation processes are energetically feasible.
AKT was also investigated as a biological target in for pyranonaphthoquinones in 2014 by Ellis and coworkers, who published an SAR study based on 7-deoxykalafungin (106b) using the method of Eid and et al. to access the parent compound.46 Compound 106b, together with “deconstruction analogues” 118, 119a–c and 107 were then tested for their ability to inhibit AKT1 and PKAα (Fig. 24). As previously reported, 118 exhibited potent inhibition of AKT1 but was inactive against PKAα. The compounds each exhibited similar profiles, although none displayed as much inhibitory activity as the parent pyranonaphthoquinone. Interestingly, compound 118 showed greater inhibition of PKAα than AKT1. These results indicate that the lactone ring, quinone and pyran are all essential to effect inhibition of AKT1.46
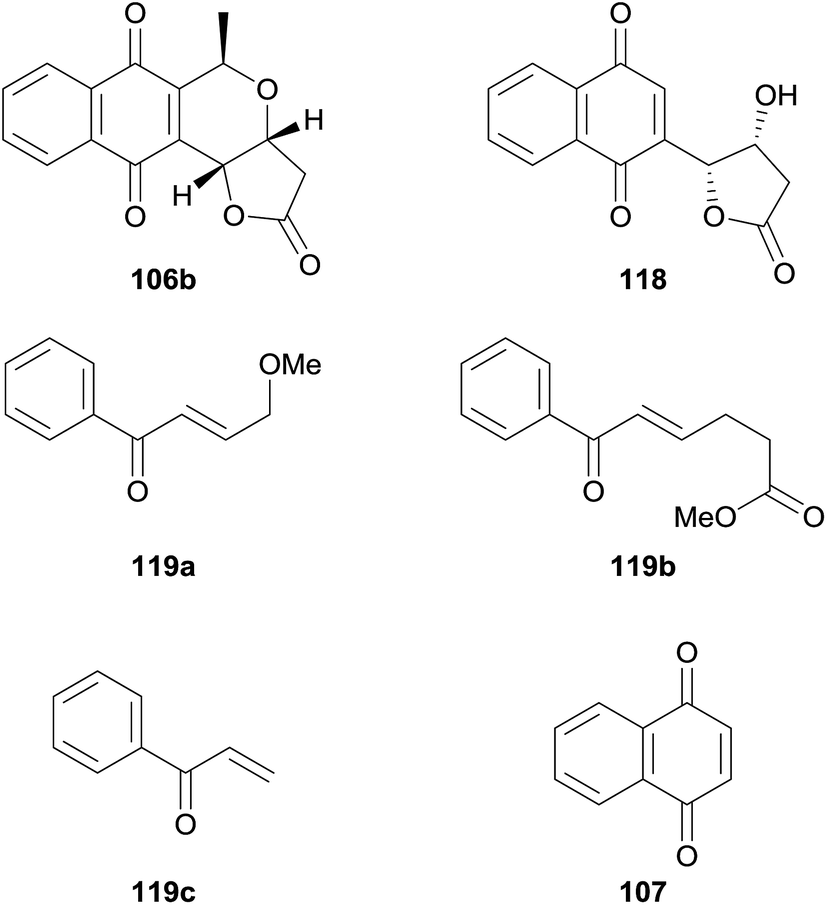 | ||
| Fig. 24 Compounds tested against AKT1/PKAα. | ||
In 2010, Fernandes and Mulay used a double OPS reaction to form the regioisomeric core of cardinalin 3 (126, Scheme 10).47 Following considerable optimisation, Fischer carbene 121 was obtained in 55% yield from dibromide 120. Double Dötz benzannulation with alkyne 122 afforded dimer 123a, which was converted to OPS precursor 123bvia methylation and TBS deprotection. Reaction of 123b with acetaldehyde dimethylacetal and boron trifluoride in dichloromethane for twelve hours at 0 °C gave a 45% yield of trans/trans-124, in addition to a 25% yield of undesired cis/trans-124. Alternatively, the use of HCl gas gave a 56% yield of cis/cis-124 and a 16% yield of cis/trans-124. CAN-mediated oxidative demethylation of the dimers provided the corresponding pyranonaphthoquinones trans/trans-125 and cis/cis-125 in high yield.47
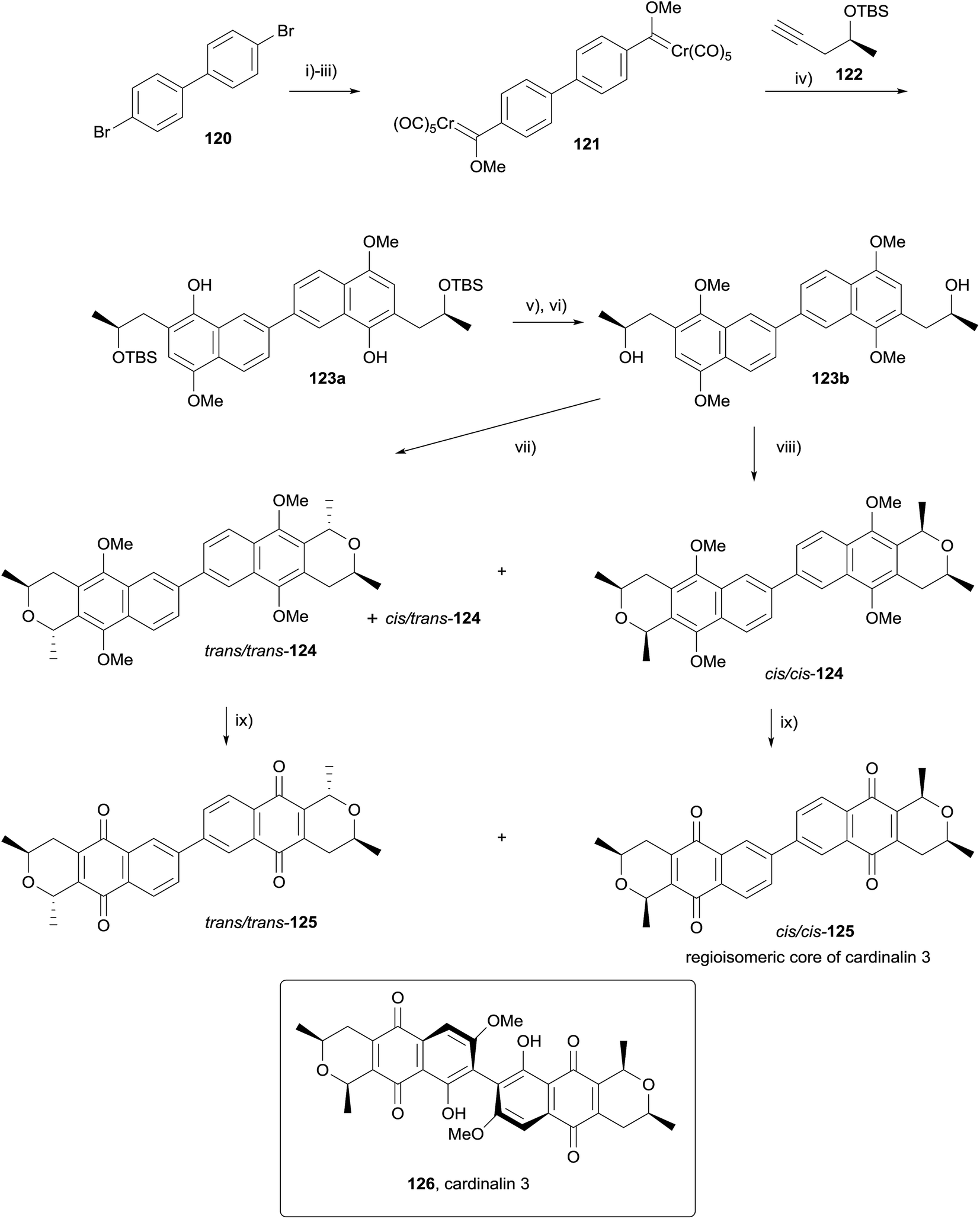 | ||
| Scheme 10 Reagents and conditions: (i) n-BuLi, Et2O, rt, 45 min; (ii) Cr(CO)6, Et2O, 0 °C, 1 h, rt, 3 h; (iii) Me3OBF4, CH2Cl2, 0 °C, 1 h, rt, 2 h, 55% over 3 steps; (iv) THF, 45 °C, 15 h, 65%; (v) NaH, MeI, DMF, 0 °C to rt, 4 h, 82%; (vi) TBAF, THF, rt, 4 h, 93%; (vii) BF3·OEt2, (MeO)2CHMe, CH2Cl2, 0 °C, 12 h, 45% trans/trans-124, 25% cis/trans-124; (viii) HCl(g), (MeO)2CHMe, Et2O, rt, 36 h, 56% cis/cis-124, 16% cis/trans-124; (ix) CAN, MeCN–H2O, rt, 30 min, 70% trans/trans-125, 75% cis/cis-125. | ||
The authors also applied this method to the synthesis of (+)-7,7′-demethoxycardinalin 3 (131, Scheme 11).48 Formation of Fischer carbene 128, followed by double Dötz benzannulation, methylation and silyl deprotection gave binaphthalene 129. The OPS reaction proceeded as before, requiring HCl gas to form cis/cis-130 as the major product (once more, boron trifluoride afforded the trans/trans-130 as the major product). Finally, oxidative demethylation and demethylation of the remaining phenols using aluminium chloride gave (+)-7,7′-demethoxycardinalin 3 (131).48
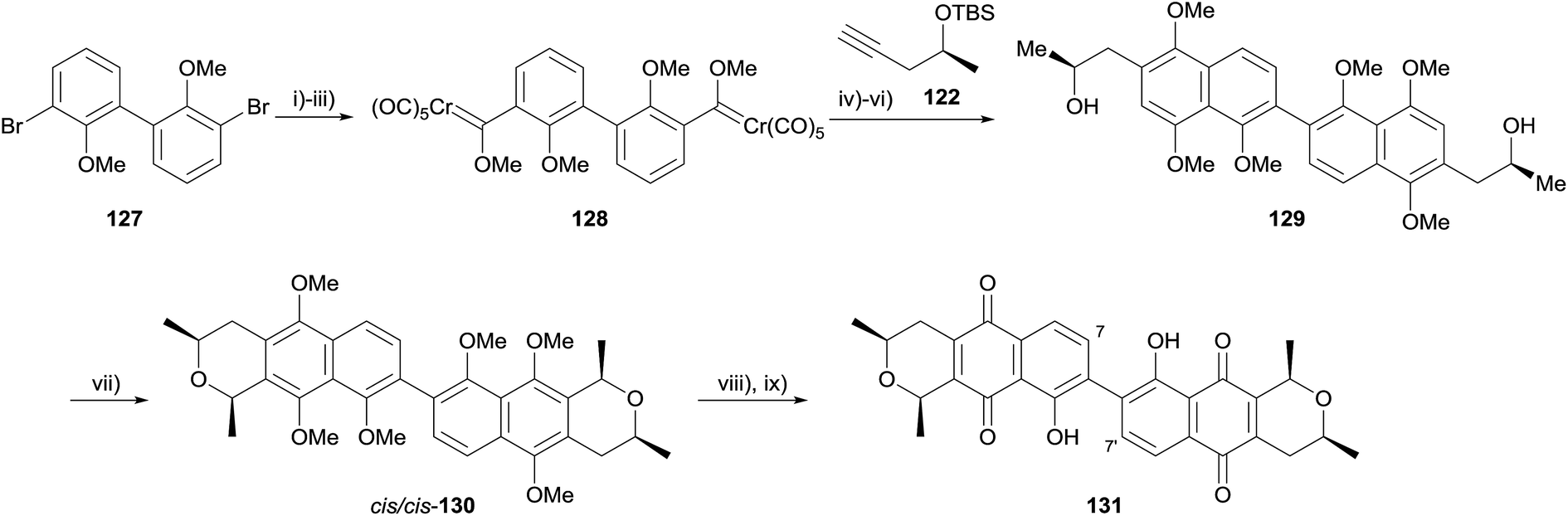 | ||
| Scheme 11 Reagents and conditions: (i) n-BuLi, Et2O, −78 °C, 30 min; (ii) Cr(CO)6, Et2O, 0 °C, 1 h, rt, 3 h; (iii) Me3OBF4, CH2Cl2, 0 °C, 1 h, rt, 3 h, 58% over 3 steps; (iv) THF, 45 °C, 15 h, 65%; (v) NaH, MeI, THF, 0 °C to rt, 4 h, 88%; (vi) TBAF, THF, rt, 4 h, 93%; (vii) HCl(g), (MeO)2CHMe, Et2O, rt, 24 h, 55% cis/cis-130, 22% cis/trans-130; (viii) CAN, MeCN–H2O, rt, 30 min, 74%; (ix) AlCl3, CH2Cl2, 0 °C, 15 min, rt, 30 min, 77%. | ||
A similar synthetic method was also used by Fernandes and coworkers for the asymmetric synthesis of kalafungin (35), frenolicin B (36) and deoxyfrenolicin (139, Scheme 12).49 The aim of this synthesis was to design a common intermediate that could be used to easily access a number of pyranonaphthoquinones. With this in mind, alkyne 133 was synthesised in six steps from D-glucono-δ-lactone (132). Elaboration via Dötz benzannulation, methylation and deprotection/lactonisation provided naphthalene intermediate 135, which was converted to kalafungin (35), frenolicin B (36), and deoxyfrenolicin (139) via OPS reaction with appropriate aldehydes followed by standard transformations.50
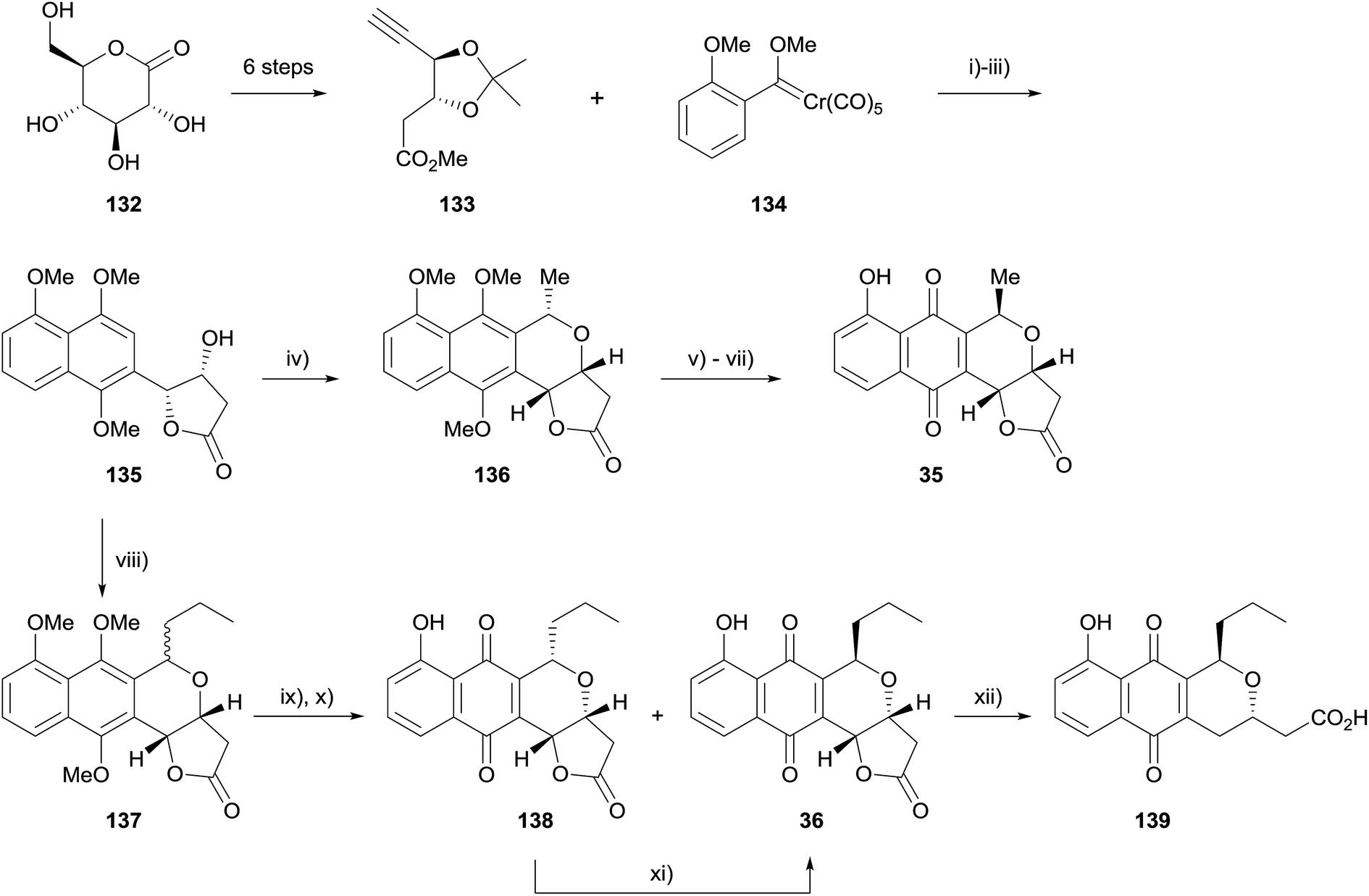 | ||
| Scheme 12 Reagents and conditions: (i) benzene, 45 °C, 12 h, 51%; (ii) NaH, MeI, DMF, 0 °C to rt, 3 h, 85%; (iii) TFA–H2O, HCl, CH2Cl2, rt, 24 h, 85%; (iv) MeCH(OMe)2, BF3·OEt2, CH2Cl2, 0 °C to rt, 17 h, 78%; (v) CAN, MeCN–H2O, rt, 15 min, 88%; (vi) BBr3, CH2Cl2, −50 °C, 25 min, then rt, 2 h, 82%; (vii) conc. H2SO4, rt, 30 min, 87%; (viii) butanal, TMSOTf, CH2Cl2, 0 °C, 2 h, 72%; (ix) CAN, MeCN–H2O, rt, 15 min, 88%; (x) BBr3, CH2Cl2, −50 °C, 30 min, rt, 2 h, 24% 138, 58% 36; (xi) conc. H2SO4, rt, 25 min, 60%; (xii) H2, Pd/C, EtOAc, rt, 6 h, 80%. | ||
In 2013, Fernandes and Mulay achieved a concise synthesis of (+)-astropaquinones B (6) and C (7) beginning with Dötz benzannulation of Fischer carbene 140 and alkyne 122 to give naphthalene 141 (Scheme 13).51 Treatment of 141 with two equivalents of sodium hydride and methyl iodide effected methylation of the free phenol together with cleavage of the phenolic TBS ether, followed by methylation to give naphthalene 142. Removal of the remaining TBS group was achieved by treatment with TBAF and subsequent OPS reaction with trimethyl orthoformate afforded pyran 144 as a single diastereoisomer. Oxidative demethylation of 144 using PIFA then provided (+)-astropaquinone B (6) in 80% yield. Interestingly, dimer 145 was also isolated from the reaction mixture in small quantities. Finally, (+)-astropaquinone C (7) was available from acid hydrolysis of (+)-astropaquinone B (6).
 | ||
| Scheme 13 Reagents and conditions: (i) THF, 45 °C, 12 h, 48%; (ii) MeI, NaH, DMF, 0 °C to rt, 6 h, 70%; (iii) TBAF, THF, rt, 6 h, 96%; (iv) CH(OMe)3, PTSA, CH2Cl2, rt, 1 h, 92%; (v) PIFA, MeCN–H2O, 0 °C, 5 min, 80% 6, 10% 145; (vi) PTSA, H2O, benzene, reflux, 4 h, 75%. | ||
Fernandes and Mulay used the same approach in a formal synthesis of thysanone (14, Scheme 14).52 The synthesis began from compound 141, an intermediate in their previous synthesis of (+)-astropaquinones B and C (see Scheme 13). Methylation, TBS deprotection and OPS reaction then gave naphthopyran 146. Unfortunately, compound 146 could not be directly oxidised to pyranonaphthoquinone 147. The free alcohol was therefore protected as an isopropyl ether and the compound oxidised to a mixture of quinones 148a/b using PIFA. Unfortunately, attempted deprotection of the 9-methoxy ether using aluminium trichloride was unsuccessful.
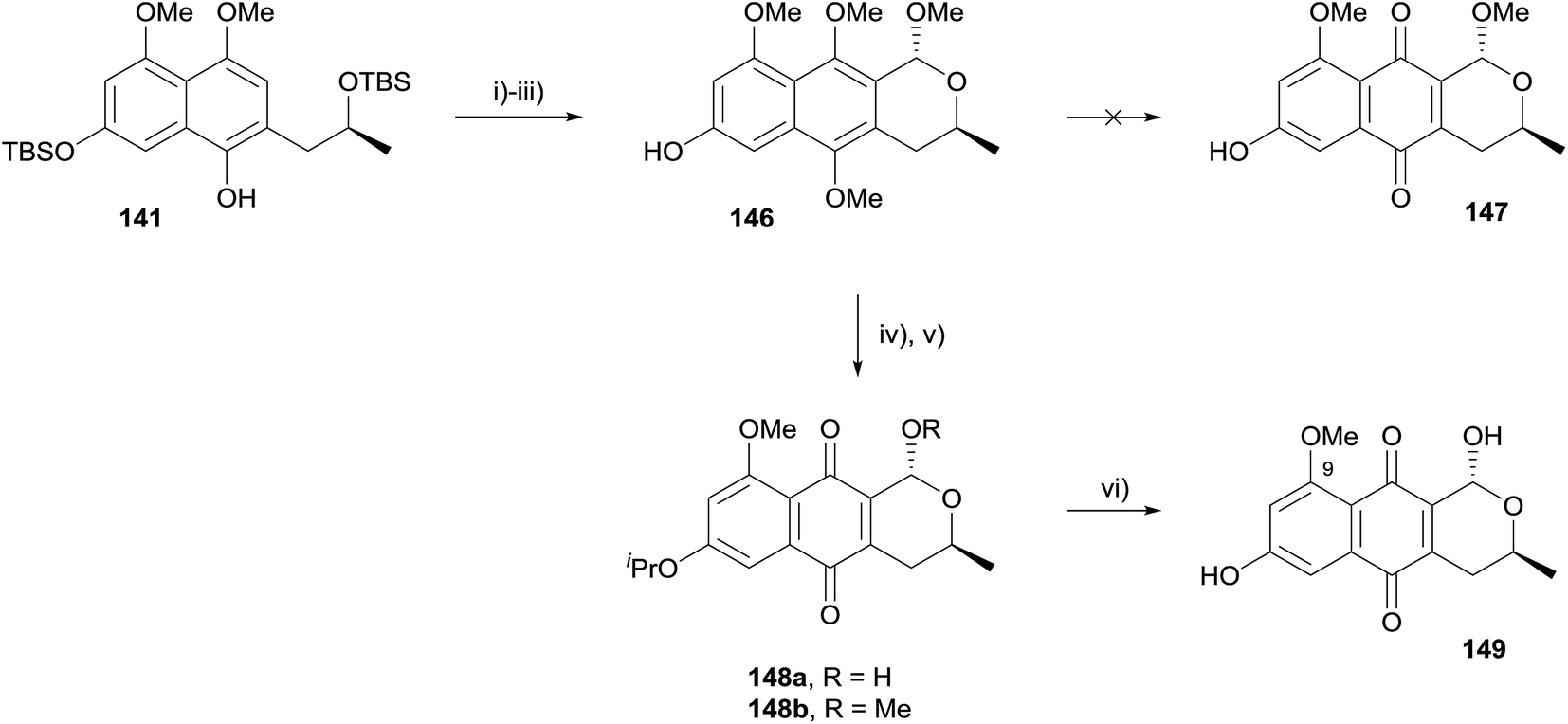 | ||
| Scheme 14 Reagents and conditions: (i) K2CO3, MeI, acetone, rt, 36 h, 92%; (ii) 2 M HCl, MeOH, rt, 4 h, 96%; (iii) CH(OMe)3, PTSA, CH2Cl2, rt, 2 h, 88%; (iv) K2CO3, i-PrBr, TBAI, acetone, reflux, 18 h, 89%; (v) PIFA, MeCN–H2O, 0 °C, 5 min, 36% 148a, 54% 148b; (vi) AlCl3, 0 °C, 15 min, rt, 2 h, 69% from 148a, 63% from 148b. | ||
The synthesis was then reattempted using a benzyl protecting group (Scheme 15). Conversion of 150 to naphthopyran 151 followed an analogous route to that of the previous synthesis. Removal of the benzyl groups in 151 resulted in unexpected demethoxylation at the C1 position. As pyranonaphthoquinone 152 is an intermediate in synthesis of thysanone (14) by Donner,53 this sequence constituted a formal synthesis of the natural product.
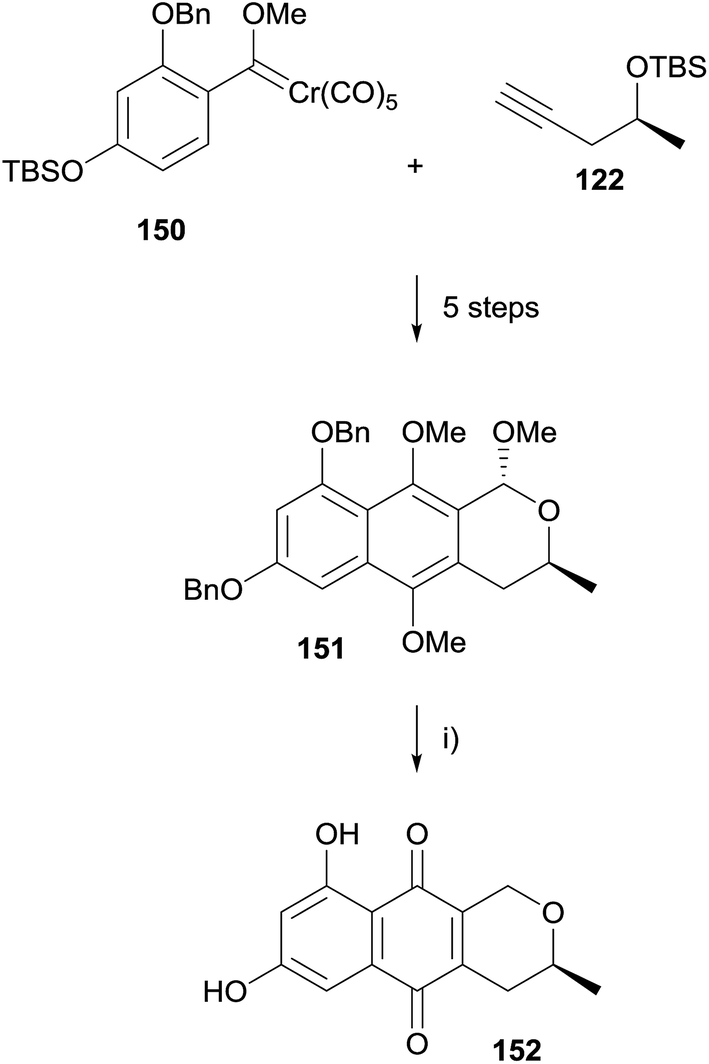 | ||
| Scheme 15 Reagents and conditions: (i) H2, Pd/C, EtOAc, rt, 3 h, 79%. | ||
Also in 2015, Mulay and Fernandes described synthetic efforts towards the synthesis of dimeric pyranonaphthoquinones related to γ-actinorhodin and crisamicin A.54 The paper detailed several unsuccessful attempts, before arriving at a successful route (Scheme 16). Using Fischer carbene 153, the familiar Dötz benzannulation sequence was used to obtain alcohol 155, which underwent OPS reaction mediated by TMS-triflate to give inseparable mixture of diastereomers (156). CAN-mediated oxidation of the mixture enabled separation of the isomers, which were identified as the desired isomer trans/trans-157 and the undesired cis/trans-157. The desired isomer was deprotected to give pyranonaphthoquinone 158. Similarly, deprotection and epimerisation of cis/trans-157 gave the same product. Finally, ester hydrolysis and lactonisation under aerobic conditions furnished deoxy-γ-actinorhodin (159), which is also an isomer of crisamicin A.54
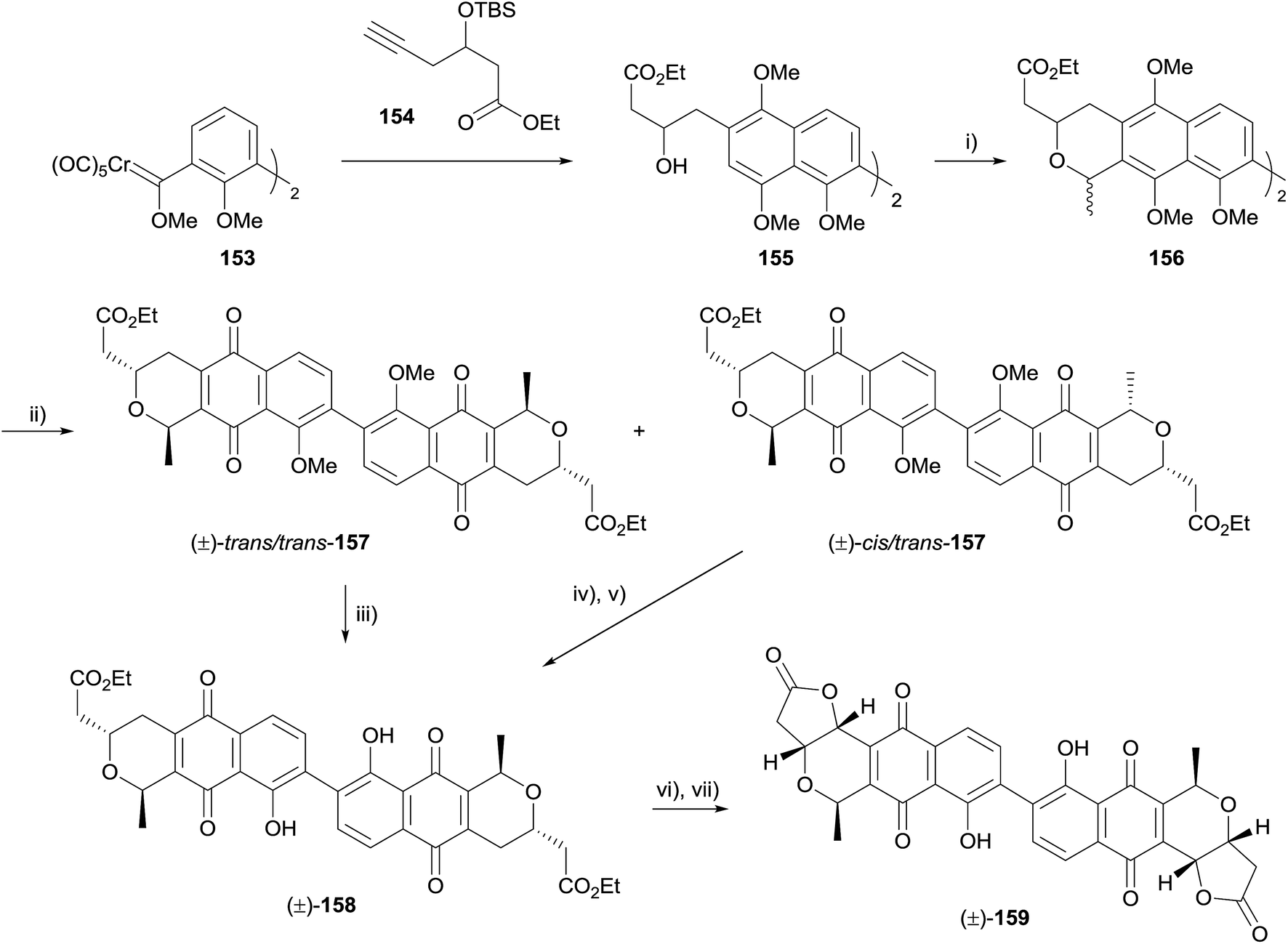 | ||
| Scheme 16 Reagents and conditions: (i) MeCH(OEt)2, TMSOTf, CH2Cl2, 0 °C to rt, 2 h, 81%; (ii) CAN, MeCN–H2O, rt, 45 min, 62% trans/trans-157, 18% cis/trans-157; (iii) AlCl3, CH2Cl2, 0 °C, 10 min, rt, 45 min, 79%; (iv) AlCl3, CH2Cl2, 0 °C, 10 min, rt, 40 min; (v) conc. H2SO4, benzene, 5 °C to rt, 1 h, 35% over two steps; (vi) LiOH, THF, rt, 6 h; (vii) air, MeOH, rt, 1 d, 34% over two steps. | ||
In 2015 Fernandes published a further synthetic study towards actinorhodin (Scheme 17). Conversion of carbene 160 to naphthopyran 161 was achieved in 4 steps.55 However, attempted oxidative homocoupling of 161 using a variety of conditions failed to give the desired dimer 163. Instead, reaction with PIFA gave a mixture of quinones 162a/b, both of which could be elaborated to deprotected pyranonaphthoquinone 164. Epimerisation gave the γ-actinorhodin monomer 165, after which the lactone was opened to provide actinorhodin monomer 49.55
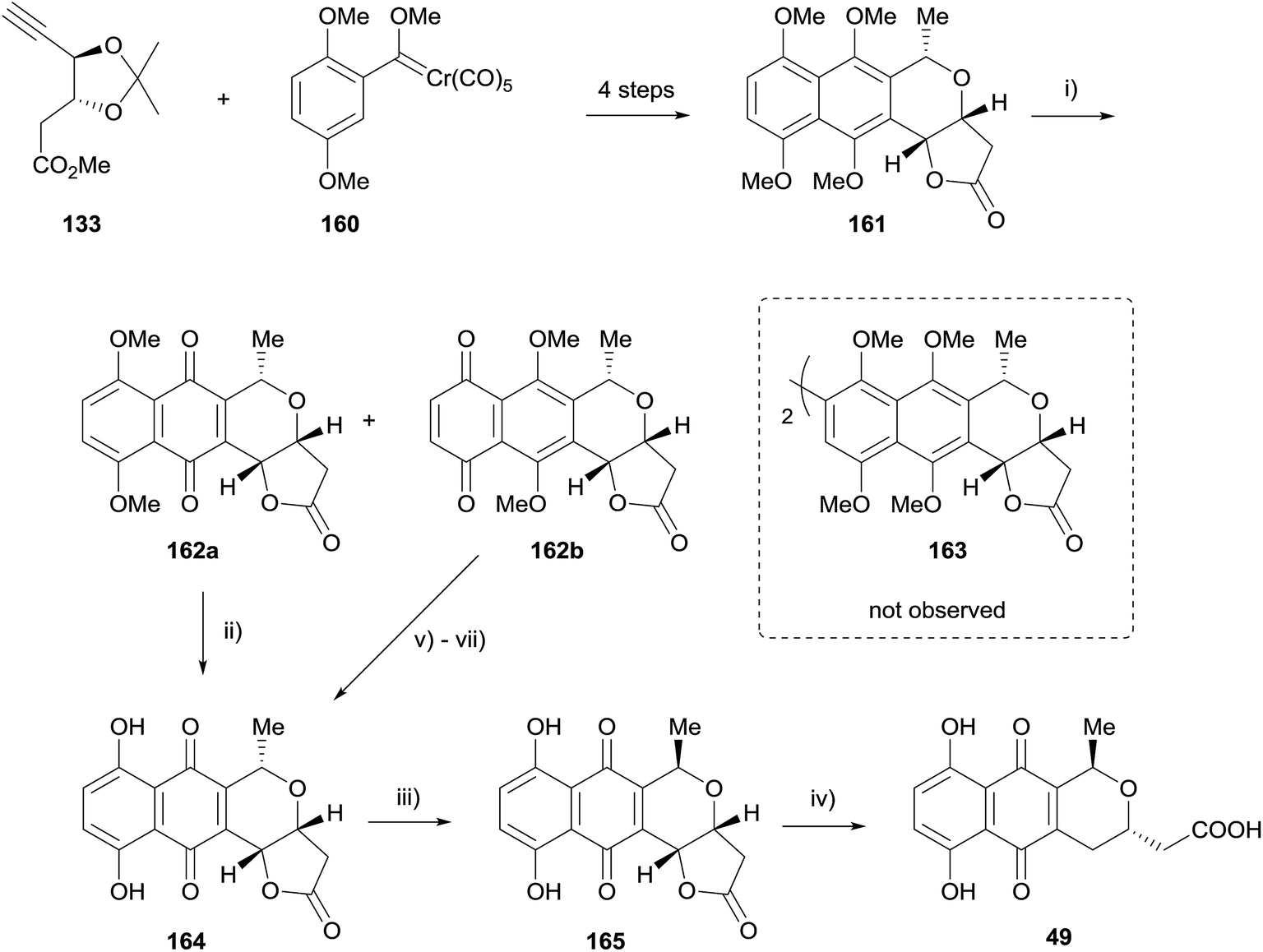 | ||
| Scheme 17 Reagents and conditions: (i) PIFA, MeCN–H2O, 0 °C, 30 min, 49% 162a, 38% 162b; (ii) AlCl3, CH2Cl2, 0 °C, 15 min, rt, 1.5 h, 68%; (iii) conc. H2SO4, PhH, 5 °C to rt, 45 min, recryst., 62%; (iv) H2, Pd/C, EtOAc, rt, 3 h, 83%; (v) Zn, Ac2O, Et3N, DMAP, 0 °C to rt, 2 h, 90%; (vi) CAN, MeCN–H2O, rt, 45 min, 78%; (vii) K2CO3, MeOH, rt, 4 h, 75%. | ||
In 2013, Thorson and coworkers published the synthesis of frenolicin B (36) and epi-frenolicin B (138) using an OPS strategy (Scheme 18).56 Heck coupling of bromonaphthalene 166 and alkene 102 followed by transesterification and Sharpless asymmetric dihydroxylation with concomitant lactonisation afforded lactone 135. The OPS reaction of 135 with butanal could be tuned to give the cis or trans isomer as the major product. The cis-naphthopyran 137a was obtained as the major product when tin chloride was used as the Lewis acid, while the use of iron trichloride gave the trans-isomer 137b as the major product. Completion of the synthesis of frenolicin B (36) and epi-frenolicin B (138) was achieved by CAN oxidation and demethylation of the remaining phenolic oxygen. The authors also described the OPS reaction of naphthalene 135 with a number of other aldehydes, targeting the cis-isomer.56
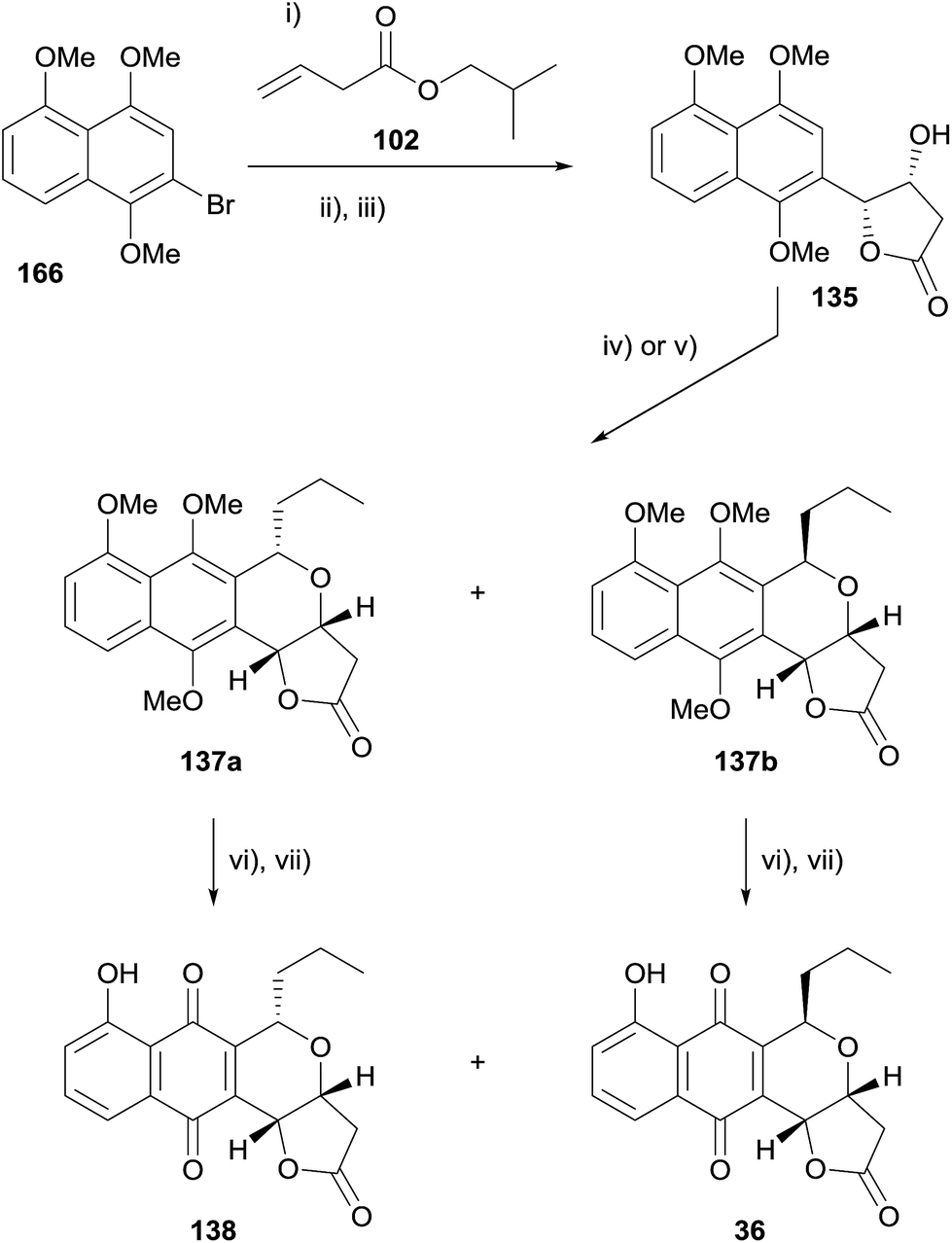 | ||
| Scheme 18 Reagents and conditions: (i) Pd(t-Bu3P)2, (Cy)2NMe, toluene, 100 °C, 84%; (ii) PTSA, MeOH, rt, 75%; (iii) K3Fe(CN)6, K2OsO4, K2CO3, NaHCO3, (DHQD)2PHAL, MeSO2NH2, t-BuOH–H2O, 0 °C to rt, 40 h, 63%; (iv) butanal, SnCl4, CH2Cl2, 0 °C to rt, 100%, 60/40 cis:trans; (v) FeCl3, THF, 0 °C to rt, 50%, 34:66 cis:trans; (vi) CAN, MeCN; (vii) BCl3, CH2Cl2, −78 °C, 61% 36, 60% 138 over two steps. | ||
Brimble and coworkers synthesised racemic 7-deoxythysanone (173) in order to probe the structural characteristics of the natural product that are vital for human rhinovirus 3C (HRV-3C) protease activity (Scheme 19).57 5-Methoxynaphthalen-1-ol (167) was elaborated to alcohol 168 over six steps. OPS reaction proceeded in high yield using dimethoxymethane and boron trifluoride diethyl etherate, affording naphthopyran 169. Oxidative demethylation gave pyranonaphthoquinone 170, which could be converted to a further three analogues; 7-deoxy-9-methoxythysanone (171), 1,7-deoxythysanone (172) and 7-deoxythysanone (173). None of the four pyranonaphthoquinones showed any activity in HRV-3C protease and cytotoxicity assays, suggesting that the 7-OH present in the natural product thysanone is essential for HRV-3C protease activity.57
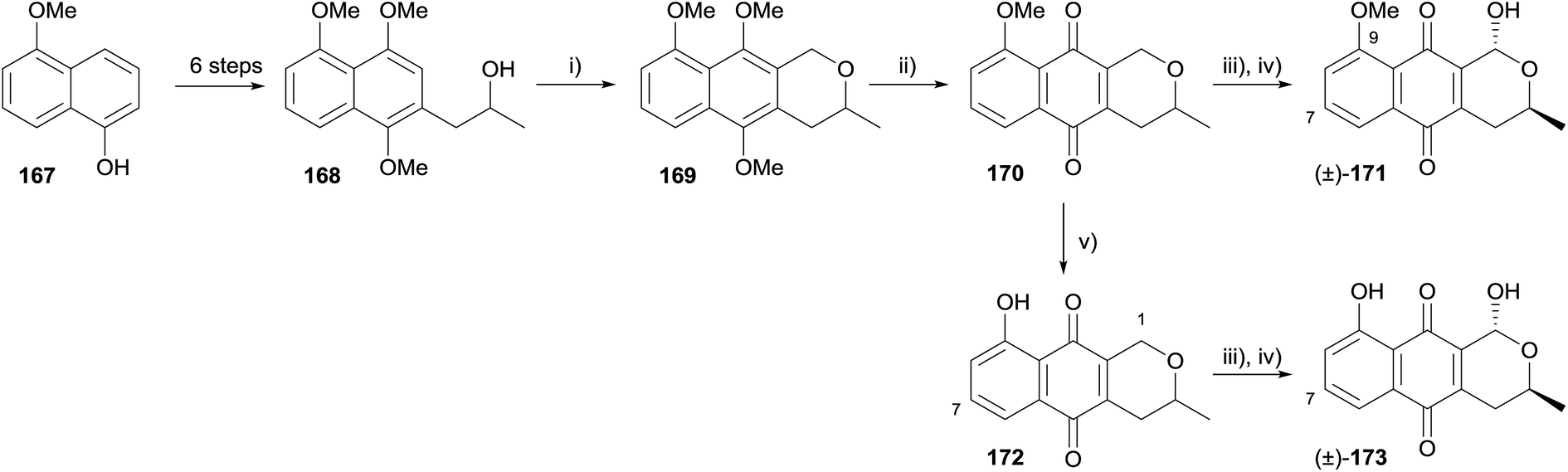 | ||
| Scheme 19 Reagents and conditions: (i) CH2(OMe)2, BF3·OEt2, CH2Cl2, 89%; (ii) AgO, HNO3, THF–H2O, 97%; (iii) Br2, (BzO)2, CCl4; (iv) H2O–THF, 56% for 171, 84% for 173 over two steps; (v) BCl3, CH2Cl2, 0 °C to rt, 98%. | ||
Intermediate 169 was used in a synthesis of (±)-thysanone (14), which hinged on a regioselective iridium-catalysed C–H borylation to install the 7-hydroxyl group (Scheme 20).58 The boronate generated from compound 169 underwent oxidation–hydrolysis, furnishing naphthol 174 which was elaborated to pyranonaphthoquinone 152. Finally, installation of the C1-hydroxyl group was achieved as previously reported, affording (±)-thysanone (14).58
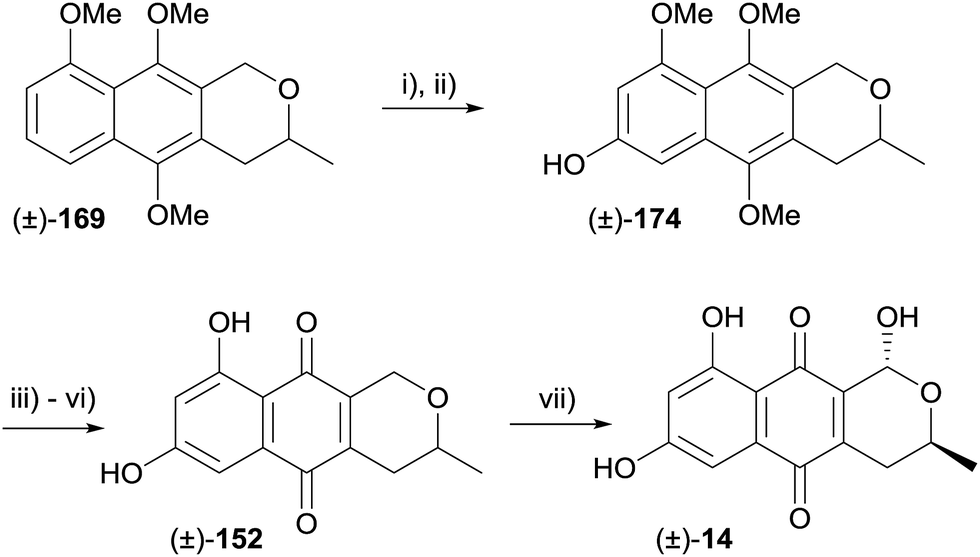 | ||
| Scheme 20 Reagents and conditions: (i) [Ir(COD)OMe]2, HBpin, dtbpy, THF, 80 °C; (ii) NMO, DCE, 100 °C, sealed tube, 30 min, 72% over two steps; (iii) NaH, TBSCl, THF, 73%; (iv) AgO, HNO3, THF–H2O, 75%; (v) BCl3, CH2Cl2, −78 °C to rt, 80%; (vi) TBAF, THF, quant.; (vii) Br2, (BzO)2, CCl4 then H2O–THF, 71%. | ||
In 2014, Paradkar and coworkers synthesised racemic 9-demethoxyeleutherin (11), 9-demethoxyisoeleutherin (180) and pentalongin (51) using a Nef reaction followed by an OPS reaction (Scheme 21).59 The aldehyde functionality was introduced to 1,4-dimethoxynaphthalene (175) by Vilsmeier–Haack formylation, followed by Henry reaction with nitroethane to form nitroalkene 177. The Nef reaction was conducted in a biphasic system, providing the corresponding ketone, which was immediately reduced to alcohol 178. OPS reaction with acetaldehyde followed by oxidative demethylation afforded racemic 9-demethoxyeleutherin (11) and 9-demethoxyisoeleutherin (180). A formal synthesis of pentalongin (51, see Fig. 13) was also completed. Aldehyde 176 was converted to a nitroalkane which underwent oxidative Nef reaction to carboxylic acid 179, an intermediate in the synthesis of pentalongin (51) by Deshpande.60
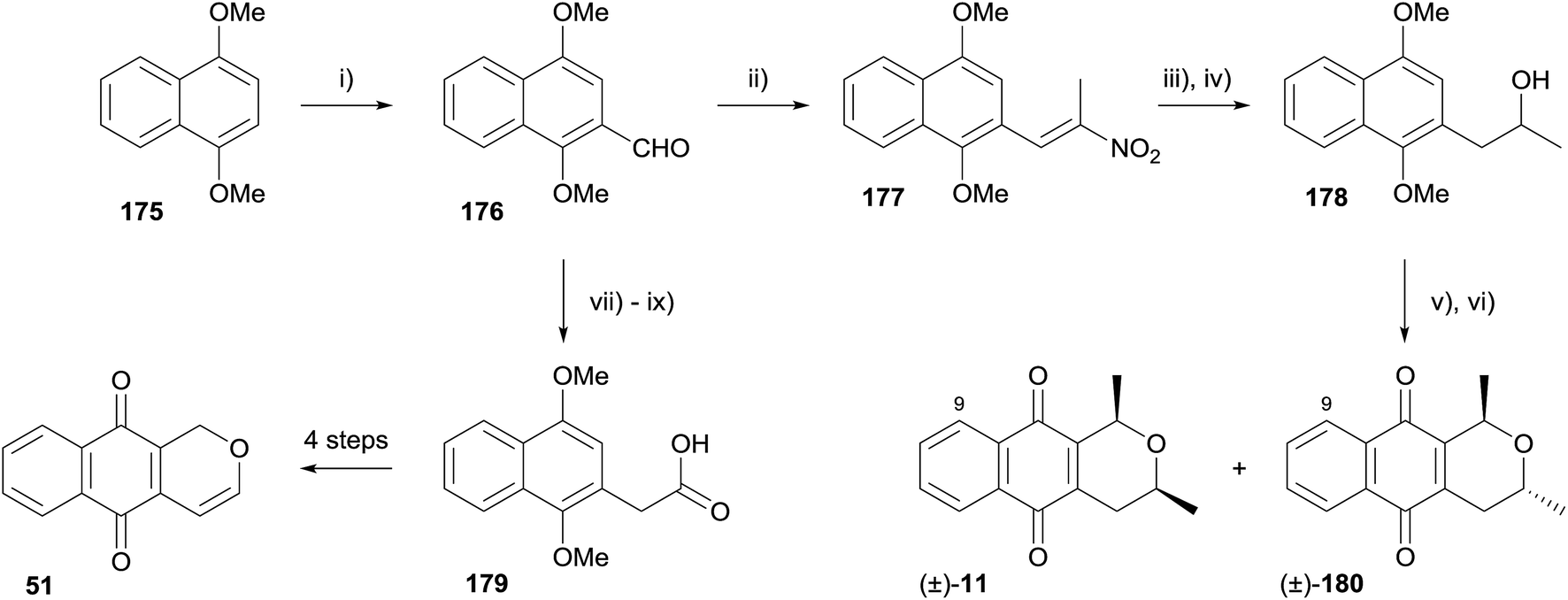 | ||
| Scheme 21 Reagents and conditions: (i) DMF–POCl3, 8 h, 94%; (ii) C2H5NO2, NH4OAc, AcOH, 20 min, 80 °C, 86%; (iii) Fe–FeCl3–HCl/toluene, 30 min, 80 °C, 86%; (iv) NaBH4/MeOH, rt, 96%; (v) excess MeCHO, conc. HCl, rt, 14 h; (vi) CAN, MeCN, 0–5 °C, 45 min, 41%, 11, 37% 180; (vii) MeNO2, NH4OAc, AcOH, 20 min, 80 °C, 84%; (viii) NaBH4, EtOH–EtOAc, rt, 2 h 80%; (ix) NaNO2, AcOH, DMSO, 35 °C, 2 h, 79%. | ||
3.2 Hauser–Kraus annulation
The reaction of stabilised phthalide anions with Michael acceptors to form 1,4-dihydroxynaphthalenes, known as the Hauser–Kraus (HK) annulation,61–63 is one of the most prevalent methods used to construct the pyranonaphthoquinone framework. In 2008, Brimble reported progress towards the dimeric pyranonaphthoquinone crisamicin A (188) that featured a complex application of the HK reaction.64 The initial goal of the strategy was to first establish a synthesis of monomer 187. Although compound 187 possesses the opposite stereochemistry to that of crisamicin A, it was used in order to establish a viable synthetic sequence using the inexpensive D-mannose as a starting material as opposed to the more expensive L-sugar. However, HK annulation of cyanophthalide 181 and either benzyl-protected enone 182 or TBDPS-protected enone 183 resulted in the formation of a non-aromatised product (Scheme 22). Compounds 184 and 185 could not be converted to pyranonaphthoquinone 187 despite investigation of a range of reaction conditions. Treatment of 185 with acid promoted the formation of dihydroxynaphthalene 186via an elimination pathway. This was attributed to the labile methoxy group present in the acetal moiety enabling elimination to occur faster than the desired aromatisation to 187.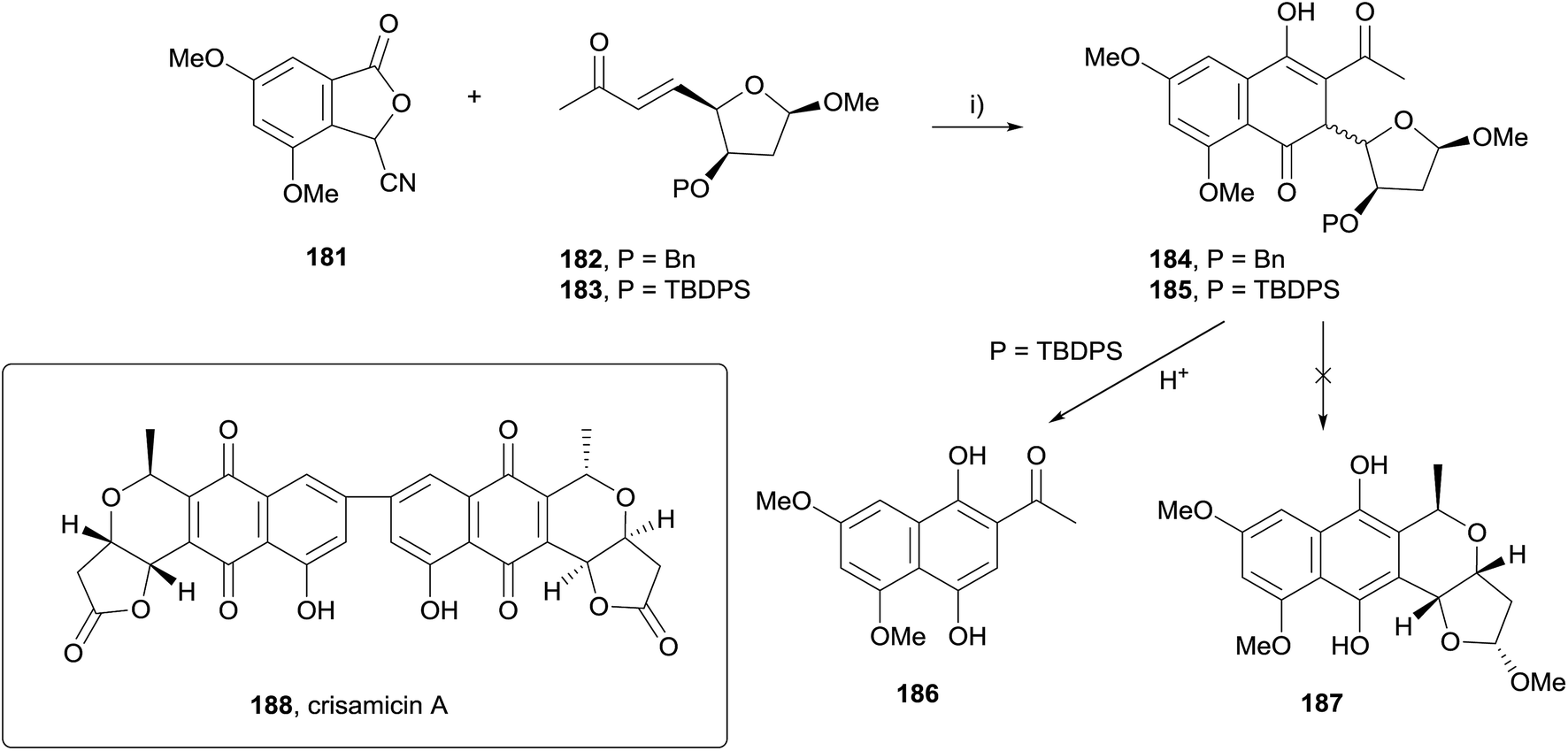 | ||
| Scheme 22 Reagents and conditions: (i) t-BuOK, DMSO, rt, 78% (P = Bn), 66% (P = TBDPS). | ||
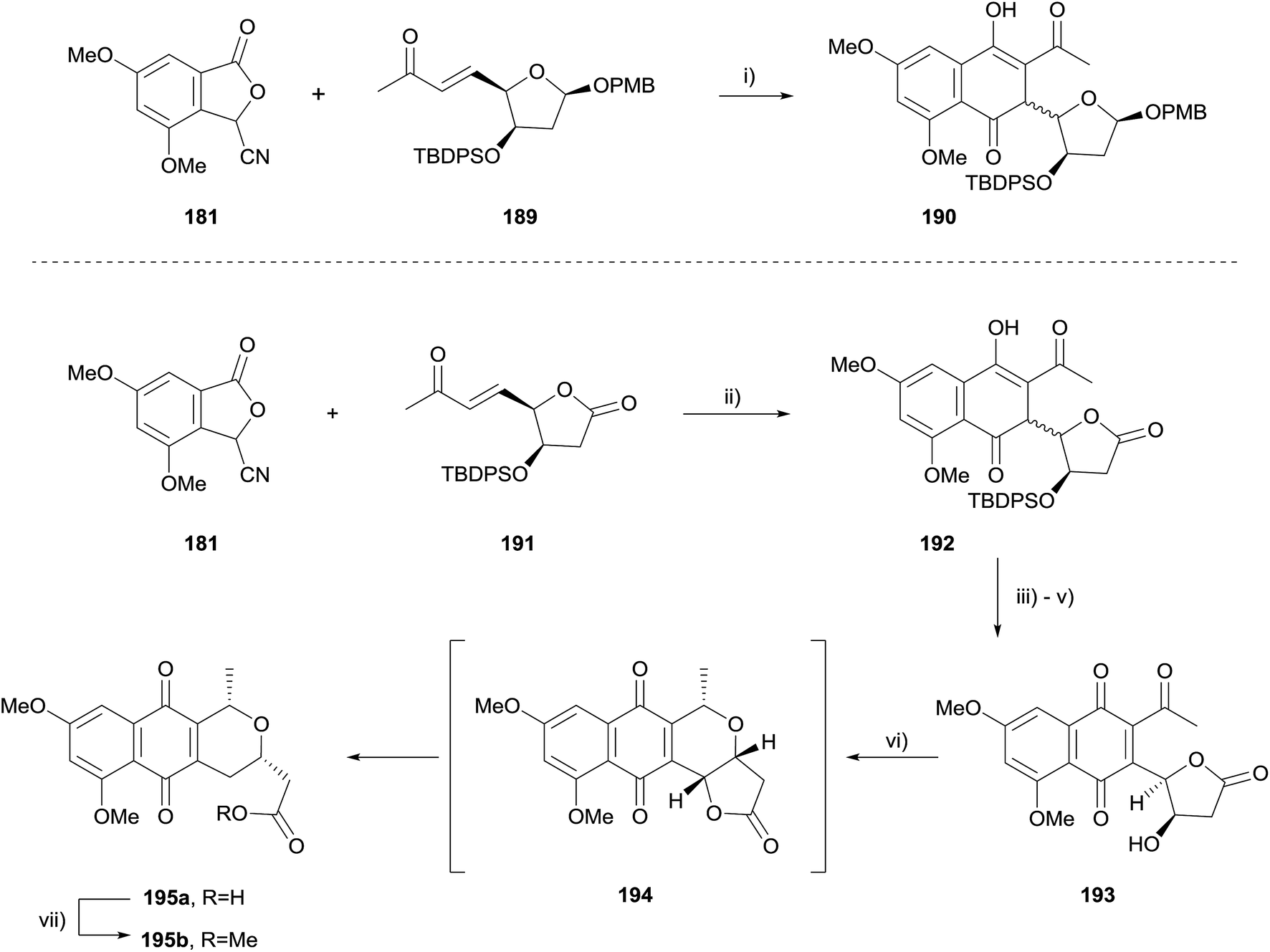
The corresponding para-methoxybenzyl (PMB) acetal 189, which could be cleaved under non-acidic conditions was therefore prepared (Scheme 23). Unfortunately, attempted HK annulation of enone 189 also resulted in non-aromatised product 190 that could not be further elaborated. The use of lactone 191 gave similar results, providing compound 192. However protection of the phenolic oxygen, TBDPS deprotection and oxidation with CAN afforded quinone 193. Lewis acid mediated cyclisation and concomitant reduction with triethylsilane did not afford the desired product 194, instead providing ring-opened carboxylic acid 195a. Attempted oxidative cyclisation of acid 195a in the presence of methanol was unsuccessful, resulting only in the formation of methyl ester 195b. However, with a model pyranonaphthoquinone framework successfully established, efforts moved towards the total synthesis of crisamicin A.
 | ||
| Scheme 23 Reagents and conditions: (i) t-BuOK, DMSO, rt, 53%; (ii) t-BuOK, DMSO, rt, 80%; (iii) BF3·OEt2, CH2Cl2, −78 °C to 0 °C then Ac2O, py, −10 °C to rt, 53% over two steps; (iv) HF·Py, THF, rt, then SiO2; (v) CAN, MeCN–H2O (2:1), 38% over 2 steps; (vi) BF3·OEt2, Et3SiH, CH2Cl2, −78 °C to rt, 63%; (vii) MeOH, O2, dark, 32%. | ||
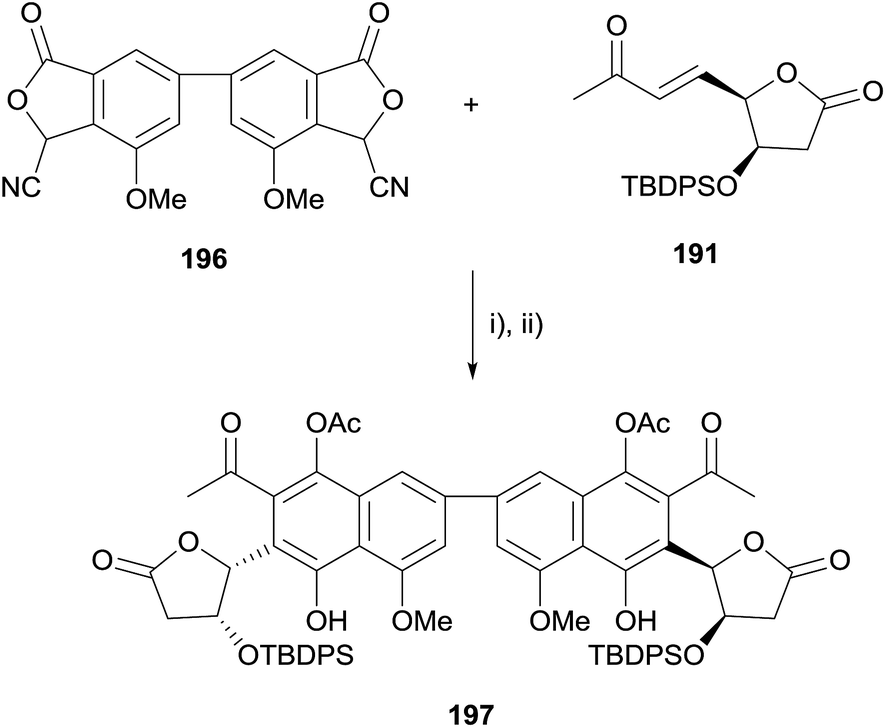
Unfortunately, the HK annulation between biscyanophthalide 196 and enone 191 provided an unstable non-aromatised adduct which, although it could be trapped as diacetate 197, could not be converted to crisamicin A (Scheme 24).
 | ||
| Scheme 24 Reagents and conditions: (i) t-BuOK, DMSO, rt; (ii) BF3·OEt2, CH2Cl2, −78 °C to 0 °C then Ac2O, py, −10 °C to rt, 23% over 2 steps. | ||
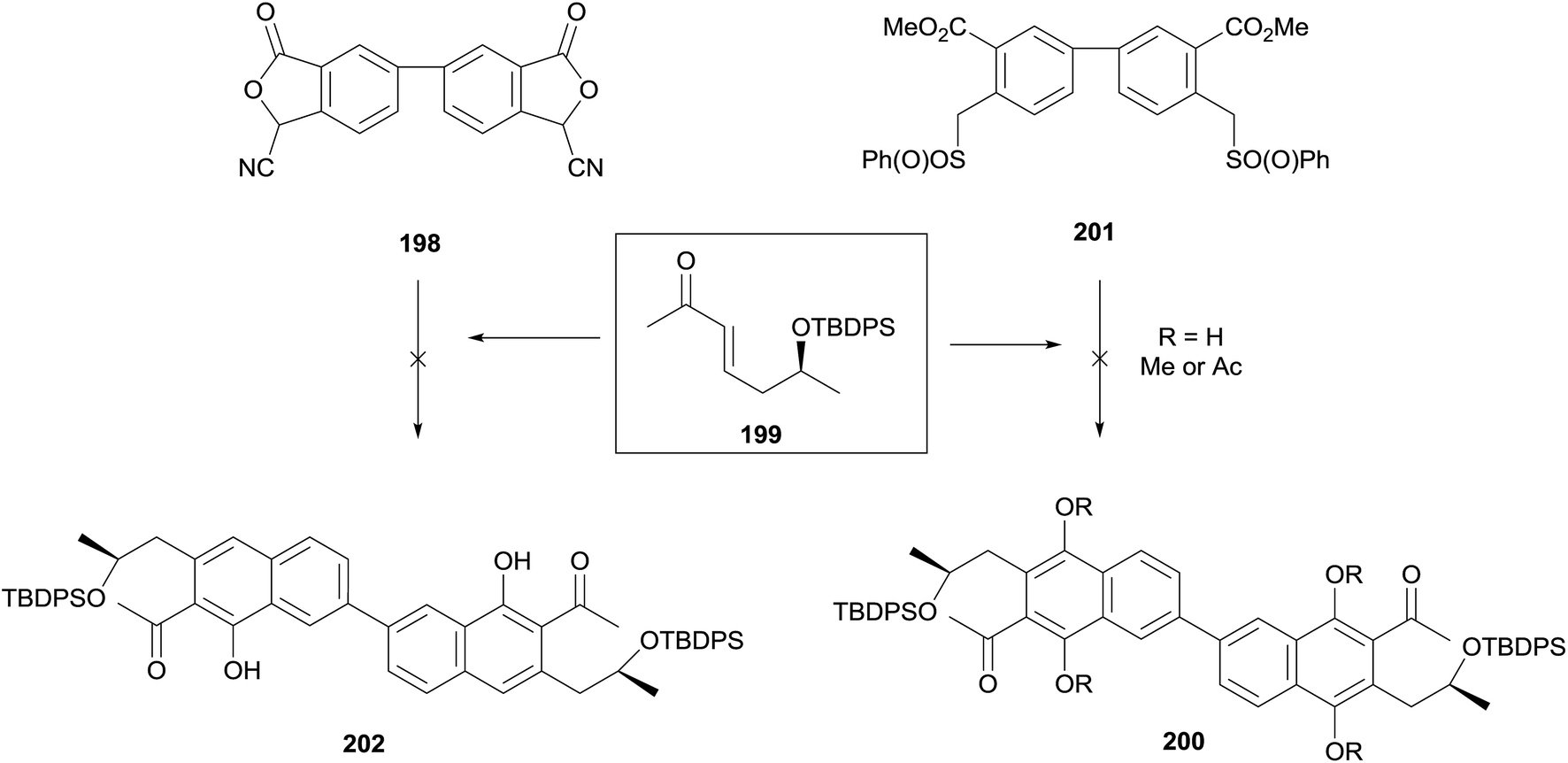
The same double annulation strategy was applied to the synthesis of the dimeric core of the cardinalins (Scheme 25).65,66 Annulation of phthalide 198 and enone 199 with potassium tert-butoxide in THF–DMSO produced a bright red solution, indicative of successful dianion formation. The colour disappeared on addition of the enone, suggesting consumption of the dianion and formation of the hydroquinone product. However all attempts to isolate the hydroquinone, trap it as a protected species or oxidise it to the naphthoquinone were unsuccessful. An alternative annulation utilising bis-sulfone 201 also failed to provide annulated product.
 | ||
| Scheme 25 Unsuccessful double annulation strategies towards the dimeric core of the cardinalins. | ||
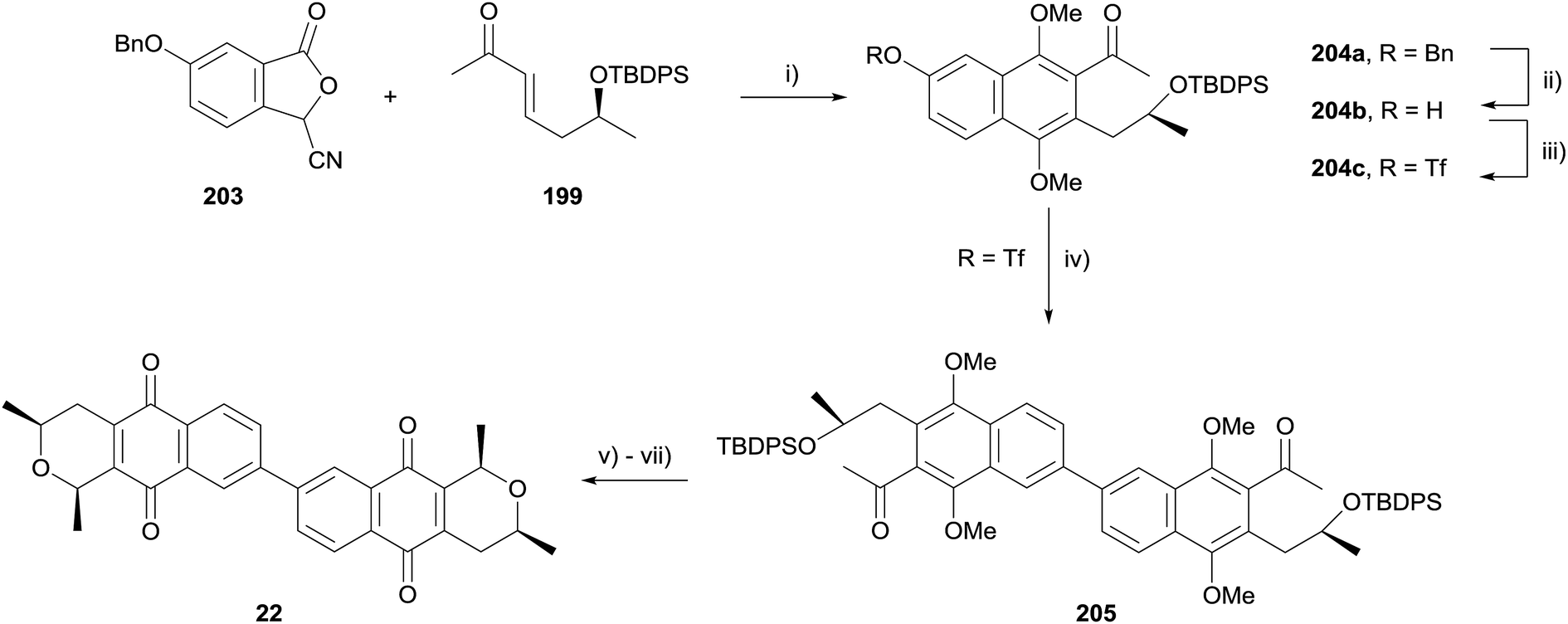
A late stage homocoupling strategy was therefore devised, where the cardinalin core was assembled by Suzuki–Miyaura cross-coupling (Scheme 26). Reaction of enone 199 with cyanophthalide 203 under standard conditions afforded naphthalene 204a. The benzyl protecting group was cleaved and the resultant phenol converted to triflate 204c. After optimisation, homocoupling was achieved in 51% yield, furnishing dimeric naphthalene 205. Silyl deprotection, reductive cyclisation and oxidative demethylation afforded 22, the core structure of the cardinalin natural products.
 | ||
| Scheme 26 Reagents and conditions: (i) t-BuOK, DMSO, rt, 30 min then K2CO3, Me2SO4, TBAI, THF–H2O, rt, 2 h, 87%; (ii) H2, Pd/C, MeOH, rt, 18 h, 97%; (iii) PhN(OTf)2, DMAP, Et3N, CH2Cl2, rt, 1 h, 95%; (iv) Cl2Pd(dppf) (10 mol%), dppf, K2CO3, bis(pinacolato)diboron, dioxane, μw, 150 °C, 1 h, 51%; (v) TBAF, THF, rt, 72 h; (vi) TFA, CH2Cl2, −78 °C, 30 min then Et3SiH, rt, 16 h, 70% (2 steps); (vii) CAN, MeCN–H2O, rt, 45 min, 63%. | ||
In 2009, Brimble and coworkers published a synthesis of the cardinalin monomer, ventiloquinone L (12, Scheme 27) and efforts towards biomimetic coupling to give cardinalin 3 itself.67 The hydroquinone formed by HK annulation was immediately trapped as dimethoxynaphthalene 207. Silyl deprotection with concomitant cyclisation followed by stereoselective reduction afforded naphthopyran 208. Oxidative demethylation and selective monodeprotection gave ventiloquinone L (12). Unfortunately, attempts to effect oxidative dimerization of 12 and 209 using basic ferricyanide, FeCl3, PbIV salts, CuI/O2, hν/O2 and CAN were all unsuccessful. Attempted dehydrogenative coupling using methylated ventiloquinone was also unsuccessful.
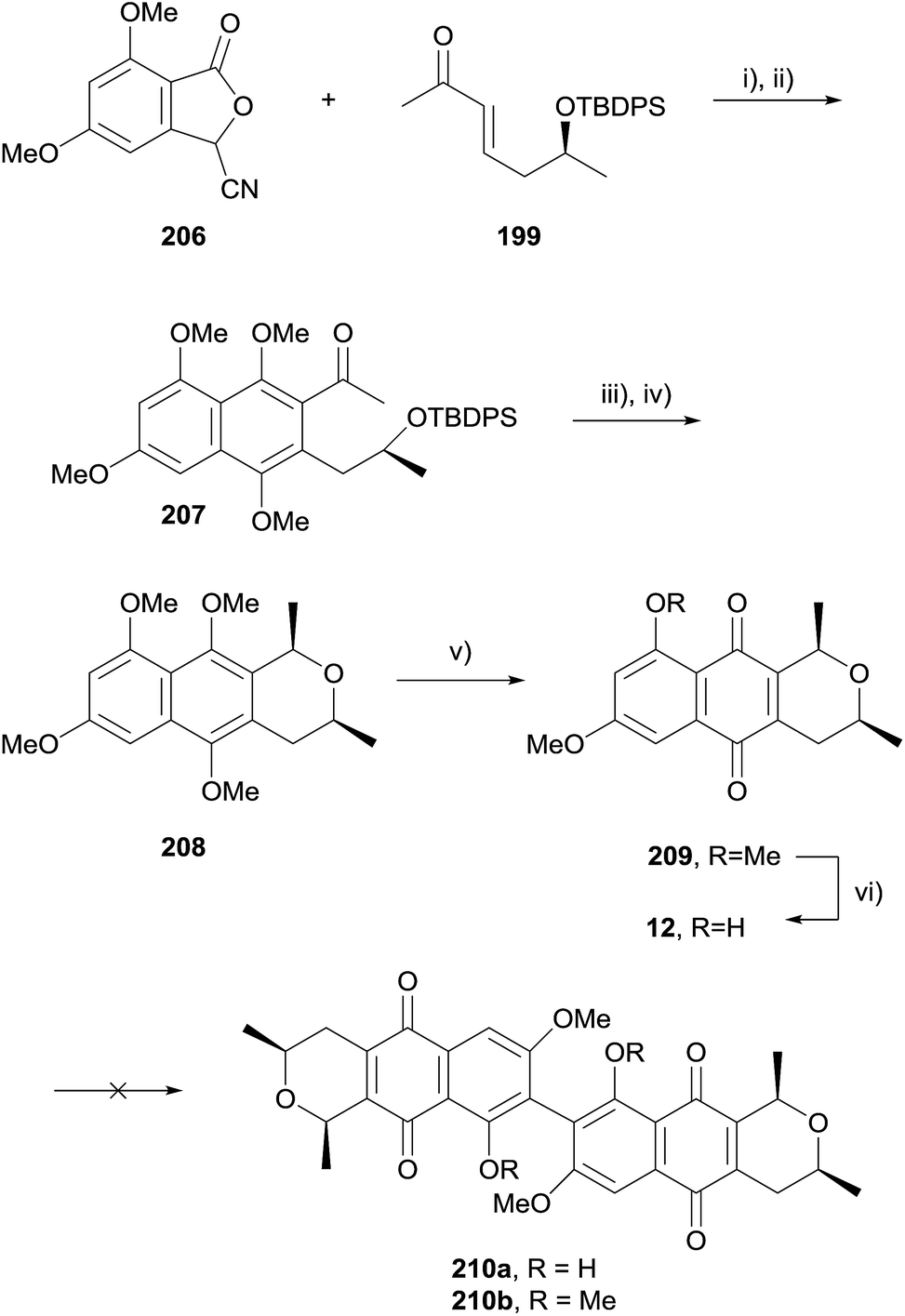 | ||
| Scheme 27 Reagents and conditions: (i) t-BuOK, DMSO, rt, 3–4 min; (ii) H2, Na2S2O4, THF–H2O then NaOH, TBABr, Me2SO4, rt, 3 h, 52%; (iii) TBAF, THF, rt, 4 d; (iv) TFA, Et3SiH, CH2Cl2, −78 °C to rt, 16 h, 65% over 2 steps; (v) PIFA, MeCN–H2O, rt, 2 h, 81%; (vi) BCl3, CH2Cl2, −78 °C to rt, 1 h, 84%. | ||
Oxidative dimerization of naphthopyran 208 using CAN was then attempted (Scheme 28). Interestingly, the product isolated was not the C8–C8′ dimer, but a dimer linked through C6–C6′ as a 1:1 mixture of separable atropisomers. Studies showed that dimerization using CAN did not occur with naphthoquinone 209 (see Scheme 27), indicating that dimerization of 208 occurred prior to oxidative demethylation. Dimer 211a was subsequently selectively demethylated using boron trichloride to give a C6 regioisomer of cardinalin 3 (211b).
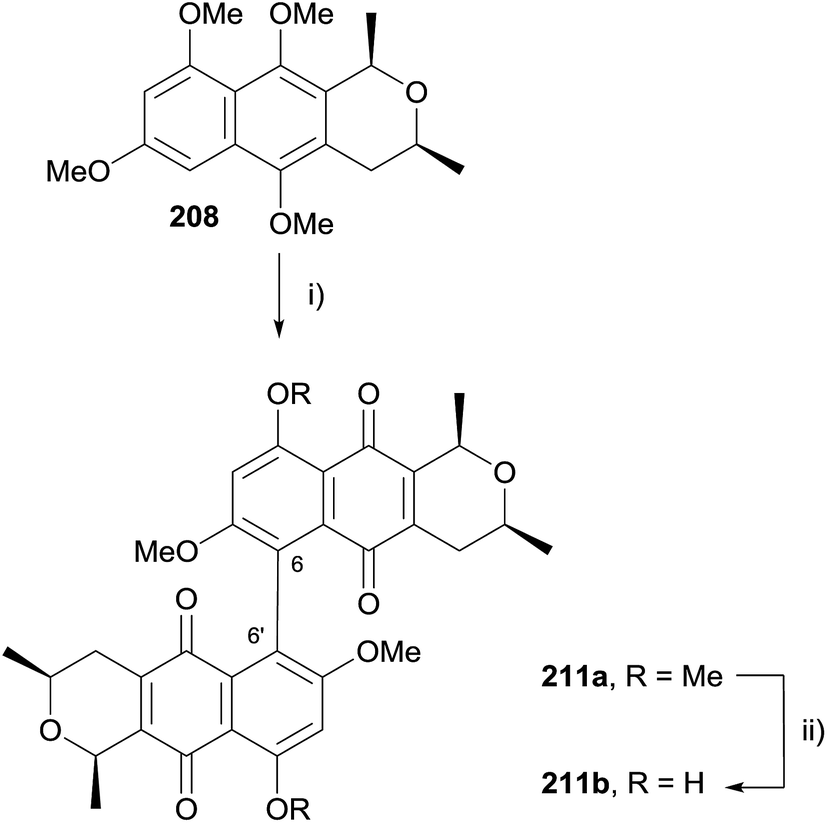 | ||
| Scheme 28 Reagents and conditions: (i) CAN (3 eq.), MeCN–H2O, rt, 30 min, 71%, 1:1; (ii) BCl3, CH2Cl2, −78 °C to 0 °C, 68% (1:1 mixture of atropisomers). | ||
In 2010, Brimble and coworkers used the HK annulation to prepare of a series of triazole analogues of the nanaomycins (Scheme 29).68 The triazole group was chosen as an enzymatically stable mimic of the amide containing nanaomycin C, in an effort to improve upon the biological activity of the natural product. Being a rigid linker, the triazole ring system holds the substituents in a similar geometry and distance to that of an amide, as well as providing a comparable dipole moment. However, unlike the amide counterpart, triazoles are stable towards hydrolytic cleavage, oxidation and reduction and thus display ideal characteristics to be successful therapeutic candidates in vivo.
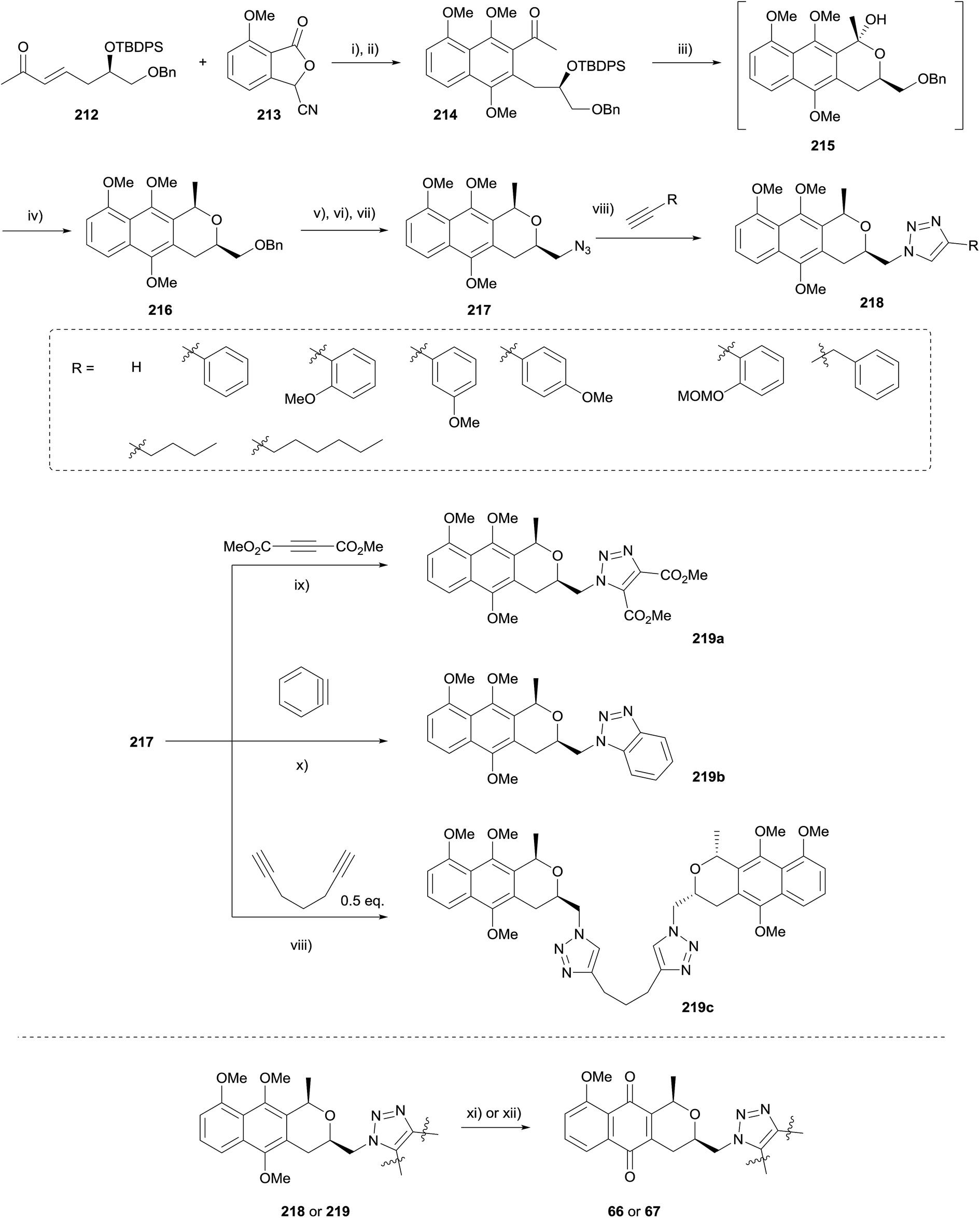 | ||
| Scheme 29 Reagents and conditions: (i) t-BuOK, DMSO, rt, 10 min; (ii) H2, Na2S2O4, THF–H2O then NaOH, TBABr, Me2SO4, rt, 2 h, 37–51% (over 2 steps); (iii) TBAF, THF, rt, 3 d; (iv) TFA, Et3SiH, CH2Cl2, −78 °C to rt, 1 h, 81% (over 2 steps); (v) Pd(OH)2, H2, EtOH, rt, 1.5 h; (vi) TsCl, Et3N, rt, 6 h, 60% (over 2 steps); (vii) NaN3, DMF, 110 °C, 5 h, 87%; (viii) CuI[P(OEt)3], alkyne, toluene, 85 °C 60–94%; (ix) neat, 100 °C, 1.25 h, 86%; (x) CsF, 18-crown-6, MeCN, 2-trimethylsilylphenyl triflate, 74%; (xi) AgO, HNO3, dioxane, rt; (xii) CAN, MeCN–H2O, rt. | ||
HK annulation employing enone 212 and cyanophthalide 213 was initially problematic, but the use of a shorter reaction time and immediate methylation of the hydroquinone formed afforded naphthalene 214. Removal of the TBDPS group and stereoselective reduction afforded naphthopyran 216. Subsequent functional group manipulations gave azide 217, which underwent copper-catalysed azide–alkyne cycloaddition (CuAAC) with various alkynes. These compounds were then subjected to oxidative demethylation with either silver oxide or CAN.68
A later publication detailed the attempted preparation of a tetrazole analogue 223via addition of azide to nitriles 220 and 222 respectively (Scheme 30).69 Benzyl ether 216 (see Scheme 29) could be converted to a mixture of cis and trans-isomers of naphthopyran 220 or naphthoquinone 222. However a number of conditions failed to convert these to the desired tetrazoles.
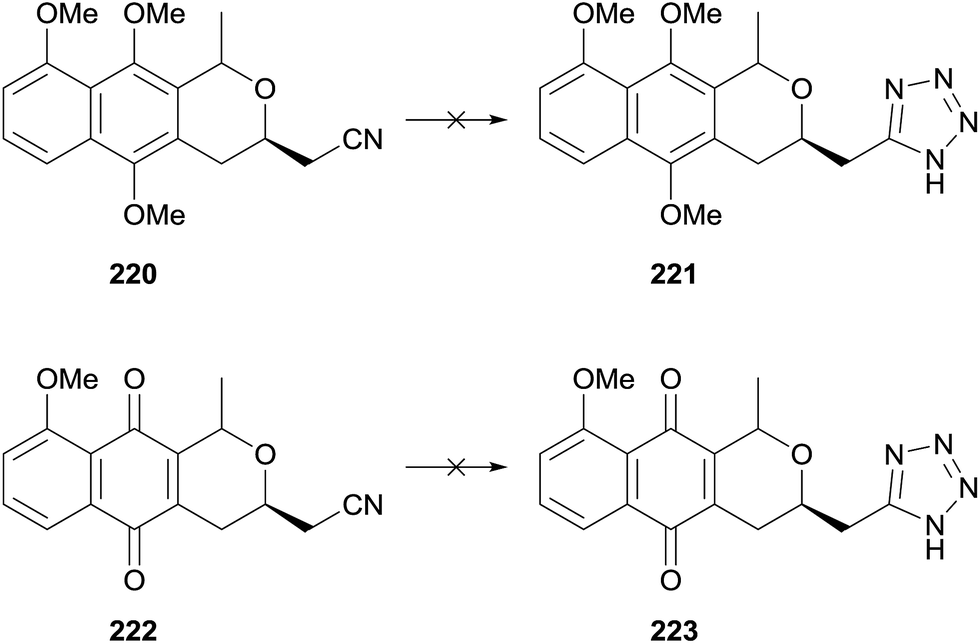 | ||
| Scheme 30 Unsuccessful synthesis of tetrazole pyranonaphthoquinone.69 | ||
An alternative route was therefore sought (Scheme 31). Tosylate 224 was converted to amide 225via a series of functional group manipulations. Tetrazole 226 was then successfully synthesised by reaction with triphenylphosphine, DIAD and trimethylsilyl azide. Finally, deprotection and CAN-mediated oxidative demethylation afforded desired tetrazole-pyranonaphthoquinone 223.
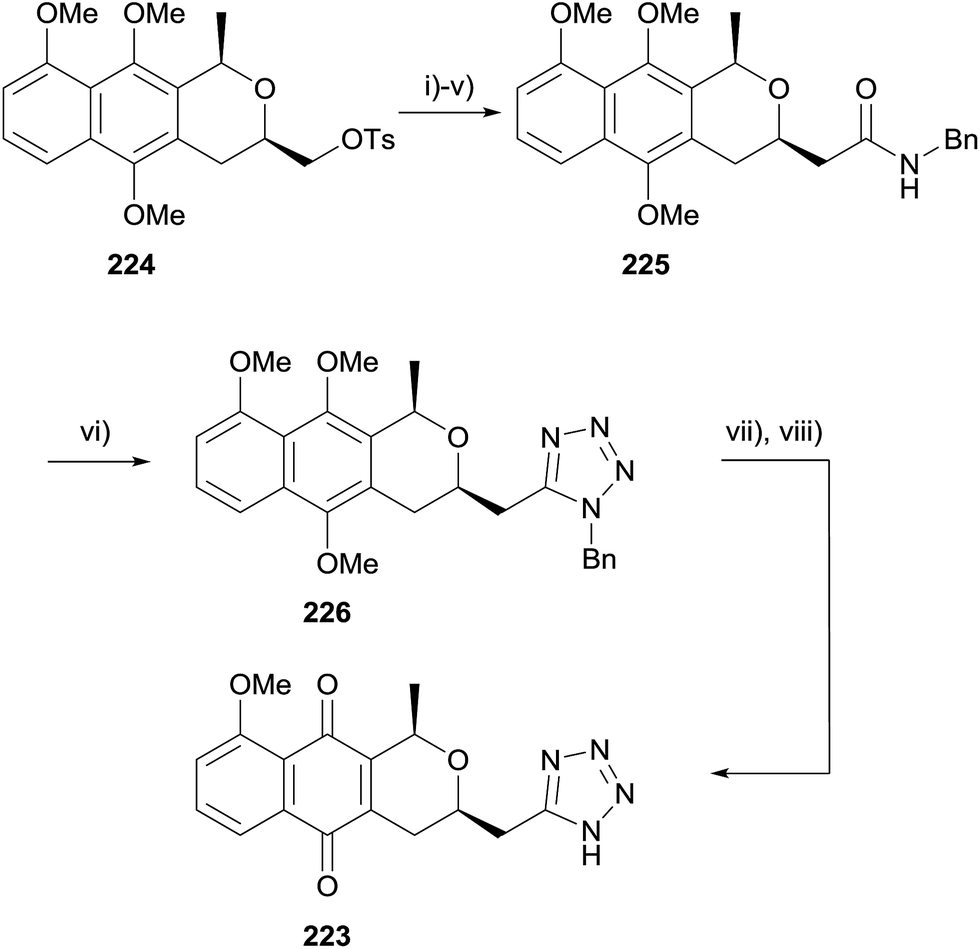 | ||
| Scheme 31 Reagents and conditions: (i) NaI, DMF, 120 °C, 6 h, 77%; (ii) vinylmagnesium bromide, CuI (10 mol%), DMPU, THF, −10 °C, 24 h, 66%; (iii) OsO4, 2,6-lutidine, NaIO4, 1,4-dioxane, H2O, 4 h, 80%; (iv) NaClO2, NaH2PO4, t-BuOH, cyclohexene, 1 h; (v) BOP, BnNH2, Et3N, CH2Cl2, 14 h, 67% over 2 steps; (vi) PPh3, DIAD then TMS-N3, THF, reflux, 17 h, 69%; (vii) H2, Pd/C, rt, 16 h; (viii) CAN, MeCN–H2O, rt, 5 min, 24% over 2 steps. | ||
Tetrazole 223 and a number of the triazole analogues 218 and 219 were tested in breast cancer cell lines overexpressing cytochrome P450 oxidoreductase (POR) under both aerobic and anoxic conditions. In general, the compounds were more active in POR over-expressing cells and also under anoxic conditions, indicating that enzymatic reduction is involved in the mechanism of action of these compounds.69
In 2012, Reutrakul and coworkers reported a synthesis of a pyranonaphthoquinone 232 isolated from Ventilago harmandiana, although no characterisation data were reported for this compound (Scheme 32).70 Enone 228 was prepared from L-rhamnose (227) in six steps. After considerable experimentation, HK annulation with arylthiophthalide 229 was performed using lithium tert-butoxide in THF followed by methylation with dimethyl sulfate and potassium carbonate in refluxing acetone to give compound 230. The benzylic ketone was reduced using cyanoborohydride and the resulting naphthopyran 231 oxidised to pyranonaphthoquinone 232 using CAN.
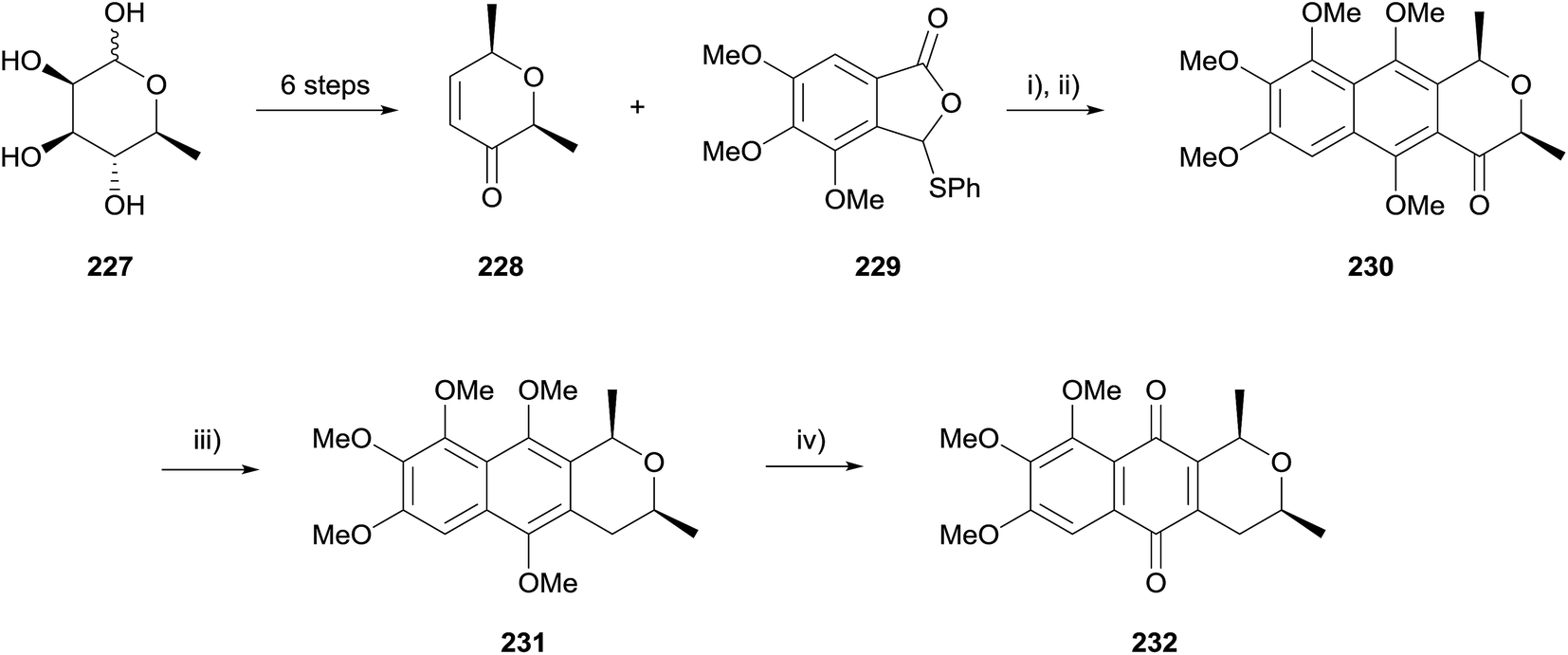 | ||
| Scheme 32 Reagents and conditions: (i) t-BuOLi, LiCl (cat.), THF, −60 °C then 228, −60 °C, 15 min to rt, 6 h, 47%; (ii) K2CO3, Me2SO4, acetone, reflux, 3 h, 81%; (iii) NaBH3CN, BF3·OEt2, MeCN, rt, 24 h, 48%; (iv) CAN, SiO2, CH2Cl2–H2O (3:1), rt, 30 min, 98%. | ||
In 2013, Brimble reported the synthesis of an analogue of the spiroketal-containing pyranonaphthoquinone griseusin B 237 (Scheme 33).71 Interestingly, a two-step annulation/reductive methylation sequence afforded the double-TBS deprotected and spirocyclised product 235 in 10% yield. Adaptation to a one-pot procedure enabled naphthopyran-spiroketal 235 to be accessed as a single isomer in 49% yield. The stereochemistry of the product was established by analysis of the nOe correlations and by comparison of the NMR data to that of 10-epi-235, which was obtained during the course of a parallel study and the structure of which was determined by X-ray crystallography. Benzyl deprotection, IBX oxidation and Pinnick oxidation then afforded carboxylic acid 236 in high yield. Oxidative demethylation utilising silver oxide afforded griseusin B analogue 237 in 58% yield.71
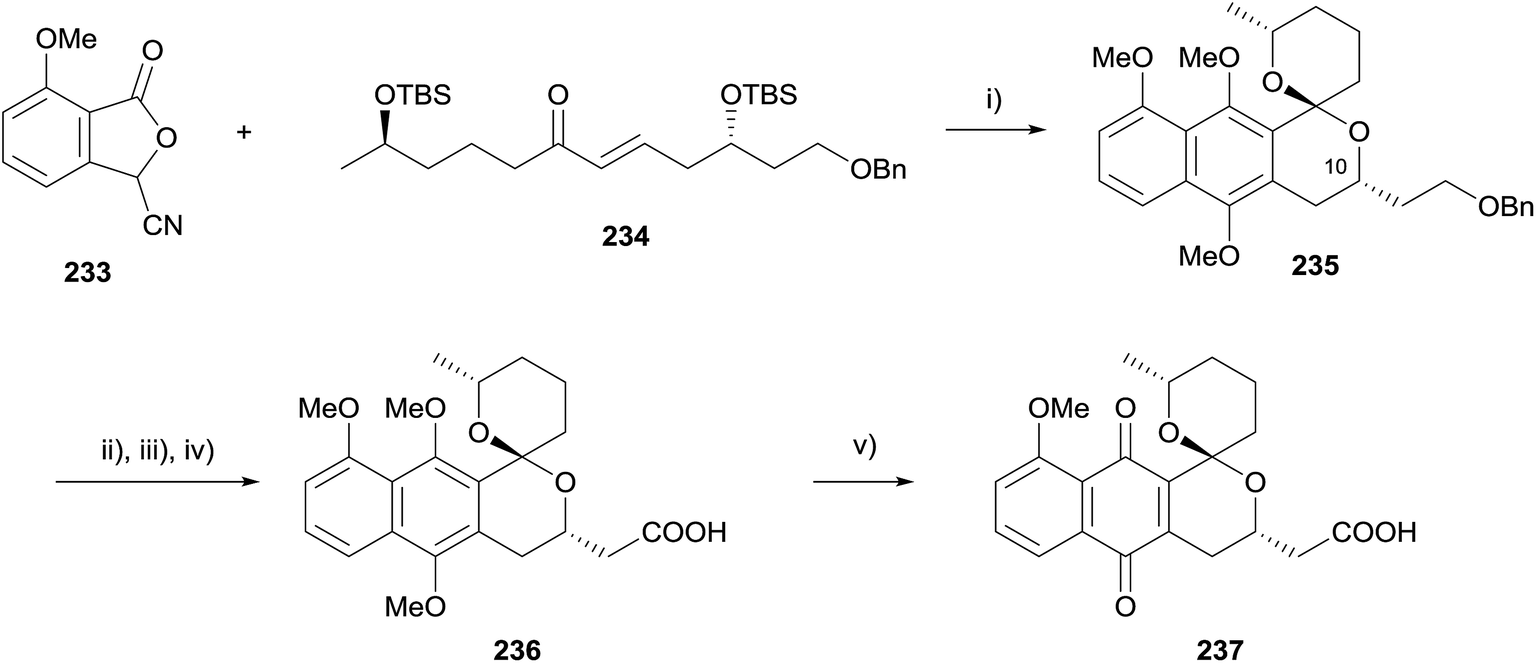 | ||
| Scheme 33 Reagents and conditions: (i) t-BuOK then NaOH, Me2SO4, THF, 49%; (ii) Pd(OH)2, H2, THF, 100%; (iii) IBX, DMSO, 94%; (iv) 2-methyl-2-butene, NaClO2, KH2PO4, t-BuOH, H2O, 83%; (v) AgO, HNO3, THF, 58%. | ||
A follow-up paper detailing attempts to synthesise griseusin B (245) was published in 2014 (Scheme 34).72 Phosphonate 239 was derived from alkyne 238 using partial hydrogenation and Sharpless asymmetric dihydroxylation. Horner–Wadsworth–Emmons (HWE) reaction of phosphonate 239 and aldehyde 240 under mild conditions gave enone 241, the substrate for the key HK annulation. The one-pot annulation/methylation conditions from the previous report were not successful. Attempts to effect the annulation using t-BuOK afforded Michael product 246 (Scheme 34, insert), which had failed to undergo Dieckmann condensation to form the naphthalene core. The stereochemical configuration of the newly generated chiral centres in compound 246 were not determined. Changing the counterion of the base to lithium resulted in successful annulation, which could be incorporated into a one-pot annulation/methylation sequence, giving naphthalene 242 in 41% yield. TBS deprotection/spirocyclisation was carried out using HF–pyridine and the resulting spiroketal 243 readily elaborated to naphthopyran 244. However, oxidative demethylation could not be achieved using AgO, CAN or PIFA.72
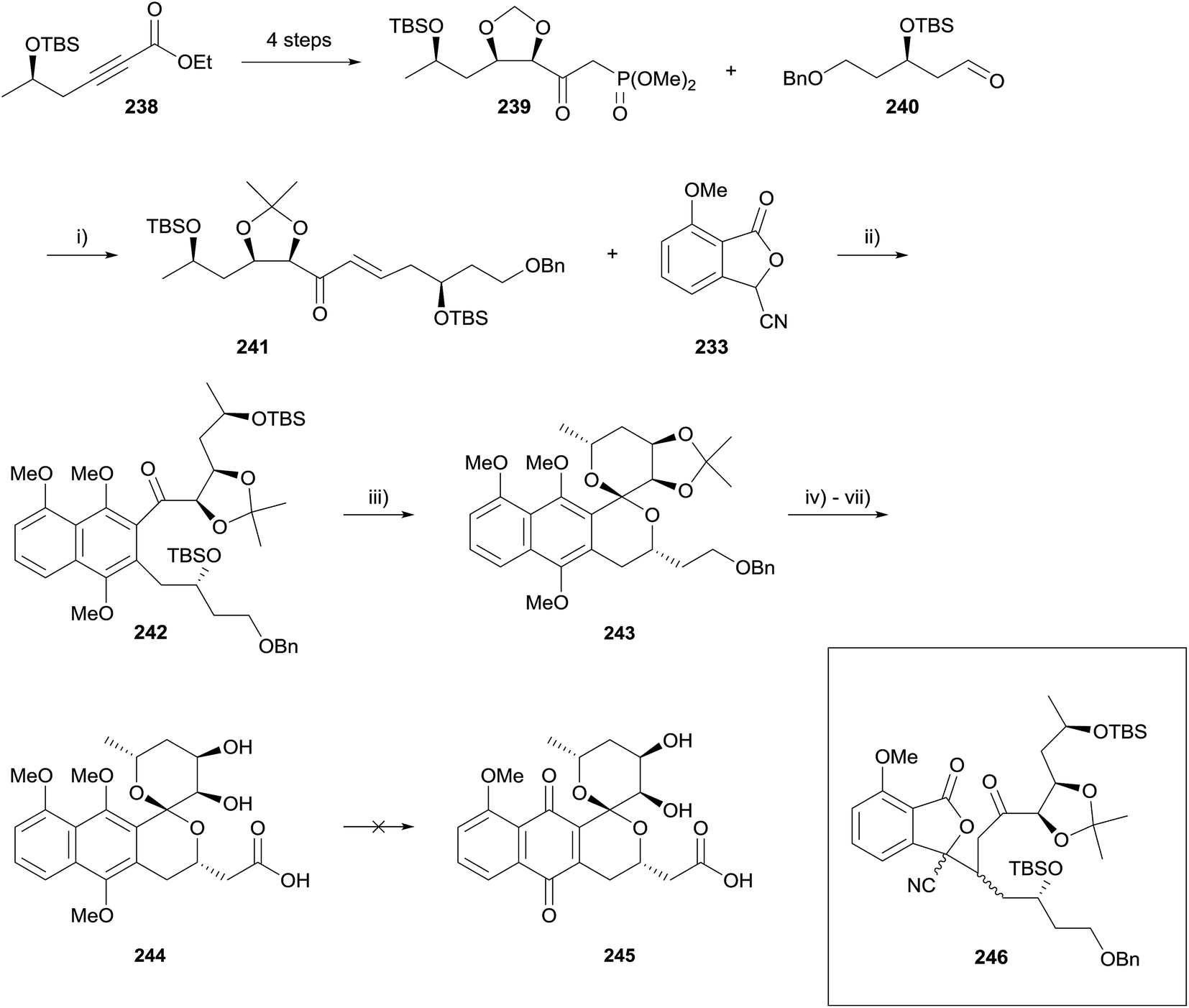 | ||
| Scheme 34 Reagents and conditions: (i) K2CO3, H2O/Et2O, 81%; (ii) t-BuOLi, THF then aq. NaOH, Me2SO4, THF, 41%; (iii) HF·pyr, THF, 73%; (iv) Pd(OH)2/C, H2 (30 psi), THF, 100%; (v) IBX, DMSO, 30 °C; (vi) 2-methyl-2-butene, NaClO2, KH2PO4, t-BuOH, H2O, 90% (2 steps); (vii) 1 M HCl, THF, 90%. | ||
In 2015, Brimble reported the formal synthesis of the enantiomer of kalafungin, nanaomycin D (ent-35) using a HK annulation as the key step for the formation of the pyranonaphthoquinone framework (Scheme 35).73 Lactone 249 was formed by Sharpless asymmetric dihydroxylation and spontaneous cyclisation of β,γ-unsaturated ester 248, which was in turn prepared from D-mannitol (247). Lactone 249 was then converted to enone 250 over 4 steps. The key HK annulation was achieved employing LDA in THF followed by reductive methylation with methyl iodide and cesium carbonate, affording naphthalene 251 in 35% yield. Acid-mediated MOM deprotection and cyclisation provided naphthopyran 252, an intermediate in the total synthesis of nanaomycin D by Brückner and coworkers.74
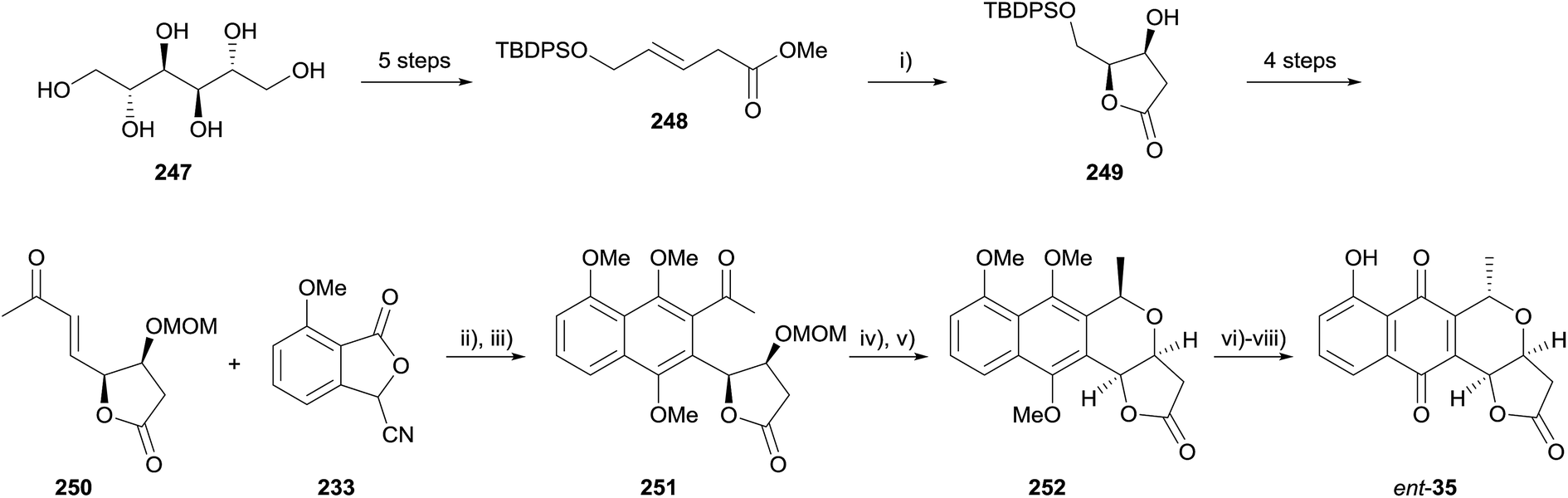 | ||
| Scheme 35 Reagents and conditions: (i) (DHQ)2PHAL, K2CO3, K3Fe(CN)6, OsO4, MeSO2NH2, H2O–t-BuOH, 0 °C, 76%, 79% ee; (ii) LDA, THF, −78 °C to rt, 30 min; (iii) Na2S2O4, Cs2CO3, MeI, rt, 15 h, 35% over 2 steps; (iv) 2 M HCl, THF, 40 °C, 2.5 h, 55%; (v) BF3·OEt2, Et3SiH, CH2Cl2, −78 °C to rt, 5 h, 26%; (vi) CAN, MeCN–H2O (1:1), rt, 15 min, 88%; (vii) BBr3, CH2Cl2, −50 °C, 25 min, then rt, 2 h, 84%; (viii) conc. H2SO4, rt, 30 min, 90%. | ||
This methodology was also investigated as a means to synthesise crisamicin A.75 The annulation/methylation sequence was first attempted using the same conditions as the nanaomycin D synthesis, however this led only to the formation of C-alkylated product 256 (Scheme 36, insert, the configuration of the newly generated stereocenter was not determined). Following extensive optimisation, the desired naphthalene 255 could be obtained in 35% yield over two steps. The benzyl ether was converted to a triflate in preparation for homocoupling. Suzuki coupling, followed by removal of the MOM groups provided dimer 259. A number of conditions for lactol formation (including concomitant deprotection/lactol formation from protected 258) failed to provide the desired lactol 260. However, it was found that treatment of 258 with acidic methanol, followed by reduction, provided diester 261 in 28% yield. Unfortunately, reformation of the lactone moiety could not be achieved despite investigation of a variety of acidic and basic conditions.
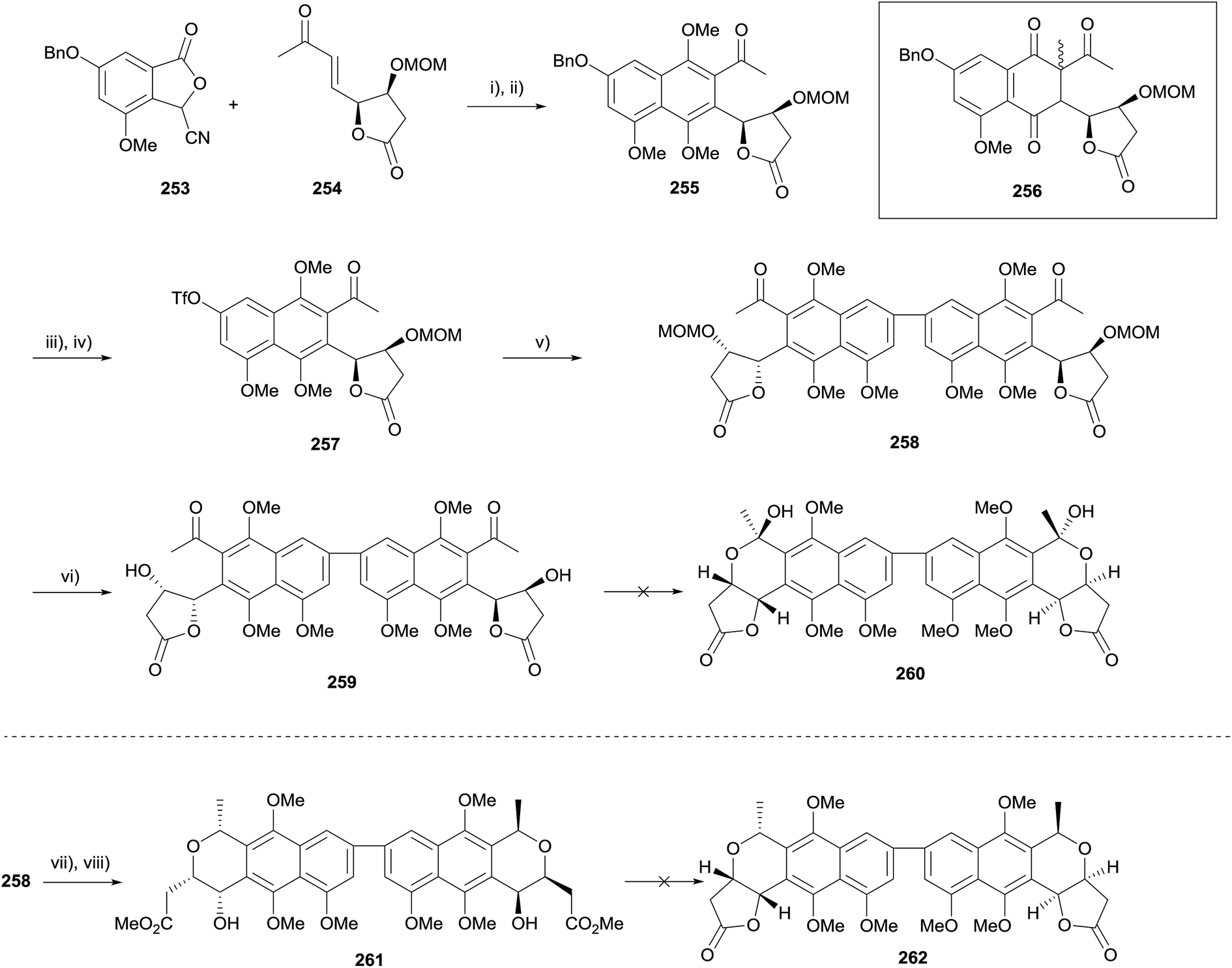 | ||
| Scheme 36 Reagents and conditions: (i) LDA, THF, −78 °C to rt; (ii) K2CO3, Me2SO4, acetone, reflux, 35% (2 steps); (iii) 10% Pd/C, H2, MeOH, rt; (iv) PhN(SO2CF3)2, NEt3, DMAP, CH2Cl2, 91% (2 steps); (v) K2CO3, bis(pinacolato)diboron, Pd(dppf)Cl2, dioxane, 85 °C, sealed tube, 55%; (vi) 2 M HCl, dioxane, 40 °C, 63%; (vii) HCl in MeOH, rt; (viii) TFA, Et3SiH, −78 °C to rt, 28% (2 steps). | ||
Following this, cyclisation of the monomer prior to Suzuki coupling was attempted. Cyclisation of benzyl protected monomer 255 was not successful, however triflate 257 could be converted to the corresponding lactol by exposure to Amberlyst® 15 (Scheme 37). Stereoselective reduction then furnished naphthopyran 263 possessing the opposite configuration to the crisamicin A monomer, nanaomycin D (ent-35) at C1. Suzuki coupling was conducted according to the previously established conditions, followed by oxidative demethylation with silver oxide. The resultant pyranonaphthoquinone 264 could not be purified but was identified by the 1H NMR spectrum of the crude reaction mixture and by mass spectral analysis. Unfortunately, the final demethylation to epi-C1/C1′-crisamicin A (265) could not be achieved using either boron trichloride or aluminium trichloride.75
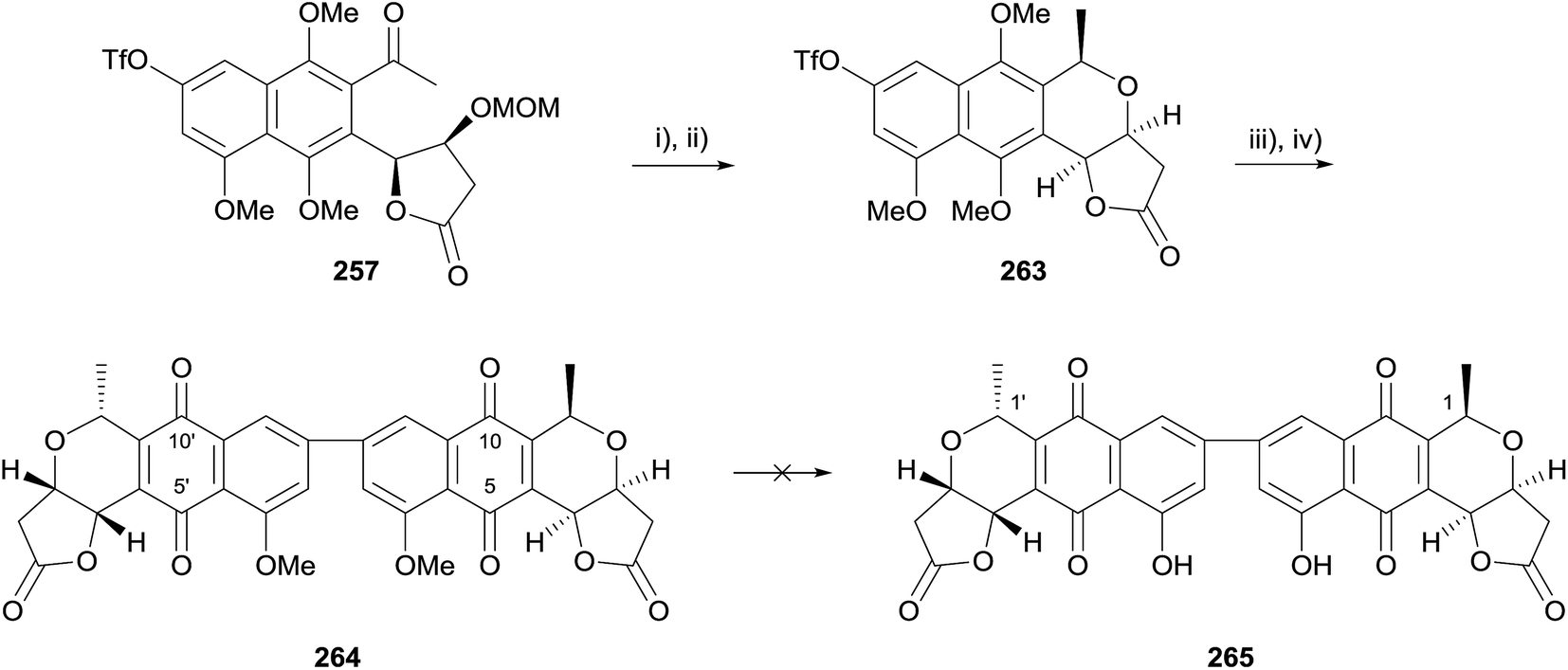 | ||
| Scheme 37 Reagents and conditions: (i) Amberlyst® 15, dioxane, rt; (ii) TFA, Et3SiH, CH2Cl2, rt to 50 °C, 31% (2 steps); (iii) K2CO3, dppf, bis(pinacolato)diboron, dioxane, 85 °C, sealed tube, 45%; (iv) AgO, dioxane, HNO3, rt. | ||
3.3 Other Michael additions
In 2011, Brimble and coworkers used an oxa-Michael strategy to make three deoxy analogues of kalafungin (Scheme 38).76 Ketone 266 was converted to enoate 267 in six steps that included a CBS reduction and radical allylation. Removal of the TBS group from 267 using buffered TBAF triggered an intramolecular oxa-Michael cyclisation, which proceeded with modest diastereoselectivity. This cis-isomer 268 was converted to cis-deoxydihydrokalafungin (20) by saponification of the methyl ester and oxidative demethylation using CAN. Interestingly, applying the same conditions to the trans naphthopyran 269 gave hydroquinone 271 as a 4:1 mixture with the expected product deoxydihydrokalafungin (270). Hydroquinone 271 could be converted to deoxydihydrokalafungin (270) using ferric chloride or to deoxykalafungin (106b) by aerial oxidation in the presence of catalytic salcomine.76
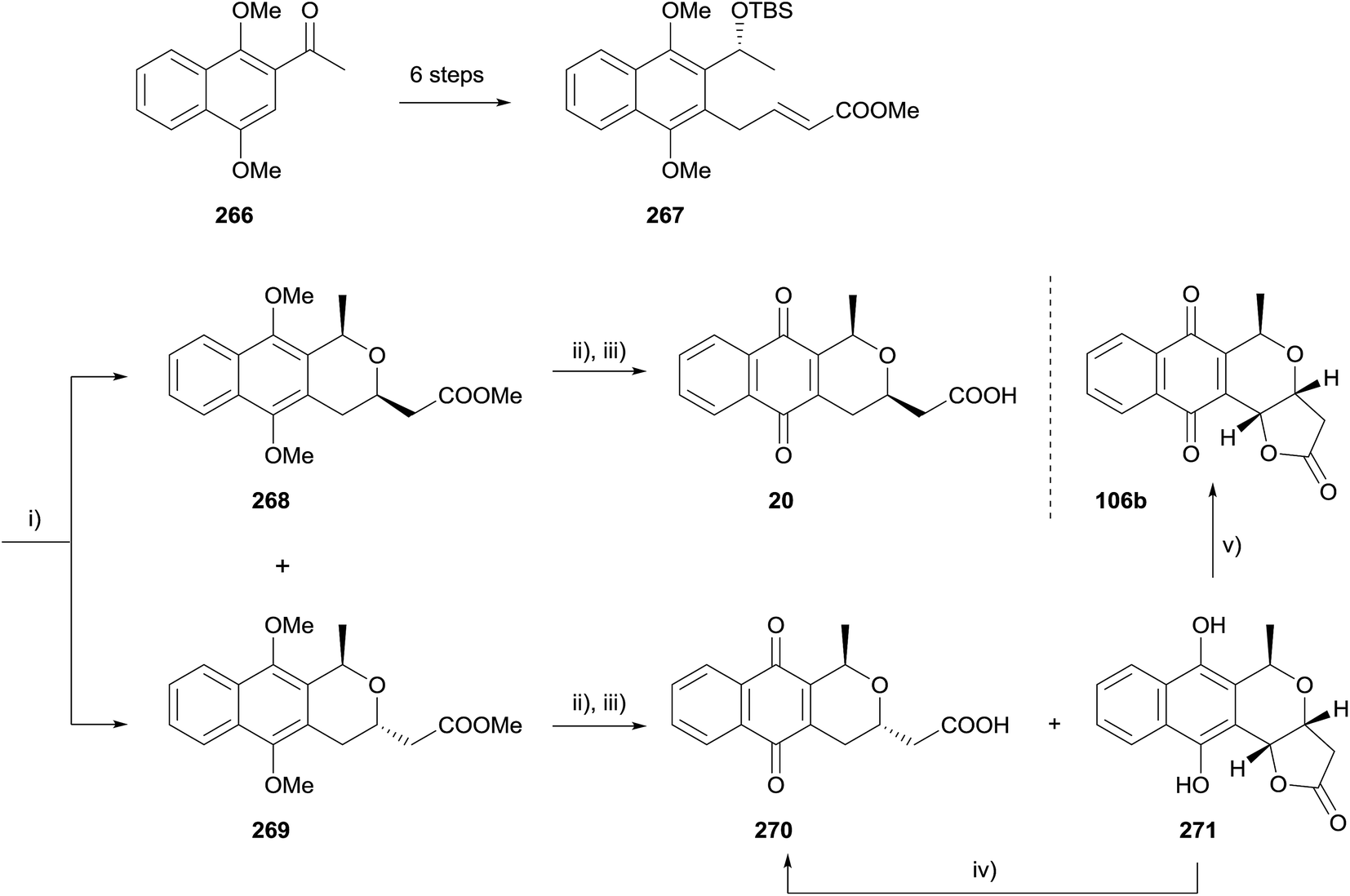 | ||
| Scheme 38 Reagents and conditions: (i) TBAF, acetic acid, THF, 40 °C, 85%, 269/268 1.3:1; (ii) LiOH, THF–H2O, rt, 94% from 269, 92% from 268; (iii) CAN, H2O–MeOH, rt, 89%, 4:1 271:270, 80%, 31; (iv) FeCl3·(H2O)6, MeOH–H2O, rt, 82%; (v) salcomine, O2, MeCN, rt, 39%. | ||
In 2015, Van Nguyen reported the synthesis of a series of herbarin analogues by Michael addition of N-acylmethylpyridinium-ylids to naphthoquinone 273 (Scheme 39).77 Naphthoic acid 272 was converted to naphthoquinone 273 in 4 steps. The key Michael addition of various N-acylmethylpyridinium-ylids was conducted in the presence of triethylamine in acetonitrile followed by dehydration catalysed by para-toluenesulfonic acid (PTSA).
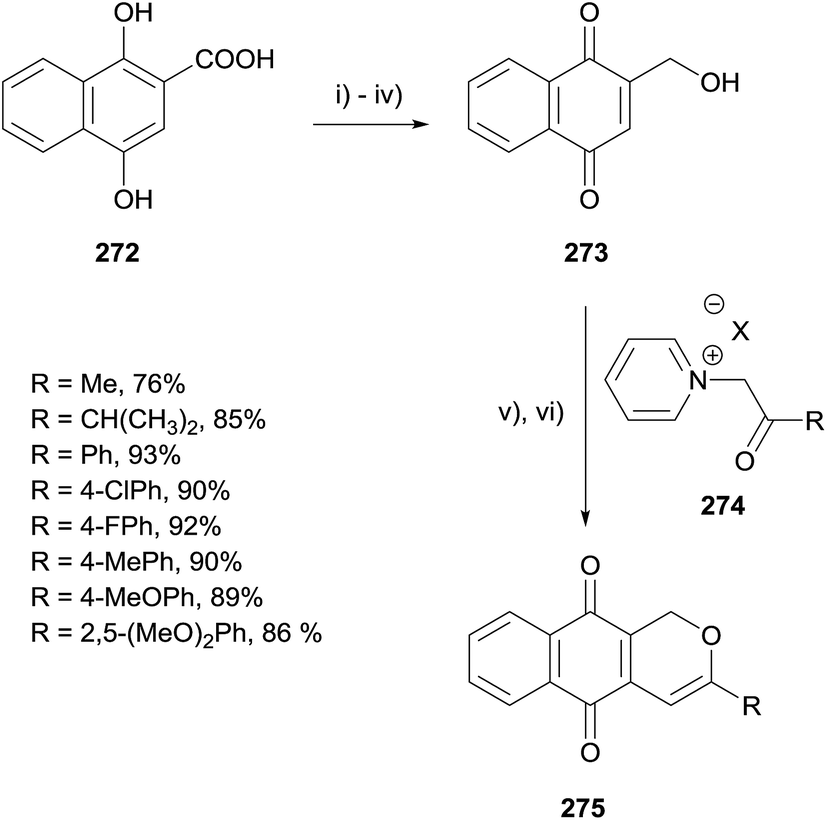 | ||
| Scheme 39 Reagents and conditions: (i) Me2SO4, K2CO3, Et2O, reflux, 12 h; (ii) KOH, Et2O, rt, 20 min, 83% over 2 steps; (iii) LiAlH4, Et2O, reflux, 2 h, 95%; (iv) CAN, MeCN–H2O, rt, 30 min, 92%; (v) Et3N, MeCN, N2, 50 °C, 2–4 h; (vi) PTSA, toluene, reflux, 10 min, 76–93%. | ||
Selected analogues were then converted to the corresponding epoxides 277a–c by reaction with hydrogen peroxide (Scheme 40). Reduction of the alkene present in compounds 275a, 275b and 275d using TFA and triethylsilane was also conducted prior to epoxidation to obtain dihydropyran analogues 278a–c. The compounds were tested against four cancer cell lines. In general, the epoxy analogues were inactive while the quinone analogues exhibited moderate to good activity, indicating that the quinone moiety is required for anticancer activity.77
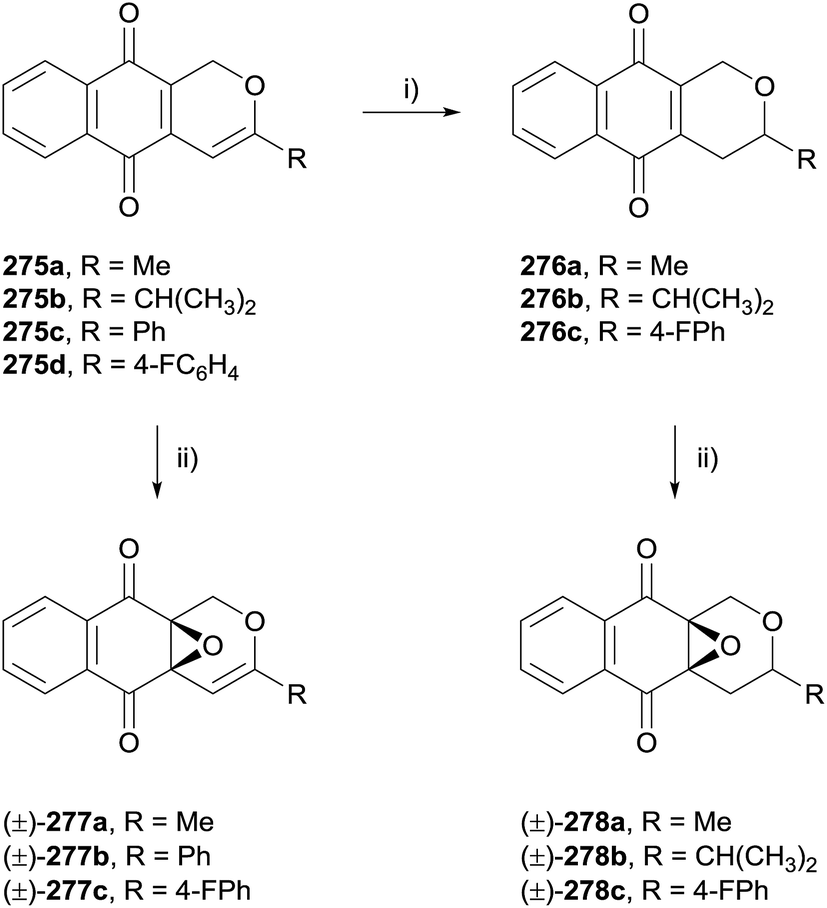 | ||
| Scheme 40 Reagents and conditions: (i) CF3COOH, CH2Cl2, rt, 12 h then Et3SiH, CH2Cl2, rt, 12 h, 61–68%; (ii) H2O2, Na2CO3, CH2Cl2, rt, 12 h, 45–52%. | ||
3.4 Toluate anion addition
The use of a tandem Michael–Dieckmann/Claisen reaction of ortho-toluates, otherwise known as the Staunton–Weinreb annulation, was recently the subject of a comprehensive review by Donner.78 The synthesis of several pyranonaphthoquinones using this strategy were discussed and as such will not be revisited here.29,79–813.5 Cycloadditions
Diels–Alder methodology is another common method that has be used to construct the pyranonaphthoquinone framework. A racemic total synthesis of the dimeric pyranonaphthoquinone crisamicin A (188) was reported in 2008 by Yang and coworkers using such an approach (Scheme 41).82 Following optimisation, palladium-catalysed carbonylative lactonisation was conducted on alkene 279 using palladium acetate, tetramethyl thiourea (TMTU), carbon monoxide and copper chloride with the addition of ammonium acetate and propylene oxide. Oxidative demethylation using CAN then furnished quinone 280. Diels–Alder reaction of quinone 280 and diene 281 under Jones' conditions afforded pyranonaphthoquinone 282. Formation of the triflate, reduction to the hydroquinone and protection gave naphthopyran 283. Compound 283 underwent Pd-catalysed borylation to give Suzuki precursor 284. Crude 284 was engaged in a homocoupling reaction catalysed by Pd–thiourea pincer complex 286, providing the dimeric framework of crisamicin A (285). Finally, deprotection, aerial oxidation of the hydroquinone and demethylation of the remaining phenol afforded racemic crisamicin A (188).82 A detailed investigation of the Diels–Alder reaction employed in this sequence was published in 2009.83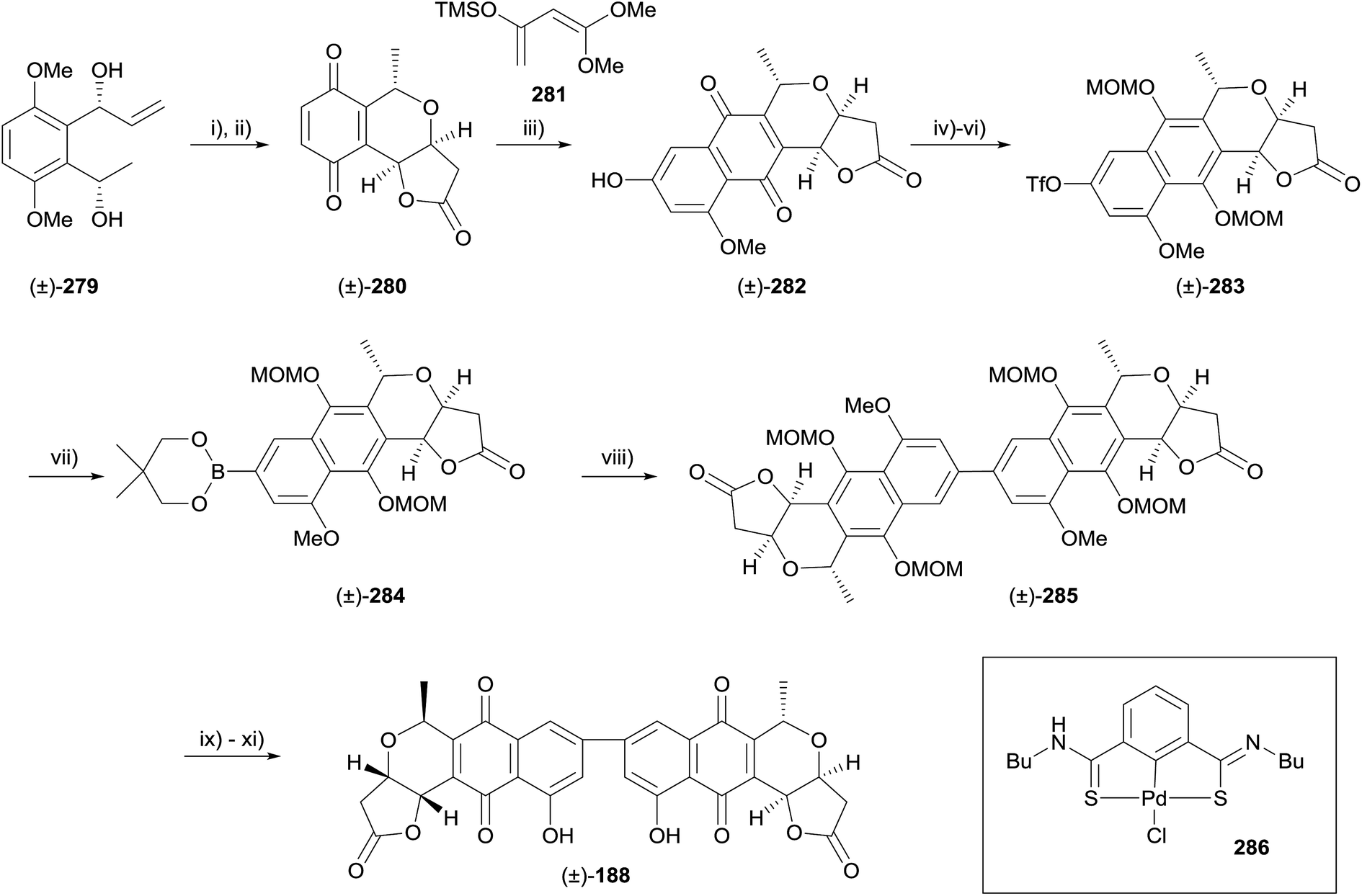 | ||
| Scheme 41 Reagents and conditions: (i) Pd(OAc)2/TMTU, CuCl2, CO, THF, NH4OAc, propylene oxide, 50 °C, 88%; (ii) CAN, MeCN/H2O, −10 °C, 89%; (iii) 281 then Jones' reagent, 85%; (iv) Tf2O, pyridine, DMAP, 78%; (v) Na2S2O4, TBABr, THF, H2O; (vi) MOMCl, DIPEA, DMAP, CH2Cl2; (vii) PdCl2(dppf), dppf, KOAc, 76%; (viii) 286, Ag2CO3, DMSO, H2O, 87% over 2 steps; (ix) TMSBr, CH2Cl2, −78 °C to −40 °C; (x) silica gel, air, rt, 93%; (xi) BCl3, CH2Cl2, −78 °C to −40 °C, 91%. | ||
3.6 Radical conjugate addition
Radical conjugate additions have emerged as a useful tool for constructing pyranonaphthoquinones following the first utilisation of this method by Donner in 2010 in a formal synthesis of racemic frenolicin B (36) and its C-5 epimer.84 In a first attempt, 4,7-dimethoxyphthalide (287) was treated with lithium aluminium hydride to give diol 288 which was further elaborated to aldehyde 289 (Scheme 42). The key intramolecular radical conjugate addition was effected using tributyltin hydride and 2,2′-azobis(isobutyronitrile) (AIBN) to give desired isochroman-lactone 291 and isochroman 290 in 13% and 46% yield respectively. Cyclisation of isochroman 290 to lactone 291 was effected by heating in the presence of potassium carbonate and 18-crown-6. Unfortunately, addition of the C5 alkyl group required to complete the synthesis of frenolicin B was unsuccessful.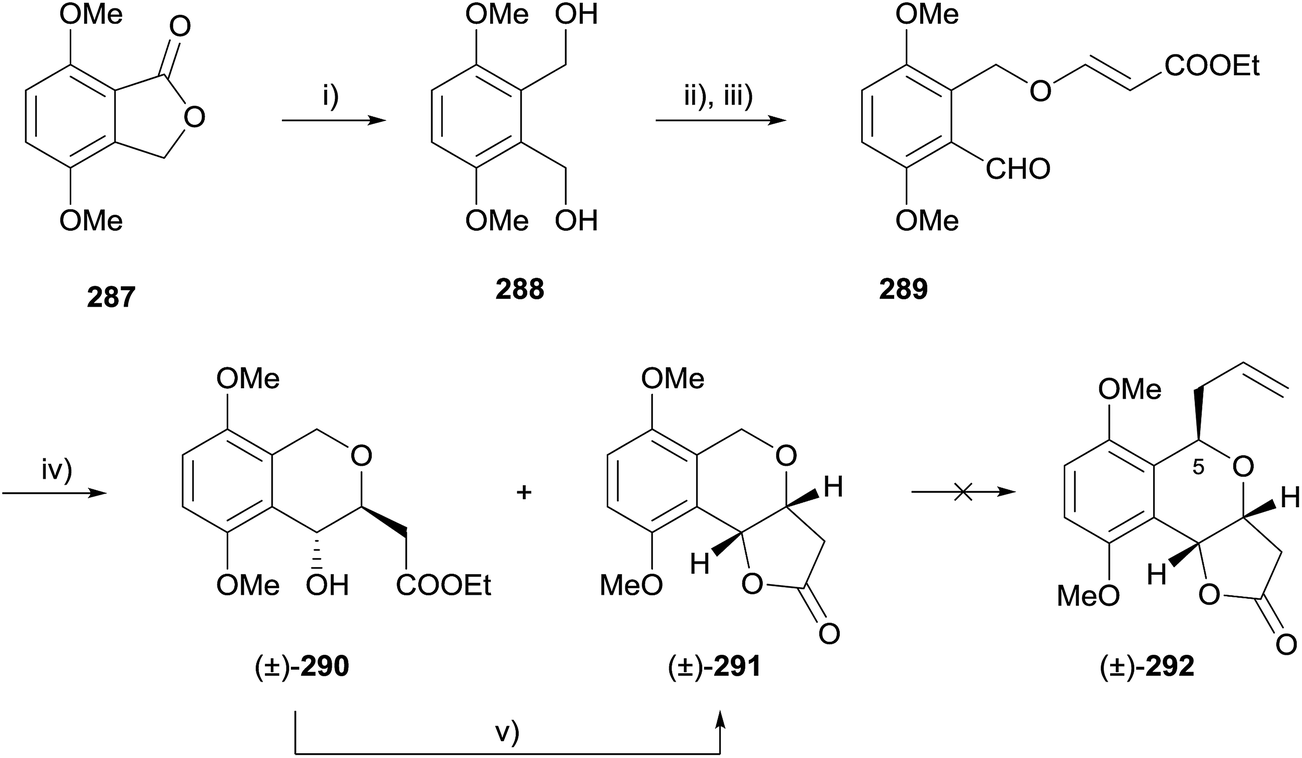 | ||
| Scheme 42 Reagents and conditions: (i) LiAlH4, THF, 0 °C, 1.5 h, 93%; (ii) ethyl propiolate, NMM, CH2Cl2, 1.5 h, 80%; (iii) TEMPO, PhI(OAc)2, CH2Cl2, 5 h, 78%; (iv) Bu3SnH, AIBN, benzene, reflux, 5 h, 46% 290, 13% 291; (v) K2CO3, 18-crown-6, DMF, 100 °C, 12 h; 66%. | ||
To circumvent the issue surrounding the introduction of the C5 alkyl group, a modified route was developed whereby the C5 alkyl group was installed prior to radical cyclisation (Scheme 43). The aldehyde (293) was masked as an acetal followed by installation of the ortho-sidechain. Unmasking of the aldehyde gave 294, which underwent radical cyclisation to the cis-cyclised product 295. C5-Epimerisation was effected with boron tribromide and CAN oxidation provided quinone 296, an intermediate in the synthesis of frenolicin B (36) reported by Kraus.85
 | ||
| Scheme 43 Reagents and conditions: (i) propane-1,3-diol, (MeO)3CH, Bu4NBr3, 45 min, 100%; (ii) BuLi, Et2O–hexane, 0 °C, then n-PrCHO, 0 °C to rt, 1.5 h, 64%; (iii) ethyl propiolate, NMM, CH2Cl2, 22 h, 75%; (iv) 2 M HCl, acetone, 16 h, 90%; (v) Bu3SnH, AIBN, benzene, 3 h, 66%; (vi) BBr3, CH2Cl2, −60 °C to rt, 1 h; (vii) CAN, MeCN–H2O, 5 min, 69% over 2 steps; (viii) 1-trimethylsilyloxybutadiene, CH2Cl2, −78 °C, 4.5 h; (ix) CrO3, aq. H2SO4, acetone, 0 °C, 40 min, 81% over two steps. | ||
Alternatively, radical cyclisation of compound 294 gave predominantly trans cyclised product 297.86 Reduction under Luche conditions then gave compound 298, also an intermediate in the synthesis by Kraus (Scheme 44).85
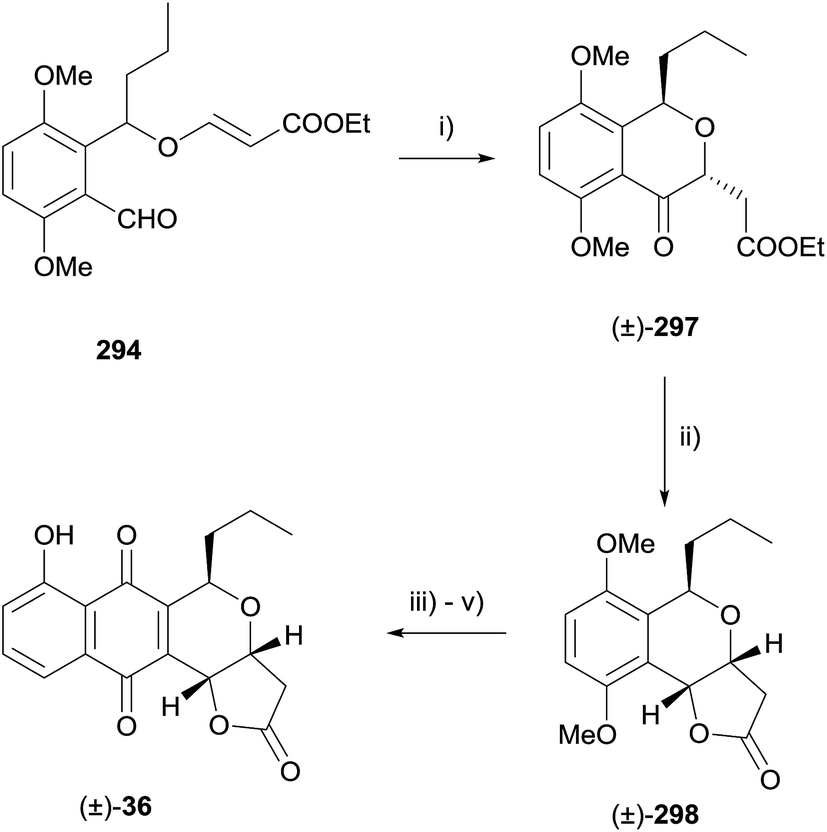 | ||
| Scheme 44 Reagents and conditions: (i) tert-dodecanethiol, ABCN, PhCl, 100 °C, 4 h, 62%; (ii) CeCl3·7H2O, NaBH4, CH2Cl2, MeOH, −78 °C, 1 h then PTSA·H2O, CHCl3, 18 h, 58%; (iii) CAN, MeCN–H2O, reaction time and yield not reported; (iv) 1-trimethylsilyloxybutadiene, CH2Cl2, −78 °C, 4.5 h; (v) CrO3, aq. H2SO4, acetone, 0 °C, 40 min, 81% over two steps. | ||
3.7 Lactonisation
Formation of the pyran ring by lactonisation is a useful but under-utilised method for the formation of the pyranonaphthoquinone ring system. All of the methods described henceforth result in a lactone or lactol product, but not all result from C–O bond formation. The resulting lactone may be present in the target structure or serve as a synthetic handle enabling further manipulations at the pyran ring.In 2014, Brimble and coworkers employed a concomitant radical alkylation-lactonisation strategy to synthesise 9-deoxythysanone (304, Scheme 45).87 1,4-Dimethoxynaphthalene (175) was converted to bromide 299 in four steps which included a regioselective iridium-catalyzed borylation reaction. Reaction with ethyl chloroformate and subsequent oxidisation provided naphthoquinone 300. Radical alkylation of 300 with (±)-301 proceeded with concomitant lactonisation, with the intermediate quinone reduced to the hydroquinone 302 in good overall yield. The lactone in hydroquinone 302 was reduced using borane-dimethylsulfide, affording pyranonaphthoquinone 303a, with the loss of intramolecular hydrogen bonding facilitating spontaneous oxidation of the hydroquinone. Finally, removal of the isopropyl protecting group gave 303b which provided racemic 9-deoxythysanone (304) upon stereoselective bromination followed by hydrolysis. Compounds 304, and 303a/b were analysed for their inhibitory activity against HRV 3C protease and all were found to be more active than the natural product thysanone (14), indicating that the C9-OH is detrimental to activity.87
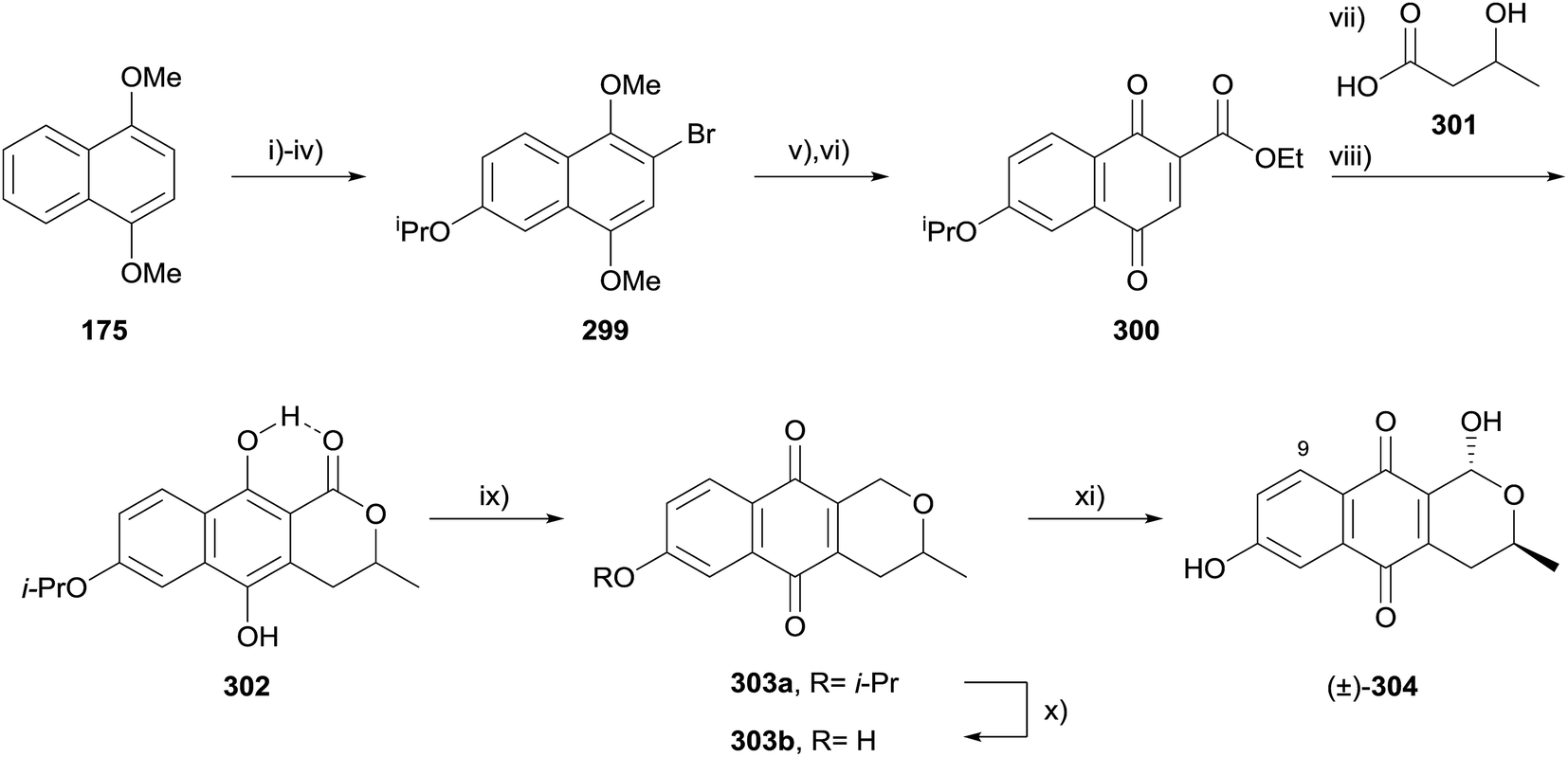 | ||
| Scheme 45 Reagents and conditions: (i) [Ir(COD)OMe]2, dtbpy, B2pin2, THF, 80 °C, 12 h; (ii) H2O2, NaOH (1 M), THF, 0 °C, 20 min, 88% over 2 steps; (iii) Br2, AcOH, rt, 30 min, 85%; (iv) TBAI, K2CO3, 2-bromopropane, acetone/DMF (12:1), reflux, 16 h, 99%; (v) ethyl chloroformate, t-BuLi, THF, −78 °C to rt, 12 h, 75%; (vi) AgO, HNO3 (6 M), THF, rt, 3 min, 88%; (vii) AgNO3, (NH4)2S2O8, MeCN–H2O (2:1), 80 °C, 5 min; (viii) aq. Na2S2O4, CH2Cl2–Et2O (1:3), rt, 60% over two steps; (ix) BH3·SMe2, THF, 0 °C to rt, 16 h, 48% (303a); (x) AlCl3, CH2Cl2, 0 °C to rt, 20 min, 94%; (xi) Br2, (BzO)2, 60 W lamp, CCl4, 40 °C, 1 h then THF–H2O (2:1), 1 h, 85%. | ||
An interesting lactonisation strategy is showcased in a report by Allin and coworkers that details the development of a dual Fries–Claisen rearrangement to assemble the lactonisation precursor during the synthesis of pyranonaphthoquinone 307 (Scheme 46).88 The anionic ortho-Fries rearrangement of compound 305 was effected using s-BuLi and TMEDA in THF. Methylation of the free phenol followed by Claisen rearrangement and further methylation gave naphthalene 306. Finally, the key acid-mediated lactonisation furnished pyranonaphthoquinone 307.88
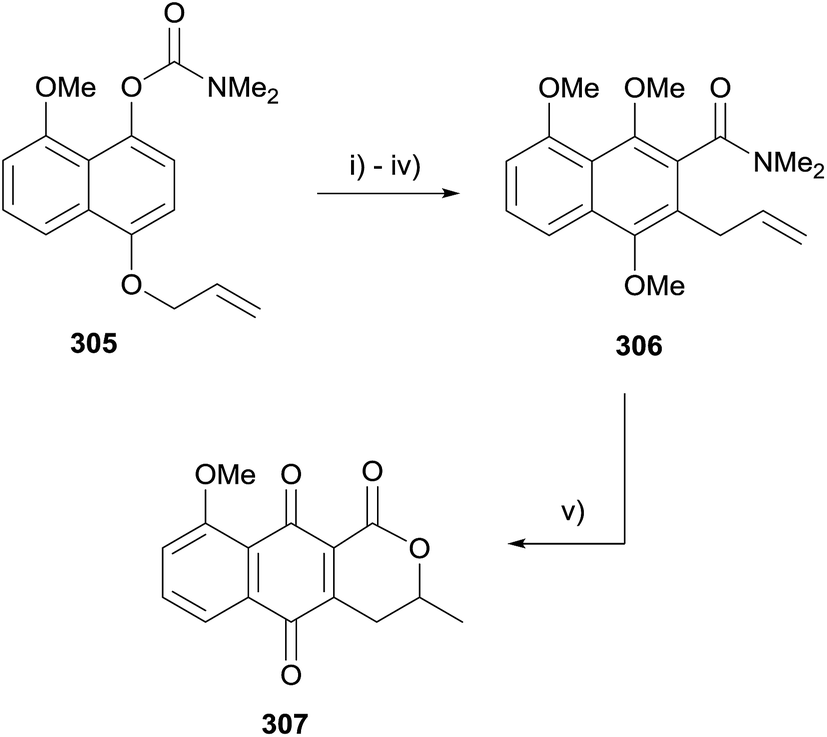 | ||
| Scheme 46 Reagents and conditions: (i) s-BuLi, TMEDA, THF, −78 °C, 92%; (ii) NaH, MeI, THF, 20 °C, 4 h, 80%; (iii) mesitylene, reflux, 3 h, 92%; (iv) K2CO3, MeI, acetone, reflux, 24 h, 83%; (v) 6 M HCl, heat, 24 h, 59%. | ||
Fries–Claisen product 306 was also used in the preparation of racemic eleutherin (9, Scheme 47).89 Iodolactonisation and subsequent dehalogenation of amide 306 afforded lactone 308. Addition of methyllithium followed by stereoselective reduction gave a 5:1 mixture of racemic cis and trans isomers of the corresponding naphthopyran. Compound 309 could be isolated by column chromatography, but the minor trans isomer could not be isolated at this stage. Oxidative demethylation using CAN afforded racemic eleutherin (9). Oxidation of the diastereomeric mixture prior to separation of the diastereomers also allowed the authors to obtain an analytical sample of racemic isoeleutherin (42).89
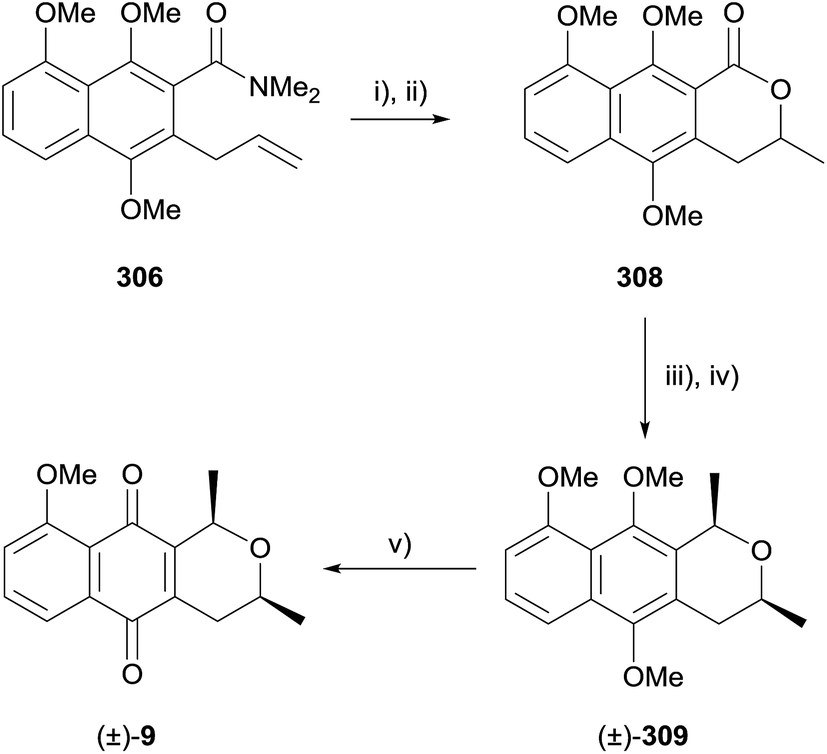 | ||
| Scheme 47 Reagents and conditions: (i) I2, NaHCO3, THF–Et2O, heat, 48 h, 83%; (ii) Bu3SnH, AIBN, toluene, heat, 2 h, 80%; (iii) MeLi, THF–Et2O (1:1), 0 °C, 12 h; (iv) Et3SiH, BF3·OEt2, CH2Cl2, −78 °C, 8 h, 85% over 2 steps; (v) CAN, MeCN–H2O (2:1), 2 h, 93%. | ||
A related example is Brimble's synthesis of monomeric pyranonaphthoquinone 21, although this involves the formation of a lactol as opposed to a lactone (Scheme 48).90 Aldehyde 311 was synthesised from naphthalene 310 in six steps. Attempted asymmetric allylation of compound 311 using either a Brown allylation or a microwave-assisted enzymatic resolution were unsuccessful.91–94 Reaction with allylmagnesium bromide furnished racemic allyl alcohol 312. Acetylation followed by intramolecular nucleophilic addition and reduction of the resultant lactol using triethylsilane provided naphthopyran 314. Modification of the side-chain and oxidative demethylation afforded racemic cis-pyranonaphthoquinone 21, which could be used as a monomer in the synthesis of dimeric pyranonaphthoquinones such as crisamicin and actinorhodin.90
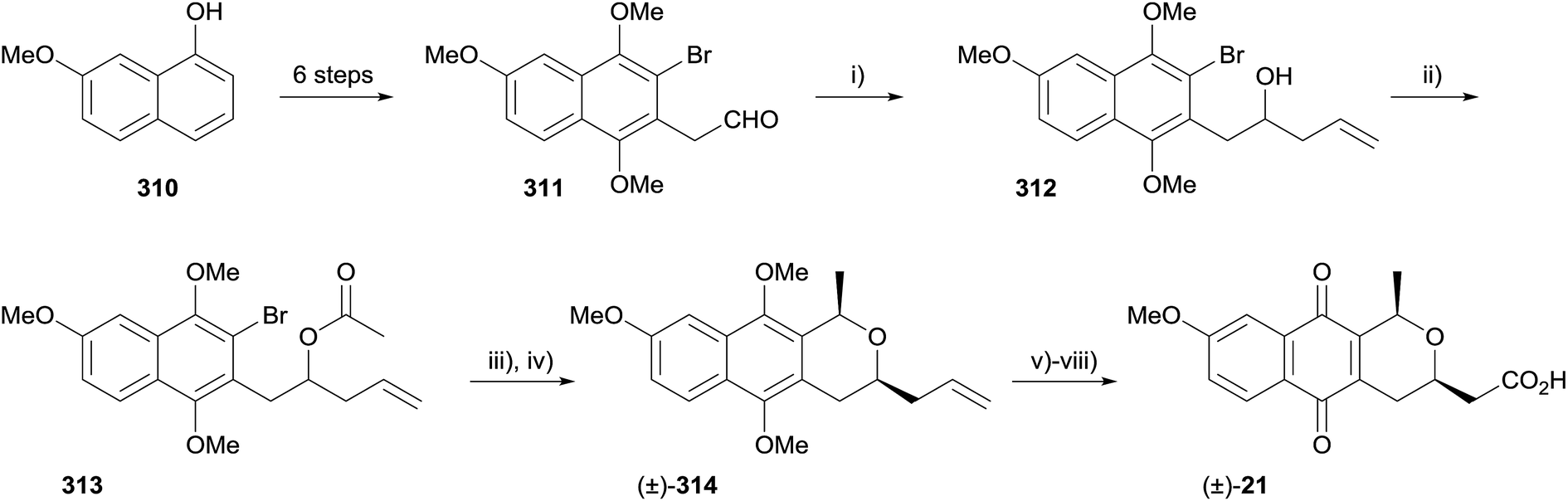 | ||
| Scheme 48 Reagents and conditions: (i) allylmagnesium bromide, Et2O, 6 h, −40 °C to rt, 89%; (ii) Ac2O, CH2Cl2, HClO4, 2 h, rt, 90%; (iii) t-BuLi, THF, 1 h, −78 °C; (iv) Et3SiH, TFA, CH2Cl2, 2 h, −78 °C to rt, 65% over 2 steps; (v) OsO4, NMO, acetone–water, 10 min, 0 °C to rt, 86%; (vi) NaIO4, MeOH–H2O, 2 h, rt, 70%; (vii) NaClO2, t-BuOH, NaH2PO4·2H2O, cyclohexene, 2 h, rt; (viii) CAN, MeCN–H2O, 10 min, 0 °C to rt, 76% over 2 steps. | ||
Brimble and coworkers also applied this method to the synthesis of the deoxy analogues of eleutherin (11 and ent-11) and thysanone (15, Scheme 49).95 The chiral precursors were prepared using a microwave-assisted kinetic resolution of alcohol 315 to give (R)-316 and (S)-315. Deoxyeleutherin (11) was prepared from acetate (S)-316 by intramolecular lactolisation and reduction with triethylsilane to give 317, followed by CAN-oxidation. The enantiomer ent-11 was prepared from (R)-316 by an analogous procedure. Finally intramolecular cyclisation of formate 318 (derived from (S)-315), followed by CAN oxidation afforded deoxythysanone (15).
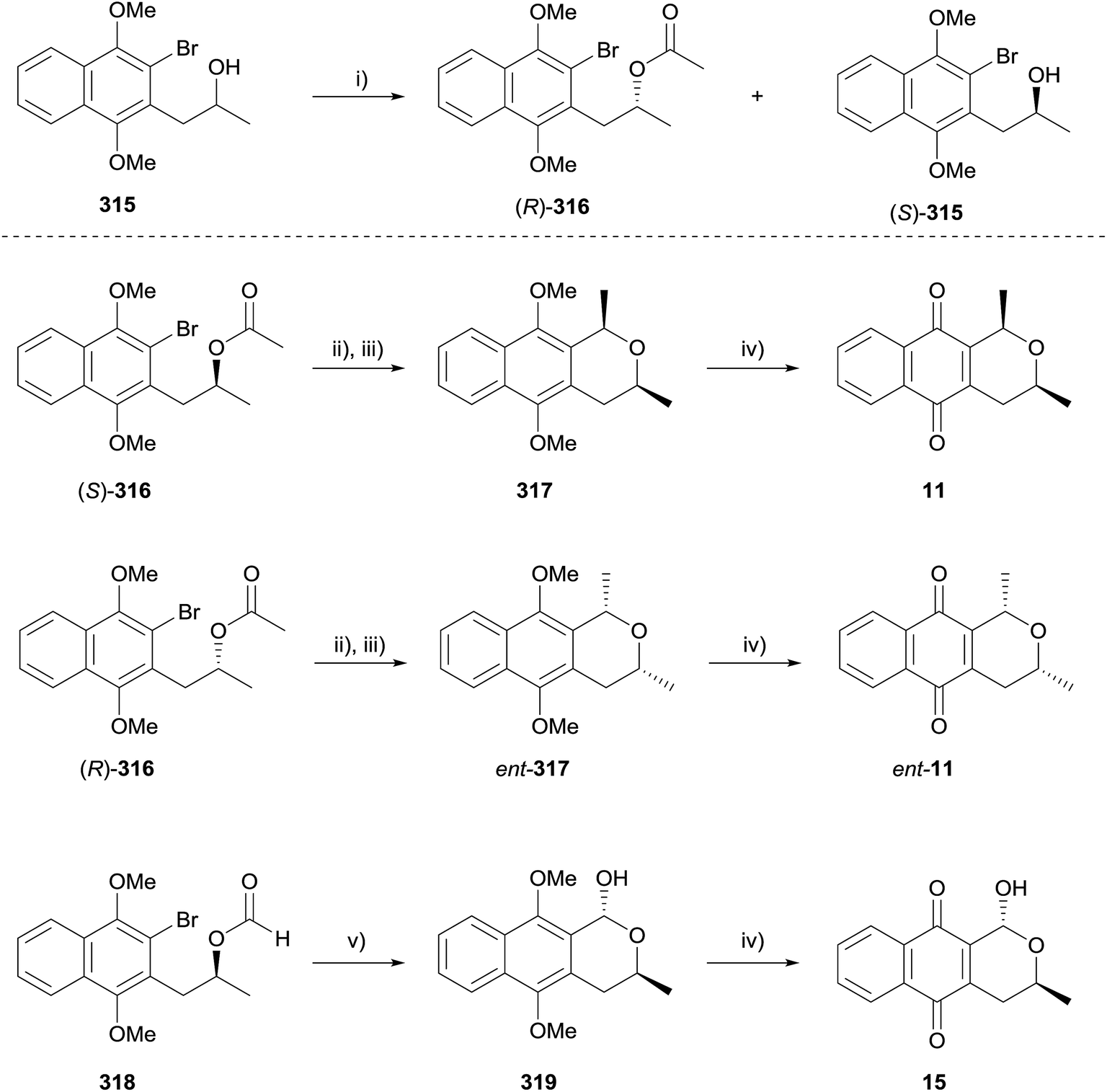 | ||
| Scheme 49 Reagents and conditions: (i) Novozyme 435®, p-chlorophenyl acetate, toluene, 60 °C, 70 W, microwave, 6 h, 45%, 99% ee. (R)-316, 40%, 93% ee. (S)-315; (ii) t-BuLi, THF, 2 h, −78 °C to rt; (iii) Et3SiH, CF3CO2H, CH2Cl2, 2 h, −78 °C to rt, 82% 317, 70%, ent-317, over two steps; (iv) CAN, MeCN–H2O, 10 min, rt, 72% 11, 75% ent-11, 74% 15; (v) t-BuLi, THF, 1 h, −78 °C, 76%. | ||
3.8 C–H olefination
In 2015, Thorson and coworkers published the first enantioselective total synthesis of griseusin A (330), griseusin C (329), and 4′-deacetylgriseusin A (29) using a novel C–H olefination strategy to construct the pyran ring of the pyranonaphthoquinone framework (Scheme 50).96 The substrate for the key C–H olefination (321) was prepared on a multigram scale in high yield, with a Sharpless asymmetric dihydroxylation employed to install chirality (97% ee). Enone 323 was synthesised in three steps in 69% yield from ethyl (R)-hydroxybutyrate (322). Following optimisation, the palladium-catalysed C–H olefination reaction of 321 with 323 gave a 48% yield of 324 and a 40% yield of desired naphthopyran 325. Compound 324 was readily converted to naphthopyran 325 in 45% yield. TBS deprotection and epoxidation followed by spirocyclisation gave the pentacyclic griseusin A core structure 326. Manipulation of the oxidation state at C-4′ and oxidative deprotection gave three pyranonaphthoquinone natural products: griseusin A (330), 4′-deacetylgriseusin A (29) and griseusin C (329).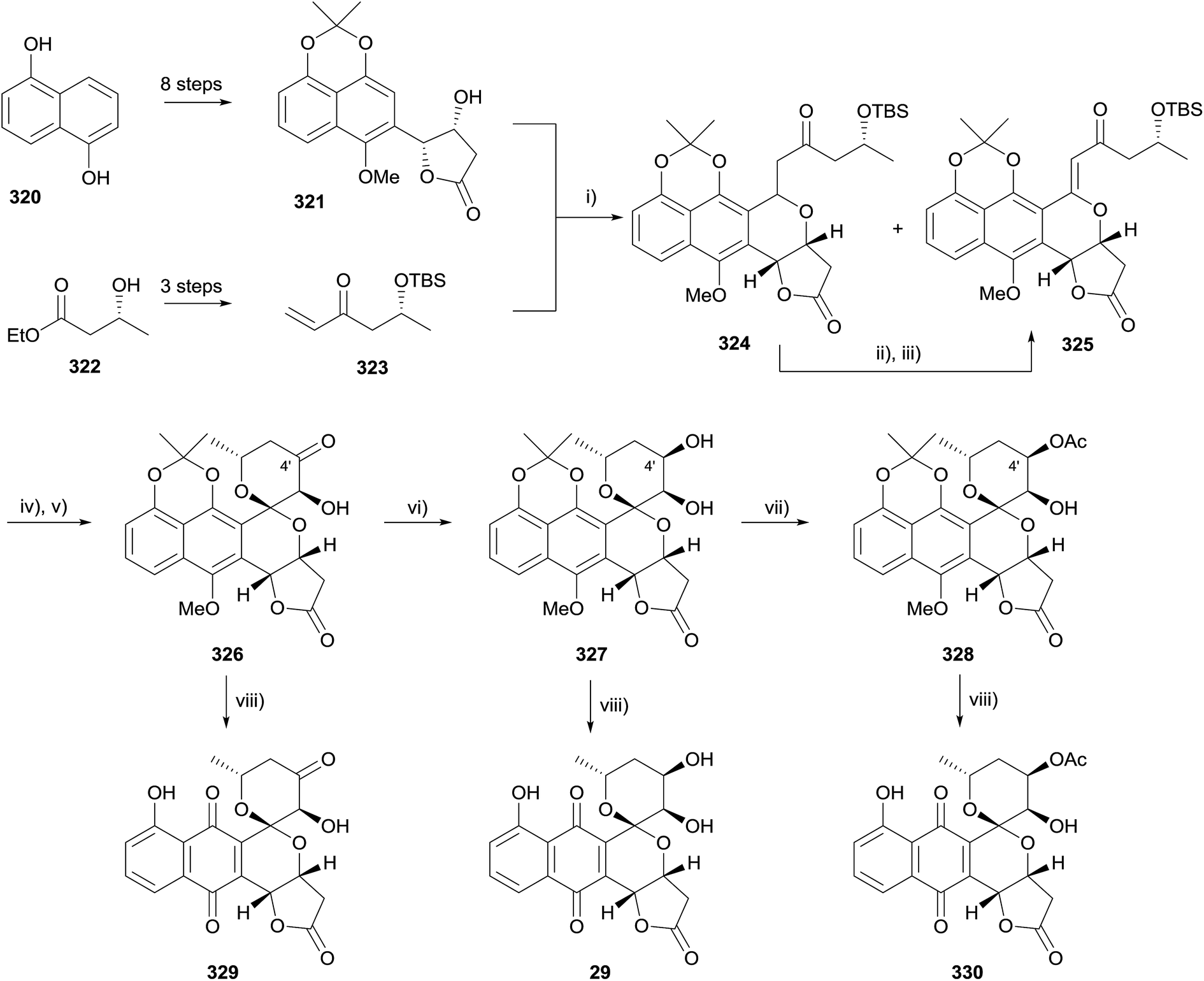 | ||
| Scheme 50 Reagents and conditions: (i) 20 mol% Pd(OAc)2, Li2CO3, Ag2CO3 on Celite®, DCE, 80 °C, 324 48%, 325, 40%; (ii) DBU, TESCl, CH2Cl2; (iii) Pd(OAc)2, O2, DMSO, 45%; (iv) HF·pyr, Et3N, dioxane, 85%; (v) TfOH, DMDO, CH2Cl2, −78 °C to 0 °C, 80%, >98:2 dr; (vi) LiBH4, THF, −78 °C, 85%; (vii) (C6H11)2NMe, Ac2O, CH2Cl2, 70%; (viii) AgO, HNO3, THF, 63% 329, 85% 29, 81% 330. | ||
3.9 Oxidation-cyclisation
De Koning and coworkers reported the synthesis of dehydroherbarin (47) and analogues using a Wacker oxidation to form the isochromene ring. The initial strategy aimed to oxidise naphthopyran 334 (Scheme 51).97 Key intermediate 332 was synthesised from known tetrasubstituted naphthalene 331 by allylation followed by Claisen rearrangement. Intermediate 332 was converted to alcohol 333 by protection of the phenol as a TBS ether and ester reduction. The key Wacker oxidation step proceeded in moderate yield using copper chloride and catalytic palladium chloride. Cyclisation of the resulting ketone and removal of the phenolic TBS group then gave naphthopyran 334. The final step required oxidation to the quinone; unfortunately, salcomine-mediated oxidation gave dicarbonyl 335, while CAN-mediated oxidation afforded hemiacetal 336. The formation of hemiacetal 336 was proposed to proceed through radical cation 337 (Scheme 51, insert).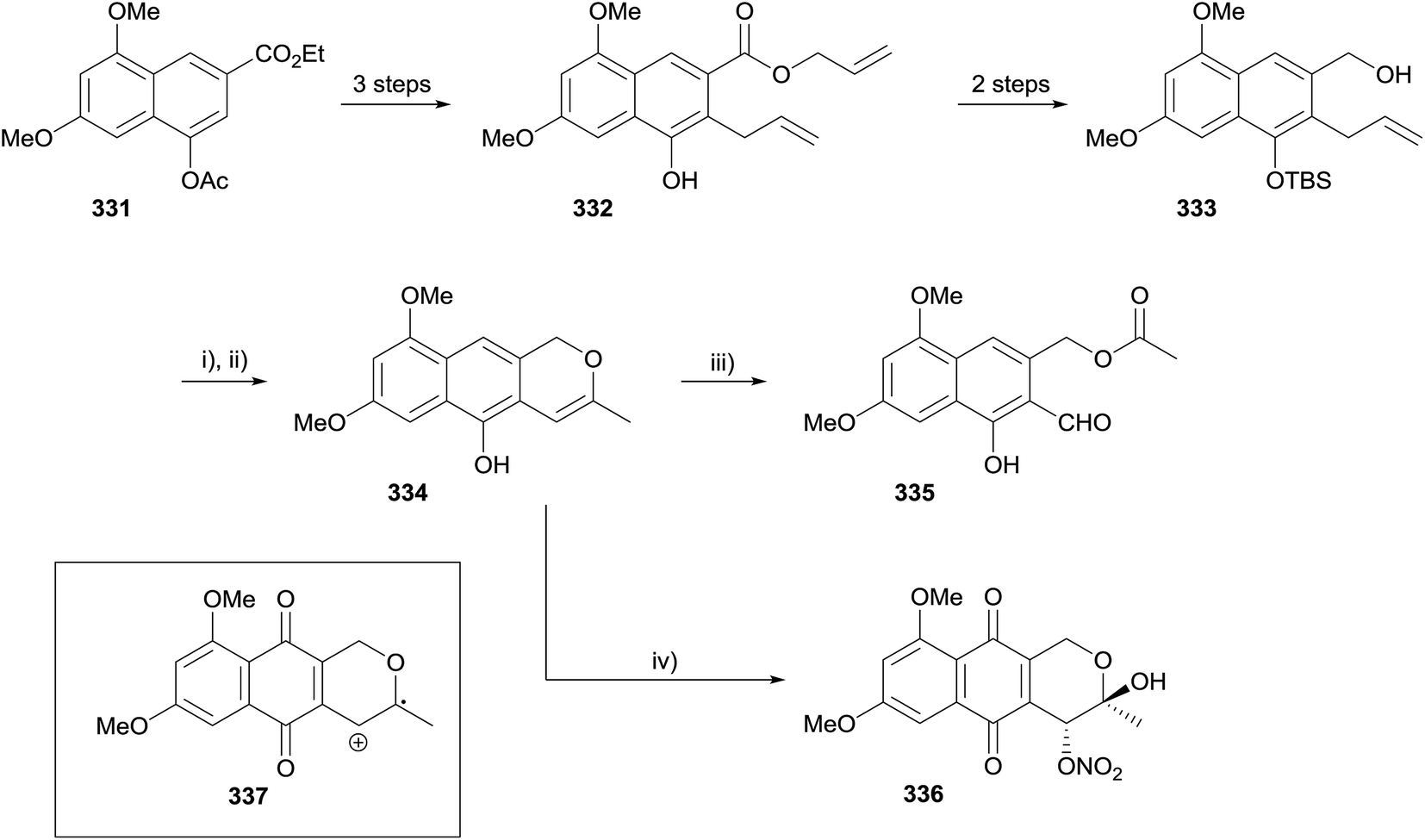 | ||
| Scheme 51 Reagents and conditions: (i) cat. PdCl2, CuCl2·2H2O, DMF–H2O, O2, rt, 24 h, 52%; (ii) TBAF, THF, rt, 18 h, 73%; (iii) salcomine, MeCN, O2 atm, rt, 24 h, 30%; (iv) CAN, MeCN–H2O, rt, 30 min, 54%. | ||
The synthetic method was then modified in order to install the correct substitution of the naphthalene ring at an earlier stage (Scheme 52). Oxidation of Claisen product 332 with PIFA, followed by aromatisation, protection and ester reduction gave naphthalene 338. Use of the same Wacker oxidation conditions as previously afforded naphthopyran 339. Oxidative demethylation with PIFA then gave the desired product, dehydroherbarin (47), albeit in a low 25% yield. This method was also applied to the synthesis of some non-pyranonaphthoquinone herbarin analogues.97
 | ||
| Scheme 52 Reagents and conditions: (i) cat. PdCl2, CuCl2·2H2O, DMF–H2O, O2, rt, 2 h, 63%; (ii) PIFA, MeCN–H2O, rt, 2 h, 25%. | ||
4. Conclusions
This review has provided a comprehensive overview of the isolation, biological activity and synthesis of pyranonaphthoquinones and other closely related compounds, updating and expanding the earlier reports.1,2 It is readily apparent that the pyranonaphthoquinone natural products remain a fertile ground for the development of new synthetic methodology. Several significant advances have been made since our previous review in 2008, in particular towards structurally complex targets such as the griseusins and asymmetric syntheses of several dimeric pyranonaphthoquinones. Several new pyranonaphthoquinones have been reported, with the discovery of the structurally unprecedented yoropyrazone and xiakemyicin A, along with the expansion of the frenolicin and griseusin family, of particular note. The ever expanding array of biological properties displayed by this natural product family provides medicinal chemists with a plethora of potential lead compounds for the development of novel therapeutics for a variety of diseases.5. Abbreviations
| ABCN | Azobiscyclohexanecarbonitrile |
| AIBN | Azobisisobutyronitrile |
| BOP | (Benzotriazol-1-yloxy)tris(dimethylamino)phosphonium hexafluorophosphate |
| CAN | Cerium ammonium nitrate |
| CoA | Coenzyme A |
| COD | 1,5-Cyclooctadiene |
| DCE | Dichloroethane |
| DHQD | Dihydroquinidine |
| DIAD | Diisopropyl azodicarboxylate |
| DIPEA | N,N-Diisopropylethylamine |
| DMAP | N,N-Dimethylaminopyridine |
| DMDO | Dimethydioxirane |
| DMF | N,N-Dimethylformamide |
| DMPU | 1,3-Dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone |
| DNPA | 4-Dihydro-9-hydroxy-1-methyl-10-oxo-3H-naphtho[2,3-c]pyran-3-acetic acid |
| DMSO | Dimethylsulfoxide |
| dtbpy | 4,4′-Di-tert-butyl-2,2′-bipyridiyl |
| IBX | 2-Iodoxybenzoic acid |
| MOM | Methoxymethyl ether |
| NDP | Nucleoside diphosphate |
| NMM | N-Methylmorpholine |
| PHAL | Phthalazine |
| PIFA | [Bis(trifluoroacetoxy)iodo]benzene |
| pin | Pinacolato |
| PTSA | para-Toluenesulfonic acid |
| pyr | Pyridine |
| SAR | Structure–activity relationship |
| TBABr | Tetrabutylammonium bromide |
| TBAF | Tetrabutylammonium fluoride |
| TBAI | Tetrabutylammonium iodide |
| TBDPS | tert-Butyldiphenylsilyl |
| TBS | tert-Butyldimethylsilyl |
| TEMPO | (2,2,6,6-Tetramethyl-piperidin-1-yl)oxyl |
| TES | Triethylsilyl |
| Tf | Trifluoromethylsulfonyl |
| TFA | Trifluoroacetic acid |
| THF | Tetrahydrofuran |
| TMEDA | Tetramethylethylenediamine |
| TMS | Trimethylsilyl |
| TMTU | N,N,N′,N′-Tetramethyl thiourea |
6. References
- M. A. Brimble, L. J. Duncalf and M. R. Nairn, Nat. Prod. Rep., 1999, 16, 267 RSC.
- J. Sperry, P. Bachu and M. A. Brimble, Nat. Prod. Rep., 2008, 25, 376 RSC.
- R. A. Fernandes, P. H. Patil and D. A. Chaudhari, Eur. J. Org. Chem., 2016 DOI:10.1002/ejoc.201600544.
- B. Hosamani, M. F. Ribeiro, E. N. da Silva Júnior and I. N. N. Namboothiri, Org. Biomol. Chem., 2016, 14, 6913 CAS.
- T. Opatz, H. Kolshorn, E. Thines and H. Anke, J. Nat. Prod., 2008, 71, 1973 CrossRef CAS PubMed.
- A. Schüffler, J. C. Liermann, H. Kolshorn, T. Opatz and H. Anke, Z. Naturforsch., C: J. Biosci., 2014, 64, 25 Search PubMed.
- L. Wang, J. Y. Dong, H.-C. Song, K. Z. Shen, L. M. Wang, R. Sun, C. R. Wang, Y. X. Gao, G. H. Li, L. Li and K. Q. Zhang, Planta Med., 2009, 75, 1339 CrossRef CAS PubMed.
- J. Sperry, I. Lorenzo-Castrillejo, M. A. Brimble and F. Machín, Bioorg. Med. Chem., 2009, 17, 7131 CrossRef CAS PubMed.
- S. Cao and J. Clardy, Tetrahedron Lett., 2011, 52, 2206 CrossRef CAS PubMed.
- M. S. Abdelfattah, T. Kazufumi and M. Ishibashi, J. Antibiot., 2011, 64, 729 CrossRef CAS PubMed.
- J. T. Fitzgerald, C. P. Ridley and C. Khosla, J. Antibiot., 2011, 64, 759 CrossRef CAS PubMed.
- H. Brockmann and H. Pini, Naturwissenschaften, 1947, 34, 190 CrossRef CAS PubMed.
- H. Brockmann, A. Zeeck, K. van der Merwe and W. Müller, Justus Liebigs Ann. Chem., 1966, 698, 209 CrossRef CAS.
- J.-H. Shin, A. K. Singh, D.-J. Cheon and J.-H. Roe, J. Bacteriol., 2011, 193, 75 CrossRef CAS PubMed.
- P. B. Tessele, F. D. Monache, N. L. M. Quintão, G. F. da Silva, L. W. Rocha, G. M. R. S. Lucena, V. M. M. Ferreira, R. D. S. Prediger and V. C. Filho, Planta Med., 2011, 77, 1035 CrossRef CAS PubMed.
- A. Campos, D. B. Vendramini-Costa, G. F. Fiorito, A. L. T. G. Ruiz, J. E. de Carvalho, G. M. R. de Souza, F. Delle-Monache and V. C. Filho, Pharm. Biol., 2016, 54, 1022 CrossRef CAS PubMed.
- W.-L. Geng, X.-Y. Wang, T. Kurtán, A. Mándi, H. Tang, B. Schulz, P. Sun and W. Zhang, J. Nat. Prod., 2012, 75, 1828 CrossRef CAS PubMed.
- T. Taguchi, T. Ebihara, A. Furukawa, Y. Hidaka, R. Ariga, S. Okamoto and K. Ichinose, Bioorg. Med. Chem. Lett., 2012, 22, 5041 CrossRef CAS PubMed.
- M. Endale, J. P. Alao, H. M. Akala, N. K. Rono, F. L. Eyase, S. Derese, A. Ndakala, M. Mbugua, D. S. Walsh, P. Sunnerhagen, M. Erdelyi and A. Yenesew, Planta Med., 2012, 78, 31 CrossRef CAS PubMed.
- R. L. Fabri, R. M. Grazul, L. O. de Carvalho, E. S. Coimbra, G. M. M. Cardoso, E. M. de Souza-Fagundes, A. D. da Silva and E. Scio, An. Acad. Bras. Cienc., 2012, 84, 1081 CrossRef CAS PubMed.
- M. S. Abdelfattah, K. Toume and M. Ishibashi, J. Antibiot., 2012, 65, 245 CrossRef CAS PubMed.
- Z.-G. Ding, J.-Y. Zhao, M.-G. Li, R. Huang, Q.-M. Li, X.-L. Cui, H.-J. Zhu and M.-L. Wen, J. Nat. Prod., 2012, 75, 1994 CrossRef CAS PubMed.
- K. Nakagawa, Y. Hiraoka and N. Imamura, J. Antibiot., 2013, 66, 295 CrossRef CAS PubMed.
- Z. Yang, J. Ding, K. Ding, D. Chen, S. Cen and M. Ge, Phytochem. Lett., 2013, 6, 257 CrossRef CAS.
- X. Wang, K. A. Shaaban, S. I. Elshahawi, L. V. Ponomareva, M. Sunkara, Y. Zhang, G. C. Copley, J. C. Hower, A. J. Morris, M. K. Kharel and J. S. Thorson, J. Nat. Prod., 2013, 76, 1441 CrossRef CAS PubMed.
- D. J. A. Bridewell, J. Sperry, J. R. Smith, P. Kosim-Satyaputra, L.-M. Ching, J. F. Jamie and M. A. Brimble, Aust. J. Chem., 2013, 66, 40 CrossRef CAS.
- B. Jiang, S. Li, W. Zhao, T. Li, L. Zuo, Y. Nan, L. Wu, H. Liu, L. Yu, G. Shan and L. Zuo, J. Nat. Prod., 2014, 77, 2130 CrossRef CAS PubMed.
- E. Stodůlková, P. Man, M. Kuzma, J. Černý, I. Císařová, A. Kubátová, M. Chudíčková, M. Kolařík and M. Flieger, Folia Microbiol., 2015, 60, 259 CrossRef PubMed.
- J. Sperry, T. Y. Yuen and M. A. Brimble, Synthesis, 2009, 2561 CAS.
- S. Saepua, J. Kornsakulkarn, W. Choowong, S. Supothina and C. Thongpanchang, Tetrahedron, 2015, 71, 2400 CrossRef CAS.
- Z.-K. Jiang, L. Guo, C. Chen, S.-W. Liu, L. Zhang, S.-J. Dai, Q.-Y. He, X.-F. You, X.-X. Hu, L. Tuo, W. Jiang and C.-H. Sun, J. Antibiot., 2015, 68, 771 CrossRef CAS PubMed.
- J. Lü, Q. He, L. Huang, X. Cai, W. Guo, J. He, L. Zhang and A. Li, PLoS One, 2015, 10, e0117690 Search PubMed.
- R. T. Sawant and S. B. Waghmode, Tetrahedron, 2009, 65, 1599 CrossRef CAS.
- C. D. Donner and M. Gill, J. Chem. Soc., Perkin Trans. 1, 2002, 938 RSC.
- R. T. Sawant, S. G. Jadhav and S. B. Waghmode, Eur. J. Org. Chem., 2010, 4442 CAS.
- T. Kometani and E. Yoshii, J. Chem. Soc., Perkin Trans. 1, 1981, 1191 RSC.
- C. N. Eid, J. Shim, J. Bikker and M. Lin, J. Org. Chem., 2009, 74, 423 CrossRef CAS PubMed.
- E. J. Salaski, G. Krishnamurthy, W.-D. Ding, K. Yu, S. S. Insaf, C. Eid, J. Shim, J. I. Levin, K. Tabei, L. Toral-Barza, W.-G. Zhang, L. A. McDonald, E. Honores, C. Hanna, A. Yamashita, B. Johnson, Z. Li, L. Laakso, D. Powell and T. S. Mansour, J. Med. Chem., 2009, 52, 2181 CrossRef CAS PubMed.
- N. Tanaka, T. Okabe, F. Isono, M. Kashiwagi, K. Nomoto, M. Takahashi, A. Shimazu and T. Nishimura, J. Antibiot., 1985, 38, 1327 CrossRef CAS PubMed.
- L. E. Johnson and A. Dietz, Appl. Microbiol., 1968, 16, 1815 CAS.
- Y. Iwai, A. Kora, Y. Takahashi, T. Hayashi, J. Awaya, R. Masuma, R. Oiwa and S. Omura, J. Antibiot., 1978, 31, 959 CrossRef CAS PubMed.
- M. A. Brimble and M. R. Nairn, Perkin 1, 2000, 317 RSC.
- H. Moore, Science, 1977, 197, 527 CAS.
- H. W. Moore and R. Czerniak, Med. Res. Rev., 1981, 1, 249 CrossRef CAS PubMed.
- P. A. Hume, M. A. Brimble and J. Reynisson, Aust. J. Chem., 2012, 65, 402 CAS.
- S. Korwar, T. Nguyen and K. C. Ellis, Bioorg. Med. Chem. Lett., 2014, 24, 271 CrossRef CAS PubMed.
- R. A. Fernandes and S. V. Mulay, Synlett, 2010, 2667 CrossRef CAS.
- R. A. Fernandes and S. V. Mulay, J. Org. Chem., 2010, 75, 7029 CrossRef CAS PubMed.
- R. A. Fernandes, V. P. Chavan, S. V. Mulay and A. Manchoju, J. Org. Chem., 2012, 77, 10455 CrossRef CAS PubMed.
- R. A. Fernandes and R. Brückner, Synlett, 2005, 1281 CAS.
- R. A. Fernandes and S. V. Mulay, Tetrahedron: Asymmetry, 2013, 24, 1281 CrossRef CAS.
- S. V. Mulay, S. P. Gholap and R. A. Fernandes, Asian J. Org. Chem., 2015, 4, 560 CrossRef CAS.
- C. D. Donner and M. Gill, Tetrahedron Lett., 1999, 40, 3921 CrossRef CAS.
- S. V. Mulay and R. A. Fernandes, Chem.–Eur. J., 2015, 21, 4842 CrossRef CAS PubMed.
- S. V. Mulay, A. Bhowmik and R. A. Fernandes, Eur. J. Org. Chem., 2015, 4931 CrossRef CAS.
- Y. Zhang, X. Wang, M. Sunkara, Q. Ye, L. V. Ponomereva, Q.-B. She, A. J. Morris and J. S. Thorson, Org. Lett., 2013, 15, 5566 CrossRef CAS PubMed.
- K. Schünemann, D. P. Furkert, S. Connelly, J. D. Fraser, J. Sperry and M. A. Brimble, Eur. J. Org. Chem., 2014, 122 CrossRef.
- K. Schünemann, D. P. Furkert, S. Connelly, J. D. Fraser, J. Sperry and M. A. Brimble, Synlett, 2014, 25, 556 CrossRef.
- R. A. Limaye, A. D. Natu and M. V. Paradkar, Synth. Commun., 2014, 44, 2503 CrossRef CAS.
- V. J. Bulbule, P. S. Koranne, Y. S. Munot, H. B. Borate and V. H. Deshpande, Synth. Commun., 2003, 33, 587 CrossRef CAS.
- K. Rathwell and M. A. Brimble, Synthesis, 2007, 643 CAS.
- D. Mal and P. Pahari, Chem. Rev., 2007, 107, 1892 CrossRef CAS PubMed.
- R. Karmakar, P. Pahari and D. Mal, Chem. Rev., 2014, 114, 6213 CrossRef CAS PubMed.
- O. Andrey, J. Sperry, U. S. Larsen and M. A. Brimble, Tetrahedron, 2008, 64, 3912 CrossRef CAS.
- M. A. Brimble, J. S. Gibson, J. J. P. Sejberg and J. Sperry, Synlett, 2008, 867 CrossRef CAS.
- J. Sperry, J. S. Gibson, J. J. P. Sejberg and M. A. Brimble, Org. Biomol. Chem., 2008, 6, 4261 CAS.
- J. Sperry, J. J. P. Sejberg, F. M. Stiemke and M. A. Brimble, Org. Biomol. Chem., 2009, 7, 2599 CAS.
- K. Rathwell, J. Sperry and M. A. Brimble, Tetrahedron, 2010, 66, 4002 CrossRef CAS.
- A. M. Heapy, A. V. Patterson, J. B. Smaill, S. M. F. Jamieson, C. P. Guise, J. Sperry, P. A. Hume, K. Rathwell and M. A. Brimble, Bioorg. Med. Chem., 2013, 21, 7971 CrossRef CAS PubMed.
- S. Kongsriprapan, C. Kuhakarn, P. Deelertpaiboon, K. Panthong, P. Tuchinda, M. Pohmakotr and V. Reutrakul, Pure Appl. Chem., 2012, 84, 1435 CrossRef CAS.
- B. J. Naysmith and M. A. Brimble, Org. Lett., 2013, 15, 2006 CrossRef CAS PubMed.
- B. J. Naysmith, D. Furkert and M. A. Brimble, Tetrahedron, 2014, 70, 1199 CrossRef CAS.
- N. P. S. Hassan, B. J. Naysmith, J. Sperry and M. A. Brimble, Tetrahedron, 2015, 71, 7137 CrossRef CAS.
- R. A. Fernandes and R. Brückner, Synlett, 2005, 1281 CAS.
- M. A. Brimble, N. P. S. Hassan, B. J. Naysmith and J. Sperry, J. Org. Chem., 2014, 79, 7169 CrossRef CAS PubMed.
- P. A. Hume, J. Sperry and M. A. Brimble, Org. Biomol. Chem., 2011, 9, 5423 CAS.
- T. A. Dang Thi, T. H. Vu Thi, H. Thi Phuong, T. Ha Nguyen, C. Pham The, C. Vu Duc, Y. Depetter, T. Van Nguyen and M. D’hooghe, Bioorg. Med. Chem. Lett., 2015, 25, 3355 CrossRef CAS PubMed.
- C. D. Donner, Tetrahedron, 2013, 69, 3747 CrossRef CAS.
- J. Sperry and M. A. Brimble, Synlett, 2008, 1910 CAS.
- A. D. Wadsworth, J. Sperry and M. A. Brimble, Synthesis, 2010, 2604 CAS.
- C. D. Donner, Tetrahedron, 2013, 69, 377 CrossRef CAS.
- Z. Li, Y. Gao, Y. Tang, M. Dai, G. Wang, Z. Wang and Z. Yang, Org. Lett., 2008, 10, 3017 CrossRef CAS PubMed.
- Y. Cui, H. Jiang, Z. Li, N. Wu, Z. Yang and J. Quan, Org. Lett., 2009, 11, 4628 CrossRef CAS PubMed.
- C. D. Donner, Synthesis, 2010, 415 CrossRef CAS.
- G. A. Kraus, J. Li, M. S. Gordon and J. H. Jensen, J. Org. Chem., 1995, 60, 1154 CrossRef CAS.
- C. D. Donner and M. I. Casana, Tetrahedron Lett., 2012, 53, 1105 CrossRef CAS.
- J. Young Jeong, J. Sperry, J. A. Taylor and M. A. Brimble, Eur. J. Med. Chem., 2014, 87, 220 CrossRef CAS PubMed.
- L. J. Duffy, J. Garcia-Torres, R. C. F. Jones and S. M. Allin, Synlett, 2012, 23, 1821 CrossRef CAS.
- L. J. Duffy, J. Garcia-Torres, R. C. F. Jones, M. R. J. Elsegood and S. M. Allin, Synlett, 2013, 24, 185 CAS.
- P. Bachu, J. Sperry and M. A. Brimble, Tetrahedron, 2008, 64, 3343 CrossRef CAS.
- H. C. Brown and P. K. Jadhav, J. Am. Chem. Soc., 1983, 105, 2092 CrossRef CAS.
- H. C. Brown and K. S. Bhat, J. Am. Chem. Soc., 1986, 108, 5919 CrossRef CAS PubMed.
- H. C. Brown, P. K. Jadhav and K. S. Bhat, J. Am. Chem. Soc., 1988, 110, 1535 CrossRef CAS.
- P. Bachu, J. S. Gibson, J. Sperry and M. A. Brimble, Tetrahedron: Asymmetry, 2007, 18, 1618 CrossRef CAS.
- P. Bachu, J. Sperry and M. A. Brimble, Tetrahedron, 2008, 64, 4827 CrossRef CAS.
- Y. Zhang, Q. Ye, X. Wang, Q.-B. She and J. S. Thorson, Angew. Chem., Int. Ed., 2015, 54, 11219 CrossRef CAS PubMed.
- A. Pillay, A. L. Rousseau, M. A. Fernandes and C. B. de Koning, Org. Biomol. Chem., 2012, 10, 7809 CAS.
| This journal is © The Royal Society of Chemistry 2017 |