
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Luminescent coordination polymers based on Ca2+ and octahedral cluster anions [{M6Cli8}Cla6]2− (M = Mo, W): synthesis and thermal stability studies†
Darya V.
Evtushok
ab,
Natalya A.
Vorotnikova
 ab,
Vladimir A.
Logvinenko
ac,
Anton I.
Smolentsev
ac,
Konstantin A.
Brylev
abc,
Pavel E.
Plyusnin
ac,
Denis P.
Pishchur
a,
Noboru
Kitamura
d,
Yuri V.
Mironov
abc,
Anastasiya O.
Solovieva
b,
Olga A.
Efremova
*e and
Michael A.
Shestopalov
*abc
ab,
Vladimir A.
Logvinenko
ac,
Anton I.
Smolentsev
ac,
Konstantin A.
Brylev
abc,
Pavel E.
Plyusnin
ac,
Denis P.
Pishchur
a,
Noboru
Kitamura
d,
Yuri V.
Mironov
abc,
Anastasiya O.
Solovieva
b,
Olga A.
Efremova
*e and
Michael A.
Shestopalov
*abc
aNikolaev Institute of Inorganic Chemistry SB RAS, 3 Acad. Lavrentiev Prospect, 630090 Novosibirsk, Russia. E-mail: shtopy@niic.nsc.ru; Tel: +73833309253
bResearch Institute of Clinical and Experimental Lymphology – Branch of the ICG SB RAS, 2 Timakova Str., 630060 Novosibirsk, Russia
cNovosibirsk State University, 2 Pirogova st., 630090 Novosibirsk, Russia
dDepartment of Chemistry, Faculty of Science, Hokkaido University, 060-0810 Sapporo, Japan
eSchool of Mathematics and Physical Sciences, University of Hull, Cottingham Road, HU6 7RX, Hull, UK. E-mail: o.efremova@hull.ac.uk; Tel: +44 (0)1482 465417
First published on 13th October 2017
Abstract
Luminescent coordination polymers (CPs) based on inexpensive stable precursors are attractive materials for applications. Here we report the synthesis and evaluation of the stability and photophysical characteristics of the first examples of phosphorescent CPs based on octahedral molybdenum and tungsten cluster anions. In particular, 1D CP trans-[{Ca(OPPh3)4}{{M6Cli8}Cla6}]∞ (M = Mo, W) can be obtained either directly at increased temperature or via intermediate phases [cis-Ca(OPPh3)4(H2O)2][{M6Cli8}Cla6]·2CH3CN that are stable at room-temperature, but convert to the titled CPs at temperatures above 100 °C.
Introduction
Coordination polymers (CPs) are a versatile class of compounds researched worldwide due to their potential in many areas including gas storage, sensing, photonics, electronics, drug delivery, etc. These compounds are typically assembled from simple building units – metal cations or cationic complexes and bridging ligands, where both the nature of the building units and the final structure determine the properties of the CPs. The predominant majority of the CPs currently developed employ organic polydentate ligands as linkers between the metal cationic centres, while those that employ inorganic linkers are relatively scarce, due to the limited number of such linkers. In the past two decades some members of our team and other groups have succeeded in employing a range of inorganic metal clusters as such linkers to develop metal-cluster based CPs that demonstrated properties such as homochirality,1,2 high porosity,3–5 ion conductivity,6 luminescence7 and magnetism.8–10For the development of luminescent materials octahedral cluster anions of molybdenum and tungsten of the general formula [{M6Xi8}La6]2− (where M is Mo or W; X is an inner halogen ligand; L is an apical inorganic or organic apical ligand) are extremely attractive building blocks. Indeed, depending on X and L, some members of this family demonstrate intensive luminescence in the red/near-infrared region with emission lifetimes of hundreds of microseconds and often with significant quantum yields.11–17 Due to these properties, materials based on these clusters have been demonstrated to have potential in solar energy harvesting,18,19 bioimaging, photodynamic therapy,20–22 photo-catalysis,23–27 oxygen sensing,28–31 lighting,32–34etc.
In order to work as a linker with another metal centre, the apical ligands L of a metal cluster should be ambidentate. On the other hand, in order to develop a photoluminescent material based on a CP of a metal cluster, the cluster should not only be highly luminescent, but ideally also be stable in the presence of water and upon heating. Unfortunately, among the known molybdenum and tungsten anions there are only a handful of them that satisfy all of the above criteria and [{M6Xi8}Xa6]2− (M is Mo or W; X is a halogen atom) are among them. Indeed these cluster anions are well-known for their luminescence properties35 and they are stable over a wide temperature range and in aqueous solution.29
The choice of the cationic part and any of the supporting ligands for a CP should be driven by their coordination ability, electronic state that would not quench the luminescence of the cluster, transparency in the visible region of the spectra, and cost. Therefore, here we present the synthesis, crystal structures, stability studies and luminescence properties of new cluster compounds based on the cationic complexes of calcium coordinated by triphenylphosphine oxide (OPPh3) and cluster anions [{M6Cli8}Cla6]2−. Namely, ionic compounds [cis-Ca(OPPh3)4(H2O)2][{M6Cli8}Cla6]·2CH3CN, where M is Mo (1) or W (2), were obtained by direct crystallisation from water/acetonitrile solution. Upon heating, these compounds converted into 1D CPs trans-[{Ca(OPPh3)4}{{M6Cli8}Cla6}]∞ (M = Mo (3), W (4)). Compounds 3 and 4 were also obtained directly by crystallisation at high temperature.
Experimental
Materials and methods
Cluster compound (H3O)2[{Mo6Cli8}Cla6]·6H2O was prepared as described in ref. 36, while (H3O)2[{W6Cli8}Cla6]·7H2O was obtained according to ref. 37. All other reagents and solvents were purchased and used without further purification. The CHNS elemental analyses were performed on a EuroVector EA3000 Elemental Analyser. Energy dispersive spectroscopy (EDS) was performed on a Hitachi TM3000 Tabletop SEM with Bruker QUANTAX 70 EDS equipment. Infrared spectra were measured on KBr pellets with a Scimitar FTS 2000 spectrometer. X-ray powder diffraction data were collected on a Philips PW 1700 diffractometer using CuKα radiation (λ = 1.5406 Å) and a graphite monochromator. Optical diffuse reflectance spectra were measured at room temperature on a Shimadzu UV-Vis-NIR 3101 PC spectrophotometer (Shimadzu Corporation, Kyoto, Japan) equipped with an integrating sphere and reproduced in the form of Kubelka–Munk theory.For emission measurements, powdered samples were placed between two non-fluorescent glass plates. The measurements were carried out at 298 K. The samples were excited by 355 nm laser pulses (6 ns duration, LOTIS TII, LS-2137/3). Corrected emission spectra were recorded on a red-light-sensitive multichannel photodetector (Hamamatsu Photonics, PMA-11). For emission decay measurements, the emission was analysed using a streakscope system (Hamamatsu Photonics, C4334 and C5094). The emission quantum yields were determined using an Absolute Photo-Luminescence Quantum Yield Measurement System (Hamamatsu Photonics, C9920-03), which comprised an excitation xenon light source (the excitation wavelength was set at 400 nm), an integrating sphere, and a red-sensitive multichannel photodetector (Hamamatsu Photonics, PMA-12).
Synthetic procedures
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4). After that (H3O)2[{M6Cli8}Cla6]·nH2O (0.14 mmol, M = Mo or W) was added to the reaction mixture and stirred for 5 min. The yellow solution was slowly cooled to room temperature and was left to stand overnight in air for crystallisation. Yellow crystals of [cis-Ca(OPPh3)4(H2O)2][{M6Cli8}Cla6]·2CH3CN were collected by filtration. Single crystals for X-ray structural analysis were separated manually from the precipitate (see the ESI,† Table S1).
:Ca:Cl:P ratio of 6:1.1:13.6:3.9 for C76H70CaCl14Mo6N2O6P4. IR (KBr, cm−1): 539s, 693s, 719s, 995m, 1118s (νPO), 1167s, 1434s, 1652m, 2260w (νCN), 3053m, 3495m (νH2O) (Fig. S1, ESI†).
:Ca:Cl:P ratio of 6:1.1:14.2:3.9 for C76H70CaCl14N2O6P4W6. IR (KBr, cm−1): 537s, 696s, 721s, 995m, 1114s (νPO), 1169s, 1434s, 1650m, 2263w (νCN), 3052m, 3501m (νH2O) (Fig. S2, ESI†).
:4) for 30 min at 60 °C, which resulted in the formation of a yellow crystalline precipitate. Yield: 272 mg (99%). Single crystals for X-ray structural analysis were separated manually from the precipitate (Table S1, ESI†).
:Ca:Cl:P ratio of 6:0.9:14.0:4.1 for C72H60CaCl14Mo6O4P4. IR (KBr, cm−1): 539s, 689s, 724s, 995m, 1122s (νPO), 1178s, 1438s, 3449m (νH2O) (Fig. S1, ESI†).
:Ca:Cl:P ratio of 6:1.1:14.3:3.9 for C72H60CaCl14Mo6O4P4. IR (KBr, cm−1): 544s, 688s, 724s, 995m, 1120s (νPO), 1175s, 1431s, 3439m (νH2O) (Fig. S2, ESI†).
:4). After that (H3O)2[{M6Cli8}Cla6]·nH2O (0.14 mmol, M = Mo or W) was added to the reaction mixture and stirred for 5 min. The yellow solution was slowly cooled to room temperature and was left to stand overnight in air for crystallisation. Yellow crystals of [cis-Ca(OPPh3)4(H2O)2][{M6Cli8}Cla6]·2CH3CN were collected by filtration. Single crystals for X-ray structural analysis were separated manually from the precipitate (see the ESI,† Table S1).
:Ca:Cl:P ratio of 6:1.1:13.6:3.9 for C76H70CaCl14Mo6N2O6P4. IR (KBr, cm−1): 539s, 693s, 719s, 995m, 1118s (νPO), 1167s, 1434s, 1652m, 2260w (νCN), 3053m, 3495m (νH2O) (Fig. S1, ESI†).
:Ca:Cl:P ratio of 6:1.1:14.2:3.9 for C76H70CaCl14N2O6P4W6. IR (KBr, cm−1): 537s, 696s, 721s, 995m, 1114s (νPO), 1169s, 1434s, 1650m, 2263w (νCN), 3052m, 3501m (νH2O) (Fig. S2, ESI†).
:4) for 30 min at 60 °C, which resulted in the formation of a yellow crystalline precipitate. Yield: 272 mg (99%). Single crystals for X-ray structural analysis were separated manually from the precipitate (Table S1, ESI†).
:Ca:Cl:P ratio of 6:0.9:14.0:4.1 for C72H60CaCl14Mo6O4P4. IR (KBr, cm−1): 539s, 689s, 724s, 995m, 1122s (νPO), 1178s, 1438s, 3449m (νH2O) (Fig. S1, ESI†).
:Ca:Cl:P ratio of 6:1.1:14.3:3.9 for C72H60CaCl14Mo6O4P4. IR (KBr, cm−1): 544s, 688s, 724s, 995m, 1120s (νPO), 1175s, 1431s, 3439m (νH2O) (Fig. S2, ESI†).
X-ray crystal structure analysis
Single-crystal X-ray diffraction data were collected at 150 K on a Bruker Nonius X8 Apex 4K CCD diffractometer with graphite monochromatised MoKα radiation (λ = 0.71073 Å). All crystallographic information is summarised in Table S1 (ESI†). Absorption corrections were made semi-empirically using the SADABS program.38 The structures were solved using direct methods and refined using the full-matrix least-squares method using the SHELXTL program package.38 All non-hydrogen atoms were refined anisotropically. The H atoms of the phenyl groups of OPPh3 ligands and the methyl groups of solvate acetonitrile molecules were positioned geometrically and refined by using a riding model. The H atoms of coordinated water molecules were located from difference Fourier maps and refined in an isotropic approximation with fixed Uiso = 0.05 Å2. The main crystallographic data and structure refinement results are presented in Table S1 (ESI†). The selected bond lengths are listed in Table S2 (ESI†). The atomic positional and thermal parameters, and full lists of bond lengths and angles were deposited at CCDC 1565898–1565900 for compounds 1, 2 and 4, respectively.Thermal analysis
Differential scanning calorimetry (DSC) measurements were performed using a heat flow measurement method on a Netzsch DSC 204 F1 Phoenix calorimeter over the 20–150 °C temperature range, at a heating rate of 6 °C min−1, in an Ar flow of 25 mL min−1. Simultaneous TG-DSC/EGA-MS measurements were performed in an apparatus consisting of an STA 449 F1 Jupiter thermal analyser and a QMS 403D Aëolos quadrupole mass spectrometer (NETZSCH, Germany). The spectrometer was connected online to a thermal analyser (STA) instrument by a quartz capillary heated to 280 °C. The QMS was operated with an electron impact ioniser with an energy of 70 eV. The ion currents of the selected mass/charge (m/z) numbers were monitored in multiple ion detection (MID) mode with a collection time of 1 s for each channel. The measurements were made in a helium flow in the 30–500 °C temperature range at the heating rate of 10 °C min−1, the gas flow rate of 30 mL min−1, in open Al2O3 crucibles. Thermogravimetric analyses (TGA) and kinetic experiments were carried out on a TG 209 F1 Iris thermobalance (NETZSCH, Germany). The measurements were made in a helium flow in the 30–800 °C temperature range at the heating rates of 5, 10 and 20 °C min−1, the gas flow rate of 60 mL min−1, in open Al2O3 crucibles. The sample masses were in the 10–15 mg range.Thermogravimetric curves obtained with different heating rates were processed with the use of the ‘Model-free’ module of Netzsch Thermokinetics 2 (Version 2004.05) software to calculate the activation energy without preliminary information about the kinetic topochemical equation. The Friedman method was used to calculate the activation energies for each experimental point of fractional conversion in the 0.005 < α < 0.995 range. The same set of experimental data was then used to search for the corresponding topochemical equation. The selection was made from 16 equations: chemical reaction at the interface, nucleation and diffusion. For the initial calculation, a differential method with linear regression was used to calculate kinetic parameters, which were then refined using the nonlinear regression method to allow the kinetic analysis of multistage thermal decomposition reactions. The F test was used to search for the best kinetic description and for the statistical control of the obtained equation.
Results and discussion
Syntheses and characterisation
The interaction of the cluster (H3O)2[{M6Cli8}Cla6] (M = Mo, W) with CaCl2 and OPPh3 in a water/acetonitrile solution at room temperature produces ionic compounds [cis-Ca(OPPh3)4(H2O)2][{M6Cli8}Cla6]·2CH3CN, where M = Mo (1) and W (2), in good yields. When the same reactions were carried out at 50 °C or above the CP cluster compounds trans-[{Ca(OPPh3)4}{{M6Cli8}Cla6}]∞ (M = Mo (3), W (4)) were formed. Upon heating at 110 °C for 10 min, 1 and 2 quantitatively converted into 3 and 4, respectively. Notably, in an earlier study a similar reaction in excess of OPPh3 under hydrothermal conditions led to [Ca(OPPh3)5][{Mo6Cli8}Cla6]·OPPh3 as a by-product (Scheme 1).39 | ||
| Scheme 1 | ||
Compounds 1, 2 and 4 were characterised using single-crystal X-ray diffraction analysis and the details of the experiment are summarised in Table S1, ESI.† Note that the crystal structure and the direct synthesis of compound 3 in boiling solvent were described earlier.36
Compounds 1 and 2 are isostructural and have ionic structures (monoclinic symmetry, space group: C2/c, Z = 4) composed of the discrete cis-[Ca(OPPh3)4(H2O)2]2+ cationic units and molybdenum or tungsten cluster anions [{M6Cli8}Cla6]2−, respectively. The central calcium atom of the complex cation is located on the 2-fold axis (0, y, ¼), while the centre of the cluster coincides with the centrosymmetric special position (0, 0, 0). All other atoms are located in general positions. In complexes 1 and 2 Ca2+ has a typical distorted octahedral geometry40 formed by the oxygen atoms of four OPPh3 and two H2O ligands in the cis-position to one another. Ca–OOPPh3 distances are in the 2.260(2)–2.337(2) Å range, while Ca–OH2O bonds are equal to 2.409(2) Å for 1 and 2.378(8) Å for 2. The cluster anion consists of a nearly perfect M6 octahedron having eight μ3-capping Cli and six apical Cla ligands. The bond lengths in the cluster anion are similar to those in the starting cluster complexes and related complexes (summarised in Table S2, ESI†).36,37,39,41
Apart from ionic bonding, the structures of 1 and 2 are supported by an extensive network of hydrogen bonds (Fig. 1). In particular, two solvate CH3CN molecules are bound to each cis-[Ca(OPPh3)4(H2O)2]2+ cation by hydrogen bonds N⋯H–O (2.77(2) Å for 1, 2.83(3) Å for 2). The cation also interacts with two cluster anions through hydrogen bonds O–H⋯Cl (3.33(5) Å for both 1 and 2), which give rise to pseudo-polymeric zigzag chains running along the c axis. Moreover, each chain is connected to two adjacent chains by π–π stacking interactions between the phenyl rings of OPPh3 ligands (average interplanar distances are about 3.8 Å). These interactions lead to the formation of infinite layers in the bc plane.
 | ||
| Fig. 1 Fragment of the structures of 1 and 2, showing hydrogen bonding (dashed lines) between the [cis-Ca(OPPh3)4(H2O)2]2+ cations, [{M6Cli8}Cla6]cluster anions and CH3CN molecules. The Ph groups of the OPPh3 are omitted for clarity. | ||
Compound 4 also belongs to the monoclinic symmetry with space group C2/c and Z = 4, and is isostructural to compound 3.36 The structure of 4 is built up from linear polymeric chains directed along the b axis and has 2-fold axis symmetry. The chains are formed by nearly square-planar cationic units {Ca(OPPh3)4}2+ connected to the {{W6Cli8}Cla6}2+ cluster anions via the apical trans-chlorine ligands (Fig. 2). The W–W, W–Cli and W–Cla distances (Table S2, ESI†) are comparable to those found in 2 and the related compounds.37,41 At a supramolecular level, the chains are interconnected through π–π stacking interactions between the phenyl rings of OPPh3 ligands with interplanar distances of about 3.7 Å.
 | ||
| Fig. 2 The structure of trans-[{Ca(OPPh3)4}{{M6Cli8}Cla6}]∞. Ph groups are omitted for clarity. | ||
Apart from the complexes described herein, there are only a few other Ca2+ complexes with triphenylphosphine oxide ligands in the Cambridge crystallographic database, including [Cp*Ca(OPPh3)3]I·CH3C6H5,42 [Ca(OPPh3)2(N(SiMe3)2)2]43 and [Ca(OPPh3)2(CH(SiMe3)2)2].44 The Ca–OOPPh3 distances in 1, 2 and 4 are close to the analogous distances in these complexes.
Transition of compounds 1 and 2 into 3 and 4 upon heating at 110 °C was confirmed by X-ray powder diffraction analysis. Indeed, the powder diffraction patterns of 3 and 4 generated by heating of 1 and 2 are in a good agreement with those calculated from the corresponding single-crystal data (Fig. S4 and S5, ESI†). Moreover, compounds 1–4 are stable under ambient conditions for a prolonged period of time (Fig. S6, ESI†). From the structural point of view, the phase transition involves the elimination of coordinated water molecules and the solvate acetonitrile, and the formation of new covalent bonds Cla–Ca–Cla combined with the cis- to trans-isomerisation in the coordination environment of Ca2+. These processes lead to an overall reduction of the unit cell volumes of 1 and 2 by about 10%.
Thermal properties and kinetic analysis
To establish the transformation of 1 and 2 into CPs 3 and 4, respectively, compounds 1 and 2 were characterised using TG-DTA at heating rates of 5, 10 and 20 K min−1. According to TG measurements (Fig. 3 and Fig. S7, S8, ESI†) there is a one-step weight loss of 4.6% for 1 and 3.8% for 2 in the 70–120 °C temperature range. The values of the weight losses are close to those calculated for the elimination of two coordinated water molecules and two acetonitrile solvate molecules from compounds 1 (5.0%) and 2 (4.1%). The subsequent heating of the cluster complex (>340 °C) led to OPPh3 ligand destruction followed by the decomposition of the cluster complexes. | ||
| Fig. 3 TGA curves of 1 and 2. Heating rates of 10 °C min−1. | ||
Both DTA and mass spectrometry modalities of the method showed that there is only a very small (if any) temperature gap between the elimination of water and acetonitrile molecules (Fig. S9 and S10, ESI†). Calculation of the activation energies using the Friedman method of calculations applicable for a one-step process has not resulted in a constant value of the activation energies for each conversion point (Fig. S11 and S12, ESI†). The kinetic analysis of both molybdenum and tungsten systems was therefore carried out assuming the two-stage process (Fig. S13 and S14, ESI†): (I) A → B and (II) B → C. For molybdenum compound 1 the best fit of the modelled thermograms with the experiment was achieved when the first stage of the reaction was modelled by the Avrami–Erofeev equation, while the second one with the nth order equation:
| I: f(α)1 = (1 − α)[−ln(1 − α)]0.62 |
| II: f(α)2 = (1 − α)0.58 |
| I: E1 = 167 ± 2 kJ mol−1, lgA1 = 23.1 ± 0.3. |
| II: E2 = 44 ± 3 kJ mol−1, lgA2 = 4.8 ± 0.1. |
| I: f1(α) = (1 − α)[−ln(1 − α)]0.17, E1 = 212 ± 10 kJ mol−1, lgA1 = 28.5 ± 0.1. |
| II: f2(α) = (1 − α)[−ln(1 − α)]0.41, E2 = 197 ± 10 kJ mol−1, lgA2 = 27.8 ± 0.2. |
In summary, the activation energies for the decomposition of the tungsten compound are higher than those of the molybdenum compound, while the enthalpies of the reactions are nearly the same. The possible explanation could be that the {W6Cli8}4+ (Mw = 1383) cluster core is heavier and larger than {Mo6Cli8}4+ (Mw = 867) that presumably would lead to increase in the activation energies associated with the transport of gas through the solid and rearrangement of the solid.
Optical properties
To analyse the optical properties of compounds 1–4, we studied both absorption and luminescence properties. The diffuse reflectance spectra of 1–4 and the corresponding parent cluster compounds presented in the form of Kubelka–Munk theory are given in Fig. S17 and S18 (ESI†). Our data show that the replacement of the Bu4N+ by {Ca(OPPh3)4}2+ has little effect on both the profile of the adsorption and the band-gap in the tungsten compounds, while in the case of molybdenum compounds 1 and 3 some shortening of the optical band gap by about 0.2 eV was observed.Room temperature luminescence spectroscopic studies were performed for solid-state powder samples of 1–4 and for related compounds (Bu4N)2[{W6Cli8}Cla6]. Luminescence characteristics emission maximum wavelengths (λem), emission quantum yields (Φem) and lifetimes (τem) of the compounds are summarised in Table 1, together with those previously reported for the solid samples of (Bu4N)2[{M6Cli8}Cla6]. Upon 355 nm excitation, the compounds show emission similar to other metal cluster compounds based on the {M6X8}4+ (M = Mo or W, X = Cl or Br) cluster core (Fig. 4). λem observed within the current study for (Bu4N)2[{M6Cli8}Cla6] (M = Mo or W) and τem determined for (Bu4N)2[{Mo6Cli8}Cla6] are comparable to those reported earlier35 and thus can be used for validation of our results. It is worth noting that, to the best of our knowledge, Φem for (Bu4N)2[{M6Cli8}Cla6] (M = Mo or W) and τem for (Bu4N)2[{W6Cli8}Cla6] in the solid state have never been reported before. The emission maxima of the calcium-based compounds are slightly red-shifted for the hexamolybdenum complexes and blue-shifted for the tungsten compounds. Also, opposite tendencies are observed for changes in τem and Φem values: for 1 and 3 they are considerably higher, while for 2 and 4 they are appreciably lower than those for (Bu4N)2[{Mo6Cli8}Cla6] and (Bu4N)2[{W6Cli8}Cla6], respectively. However, there is a similarity in the changes in the luminescence properties of the studied compounds: τem and Φem determined for CPs trans-[{Ca(OPPh3)4}{{M6Cli8}Cla6}]∞ are higher than those of the corresponding parent ionic compounds [cis-Ca(OPPh3)4(H2O)2][{M6Cli8}Cla6]·2CH3CN for both M = Mo and M = W (Table 1). The lower values of luminescence quantum yields and lifetimes for aqua-complexes 1 and 2 in comparison with the corresponding CPs are likely due to energy dissipation through vibrations of H2O.
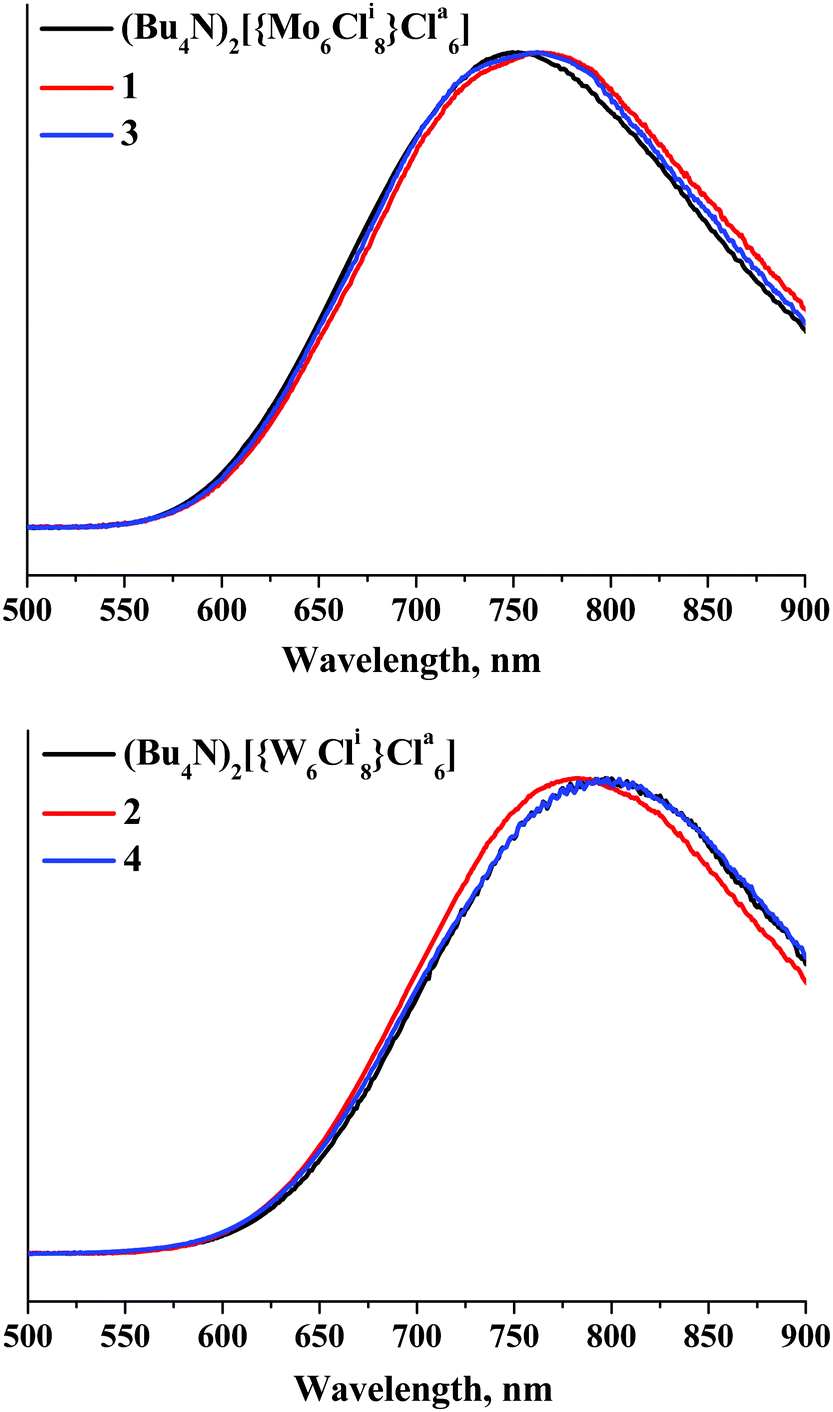 | ||
| Fig. 4 Normalized emission spectra of 1, 3 and (Bu4N)2[{Mo6Cli8}Cla6] (top) and 2, 4 and (Bu4N)2[{W6Cli8}Cla6] (bottom) in the solid state. | ||
The effect of the cationic part on the luminescence properties of octahedral anionic clusters have been discussed in several earlier works, where reasons such as energy transfer from the excited metal cluster to the cationic unit, in particular to the aromatic part of the cationic unit and self-absorption by the material, were discussed.46–50 In the present system, however, we believe that the molecular packing structures in the solid state have effects on the luminescence. Indeed, it has recently been reported that the emission lifetime of [{Mo6Cli8}Cla6]2− is very sensitive to the packing structures in the crystalline phase through the intermolecular interactions between the inner-Cl and apical-Cl ligands of the nearest neighbour cluster anion. Since the emissive excited state of [{Mo6Cli8}Cla6]2− is localised primarily on the {Mo6Cli8}4+-core, the perturbation to the cluster core influences strongly the emission properties.51 The careful evaluation of the distances between the centers of cluster anions shows that in the case of the Ca compounds the shortest distances between neighbouring cluster anions (12.466 Å for 1 and 13.051 Å for 3) are significantly longer than that for the Bu4N+ compounds (8.645 Å). This “isolation” of cluster anions would explain the longer τem and larger Φem for 1 and 3 in comparison with the corresponding Bu4N+ salt. We expected similar situations to those in the molybdenum crystalline phases to also emerge in the tungsten clusters. However, the tendency is opposite, i.e. although cluster anions are located further from each other, the emission quantum yields and the lifetimes of 2 and 4 are smaller and shorter, respectively. The opposite trend might be due to the more dense molecular packings of the tungsten compounds. Indeed, although the tungsten cluster anion is bigger than the molybdenum one, both the volumes of unit cells in 2 and 4 are smaller and Ca–Cla contacts are shorter than those in 1 and 3. These intermolecular interactions can facilitate efficient non-radiative decay to the ground state or excitation energy migration between cluster anions and subsequent energy trapping by crystal defects.
Conclusions
In summary, we demonstrate the preparation of one-dimensional photoluminescent CPs based on cationic units {Ca(OPPh3)4}2+ and photoluminescent cluster anions [{M6Cli8}Cla6]2− (M = Mo or W). In particular, we demonstrate that CPs trans-[{Ca(OPPh3)4}{{M6Cli8}Cla6}]∞ are formed at increased temperature in a water/acetonitrile mixture and remain stable at room temperature and up to 340 °C. However, if the reaction is carried out at room temperature, ionic compounds [cis-Ca(OPPh3)4(H2O)2][{M6Cli8}Cla6]·2CH3CN are formed. The latter can be viewed as an intermediate phase, since these compounds convert into more thermodynamically stable CP phases at increased temperature. Indeed, although the phase transition according to DSC studies has a large positive enthalpy contribution of around 205–206 kJ mol−1, the conversation has a significant increase of the total entropy of the system that would not depend on the cluster composition. The kinetic parameters of the phase transition do, however, depend on the cluster core: the tungsten system is significantly more kinetically inert than the molybdenum one, as it is expressed by noticeably higher values of activation energy.All four of the compounds studied demonstrate luminescence in the visible region. Notably, the photoluminescence lifetimes and quantum yields of the [{Mo6Cli8}Cla6]2−-based CPs almost doubled in comparison with those of (Bu4N)2[{Mo6Cli8}Cla6], while for the hexatungsten compounds the opposite tendency was observed.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by RFBR according to research projects No. 17-03-00140 and No. 16-34-00542.Notes and references
- Y. V. Mironov, N. G. Naumov, K. A. Brylev, O. A. Efremova, V. E. Fedorov and K. Hegetschweiler, Angew. Chem., Int. Ed., 2004, 43, 1297–1300 CrossRef CAS PubMed.
- Y. V. Mironov, N. G. Naumov, K. A. Brylev, O. A. Efremova, V. E. Fedorov and K. Hegetschweiler, Russ. J. Coord. Chem., 2005, 31, 269–281 CrossRef CAS.
- N. G. Naumov, A. V. Virovets and V. E. Fedorov, Inorg. Chem. Commun., 2000, 3, 71–72 CrossRef CAS.
- M. A. Shestopalov, A. Y. Ledneva, S. Cordier, O. Hernandez, M. Potel, T. Roisnel, N. G. Naumov and C. Perrin, Angew. Chem., Int. Ed., 2011, 50, 7300–7303 CrossRef CAS PubMed.
- N. Ding, G. S. Armatas and M. G. Kanatzidis, J. Am. Chem. Soc., 2010, 132, 6728–6734 CrossRef CAS PubMed.
- O. A. Efremova, E. O. Golenko, Y. V. Mironov, N. K. Moroz, C.-C. Wang and V. E. Fedorov, J. Phys. Chem. C, 2007, 111, 11008–11011 CAS.
- Y. V. Mironov, V. E. Fedorov, H. Bang and S.-J. Kim, Eur. J. Inorg. Chem., 2006, 553–557 CrossRef CAS.
- S. Kim, Y. Kim, J. Lee, W. Shin, M. Lee and S.-J. Kim, Inorg. Chim. Acta, 2007, 360, 1890–1894 CrossRef CAS.
- Y. M. Litvinova, Y. M. Gayfulin, D. G. Samsonenko, A. S. Bogomyakov, W. H. Shon, S.-J. Kim, J.-S. Rhyee and Y. V. Mironov, Polyhedron, 2016, 115, 174–179 CrossRef CAS.
- Y. M. Litvinova, Y. M. Gayfulin, A. S. Bogomyakov, D. G. Samsonenko and Y. V. Mironov, J. Cluster Sci., 2017 DOI:10.1007/s10876-10017-11276-z.
- M. N. Sokolov, M. A. Mihailov, E. V. Peresypkina, K. A. Brylev, N. Kitamura and V. P. Fedin, Dalton Trans., 2011, 40, 6375–6377 RSC.
- K. Kirakci, P. Kubát, M. Dušek, K. Fejfarová, V. Šícha, J. Mosinger and K. Lang, Eur. J. Inorg. Chem., 2012, 3107–3111 CrossRef CAS.
- O. A. Efremova, M. A. Shestopalov, N. A. Chirtsova, A. I. Smolentsev, Y. V. Mironov, N. Kitamura, K. A. Brylev and A. J. Sutherland, Dalton Trans., 2014, 43, 6021–6025 RSC.
- M. A. Mikhailov, K. A. Brylev, P. A. Abramov, E. Sakuda, S. Akagi, A. Ito, N. Kitamura and M. N. Sokolov, Inorg. Chem., 2016, 55, 8437–8445 CrossRef CAS PubMed.
- O. A. Efremova, Y. A. Vorotnikov, K. A. Brylev, N. A. Vorotnikova, I. N. Novozhilov, N. V. Kuratieva, M. V. Edeleva, D. M. Benoit, N. Kitamura, Y. V. Mironov, M. A. Shestopalov and A. J. Sutherland, Dalton Trans., 2016, 45, 15427–15435 RSC.
- D. V. Evtushok, A. R. Melnikov, N. A. Vorotnikova, Y. A. Vorotnikov, A. A. Ryadun, N. V. Kuratieva, K. V. Kozyr, N. R. Obedinskaya, E. I. Kretov, I. N. Novozhilov, Y. V. Mironov, D. V. Stass, O. A. Efremova and M. A. Shestopalov, Dalton Trans., 2017, 46, 11738–11747 RSC.
- S. Akagi, S. Fujii, T. Horiguchi and N. Kitamura, J. Cluster Sci., 2017, 28, 757–772 CrossRef CAS.
- Y. Zhao and R. R. Lunt, Adv. Energy Mater., 2013, 3, 1143–1148 CrossRef CAS.
- A. Renaud, F. Grasset, B. Dierre, T. Uchikoshi, N. Ohashi, T. Takei, A. Planchat, L. Cario, S. Jobic, F. Odobel and S. Cordier, ChemistrySelect, 2016, 1, 2284–2289 CrossRef CAS.
- A. O. Solovieva, Y. A. Vorotnikov, K. E. Trifonova, O. A. Efremova, A. A. Krasilnikova, K. A. Brylev, E. V. Vorontsova, P. A. Avrorov, L. V. Shestopalova, A. F. Poveshchenko, Y. V. Mironov and M. A. Shestopalov, J. Mater. Chem. B, 2016, 4, 4839–4846 RSC.
- A. Beltrán, M. Mikhailov, M. N. Sokolov, V. Pérez-Laguna, A. Rezusta, M. J. Revillo and F. Galindo, J. Mater. Chem. B, 2016, 4, 5975–5979 RSC.
- C. Felip-León, C. A. del Valle, V. Pérez-Laguna, M. I. Millán-Lou, J. F. Miravet, M. Mikhailov, M. N. Sokolov, A. Rezusta-López and F. Galindo, J. Mater. Chem. B, 2017, 5, 6058–6064 RSC.
- A. Barras, S. Cordier and R. Boukherroub, Appl. Catal., B, 2012, 123-124, 1–8 CrossRef CAS.
- P. Kumar, S. Kumar, S. Cordier, S. Paofai, R. Boukherroub and S. L. Jain, RSC Adv., 2014, 4, 10420–10423 RSC.
- P. Kumar, H. P. Mungse, S. Cordier, R. Boukherroub, O. P. Khatri and S. L. Jain, Carbon, 2015, 94, 91–100 CrossRef CAS.
- S. Kumar, O. P. Khatri, S. Cordier, R. Boukherroub and S. L. Jain, Chem. – Eur. J., 2015, 21, 3488–3494 CrossRef CAS PubMed.
- M. Feliz, M. Puche, P. Atienzar, P. Concepción, S. Cordier and Y. Molard, ChemSusChem, 2016, 9, 1963–1971 CrossRef CAS PubMed.
- R. N. Ghosh, G. L. Baker, C. Ruud and D. G. Nocera, Appl. Phys. Lett., 1999, 75, 2885–2887 CrossRef CAS.
- D. J. Osborn III, G. L. Baker and R. N. Ghosh, J. Sol-Gel Sci. Technol., 2005, 36, 5–10 CrossRef.
- L. Riehl, M. Ströbele, D. Enseling, T. Jüstel and H.-J. Meyer, Z. Anorg. Allg. Chem., 2016, 642, 403–408 CrossRef CAS.
- R. N. Ghosh, P. A. Askeland, S. Kramer and R. Loloee, Appl. Phys. Lett., 2011, 98, 221103 CrossRef.
- Y. Molard, Acc. Chem. Res., 2016, 49, 1514–1523 CrossRef CAS PubMed.
- T. G. Truong, B. Dierre, F. Grasset, N. Saito, N. Saito, T. K. N. Nguyen, K. Takahashi, T. Uchikoshi, M. Amela-Cortes, Y. Molard, S. Cordier and N. Ohashi, Sci. Technol. Adv. Mater., 2016, 17, 443–453 CrossRef CAS PubMed.
- O. A. Efremova, K. A. Brylev, Y. A. Vorotnikov, L. Vejsadová, M. A. Shestopalov, G. F. Chimonides, P. Mikes, P. D. Topham, S.-J. Kim, N. Kitamura and A. J. Sutherland, J. Mater. Chem. C, 2016, 4, 497–503 RSC.
- A. W. Maverick, J. S. Najdzionek, D. MacKenzie, D. G. Nocera and H. B. Gray, J. Am. Chem. Soc., 1983, 105, 1878–1882 CrossRef CAS.
- Z. S. Kozhomuratova, Y. V. Mironov, M. A. Shestopalov, Y. M. Gaifulin, N. V. Kurat'eva, E. M. Uskov and V. E. Fedorov, Russ. J. Coord. Chem., 2007, 33, 1–6 CrossRef CAS.
- P. A. Abramov, A. V. Rogachev, M. A. Mikhailov, A. V. Virovets, E. V. Peresypkina, M. N. Sokolov and V. P. Fedin, Russ. J. Coord. Chem., 2014, 40, 259–267 CrossRef CAS.
- Bruker, APEX2 (Version 1.08), SAINT (Version 07.03), SADABS (Version 02.11), SHELXTL (Version 06.12), Bruker AXS Inc., Madison, WI, USA, 2004 Search PubMed.
- M. A. Shestopalov, A. I. Smolentsev, Z. S. Kozhomuratova, V. E. Fedorov and Y. V. Mironov, Russ. J. Coord. Chem., 2012, 38, 50–54 CrossRef CAS.
- A. K. Katz, J. P. Glusker, S. A. Beebe and C. W. Bock, J. Am. Chem. Soc., 1996, 118, 5752–5763 CrossRef CAS.
- D. H. Johnston, C. M. Brown, A. S. Yu and J. C. Gallucci, Acta Crystallogr., 2010, C66, m303–m306 Search PubMed.
- K. C. Jayaratne, L. S. Fitts, T. P. Hanusa and V. G. Young, Organometallics, 2001, 20, 3638–3640 CrossRef CAS.
- A. G. M. Barrett, M. R. Crimmin, M. S. Hill, G. Kociok-Köhn, D. J. MacDougall, M. F. Mahon and P. A. Procopiou, Organometallics, 2008, 27, 3939–3946 CrossRef CAS.
- M. S. Hill, M. F. Mahon and T. P. Robinson, Chem. Commun., 2010, 46, 2498–2500 RSC.
- A. W. Maverick and H. B. Gray, J. Am. Chem. Soc., 1981, 103, 1298–1300 CrossRef CAS.
- Y. A. Vorotnikov, M. Mikhailov, K. A. Brylev, D. A. Piryazev, N. V. Kuratieva, M. N. Sokolov, Y. V. Mironov and M. A. Shestopalov, Russ. Chem. Bull., 2015, 64, 2591–2596 CrossRef CAS.
- B. Dierre, K. Costuas, N. Dumait, S. Paofai, M. Amela-Cortes, Y. Molard, F. Grasset, Y. Cho, K. Takahashi, N. Ohashi, T. Uchikoshi and S. Cordier, Sci. Technol. Adv. Mater., 2017, 18, 458–466 CrossRef PubMed.
- Y. A. Vorotnikov, O. A. Efremova, N. A. Vorotnikova, K. A. Brylev, M. V. Edeleva, A. R. Tsygankova, A. I. Smolentsev, N. Kitamura, Y. V. Mironov and M. A. Shestopalov, RSC Adv., 2016, 6, 43367–43375 RSC.
- K. Kirakci, P. Kubát, M. Kučeráková, V. Šícha, H. Gbelcová, P. Lovecká, P. Grznárová, T. Ruml and K. Lang, Inorg. Chim. Acta, 2016, 441, 42–49 CrossRef CAS.
- M. S. Tarasenko, A. Y. Ledneva, N. V. Kurat'eva, D. Y. Naumov, S.-J. Kim, V. E. Fedorov and N. G. Naumov, Russ. J. Coord. Chem., 2007, 33, 876–885 CrossRef CAS.
- N. Kitamura, Y. Kuwahara, Y. Ueda, Y. Ito, S. Ishizaka, Y. Sasaki, K. Tsuge and S. Akagi, Bull. Chem. Soc. Jpn., 2017, 50, 1164–1173 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Tables with crystallographic data; and FTIR, XRPD, TGA, DTG, and DSC curves and related data. CCDC 1565898–1565900 (1, 2 and 4). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7nj02858j |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2017 |