
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA novel family of homoleptic copper(I) complexes featuring disubstituted cyanamides: a combined synthetic, structural, and theoretical study†
Anna A.
Melekhova
,
Alexander S.
Novikov
 ,
Taras L.
Panikorovskii
,
Nadezhda A.
Bokach
* and
Vadim Yu.
Kukushkin
*
,
Taras L.
Panikorovskii
,
Nadezhda A.
Bokach
* and
Vadim Yu.
Kukushkin
*
Saint Petersburg State University, 7/9 Universitetskaya Nab., 199034 Saint Petersburg, Russian Federation. E-mail: n.bokach@spbu.ru; v.kukushkin@spbu.ru
First published on 16th October 2017
Abstract
The homoleptic copper(I) complexes [Cu(NCNRR′)4](BF4) (R/R′ = Me/Me 1, Et/Et 2, C5H103, C4H8O 4, C4H85, C3H6C6H46, CH2Ph/CH2Ph 7, Me/Ph 8) featuring disubstituted cyanamides were obtained in excellent (92–97%) yields by the reaction of [Cu(NCMe)4](BF4) and 4 equivalents of NCNRR′. Complexes 1–8 were characterized by atomic absorption spectrometry (Cu%), high resolution ESI+-MS, molar conductivities, TG/DTA, and 1H, 13C{1H} NMR, FTIR spectroscopic techniques, and also by single-crystal X-ray diffraction (1, 3, and 4). Results of DFT calculations and X-ray structure determinations reveal that equilibrium geometries of [Cu(NCMe)4]+ and [Cu(NCNMe2)4]+ in the gas phase are normal tetrahedral (Td) and significantly distorted, respectively. Effects of crystal packing influence the values of the Cu–N–C angles in [Cu(NCNRR′)4]+, which points out to the noticeable contribution of the heterocumulene mesomeric form for the dialkylcyanamide copper(I) complexes. The QTAIM and NBO analyses indicate that relatively weak Cu–N contacts (15–31 kcal mol−1) in both cases exhibit single bond character and clearly polarized toward the N atom (by 91–95%). The CDA shows that the {M} ← L σ-donation substantially prevails over the {M} → L π-back-donation in both [Cu(NCMe)4]+ and [Cu(NCNMe2)4]+. The orbital, charge, and vibrational frequency arguments as well as inspection of the FTIR data suggest that the electrophilic activation of the N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C group in homoleptic nitrile and dialkylcyanamide copper(I) complexes is similar, and the different behavior of nitriles and cyanamides in the 1,3-dipolar cycloaddition of ketonitrones is mainly due to the difference in the atomic charges.
C group in homoleptic nitrile and dialkylcyanamide copper(I) complexes is similar, and the different behavior of nitriles and cyanamides in the 1,3-dipolar cycloaddition of ketonitrones is mainly due to the difference in the atomic charges.
Introduction
Recently we reported on the CuI-catalyzed 1,3-dipolar cycloaddition of ketonitrones, Ph2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N+(R′)O−, to cyanamides, NCNRR′,1 that gives corresponding 5-amino-substituted 2,3-dihydro-1,2,4-oxadiazoles (Scheme 1).2 The reaction proceeds under mild conditions and requires 10 mol% of [Cu(NCMe)4](BF4) as the catalyst. This synthesis exemplifies the first catalytic system for the generation of less explored metal-free 2,3-dihydro-1,2,4-oxadiazoles.
N+(R′)O−, to cyanamides, NCNRR′,1 that gives corresponding 5-amino-substituted 2,3-dihydro-1,2,4-oxadiazoles (Scheme 1).2 The reaction proceeds under mild conditions and requires 10 mol% of [Cu(NCMe)4](BF4) as the catalyst. This synthesis exemplifies the first catalytic system for the generation of less explored metal-free 2,3-dihydro-1,2,4-oxadiazoles.
 | ||
| Scheme 1 CuI-catalyzed cycloaddition of ketonitrones. | ||
High resolution ESI+-MS monitoring of the reaction mixture revealed that, at the initial stage, the acetonitrile ligands at [Cu(NCMe)4]+ are displaced by NCNRR′ forming yet unreported homoleptic [Cu(NCNRR′)4]+ species. This observation and our general interest in the chemistry of metal-activated substrates featuring a CN triple bond (for our reviews see ref. 3) stimulated the current study on the synthesis of [Cu(NCNRR′)4]+ complexes and verification of activation modes of NCNRR′ ligands in these species.
In general, homoleptic solvento-complexes bearing the so-called weak donor ligands4 attract attention due to their useful synthetic properties. These species often exhibit good solubility in a solvent that also functions as the ligand. Accordingly, they serve as starting materials for the preparation of coordination compounds in non-aqueous solvents, a convenient source of solvated cations in non-aqueous media, and also as a soluble form of catalysts/pre-catalysts in metal-catalyzed transformations.4b Among weak donor solvento-complexes, nitrile homoleptic complexes are the most widespread due to a broad application spectrum of liquid nitriles, first of all MeCN and PhCN, as aprotic solvents of moderate donor ability (e.g., DNMeCN = 144b).
In this work, we developed a high-yielding method leading to a novel family of homoleptic cationic copper(I) species [Cu(NCNRR′)4](BF4). These complexes could be interesting as precursors for the substitution chemistry and also as potential intermediates in copper-involving catalytic reactions of cyanamides.2,5 A theoretical comparative study of the nature of Cu–N coordination bonds in homoleptic nitrile- and dialkylcyanamide copper(I) complexes and a comparison of the electrophilic activation of NCR and NCNRR′ species, viz. orbital, charge, and vibrational frequencies analysis, were also conduced in this work and the obtained data provide a further insight into the mechanism of CuI-catalyzed 1,3-dipolar cycloaddition of ketonitrones to cyanamides (Scheme 1).2 All these results are given in the following sections.
Results and discussion
Synthetic approach to [Cu(NCNRR′)4](BF4)
Homoleptic [Cu(NCR)4]+ complexes featuring conventional nitrile (R = Alk, Aryl) ligands are well studied and, in particular, copper(I) [Cu(NCMe)4](X) compounds were employed as active species for non-aqueous redox flow batteries,6 a source of structure-directing cation [Cu(NCMe)4]+ in the formation of all-inorganic inverse Keggin structures,7 and also as catalyst precursors in varieties of organic transformations.8 [Cu(NCR)4](X) (X = ClO4, BF4; R = Alk, Ar, CHCH2) species have been prepared by the reaction of CuClO4 with excess NCR9 and upon treatment of Cu2O with BF3·Et2O in appropriate nitrile solvents.10 The acetonitrile complexes [Cu(NCMe)4](X) were synthesized by oxidation of metallic copper in acetonitrile using silver11 or nitrosonium salts12 or electrolytic oxidation,11a,12 by reduction of copper(II) salts in MeCN,12 and also by the reaction of copper(I) precursors (Cu2O in the presence of HClO4 or CuCl) with MeCN.8a,13
Dialkylcyanamides, as compared to conventional nitriles, are stronger σ-donors3b and consequently they are better ligands toward CuI centers.3b Therefore we expected a facile substitution of NCMe ligands in [Cu(NCMe)4](BF4) with NCNR2. Indeed, the homoleptic complexes [Cu(NCNRR′)4](BF4) (R/R′ = Me/Me 1, Et/Et 2, C5H103, C4H8O 4, C4H85, C3H6C6H4 (NCNC3H6C6H4 is 3,4-dihydroisoquinoline-2(1H) carbonitrile) 6, CH2Ph/CH2Ph 7, Me/Ph 8) were generated by the reaction of [Cu(NCMe)4](BF4) with 4 equivalents of NCNRR′. The substitution proceeds in CH2Cl2 at RT for 30 min and target cyanamide complexes 1–8 were isolated in excellent (92–97%) yields. The experimentally observed direction of the substitution is thermodynamically favorable, which is followed from the analysis of calculated total reaction energy (ΔE = −10.3 kcal mol−1), enthalpy (ΔH = −10.0 kcal mol−1), and Gibbs free energy of the reaction (ΔG = −9.6 kcal mol−1) in the gas phase (see Tables S4 and S5 in the ESI†).
The stabilities of copper(I) homoleptic complexes with NCR ligands differ considerably. Thus, if the complexes [Cu(NCR)4]X (R = Ph, p-MeOC6H4, 1-naphtyl; X = ClO4) could be stored in air for months, the other (R = Me, Pr, CHCH2; X = ClO4) should be kept under dinitrogen to prevent their degradation.9c Our complexes 3–7 (R/R′ = C5H10, C4H8O, C4H8, C3H6C6H4, CH2Ph/CH2Ph) are shelf-stable for a prolonged time in air at low temperatures (5 °C; 3–4) or even at RT (5–7), whereas complexes 1, 2, and 8 (R/R′ = Me/Me, Et/Et, Ph/Me) are rather unstable in air at RT and after a few hours start to decompose. Hence, complexes 3–7 – from a novel family of homoleptic cyanamide copper(I) complexes – can be recommended for synthetic, reactivity, and catalytic studies.
It is of note in these respects that the copper chemistry of dialkylcyanamide species is almost unexplored despite growing interest (for reviews see ref. 3b) to these specific cyanamide substrates. A very limited amount of the known examples of copper-involving synthetic transformations of cyanamides include CuCl2-catalyzed (20 mol%) reactions of NCN(H)R with amines in the presence of boronic acids R3B(OH)2 leading to N,N′,N′′-substituted guanidines (Scheme 2, a);5aN,N′-substituted guanidines were also obtained in CuI/Xantphos-catalyzed (5/5 mol%) reaction of monosubstituted cyanamides with amines (b).5b
 | ||
| Scheme 2 The known copper-catalyzed reactions of cyanamides. | ||
The reaction between NCN(H)R, amines, and boronic acids that proceeded under prolonged heating in the presence of catalytic amounts of CuCl2 (20 mol%) results in 2-aminobenzimidazoles (c) or 2-aminoquinazolines (d). The only example of Cu-catalyzed reaction of disubstituted cyanamides is represented by the 1,3-dipolar cycloaddition of ketonitrones to cyanamides in the presence of [Cu(NCMe)4](BF4) (10 mol%) (e) developed by our group.2
Analytical and spectroscopic data for 1–8
Complexes 1–8 were characterized by atomic absorption spectrometry (Cu%), high resolution ESI+-MS, molar conductivities, TG/DTA, and 1H, 13C{1H} NMR, FTIR spectroscopic techniques, and also by single-crystal X-ray diffraction (for complexes 1, 3, and 4). Complexes 2 and 6 were isolated as hygroscopic oily residues and, although all spectral data support their formulation, satisfactory elemental analyses were not obtained.The atomic absorption spectrometry data for 1–8 are in good agreement with the calculated values. The HRESI+-MS of these complexes exhibit sets of peaks corresponding to the fragmentation ions [M – (NCNRR′)2]+. The values of molar conductivities in nitromethane (69–95 Ohm−1 cm−1 mol−1) agree with the typical range for 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 electrolytes (75–95 Ohm−1 cm−1 mol−1 in MeNO214). The TGA data (Fig. S33–S38; ESI†) demonstrate that mass loss starts in the interval ca. 35–100 °C, while the final product of thermal decomposition is CuF2 that is formed at 500–560 °C. The 1H and 13C{1H} NMR spectra display one set of signals, which correspond to resonances of protons or 13C, respectively, of the substituents in the NCNRR′ ligands.
:1 electrolytes (75–95 Ohm−1 cm−1 mol−1 in MeNO214). The TGA data (Fig. S33–S38; ESI†) demonstrate that mass loss starts in the interval ca. 35–100 °C, while the final product of thermal decomposition is CuF2 that is formed at 500–560 °C. The 1H and 13C{1H} NMR spectra display one set of signals, which correspond to resonances of protons or 13C, respectively, of the substituents in the NCNRR′ ligands.
The FTIR spectra of 1–8 in KBr pellets and also in Nujol oil display two CN absorption bands in the range 2235–2243 cm−1 and 2207–2222 cm−1; the former bands are moderately high-frequency shifted (by 20 cm−1) than those in the corresponding uncomplexed cyanamides (2211–2224 cm−1).
X-ray diffraction studies
The coordination polyhedra of 1, 3, and 4 are formed by the four dialkylcyanamides resulting in a tetrahedral geometry with τ4 0.90–0.9615 (Fig. 1–3). In these homoleptic complexes, N–Cu–N bond angles around the copper(I) centers are in the interval from 100.98(6) to 117.37(6)° that is close to those in the acetonitrile analogue [Cu(NCMe)4](BF4) (105.6(2)–113.4(2)°).16 The Cu–N bond distances (1.9661(16)–2.0150(16) Å) are equal within 3σ to those in [Cu(NCMe)4](BF4) (1.972(6)–2.028(6) Å)16 and in [Cu(NCNMe2)2(DPEphos)](BF4) (DPEphos = bis[2-(diphenylphosphino)-phenyl]ether; 1.999(4)–2.023(4) Å),17 whereas they are smaller than in the copper(II) complexes bearing NCNMe2, viz. [Cu2Cl2(dppm)2(NCNMe2)]·2NCNMe2 (2.158(5) Å).18 The CN bond lengths (1.143(3)–1.155(3) Å) are close to the corresponding bond values in the [Cu(NCMe)4](BF4) (1.107(6)–1.140(7) Å) complex, the NCNMe2 ligands in [Cu(NCNMe2)2(DPEphos)](BF4) (1.143(6)–1.146(5) Å),17 and the corresponding distance in the copper(II) complex [Cu2Cl2(NCNMe2)(dppm)2]·2NCNMe2 (1.134(7) Å).18 The Cu–N–C fragment deviates from linearity with the bond angle in the range 161.87(4)–177.73(19)°, which is comparable with that in other relevant disubstituted cyanamide complexes, for instance, in [Cu(NCMe)4](BF4) (169.9(8)–179.0(7)°16), [Cu(NCNMe2)2(DPEphos)](BF4) (151.1(4)–167.3(4)°17), and [Cu2Cl2(NCNMe2)(dppm)2]·2NCNMe2 (154.2(4)°18). It is should be noted that within one cation these deviations from linearity (180°) vary significantly, from 6.5 to 15°, for different Cu–N–C angles. Thus, the value of the Cu–N–C angles is in the range 161.9(1)–168.4(1)° for 1 (where the Cu atom has the special position (8d)), 165.6(2)–177.7(2)° for 3, and 161.0(2)–176.1(1)° and 160.1(2)–170.0(2)° for two independent cations of 4, which reflects inequivalency of the Cu–N bonds in [Cu(NCR)4](BF4). In the structurally similar acetonitrile complexes [Cu(NCMe)4]X (X = CF3SO3,19 ClO4,20 BF4,16,21 PF622), deviation from the linearity of the angle Cu–N–C (by 0.5–11.5°) is smaller than that for the cyanamide complexes (by 2.3–18.2°), and the difference between the Cu–N–C angles in one cation is slightly smaller (3.5–10° vs. 6.5–15°). It is noteworthy that the observed distortions in the structures of 1–3 are smaller than those for equilibrium geometries of 1–3 in the gas phase obtained from computational studies (see the next section), which is probably due to crystal packing effects, in particular, weak interactions between the complex cation and BF4−. Thus, for all structures of 1–3 weak CH⋯F contacts between the alkyl substituents of the cynamide ligands and BF4− were detected.
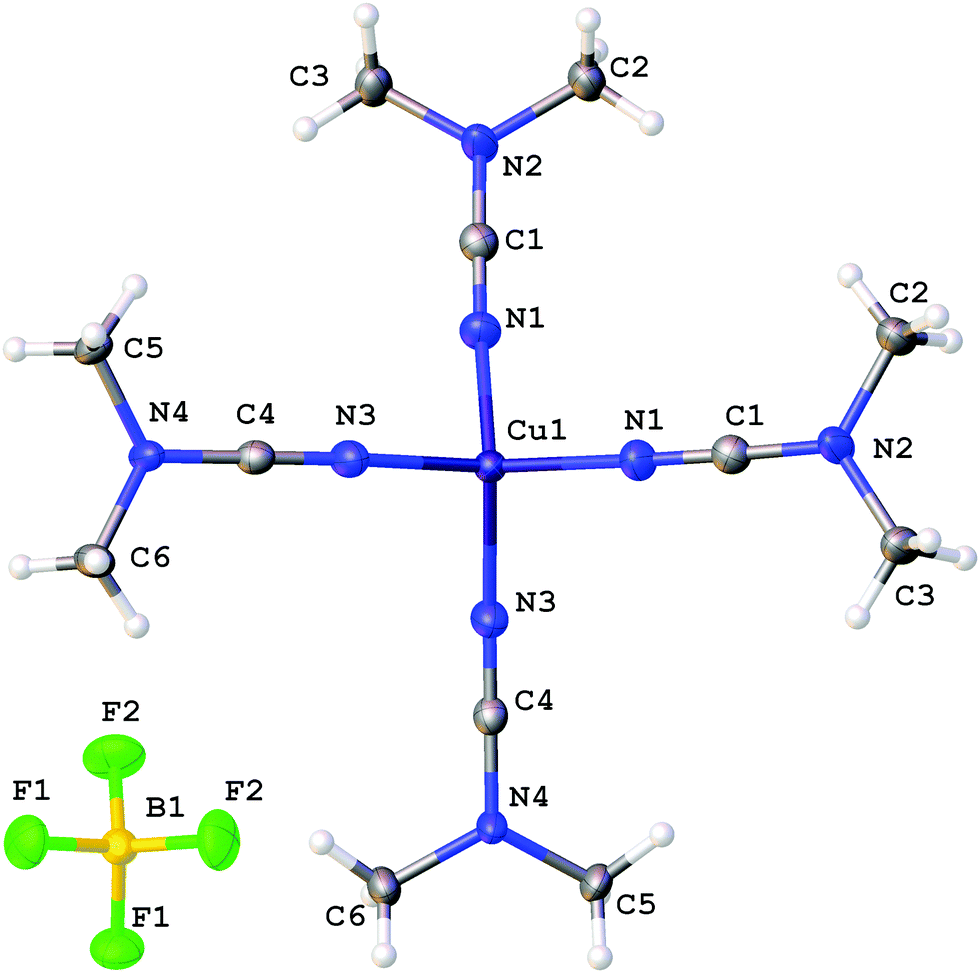 | ||
| Fig. 1 Molecular structure of 1 with the atomic numbering scheme. Thermal ellipsoids are drawn at the 50% probability level. | ||
 | ||
| Fig. 2 Molecular structure of 3 with the atomic numbering scheme. Thermal ellipsoids are drawn at the 50% probability level. | ||
 | ||
| Fig. 3 Molecular structure of 4 with the atomic numbering scheme. Thermal ellipsoids are drawn at the 50% probability level. | ||
The nature of Cu–N coordination bonds in homoleptic nitrile and dialkylcyanamide copper(I) complexes
In order to compare the nature of Cu–N coordination bonds in homoleptic nitrile and cyanamide copper(I) complexes, we carried out an integrated computational study including the full geometry optimization of the [Cu(NCMe)4]+ and [Cu(NCNMe2)4]+ model cationic species in the gas phase, the topological analysis of the electron density distribution (QTAIM),23 the natural bond orbital and charge decomposition analyses (NBO and CDA),24 and calculation of the vertical total energies for Cu–N coordination bond dissociations. This approach has already been successfully used by us for studying the bonding properties in various transition metal complexes.25 Notably the geometry optimization of [Cu(NCNMe2)4]+ was a really challenging task. We made more than ten attempts to get an alternative equilibrium geometry, and various input model structures were constructed based on the experimental X-ray geometries for similar complexes (3 and 4) as well as idealized structures with Td symmetry with equivalent Cu–N distances and Cu–N–C angles. Different input structures converged to very similar geometries during the optimization, and, in most cases, geometry oscillations without completing the optimization procedure or imaginary frequencies were observed.The full geometry optimization of [Cu(NCMe)4]+ leads to almost symmetrical tetrahedral configuration Td (bond lengths: Cu–N 2.017–2.022 Å; NC 1.156 Å; C–C 1.448–1.449 Å; angles: N–Cu–N 109.09–110.03°; Cu–N–C 179.31–179.64°; N–C–C 179.75–179.81°), whereas equilibrium geometry of [Cu(NCNMe2)4]+ in the gas phase is significantly distorted (bond lengths: Cu–N 1.964–2.246 Å; NC 1.166–1.170 Å; C–N 1.312–1.322 Å; angles: N–Cu–N 92.45–118.99°; Cu–N–C 124.46–177.69°; N–C–N 176.55–178.45°) (Fig. 4 and Table S6, ESI†). The Cu–N–C fragments in the optimized equilibrium structure of [Cu(NCNMe2)4]+ significantly deviate from linearity, even higher than those in the experimental X-ray structures of 1, 3, and 4, and this provides additional evidence of a noticeable contribution of the heterocumulene mesomeric form N(−)CN(+)R21 for such species.
 | ||
| Fig. 4 Optimized equilibrium geometries of [Cu(NCMe)4]+ and [Cu(NCNMe2)4]+ in the gas phase. | ||
The main results of the topological analysis of the electron density distribution (QTAIM)23 for the Cu–N coordination bonds in the optimized equilibrium geometries of [Cu(NCMe)4]+ and [Cu(NCNMe2)4]+ in the gas phase are presented in Table 1; appropriate contour line diagrams of the Laplacian distribution ∇2ρ(r), bond paths, and selected zero-flux surfaces are shown in Fig. 5. The Poincare–Hopf relationship in both cases is satisfied, thus all critical points have been found.
| Bond (length) | ρ(r) | ∇2ρ(r) | H b | ε |
|---|---|---|---|---|
| a ρ(r): density of all electrons, ∇2ρ(r): Laplacian of electron density, Hb: energy density, ε: ellipticity. All values are given in Hartree. | ||||
| [Cu(NCMe)4]+ | ||||
| Cu–N (2.017–2.022 Å) | 0.071–0.072 | 0.449–0.455 | 0.001 | 0.000–0.001 |
| [Cu(NCNMe2)4]+ | ||||
| Cu–N (1.964 Å) | 0.081 | 0.519 | −0.002 | 0.032 |
| Cu–N (1.968 Å) | 0.081 | 0.515 | −0.002 | 0.033 |
| Cu–N (2.011 Å) | 0.073 | 0.449 | 0.001 | 0.035 |
| Cu–N (2.246 Å) | 0.044 | 0.203 | 0.001 | 0.007 |
 | ||
| Fig. 5 Contour line diagrams of the Laplacian distribution ∇2ρ(r), bond paths and selected zero-flux surfaces for [Cu(NCMe)4]+ (top) and [Cu(NCNMe2)4]+ (bottom). Bond critical points (3, −1) are shown in blue, nuclear critical points (3, −3) – in pale brown, ring critical points (3, +1) – in orange, length units – Å. | ||
Suitable bond critical points (3, −1) were found for all Cu–N contacts in both [Cu(NCMe)4]+ and [Cu(NCNMe2)4]+ species. The values of ρ(r), ∇2ρ(r), and Hb in these bond critical points (3, −1) are typical for closed-shell interactions. The ellipticity of the Cu–N coordination bonds is negligible in [Cu(NCMe)4]+ (0.000–0.001 Hartree) and low in [Cu(NCNMe2)4]+ (0.007–0.035 Hartree) thus confirming the single bond character of these contacts.
The NBO analysis indicates the presence of σ-type Cu–N bond orbitals for all appropriate contacts in both [Cu(NCMe)4]+ and [Cu(NCNMe2)4]+. The Cu–N bond orbitals are clearly polarized toward the N atom (by 91% in [Cu(NCMe)4]+ and 91–95% in [Cu(NCNMe2)4]+; Table 2), and values of Wiberg bond indices (WI)26 for these contacts significantly lower than 1. Indeed, the calculated vertical total energies (Ev) for the Cu–N coordination bond dissociation in [Cu(NCMe)4]+ and [Cu(NCNMe2)4]+ are only 25 and 15–31 kcal mol−1 indicating that these bonds are relatively weak (Table S2, ESI†).
a
| Bond (length) | %Cu | %N | WI |
|---|---|---|---|
| a The hybridization of the atoms is indicated with the percent contribution of the s and p orbitals as a superscript, WI – Wiberg bond indices. | |||
| [Cu(NCMe)4]+ | |||
| Cu–N (2.017–2.022 Å) | 8.90; s25.1p73.8 | 91.10; s54.7p45.3 | 0.36 |
| [Cu(NCNMe2)4]+ | |||
| Cu–N (1.964 Å) | 9.27; s28.3p70.2 | 90.73; s56.1p43.9 | 0.38 |
| Cu–N (1.968 Å) | 9.23; s28.3p70.2 | 90.77; s56.1p43.9 | 0.38 |
| Cu–N (2.011 Å) | 8.52; s26.4p72.4 | 91.48; s56.3p43.7 | 0.35 |
| Cu–N (2.246 Å) | 5.44; s16.3p83.4 | 94.56; s53.4p46.5 | 0.22 |
The results of the CDA calculations reveal that the {M} ← L σ-donation prevails over the {M} → L π-back-donation in both [Cu(NCMe)4]+ and [Cu(NCNMe2)4]+ complexes. Indeed, the values of appropriate σ-donation (d) and π-back-donation (b) terms are 0.171 vs. 0.017 for [Cu(NCMe)4]+ and 0.158–0.181 vs. 0.011–0.023 for [Cu(NCNMe2)4]+ (Table 3). The overlap population between the occupied fragment orbitals (FOs) of the two fragments in the corresponding complex orbital (term r) is negative in all cases. It implies that in this complex orbital, the electrons of occupied FOs are depleted (mainly due to the Pauli repulsion) from the overlap region between the two fragments. The negative values of r reveals that the repulsive effect dominates the overall interaction between occupied FOs, which results in the corresponding electrons moving away toward non-overlapping regions from overlap regions. The net electron transfer between the donor and acceptor fragments was estimated using extended charge decomposition analysis (ECDA) formalism.27 The net number of electrons transferred from L to {M} is 0.192 in [Cu(NCMe)4]+ and 0.174–0.207 in [Cu(NCNMe2)4]+.
a
| Bond (length) | d | b | r | N |
|---|---|---|---|---|
| a d – {M} ← L σ-donation, b – {M} → L π-back-donation, r – repulsive part, N – net electron transfer between the donor and acceptor fragments. | ||||
| [Cu(NCMe)4]+ | ||||
| Cu–N (2.017–2.022 Å) | 0.171 | 0.017 | −0.110 | 0.192 |
| [Cu(NCNMe2)4]+ | ||||
| Cu–N (1.964 and 1.968 Å) | 0.173 | 0.019 | −0.111 | 0.207 |
| Cu–N (2.011 Å) | 0.181 | 0.023 | −0.139 | 0.197 |
| Cu–N (2.246 Å) | 0.158 | 0.011 | −0.134 | 0.174 |
Thus, the results of this computational study reveal that (i) equilibrium geometries of [Cu(NCMe)4]+ and [Cu(NCNMe2)4]+ in the gas phase are normal tetrahedral and significantly distorted, respectively. This points out to the noticeable contribution of the heterocumulene mesomeric form for the dialkylcyanamide copper(I) complex; (ii) relatively weak Cu–N contacts in both cases exhibit single bond character and clearly polarized toward the N atom; (iii) the {M} ← L σ-donation substantially prevails over the {M} → L π-back-donation in both cationic complexes. One can conclude that the nature of Cu–N coordination bonds in homoleptic nitrile and dialkylcyanamide copper(I) complexes is similar, but cyanamide is a better ligand toward the copper center from the thermodynamics viewpoint (see ESI†).
Electrophilic ligand activation in homoleptic nitrile and dialkylcyanamide copper(I) complexes
C moiety.28 In this context, understanding the degree of electrophilic ligand activation is a logical task. The relative activation of ligands in [Cu(NCMe)4]+ and [Cu(NCNMe2)4]+ model cationic species has been analyzed from the orbital, charge, and vibrational frequency arguments (Table 4). The composition and energies of the frontier molecular orbitals (FMOs) centered on the NC fragments are some of the main factors determining the reactivity toward nucleophilic addition and 1,3-dipolar cycloaddition of these species. The coordination of NCMe and NCNMe2 to the copper(I) metal center results in a decrease of both HOMOπ(NC) and LUMOπ*(NC) energy levels about the same degree in both cases, and simple qualitative MO consideration suggests almost equal relative electrophilic activation of these species (Fig. 6). Another important factor affecting the reactivity of these species is the charge distribution on reacting atoms, for our case, the charges on the N and C atoms of the NC group of NCR. The Δq(N) and Δq(C) for [Cu(NCMe)4]+ ↔ NCMe and NCNMe2 ↔ [Cu(NCNMe2)4]+ pairs are well comparable (0.05 and 0.15 vs. 0.03–0.06 and 0.09–0.14); thus, from the charge factor view point, quite similar activation of NCMe and NCNMe2 ligands upon coordination to the copper(I) metal center is also expected. Despite this, the cyanamide ligands exhibit greater absolute NBO atomic charges than the NCMe ligands and this explains the difference in the reactivity between cyanamides and conventional nitriles toward CuI-catalyzed 1,3-dipolar cycloaddition of ketonitrones that starts from the nucleophilic attack of the O center of a ketonitrone to an electrophilically activated C atom of the nitrile group.2 In this cycloaddition, dialkylcyanamides react with ketonitrones in the presence of copper(I), whereas conventional nitriles are inert in this reaction under the same CuI-catalyzed conditions.
C fragments (in a.u.), NBO atomic charges (q), and calculated values of the unscaled normal mode frequencies ν(NC) (in cm−1) in NCMe, [Cu(NCMe)4]+, NCNMe2, and [Cu(NCNMe2)4]+
| Structure | NCMe | [Cu(NCMe)4]+ | NCNMe2 | [Cu(NCNMe2)4]+ |
|---|---|---|---|---|
|
First occupied MOs
involving the π(N |
HOMO
−0.34078 |
HOMO−5
−0.46695 |
HOMO
−0.25695 |
HOMO−5
−0.36109 |
|
First unoccupied MOs
involving the π*(N |
LUMO
0.04367 |
LUMO+1
−0.09601 |
LUMO
0.04952 |
LUMO+1
−0.07839 |
| q(N) | −0.35 | −0.40 | −0.40 | −0.43, −0.45, −0.46 |
| q(C) | 0.31 | 0.46 | 0.45 | 0.54, 0.57, 0.59 |
|
ν(NC) |
2389 | 2412, 2406 | 2352 | 2386, 2381, 2367, 2337 |
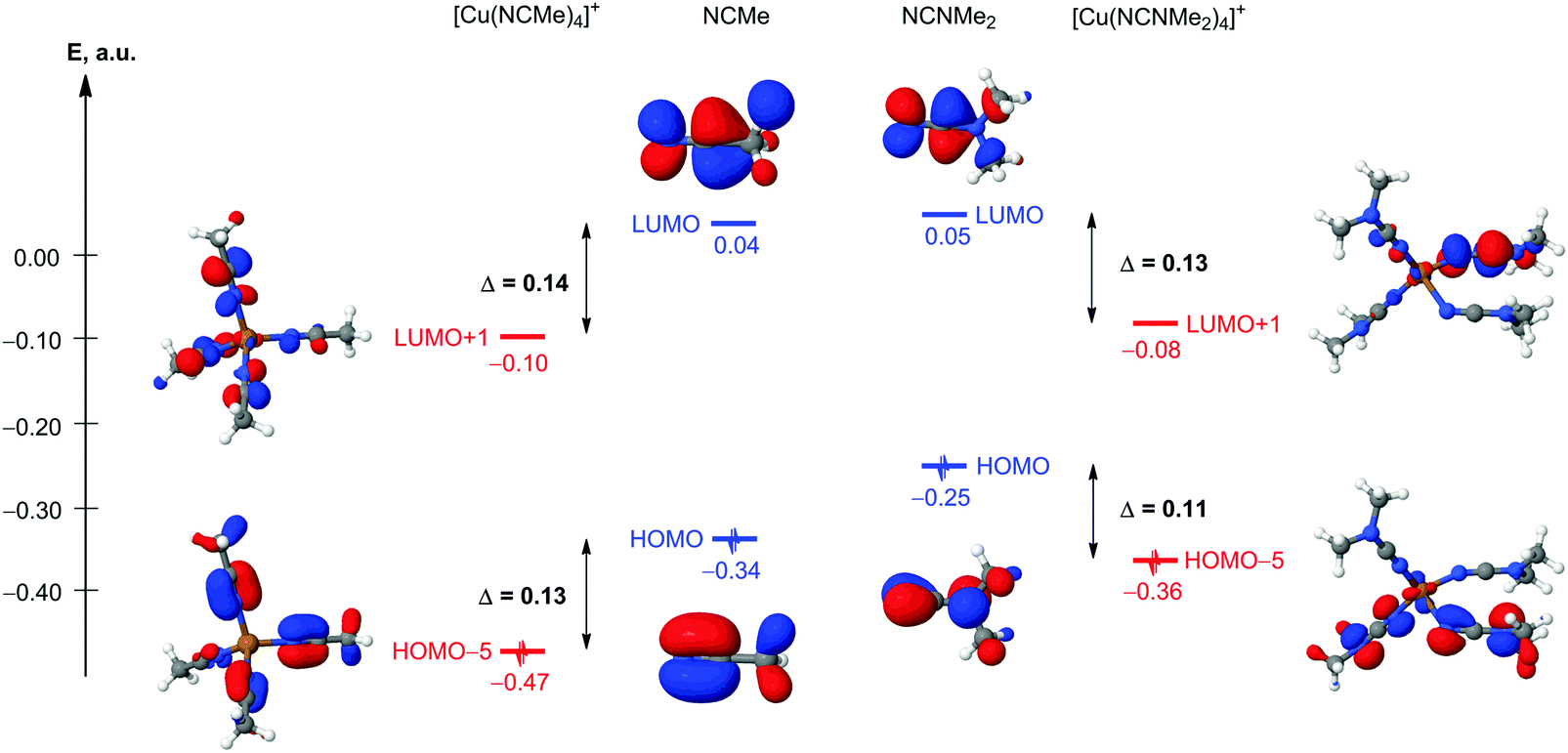 | ||
| Fig. 6 Relative energies of the FMOs centered on the NC fragments in NCMe, [Cu(NCMe)4]+, NCNMe2, and [Cu(NCNMe2)4]+. | ||
C) vibration is the most analytically important characteristic frequency in the IR spectra of [Cu(NCMe)4]+ and [Cu(NCNMe2)4]+, and a change in this frequency on going from the uncomplexed to the coordinated ligand can be used as an indicator of the activation of these substrates toward nucleophilic or electrophilic attacks (theoretical interpretations of such phenomena were discussed in a review on the topic29). The calculated Δν for [Cu(NCMe)4]+ ↔ NCMe and NCNMe2 ↔ [Cu(NCNMe2)4]+ pairs are 17–23 cm−1vs. −15–34 cm−1 (Table 4), which denote that the cyanamide ligands are activated similarly to conventional nitrile copper(I)-bound species. These data are in agreement with an experimentally observed shift (+20 cm−1) for dialkylcyanamide complexes (see above for the FTIR spectroscopy data). The values of this shift correspond to a moderate electrophilic activation upon the coordination of NCNRR′ to the copper(I) center. It is known that moderate-to-strong activation is accompanied with a +30–100 cm−1 shift and strong activation with a shift up to −50 cm−1.30
To summarize, the analysis of the orbital, charge, and vibrational frequency arguments and also inspection of the FTIR data suggest that the electrophilic nitrile group activation in homoleptic nitrile and dialkylcyanamide copper(I) complexes is similar, and different behaviors of nitriles and cyanamides in the 1,3-dipolar cycloaddition of ketonitrones is mainly due to the difference in the atomic charges.
Conclusions
Transition metal complexes bearing substituted cyanamide ligands attract significant attention due to diverse reactivity patterns of NCNRR′ (R = H, Alk, Ar; R′ = Alk, Ar), which, in many instances, differ from those known for conventional nitrile ligands NCR (R = Alk, Ar).3b,31 Metal-mediated and metal-catalyzed reactions of cyanamides have been repeatedly studied in the past few years (see reviews3b,e and recent examples5,32). In this study, we developed a high-yielding synthesis leading to a novel family of the cationic homoleptic [Cu(NCNRR′)4]X (X = BF4) species. This type of complex, viz. [Cu(NCR)4]X, was well known for conventional nitriles (R = Alk, Ar) and unknown before this work for cyanamides (R = NRR′). Our complexes 3–7 (R/R′ = C5H10, C4H8O, C4H8, C3H6C6H4, CH2Ph/CH2Ph) exhibit sufficient stability and they can be recommended for synthetic, reactivity, and catalytic studies.In the context of the copper chemistry of cyanamides, it is noteworthy that although homoleptic complexes [Cu(NCNRR′)4]+ were not reported previously, some other copper(I) mononuclear complexes featuring disubstituted cyanamides are represented by one structurally characterized complex, viz., [Cu(NCNMe2)2(DPEphos)](BF4) (DPEphos is bis[(2-diphenylphosphino)phenyl]-ether).17 Several dinuclear copper(I) species bearing coordinated dimethylcyanamide were also reported and these examples include [Cu2(μ2-Cl)2(dppm)2(NCNMe2)]·2NCNMe2, [Cu2(μ2-Cl)(dppm)2(NCNMe2)](Cl), [Cu2(μ2-X)(dppm)2(NCNMe2)2](X) (X = ClO4, NO3), and [Cu2(dppm)2(NCNMe2)3](BF4)2.18 In addition, copper(II) complexes with disubstituted cyanamides are represented by the clusters [Cu4Cl6O(NCNR2)] (R = C3H5, ½C5H10, ½C4H8, CPh2).33
Results of quantum chemical DFT calculations and X-ray structure determinations demonstrated that effects of crystal packing noticeably influence the values of the Cu–N–C angles in [Cu(NCNRR′)4]+. Equilibrium geometry of [Cu(NCNMe2)4]+ in the gas phase is distorted (the Cu–N–C angles are 125–178°), and this provides additional evidence of a substantial contribution of the heterocumulene mesomeric form N(−)CN(+)R2 for such species.
The computational study, performed for the conventional nitrile and cyanamide model complexes, [Cu(NCMe)4]+ and [Cu(NCNMe2)4]+, reveal that relatively weak Cu–N bonds (25 and 15–31 kcal mol−1, correspondingly) in both cases have a single bond character and are clearly polarized toward the N atom (by 91–95%). The {M} ← L σ-donation substantially prevails over the {M} → L π-back-donation in both cationic complexes. The relative electrophilic activation of the ligands in [Cu(NCMe)4]+ and [Cu(NCNMe2)4]+ model cationic species was analyzed from the orbital, charge, and vibrational frequency arguments and this study demonstrates similar degrees of the electrophilic activation of these species. Despite this, NCNMe2 ligands exhibit NBO atomic charges greater than those of NCMe ligands and this explains the difference in the reactivity between cyanamides and conventional nitriles toward CuI-catalyzed 1,3-dipolar cycloaddition of ketonitrones.2
Experimental section
Materials and instrumentation
The dialkylcyanamides NCNRR′ (R/R′ = Me/Me, Et/Et, C5H10, C4H8O, C4H8; Aldrich) and solvents were obtained from commercial sources and used as received. The dialkylcyanamides NCN(CH2Ph)2, NCN(Me)Ph, NCNC3H6C6H4,34 and the copper(I) complex [Cu(NCMe)4](BF4),35 were synthesized in accord with the published methods. The HRESI mass spectra were obtained on a Bruker micrOTOF spectrometer equipped with an electrospray ionization source and MeOH was employed as the solvent. The instrument was operated in positive ion mode using an m/z range of 50–3000. The capillary voltage of the ion source was set at −4500 V (ESI+ MS) and the capillary exit at ±(70–150) V. In the isotopic pattern, the most intensive peak is reported. Infrared spectra were recorded using a Bruker FTIR TENSOR 27 instrument in KBr pellets. 1H and 13C{1H} NMR spectra were measured using a Bruker Avance III 400/100 MHz spectrometer at ambient temperature. Residual solvent signals were used as the internal standard. Atomic absorption spectrometry (AAS) was carried out on a Shimadzu AA-7000 spectrometer (spectral range 189–900 nm) using a flame emission spectroscopy method. Standard Cu samples for the calibration solutions prepared of MERCK standard in 0.1 M HNO3. Calibration solutions were 0.5–2.0 mg L−1. Spectral analysis of the sample solutions was carried out without dilution. Molar conductivities of copper complexes 1–8 were measured at a Mettler Toledo meter FE30/FG3 in nitromethane solutions (molar concentrations were 2.0–5.5 × 10−4 mol L−1) at RT.X-ray structure determinations
Suitable crystals of 1, 3, and 4 were measured at Agilent Technologies Xcalibur EOS diffractometer with monochromated MoKα radiation. All structures have been solved using the direct methods by means of the SHELX program36 incorporated in the OLEX2 program package.37 For crystallographic data and refinement parameters see the ESI† (Table S1). The carbon-bound H atoms were placed in calculated positions and were included in the refinement in the ‘riding’ model approximation, with Uiso(H) set to 1.5Ueq(C) and C–H 0.98 Å for CH3 groups, with Uiso(H) set to 1.2Ueq(C) and C–H 0.99 Å for CH2 groups and with Uiso(H) set to 1.2Ueq(C), C–H 0.95 Å for CH groups. Empirical absorption correction was applied in the CrysAlisPro38 program complex using spherical harmonics, implemented in the SCALE3 ABSPACK scaling algorithm. Supplementary crystallographic data for this paper have been deposited at Cambridge Crystallographic Data Centre (CCDC 1578478 (1), 1578477 (3) and 1578479 (4)).†Computational details
The full geometry optimization of NCMe, [Cu(NCMe)4]+, NCNMe2, and [Cu(NCNMe2)4]+ has been carried out at the DFT level of theory using the M06 functional39 with the help of the Gaussian-09 program package.40 No symmetry restrictions have been applied during the geometry optimization. The calculations were carried out using the multi electron fit fully relativistic energy-consistent pseudopotential MDF10 of the Stuttgart/Cologne group that described 10 core electrons and the appropriate contracted basis set for the copper atom41 and the 6-31G(d) basis sets for other atoms. The Hessian matrix was calculated analytically for the optimized structures in order to prove the location of correct minima (no imaginary frequencies). The topological analysis of the electron density distribution with the help of the “atoms in molecules” method developed by Bader (QTAIM)23 and charge decomposition analysis developed by Dapprich and Frenking (CDA)24 have been carried out by using the Multiwfn program (version 3.3.8).42 The Cartesian atomic coordinates for optimized equilibrium structures of NCMe, [Cu(NCMe)4]+, NCNMe2, and [Cu(NCNMe2)4]+ are presented in the ESI,† Table S3.Synthetic work
N). 1H NMR in CDCl3, δ: 2.96 (s, CH3). 13C{1H} NMR in CDCl3, δ: 40.13 (CH3), NCN was not detected. Λm = 83 Ohm−1 cm−1 mol−1.
N). 1H NMR in CDCl3, δ: 1.30 (t, 6H, CH3), 3.16 (q, 4H, CH2). 13C{1H} NMR in CDCl3, δ: 12.83 (CH3), 45.87 (CH2), NCN was not detected. Λm = 87 Ohm−1 cm−1 mol−1.
N). 1H NMR in CDCl3, δ: 1.60 (m, 2H), 1.69 (m, 4H), 3.28 (t, 4H). 13C{1H} NMR in CDCl3, δ: 22.71 (CH2), 24.61 (CH2), 49.92 (CH2), NCN was not detected. Λm = 82 Ohm−1 cm−1 mol−1.
N). 1H NMR in CDCl3, δ: 3.36 (t, 4H, CH2), 3.79 (t, 4H, CH2). 13C{1H} NMR in CDCl3, δ: 48.38 (CH2), 65.64 (CH2). Λm = 95 Ohm−1 cm−1 mol−1.
N). 1H NMR in CDCl3, δ: 1.97 (t, 4H, CH2), 3.50 (t, 4H, CH2). 13C{1H} NMR in CDCl3, δ: 25.79 (CH2), 50.61 (CH2), NCN was not detected. Λm = 69 Ohm−1 cm−1 mol−1.
C). 1H NMR in CDCl3, δ: 3.01 (t, J 5.7 Hz, 2H), 3.62 (t, J 5.8 Hz, 2H), 4.53 (s, 2H), 7.09–7.11 (m, 1H), 7.16–7.18 (m, 1H), 7.22–7.24 (m, 2H). 13C{1H} NMR in CDCl3, δ: 27.70 (CH2), 46.56 (CH2), 49.48 (CH2), 126.04, 126.86, 127.42, 129.23, 130.27, 132.51 (C6H4), NCN was not detected. Λm = 85 Ohm−1 cm−1 mol−1.
C). 1H NMR in CDCl3, δ: 4.24 (s, 2H), 7.35–7.42 (m, 2H), 7.297.31 (m, 3H). 13C{1H} NMR in CDCl3, δ: 54.28 (CH2), 128.83, 128.90, 129.04, 133.70 (Ph), NCN was not detected. Λm = 94 Ohm−1 cm−1 mol−1.
C). 1H NMR in CDCl3, δ: 3.47 (s, 3H), 7.16 (dd, J 16.9, 7.9 Hz, 3H), 7.41 (t, J 7.9 Hz, 2H). 13C{1H} NMR in CDCl3, δ: 37.14 (CH3), 115.58, 124.45, 129.86, 139.25 (Ph), NCN was not detected. Λm = 81 Ohm−1 cm−1 mol−1.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was supported by the Russian Foundation for Basic Research (grant 17-03-00110). The authors thank the Center for X-ray Diffraction Studies, Center for Magnetic Resonance, Center for Chemical Analysis and Materials Research, the Center for Thermogravimetric and Calorimetric Research and Chemistry Educational Centre (all belong to Saint Petersburg State University) for the physicochemical measurements.References
- T. B. Anisimova, N. A. Bokach, I. O. Fritsky and M. Haukka, J. Mol. Struct., 2011, 1005, 141–143 CrossRef CAS.
- A. A. Melekhova, A. S. Smirnov, A. S. Novikov, T. L. Panikorovskii, N. A. Bokach and V. Y. Kukushkin, ACS Omega, 2017, 2, 1380–1391 CrossRef CAS.
- (a) V. Y. Kukushkin and A. J. L. Pombeiro, Chem. Rev., 2002, 102, 1771–1802 CrossRef CAS PubMed; (b) N. A. Bokach and V. Y. Kukushkin, Coord. Chem. Rev., 2013, 257, 2293–2316 CrossRef CAS; (c) V. P. Boyarskiy, N. A. Bokach, K. V. Luzyanin and V. Y. Kukushkin, Chem. Rev., 2015, 115, 2698–2779 CrossRef CAS PubMed; (d) N. A. Bokach, M. L. Kuznetsov and V. Y. Kukushkin, Coord. Chem. Rev., 2011, 255, 2946–2967 CrossRef CAS; (e) D. S. Bolotin, V. A. Rassadin, N. A. Bokach and V. Y. Kukushkin, Inorg. Chim. Acta, 2017, 455, 446–454 CrossRef CAS; (f) V. Y. Kukushkin and A. J. L. Pombeiro, Inorg. Chim. Acta, 2005, 358, 1–21 CrossRef CAS.
- (a) J. A. Davies and F. R. Hartley, Chem. Rev., 1981, 81, 79–90 CrossRef CAS; (b) J. A. Davies, C. M. Hockensmith, V. Y. Kukushkin and Y. N. Kukushkin, Synthetic Coordination Chemistry: Principles and Practice, World Scientific, 1996, pp. 95–156 DOI:10.1142/9789812831378_0004.
- (a) J. Li and L. Neuville, Org. Lett., 2013, 15, 6124–6127 CrossRef CAS PubMed; (b) C. J. Zeng, C. J. Chen, C. W. Chang, H. T. Chen and T. C. Chien, Aust. J. Chem., 2014, 67, 1134–1137 CrossRef CAS.
- Y. Li, J. Sniekers, J. Malaquias, X. F. Li, S. Schaltin, L. Stappers, K. Binnemans, J. Fransaer and I. F. J. Vankelecom, Electrochim. Acta, 2017, 236, 116–121 CrossRef CAS.
- J. Fielden, K. Quasdorf, L. Cronin and P. Kogerler, Dalton Trans., 2012, 41, 9876–9878 RSC.
- (a) Y. Zhang, W. Sun, C. Freund, A. M. Santos, E. Herdtweck, J. Mink and F. E. Kuhn, Inorg. Chim. Acta, 2006, 359, 4723–4729 CrossRef CAS; (b) J. M. Hoover, J. E. Steves and S. S. Stahl, Nat. Protoc., 2012, 7, 1161–1166 CrossRef CAS PubMed.
- (a) I. V. Nelson, R. T. Iwamoto and J. Kleinberg, J. Am. Chem. Soc., 1964, 86, 364 CrossRef CAS; (b) M. Kubota and D. L. Johnston, J. Am. Chem. Soc., 1966, 88, 2451–2455 CrossRef CAS; (c) M. Kubota and D. L. Johnston, J. Inorg. Nucl. Chem., 1967, 29, 769–773 CrossRef CAS.
- H. Meerwein, V. Hederich and K. Wunderlich, Arch. Pharm., 1958, 291, 541–554 CrossRef CAS.
- (a) H. H. Morgan, J. Chem. Soc., 1923, 2901–2907 RSC; (b) G. Bergerhoff, Z. Anorg. Allg. Chem., 1964, 327, 139–142 CrossRef CAS.
- B. J. Hathaway, D. G. Holah and J. D. Postlethwaite, J. Chem. Soc., 1961, 3215–3218, 10.1039/JR9610003215.
- P. Hemmerich and C. Sigwart, Experientia, 1963, 19, 488–489 CrossRef CAS.
- W. J. Geary, Coord. Chem. Rev., 1971, 7, 81–122 CrossRef CAS.
- L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955–964 RSC.
- P. G. Jones and O. Crespo, Acta Crystallogr., Sect. C: Struct. Chem., 1998, 54, 18–20 Search PubMed.
- S. M. Kuang, D. G. Cuttell, D. R. McMillin, P. E. Fanwick and R. A. Walton, Inorg. Chem., 2002, 41, 3313–3322 CrossRef CAS PubMed.
- J. K. Bera, M. Nethaji and A. G. Samuelson, Inorg. Chem., 1999, 38, 218–228 CrossRef CAS.
- D. S. Gill, R. Singh, D. S. Rana, J. R. Wagler and E. Kroke, Z. Naturforsch., 2011, 66b, 1042–1048 CrossRef.
- (a) H.-G. Hao, X.-D. Zheng and T.-B. Lu, Angew. Chem., Int. Ed., 2010, 49, 8148–8151 CrossRef CAS PubMed; (b) M. Sarma and B. Mondal, Inorg. Chem., 2011, 50, 3206–3212 CrossRef CAS PubMed.
- M. Kleinwachter, L. Vendier, C. Dinoi and M. Etienne, Dalton Trans., 2013, 42, 10102–10105 RSC.
- J. M. Bak, Effendy, S. Grabowsky, L. F. Lindoy, J. R. Price, B. W. Skelton and A. H. White, CrystEngComm, 2013, 15, 1125–1138 RSC.
- R. F. W. Bader, Chem. Rev., 1991, 91, 893–928 CrossRef CAS.
- S. Dapprich and G. Frenking, J. Phys. Chem., 1995, 99, 9352–9362 CrossRef CAS.
- (a) V. N. Mikhaylov, V. N. Sorokoumov, K. A. Korvinson, A. S. Novikov and I. A. Balova, Organometallics, 2016, 35, 1684–1697 CrossRef CAS; (b) T. B. Anisimova, M. A. Kinzhalov, M. F. C. Guedes da Silva, A. S. Novikov, V. Y. Kukushkin, A. J. L. Pombeiro and K. V. Luzyanin, New J. Chem., 2017, 41, 3246–3250 RSC; (c) A. S. Novikov, M. L. Kuznetsov and A. J. L. Pombeiro, Eur. J. Chem., 2013, 19, 2874–2888 CrossRef CAS PubMed.
- K. B. Wiberg, Tetrahedron, 1968, 24, 1083–1096 CrossRef CAS.
- S. I. Gorelsky, S. Ghosh and E. I. Solomon, J. Am. Chem. Soc., 2006, 128, 278–290 CrossRef CAS PubMed.
- (a) F. Himo, Z. P. Demko and L. Noodleman, J. Org. Chem., 2003, 68, 9076–9080 CrossRef CAS PubMed; (b) F. Himo, Z. P. Demko, L. Noodleman and K. B. Sharpless, J. Am. Chem. Soc., 2003, 125, 9983–9987 CrossRef CAS PubMed; (c) G. Wagner, Chem. – Eur. J., 2003, 9, 1503–1510 CrossRef CAS PubMed; (d) M. L. Kuznetsov, V. Y. Kukushkin, A. I. Dement'ev and A. J. L. Pombeiro, J. Phys. Chem. A, 2003, 107, 6108–6120 CrossRef CAS; (e) M. L. Kuznetsov and V. Y. Kukushkin, J. Org. Chem., 2006, 71, 582–592 CrossRef CAS PubMed; (f) M. L. Kuznetsov, A. A. Nazarov, L. V. Kozlova and V. Y. Kukushkin, J. Org. Chem., 2007, 72, 4475–4485 CrossRef CAS PubMed; (g) M. L. Kuznetsov, V. Y. Kukushkin and A. J. L. Pombeiro, J. Org. Chem., 2010, 75, 1474–1490 CrossRef CAS PubMed.
- M. L. Kuznetsov, Russ. Chem. Rev., 2002, 71, 265–282 CrossRef CAS.
- N. A. Bokach and V. Y. Kukushkin, Coord. Chem. Rev., 2013, 257, 2293–2316 CrossRef CAS.
- (a) P. V. Gushchin, M. L. Kuznetsov, M. Haukka, M.-J. Wang, A. V. Gribanov and V. Y. Kukushkin, Inorg. Chem., 2009, 48, 2583–2592 CrossRef CAS PubMed; (b) P. V. Gushchin, M. R. Tyan, N. A. Bokach, M. D. Revenco, M. Haukka, M.-J. Wang, C.-H. Lai, P.-T. Chou and V. Y. Kukushkin, Inorg. Chem., 2008, 47, 11487–11500 CrossRef CAS PubMed.
- (a) C. X. Wang, D. P. Wang, F. Xu, B. Pan and B. S. Wan, J. Org. Chem., 2013, 78, 3065–3072 CrossRef CAS PubMed; (b) G. Albertin, S. Antoniutti, A. Caia and J. Castro, Dalton Trans., 2014, 43, 7314–7323 RSC; (c) T. Hashimoto, S. Ishii, R. Yano, H. Miura, K. Sakata and R. Takeuchi, Adv. Synth. Catal., 2015, 357, 3901–3916 CrossRef CAS; (d) L. Q. Tran, J. H. Li and L. Neuville, J. Org. Chem., 2015, 80, 6102–6108 CrossRef CAS PubMed; (e) C. H. Wang, T. H. Hsieh, C. C. Lin, W. H. Yeh, C. A. Lin and T. C. Chien, Synlett, 2015, 1823–1826 Search PubMed; (f) E. Alegria, M. F. C. Guedes da Silva, M. L. Kuznetsov, L. Martins and A. J. L. Pombeiro, Electrochim. Acta, 2016, 218, 252–262 CrossRef CAS; (g) M. J. Fernandez-Rodriguez, E. Martinez-Viviente, J. Vicente and P. G. Jones, Dalton Trans., 2016, 45, 820–830 RSC; (h) J. Zheng, Z. Y. Deng, Y. Zhang and S. L. Cui, Adv. Synth. Catal., 2016, 358, 746–751 CrossRef CAS; (i) A. S. Smirnov, E. S. Butukhanova, N. A. Bokach, G. L. Starova, V. V. Gurzhiy, M. L. Kuznetsov and V. Y. Kukushkin, Dalton Trans., 2014, 43, 15798–15811 RSC; (j) E. V. Andrusenko, E. V. Kabin, A. S. Novikov, N. A. Bokach, G. L. Starova, A. A. Zolotarev and V. Y. Kukushkin, Eur. J. Inorg. Chem., 2015, 4894–4904, DOI:10.1002/ejic.201500693; (k) M. Y. Demakova, D. S. Bolotin, N. A. Bokach, G. L. Starova and V. Y. Kukushkin, Inorg. Chim. Acta, 2015, 425, 114–117 CrossRef CAS; (l) V. A. Rassadin, V. P. Boyarskiy and V. Y. Kukushkin, Org. Lett., 2015, 17, 3502–3505 CrossRef CAS PubMed; (m) A. S. Smirnov, E. S. Yandanova, N. A. Bokach, G. L. Starova, V. V. Gurzhiy, M. S. Avdontceva, A. A. Zolotarev and V. Y. Kukushkin, New J. Chem., 2015, 39, 9330–9344 RSC; (n) K. I. Kulish, A. S. Novikov, P. M. Tolstoy, D. S. Bolotin, N. A. Bokach, A. A. Zolotarev and V. Y. Kukushkin, J. Mol. Struct., 2016, 1111, 142–150 CrossRef CAS; (o) J. Vicente, J.-A. Abad, M.-J. López-Sáez, P. G. Jones and D. Bautista, Chem. – Eur. J., 2010, 16, 661–676 CrossRef CAS PubMed.
- (a) H. Tom Dieck and H. P. Brehm, Chem. Ber., 1969, 102, 3577–3583 CrossRef CAS; (b) E. A. Goreshnik and V. V. Oliinik, Zh. Neorg. Khim., 1996, 41, 206–208 CAS.
- R. M. Stolley, M. T. Maczka and J. Louie, Eur. J. Org. Chem., 2011, 3815–3824 CrossRef CAS PubMed.
- G. J. Kubas, Inorg. Synth., 1979, 19, 90–92 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., 2008, A64, 112 CrossRef PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- CrysAlisPro, Agilent technologies, Version 1.171.36.20 1.171.36.20 edn., 2012 Search PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision C.01, Gaussian, Inc., Wallingford CT, 2010 Search PubMed.
- D. Figgen, G. Rauhut, M. Dolg and H. Stoll, Chem. Phys., 2005, 311, 227–244 CrossRef CAS.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Spectra of compounds, TGA data, crystal data, and results of theoretical calculations. CCDC 1578477–1578479. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7nj02798b |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2017 |