
Synthesis and characterization of new H-shaped triphenylene discotic room-temperature liquid crystal tetramers by a copper-free click reaction†
Kan
Zhang
,
Yuefeng
Bai
 *,
Chun
Feng
,
Guanghui
Ning
,
Hailiang
Ni
,
Wenhao
Yu
,
Keqing
Zhao
,
Biqin
Wang
and
Ping
Hu
*
*,
Chun
Feng
,
Guanghui
Ning
,
Hailiang
Ni
,
Wenhao
Yu
,
Keqing
Zhao
,
Biqin
Wang
and
Ping
Hu
*
College of Chemistry and Materials Science, Sichuan Normal University, Chengdu, Sichuan Province, P. R. China. E-mail: byf610327@163.com; hp_x@163.com
First published on 15th November 2017
Abstract
Herein, four new H-shaped triphenylene discotic liquid crystal tetramers have been designed and synthesized using a copper-free [3+2] cycloaddition reaction between the triphenylene dimer and diazide compounds. To probe the molecular self-assembly and mesophase properties, the length and rigid spacers in diazide have been tailored. The tetramers 7 with a soft spacer display a room-temperature enantiotropic DLC mesophase, and X-ray diffraction confirms that they exhibit an unusual mesophase of a lamellar–hexagonal columnar phase (Colhl). The results of the photophysical tests show that all the new H-shaped DLC tetramers display relatively high intensity photoluminescence properties.
Introduction
Among various soft materials, discotic liquid crystals (DLCs) have received significant attention ever since they have been discovered by Chandrasekhar in 1977.1–5 In the LC phase, the DLC molecules can self-assemble into one-dimensional (1D) columns as a charge or energy migration channel and then further self-organize into two-dimensional (2D) lattice forms in their bulk form.6–11 Although the degree of order in the molecular alignment of DLCs is critical to liquid crystal devices, the discotic molecules possess full fluctuational, translational, and rotational freedom in their molecular alignment, and this significantly affects the mobility within the columns of DLCs.12,13 Therefore, efforts have been made to enhance the degree of order in the columnar mesophase. More generally, control over the degree of order of the molecular alignment in DLCs is usually achieved by modifying the molecular structure and/or relying on various anchoring effects such as π–π interactions,14–16 hydrogen bonding,17–21 metal complexation,22–24 donor–acceptor effects25,26 and microphase separation effects containing fluorophilic–fluorophobic effects,27–31 hydrophilic–hydrophobic effects,32–35 and other intermolecular interactions.36 Evidently, the self-assembly behavior in DLCs depends significantly on the molecular structure variations, and use of different linkages among the rigid cores to construct DLC dimers or oligomers is an efficient method.37–40In the reported DLC oligomer series, two types of materials have been prepared; one is a linear-shaped DLC in which the cores are connected one by one via a soft or rigid linkage. The other is a star-shaped DLC in which the cores are linked to a central unit. The units can be a single carbon atom,41,42 benzene,43,44 triazine,45–47 siloxane,48 or another aromatic core. The development of novel shaped DLC molecules should open new perspectives for their use as sophisticated functional materials. Therefore, we have designed and synthesised new H-shaped triphenylene DLC tetramers in which two triphenylene cores are initially connected by carbon–carbon triple bonds. Then, two of the triphenylene dimers are connected by a soft alkyl chain or a rigid biphenyl group linkage via a copper-free [3+2] cycloaddition reaction known as the click reaction.
We know that the click chemistry has been widely investigated and applied in almost every field of synthetic chemistry over the past fifteen years,49–51 and the copper(I)-catalyzed azide-terminal alkyne cycloaddition (CuAAC) is an archetypical example of the five common types of click chemistry.52–58 However, residual trace amounts of copper irons originating from the CuAAC are toxic to living cells and can affect the conduction properties of photo-electronic materials as they can form lattice defects.59–61 Our group has developed the click reaction method of strain-promoted azide–alkyne coupling (SPAAC), and the cycloheptyne substrate has been replaced by linear carbon–carbon triple bonds bearing electron-withdrawing groups.62 As part of an ongoing programme aimed at enhancing the degree of order and charge migration rate, new H-shaped DLCs were achieved under these reaction conditions via a double click reaction with moderate yields.
Experimental
General methods
1H NMR and 13C NMR spectra were obtained at room temperature using a Varian 400 spectrometer. Chemical shifts are given in ppm (δ) relative to tetramethylsilane (TMS). High-resolution mass spectroscopy (HRMS) was conducted using Varian 7.0T FTICR-MS or Waters Xevo G2-XS Tof. The liquid crystalline properties were studied using an Olympus BX41 polarising optical microscope (POM) with crossed polarizers and a Linkam T95-PE hot stage. Differential scanning calorimetry (DSC) and thermal gravimetric analysis (TGA) were performed using TA Discovery series, and X-ray diffraction (XRD) was carried out using Rigaku SmartLab (3) with a hot accessory. UV/vis absorption spectra were obtained using a Perkin Elmer Lambda 950 spectrophotometer. Fluorescence spectra were obtained using HORIBA Fluoromax-4p. Reagents were purchased from commercial suppliers and used as received. 2-Hydroxy-3,6,7,10,11-penta hexyloxy triphenylene was synthesized according to a literature procedure.632-Hydroxy-3,6,7,10,11-penta hexyloxy triphenylene 1
1H NMR (CDCl3, 400 MHz) δ (ppm): 7.96 (s, 1 H, ArH), 7.82–7.83 (m, 4 H, ArH), 7.77 (s, 1 H, ArH), 5.91 (s, 1 H, ArOH), 4.29 (t, J = 6.4 Hz, 2 H, OCH2), 4.19–4.24 (m, 8 H, OCH2), 1.92–1.96 (m, 10 H, CH2), 1.56–1.58 (m, 10 H, CH2), 1.38–1.41 (m, 20 H, CH2), 0.92–0.94 (m, 15 H, CH3). 13C NMR (CDCl3, 100 MHz) δ (ppm): 149.07, 148.93, 148.74, 148.66, 145.80, 145.19, 123.87, 123.64, 123.57, 123.50, 123.14, 122.93, 107.46, 107.27, 107.10, 106.29, 104.25, 69.86, 69.82, 69.55, 69.05, 31.70, 31.66, 31.64, 29.46, 29.41, 29.28, 25.87, 25.84, 25.82, 22.68, 22.65, 14.09. HRMS (ESI-TOF): calculated for C48H72O6 [M + H]+, 745.5402; found: 745.5423.Ethyl 2-((3,6,7,10,11-pentakis(hexyloxy)triphenylen-2-yl)oxy) acetate 2
Under an Ar atmosphere, 2-hydroxy-3,6,7,10,11-pentakis(hexyloxy)triphenylene 1 (1.6 g, 2.0 mmol) and ethyl 2-bromoacetate (720 mg, 4 mmol) were dissolved to a suspension of K2CO3 (1.10 g, 8 mmol) in 50 mL of acetone, and the mixture was stirred at reflux. After full consumption of the starting material, which was detected by TLC, the mixture was allowed to cool down to room temperature, and 100 mL of H2O was added. The resulting mixture was subsequently extracted with ethyl acetate (35 mL × 3), and the organic extracts were combined and washed with saturated brine, dried over anhydrous MgSO4, and concentrated in vacuo. Then, the resulting residue was purified by column chromatography (silica gel, eluting with dichloromethane (DCM)/petroleum (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1)) and recrystallized from methanol to obtain a pure compound 2 as a white solid (1.42 g, 80%, m.p. 81.5 °C). 1H NMR (CDCl3, 400 MHz) δ (ppm): 7.94 (s, 1 H, ArH), 7.83–7.84 (m, 4 H, ArH), 7.79 (s, 1 H, ArH), 4.87 (s, 2 H, OCH2COO), 4.28–4.34 (m, 2 H, COOCH2), 4.20–4.27 (m, 10 H, OCH2), 1.91–1.98 (m, 10 H, CH2), 1.54–1.60 (m, 10 H, CH2), 1.36–1.40 (m, 20 H, CH2), 1.33 (t, J = 7.2 Hz, 3 H, CH2), 0.93 (t, J = 6.6 Hz, 15 H, CH2). 13C NMR (CDCl3, 100 MHz) δ (ppm): 169.34, 149.22, 149.08, 149.02, 148.82, 148.79, 147.10, 125.04, 123.97, 123.40, 123.29, 123.24, 123.13, 110.31, 107.40, 107.19, 106.92, 106.74, 106.41, 69.72, 69.51, 69.30, 67.81, 61.33, 31.70, 29.40, 29.31, 25.87, 25.85, 22.69, 14.29, 14.11. HRMS (ESI-TOF): calculated for C52H78O8 [M + H]+, 831.5769; found: 831.5786.
:1)) and recrystallized from methanol to obtain a pure compound 2 as a white solid (1.42 g, 80%, m.p. 81.5 °C). 1H NMR (CDCl3, 400 MHz) δ (ppm): 7.94 (s, 1 H, ArH), 7.83–7.84 (m, 4 H, ArH), 7.79 (s, 1 H, ArH), 4.87 (s, 2 H, OCH2COO), 4.28–4.34 (m, 2 H, COOCH2), 4.20–4.27 (m, 10 H, OCH2), 1.91–1.98 (m, 10 H, CH2), 1.54–1.60 (m, 10 H, CH2), 1.36–1.40 (m, 20 H, CH2), 1.33 (t, J = 7.2 Hz, 3 H, CH2), 0.93 (t, J = 6.6 Hz, 15 H, CH2). 13C NMR (CDCl3, 100 MHz) δ (ppm): 169.34, 149.22, 149.08, 149.02, 148.82, 148.79, 147.10, 125.04, 123.97, 123.40, 123.29, 123.24, 123.13, 110.31, 107.40, 107.19, 106.92, 106.74, 106.41, 69.72, 69.51, 69.30, 67.81, 61.33, 31.70, 29.40, 29.31, 25.87, 25.85, 22.69, 14.29, 14.11. HRMS (ESI-TOF): calculated for C52H78O8 [M + H]+, 831.5769; found: 831.5786.
2-((3,6,7,10,11-pentakis(hexyloxy)triphenylen-2-yl)oxy)acetic acid 3
KOH (423 mg, 7.6 mmol) was dissolved in 20 mL of H2O, and then, 80 mL of ethanol and compound 2 (1.42 g, 1.9 mmol) were added consecutively. The mixture was stirred at reflux overnight. The reaction mixture was allowed to cool to room temperature, acidified with 2 M HCl, and filtered through a Buchner funnel to obtain the desired product 3 as a white powder (1.23 g, yield 90%). 1H NMR (400 MHz, CDCl3) δ (ppm): 8.08 (s, 1 H, ArH), 7.90–7.92 (m, 4 H, ArH), 7.84 (s, 1 H, ArH), 4.95 (s, 2 H, CH2COO), 4.32–4.37 (m, 10 H, OCH2), 2.04–2.05 (m, 10 H, CH2), 1.66–1.68 (m, 10 H, CH2), 1.47–1.50 (m, 20 H, CH2), 1.03–1.05 (m, 15 H, CH3). 13C NMR (CDCl3, 100 MHz) δ (ppm): 170.73, 149.59, 149.20, 149.06, 148.82, 148.63, 146.77, 126.02, 124.33, 123.47, 123.36, 123.07, 122.80, 112.35, 107.64, 107.15, 106.80, 106.33, 106.10, 69.87, 69.72, 69.63, 69.46, 69.36, 69.30, 31.69, 31.61, 29.43, 29.38, 29.36, 29.11, 25.86, 25.84, 25.78, 22.68, 22.61, 14.09. HRMS (ESI-TOF): calculated for C50H74O8 [M − H]−, 801.5311; found: 801.5301.But-2-yne-1,4-diylbis(2-((3,6,7,10,11-pentahexyloxytriphenylen-2-yl)oxy)acetate) 4
Compound 3 (2 g, 2.25 mmol), 1,4-butynediol (96 mg, 1.1 mmol), DCC (513 mg, 2.5 mmol), and DMAP (60 mg) were dissolved in 20 mL of dry dichloromethane, and the resulting mixture was stirred at room temperature for 12 h. After removing the by-product via filtration, the filtrate was concentrated in vacuo, and the residue was purified by column chromatography (silica gel, eluting with DCM/petroleum (2:1)) followed by recrystallization from the mixed solvents ethyl acetate/ethanol (v/v = 1:5) to obtain the pure compound 4 as a white solid (1 g, 54%, Colh 122 °C Iso). 1H NMR (400 MHz, CDCl3) δ (ppm): 7.96 (s, 2 H, ArH), 7.79–7.83 (m, 10 H, ArH), 4.87 (s, 4 H, ArOCH2COO), 4.78 (s, 4 H, COOCH2), 4.20–4.25 (m, 20 H, OCH2), 1.91–1.95 (m, 20 H, CH2), 1.54–1.57 (m, 20 H, CH2), 1.37–1.42 (m, 40 H, CH2), 0.90–0.94 (m, 30 H, CH3). 13C NMR (CDCl3, 100 MHz) δ (ppm): 168.55, 149.31, 149.11, 149.05, 148.87, 148.81, 146.87, 125.28, 124.05, 123.36, 123.29, 123.26, 123.08, 110.94, 107.50, 107.17, 106.94, 106.76, 106.49, 80.76, 69.77, 69.71, 69.50, 69.31, 69.29, 67.61, 52.56, 31.69, 31.66, 29.42, 29.39, 29.29, 25.86, 22.67, 14.08. MALDI-MS: calculated for C104H150O16 [M]+, 1655.0924; found: 1655.0924.
1,4-Diazidobutane 6a
1,4-Dibromobutane 5a (2 g, 9.3 mmol) and NaN3 (3 g, 46 mmol) were added to 2 mL of DMF, and the resulting mixture was carefully heated to 85 °C and stirred for about 24 h. The reaction mixture was allowed to cool to room temperature, 100 mL of H2O was added, and the reaction mixture was extracted with DCM (30 mL × 3); the organic extracts were combined and washed with saturated brine, dried over anhydrous MgSO4, and concentrated in vacuo. Compound 6a was obtained as a light yellow oil and used without any further purification (1.1 g, 85%). 1H NMR (400 MHz, CDCl3) δ (ppm): 3.45 (t, J = 7.0 Hz, 4 H, CH2), 2.03–2.06 (m, 4 H, CH2). 13C NMR (CDCl3) δ (ppm): 32.56, 30.92. HRMS (ESI-TOF): calculated for C4H8N6 [M]+, 140.0810; found: 140.0802.1,6-Diazidohexane 6b
The synthesis procedure of 6b is same with 6a. 1,6-Dibromohexane 5b (2 g, 8.2 mmol), NaN3 (2.7 g, 41 mmol), and DMF 2 mL. The product 6b was obtained in 87% yield (1.2 g) by the procedure analogous to that described for 6a. 1H NMR (400 MHz, CDCl3) δ (ppm): 3.27 (t, J = 7.0 Hz, 4 H, CH2), 1.59–1.62 (m, 4 H, CH2), 1.38–1.42 (m, 4 H, CH2). 13C NMR (CDCl3, 100 MHz) δ (ppm): 51.20, 28.66, 26.22. HRMS (ESI-TOF): calculated for C6H12N6 [M]+, 168.1123; found: 168.1175.Tetramer 7a
The triphenylene dimer 4 (200 mg, 0.12 mmol) and 1,4-diazidobutane 6a (24.6 mg, 0.06 mmol) were dissolved in 2 mL of xylene and heated at reflux for 24 h. After full consumption of the starting material, 4 was detected by TLC, and the mixture was allowed to cool down to room temperature. Then, it was purified by column chromatography (silica gel, eluting with DCM/ethyl acetate (50:1)) directly and recrystallized from methanol to obtain the pure compound 7a as a white powder (120 mg, 53%). 1H NMR (CDCl3, 400 MHz) δ (ppm): 7.74 (s, 2 H, ArH), 7.73 (s, 2 H, ArH), 7.69 (s, 2 H, ArH), 7.68 (s, 2 H, ArH), 7.65 (s, 2 H, ArH), 7.64 (s, 2 H, ArH), 7.62 (s, 2 H, ArH), 7.59 (s, 10 H, ArH), 5.38 (s, 4 H, ArOCH2COO), 5.22 (s, 4 H, ArOCH2COO), 4.82 (s, 4 H, COOCH2), 4.76 (s, 4 H, COOCH2), 4.04–4.20 (m, 44 H, OCH2, N–CH2), 1.80–1.90 (m, 40 H, CH2), 1.75–1.77 (m, 4 H, CH2), 1.43–1.54 (m, 44 H, CH2), 1.32–1.38 (m, 80 H, CH2), 0.88–0.93 (m, 60 H, CH2). 13C NMR (CDCl3) δ (ppm): 169.00, 168.90, 149.10, 149.07, 149.01, 148.95, 148.84, 148.72, 148.70, 148.66, 148.64, 148.51, 146.67, 146.39, 141.97, 130.34, 124.94, 124.86, 123.79, 123.21, 123.11, 123.04, 123.01, 122.92, 122.81, 109.99, 109.94, 107.13, 107.04, 106.98, 106.84, 106.75, 106.68, 106.54, 106.46, 106.36, 106.27, 69.63, 69.56, 69.51, 69.43, 69.39, 69.36, 69.28, 69.27, 69.23, 69.09, 68.98, 67.29, 67.23, 57.14, 53.13, 47.39, 31.73, 31.63, 31.60, 29.45, 29.21, 29.19, 25.88, 25.75, 25.72, 22.68, 22.62, 14.08. MALDI-MS: calculated for C212H308N6O32 [M + Na]+, 3475.2623; found: 3475.2610.
Tetramer 7b
The synthesis procedure of tetramer 7b is same with 7a. The triphenylene dimer 4 (200 mg, 0.12 mmol), 1,6-diazidohexane 5b (28 mg, 0.06 mmol), and xylene 2 mL. The product 7b was obtained as a white solid (110 mg, 48%) using a procedure analogous to that described for 7a. 1H NMR (400 MHz, CDCl3) δ (ppm): 7.75 (s, 4 H, ArH), 7.67–7.69 (m, 8 H, ArH), 7.61 (s, 12 H, ArH), 5.42 (s, 4 H, CH2COO), 5.27 (s, 4 H, CH2COO), 4.84 (s, 4 H, COOCH2), 4.79 (s, 4 H, COOCH2), 4.03–4.20 (m, 44 H, ArOCH2, N–CH2), 1.80–1.94 (m, 40 H, CH2), 1.66–1.68 (m, 4 H, CH2), 1.48–1.57 (m, 40 H, CH2), 1.33–1.40 (m, 80 H, CH2), 1.09–1.11 (m, 4 H, CH2), 0.89–0.95 (m, 60 H, CH3). 13C NMR (CDCl3) δ (ppm): 169.05, 168.75, 149.11, 149.04, 148.98, 148.93, 148.84, 148.68, 148.65, 148.61, 148.57, 146.64, 146.38, 141.89, 130.20, 124.99, 124.86, 123.81, 123.78, 123.22, 123.18, 123.11, 123.04, 123.01, 122.91, 122.81, 110.15, 109.92, 107.06, 106.97, 106.82, 106.70, 106.65, 106.48, 106.39, 106.30, 106.22, 31.74, 31.66, 31.62, 29.64, 29.45, 29.24, 29.21, 25.90, 25.79, 25.76, 22.70, 22.65, 14.11. MALDI-MS: calculated for C214H312N6O32 [M + Na]+, 3503.2936; found: 3503.2938.[1,1′-Biphenyl]-4,4′-diyl bis(4-bromobutanoate) 9a
4,4′-Dihydroxybiphenyl 8 (1.5 g, 6 mmol), 4-bromobutyric acid (3.36 g, 20 mmol), DCC (1.66 g, 20 mmol), and DMAP (200 mg) were dissolved in 25 mL of dry dichloromethane, and the resulting mixture was stirred at room temperature for 12 h. After removing the by-product via filtration, the filtrate was concentrated in vacuo, and the residue was purified by column chromatography (silica gel, eluting with DCM/petroleum (2:1)) followed by recrystallization from the mixed solvents of ethyl acetate/ethanol (v/v = 1:5) to obtain the pure compound 9a as a white solid (1.55 g, 46%). 1H NMR (400 MHz, CDCl3) δ (ppm): 7.56 (d, J = 8.4 Hz, 4 H, ArH), 7.16 (d, J = 8.4 Hz, 4 H, ArH), 3.56 (t, J = 6.2 Hz, 4 H, CH2Br), 2.81 (t, J = 7.0 Hz, 4 H, CH2COO), 2.30–2.33 (m, 4 H, CH2). 13C NMR (CDCl3) δ (ppm): 171.23, 149.96, 138.15, 128.20, 121.86, 32.67, 32.53, 27.54. HRMS (ESI-TOF): calculated for C20H20Br2O4 [M + H]+, 482.9801; found: 482.9834.
[1,1′-Biphenyl]-4,4′-diyl bis(6-bromohexanoate) 9b
The synthesis procedure of 9b is same with 9a. 4,4′-Dihydroxybiphenyl 8 (3 g, 16 mmol), 6-bromohexanoic acid (7.82 g, 40 mmol), DCC (3.32 g, 16 mmol), DMAP (400 mg), and dry DCM 50 mL. The product 9b was obtained as a white powder (4.5 g, 51%, m.p. 44 °C) using a procedure analogous to that described for 9a. 1H NMR (400 MHz, CDCl3) δ (ppm): 7.55 (d, J = 8.8 Hz, 4 H, ArH), 7.15 (d, J = 8.8 Hz, 4 H, ArH), 3.45 (t, J = 6.6 Hz, 4 H, CH2Br), 2.61 (t, J = 7.4 Hz, 4 H, CH2COO), 1.90–1.98 (m, 4 H, CH2), 1.77–1.84 (m, 4 H, CH2), 1.55–1.63 (m, 4 H, CH2). 13C NMR (CDCl3) δ (ppm): 172.05, 150.08, 138.06, 128.16, 121.92, 34.15, 33.63, 33.58, 32.36, 27.61, 24.06. HRMS (ESI-TOF): calculated for C24H28Br2O4 [M + H]+, 539.0427; found: 539.0437.[1,1′-Biphenyl]-4,4′-diyl bis(4-azidobutanoate) 10a
[1,1′-Biphenyl]-4,4′-diyl bis(4-bromobutanoate) 9a (700 mg, 1.67 mmol) and NaN3 (542 mg, 8.3 mmol) were added to 2 mL of DMF, and the resulting mixture was stirred at room temperature for 24 h. After full consumption of the starting material, 9a was detected by TLC, 100 mL of H2O was added to the mixture, and the resulting mixture was extracted with DCM (30 mL × 3); the organic extracts were combined and washed with saturated brine, dried over anhydrous MgSO4, and concentrated in vacuo, and the resulting residue was purified by column chromatography (silica gel, eluting with DCM/petroleum (2:1)) followed by recrystallization from the mixed solvents ethyl acetate and ethanol (v/v = 1:5) to obtain the pure compound 10a as a white solid (544 mg, 80%). 1H NMR (400 MHz, CDCl3) δ (ppm): 7.56 (d, J = 8.5 Hz, 4 H, ArH), 7.16 (d, J = 8.5 Hz, 4 H, ArH), 3.47 (t, J = 6.6 Hz, 4 H, CH2N3), 2.71 (t, J = 7.2 Hz, 4 H, CH2COO), 2.01–2.08 (m, 4 H, CH2). 13C NMR (CDCl3) δ (ppm): 171.38, 149.98, 138.14, 128.20, 121.85, 50.54, 31.26, 24.20. HRMS (ESI-TOF): calculated for C20H20N6O4 [M + H]+, 409.1624; found: 409.1821.
[1,1′-Biphenyl]-4,4′-diyl bis(6-azidohexanoate) 10b
The synthesis procedure of 10b is same with 10a. [1,1′-Biphenyl]-4,4′-diyl bis(6-bromohexanoate) 9b (800 mg, 1.48 mmol), NaN3 (481 mg, 7.4 mmol), and DMF 2 mL. The product 10b was obtained as a white powder (550 mg, 80%), (Cr 36 °C SB 97 °C Iso) using a procedure analogous to that described for 10a. 1H NMR (400 MHz, CDCl3) δ (ppm): 7.56 (d, J = 8.4 Hz, 4 H, ArH), 7.15 (d, J = 8.4 Hz, 4 H, ArH), 3.32 (t, J = 6.8 Hz, 4 H, CH2N3), 2.62 (t, J = 7.4 Hz, 4 H, CH2COO), 1.77–1.85 (m, 4 H, CH2), 1.65–1.72 (m, 4 H, CH2), 1.48–1.56 (m, 4 H, CH2). 13C NMR (CDCl3) δ (ppm): 172.05, 150.07, 138.07, 128.16, 121.90, 51.20, 34.16, 28.58, 26.22, 24.43. HRMS (ESI-TOF): calculated for C24H28N6O4 [M + H]+, 465.2250; found: 465.2497.Tetramer 11a
The synthesis procedure of tetramer 11a is same with 7a. The triphenylene dimer 4 (150 mg, 0.09 mmol), diazide 10a (6.3 mg, 0.045 mmol), and xylene 2 mL. The product 11a was obtained as a white solid (90 mg, 58%) using a analogous procedure analogous to that described for 7a. 1H NMR (400 MHz, CDCl3) δ (ppm): 7.78 (s, 2 H, ArH), 7.76 (s, 2 H, ArH), 7.72 (s, 6 H, ArH), 7.70 (s, 2 H, ArH), 7.67 (s, 8 H, ArH), 7.63 (s, 2 H, ArH), 7.61 (s, 2 H, ArH), 7.34 (d, J = 8.4 Hz, 4 H, ArH), 6.97 (d, J = 8.4 Hz, 4 H, ArH), 5.46 (s, 4 H, ArOCH2COO), 5.32 (s, 4 H, ArOCH2COO), 4.85 (s, 4 H, COOCH2), 4.80 (s, 4 H, COOCH2), 4.27 (t, J = 7.0 Hz, 4 H, N–CH2), 4.11–4.21 (m, 40 H, OCH2), 2.50 (t, J = 6.8 Hz, 4 H, CH2COOAr), 2.16 (t, J = 6.8 Hz, 4 H, CH2COOAr), 1.84–1.94 (m, 40 H, CH2), 1.54–1.59 (m, 44 H, CH2), 1.34–1.39 (m, 80 H, CH2), 0.90–0.94 (m, 60 H, CH2). 13C NMR (CDCl3) δ (ppm): 170.77, 169.11, 168.70, 149.76, 149.23, 149.16, 149.10, 149.05, 148.92, 148.80, 148.75, 148.72, 148.67, 146.74, 146.47, 142.02, 137.88, 130.50, 127.91, 125.10, 124.93, 123.95, 123.84, 123.30, 123.22, 123.13, 123.04, 122.96, 122.89, 121.65, 110.30, 110.07, 107.23, 107.17, 107.11, 107.05, 106.83, 106.58, 106.52, 106.28, 69.70, 69.63, 69.60, 69.45, 69.42, 69.29, 69.20, 69.08, 67.43, 57.29, 53.39, 53.37, 47.43, 31.72, 31.72, 31.65, 31.60, 30.69, 29.44, 29.26, 29.20, 25.88, 25.79, 25.75, 24.68, 22.68, 22.63, 14.08. MALDI-MS: calculated for C228H320N6O36 [M + Na]+, 3743.3359; found: 3743.3355.Tetramer 11b
The synthesis procedure of tetramer 11b is same with 7a. Triphenylene dimer 4 (200 mg, 0.12 mmol), diazide 10b (10.2 mg, 0.06 mmol) and xylene 2 mL. The product 11a was obtained as a white solid (95 mg, 45%) using a procedure analogous to that described for 7a. 1H NMR (400 MHz, CDCl3) δ (ppm): 7.79 (s, 2 H, ArH), 7.77 (s, 2 H, ArH), 7.72 (s, 8 H, ArH), 7.68 (s, 8 H, ArH), 7.64 (s, 2 H, ArH), 7.61 (s, 2 H, ArH), 7.44 (d, J = 8.8 Hz, 4 H, ArH), 7.02 (d, J = 8.4 Hz, 4 H, ArH), 5.45 (s, 4 H, ArOCH2COO), 5.28 (s, 4 H, ArOCH2COO), 4.86 (s, 4 H, COOCH2), 4.82 (s, 4 H, COOCH2), 4.10–4.21 (m, 40 H, OCH2), 4.05 (t, J = 7.0 Hz, 4 H, N–CH2), 2.37 (t, J = 7.2 Hz, 4 H, CH2COOAr), 1.82–1.93 (m, 40 H, CH2), 1.73–1.76 (m, 4 H, CH2), 1.55–1.56 (m, 44 H, CH2), 1.37–1.38 (m, 80 H, CH2), 1.20–1.25 (m, 4 H, CH2), 0.91–0.94 (m, 60 H, CH3). 13C NMR (CDCl3) δ (ppm): 171.69, 169.12, 168.77, 149.23, 149.14, 149.08, 149.03, 148.93, 148.77, 148.69, 148.64, 146.70, 146.44, 141.87, 137.89, 130.15, 127.96, 125.08, 124.90, 123.91, 123.84, 123.30, 123.19, 123.12, 123.10, 123.02, 122.93, 122.87, 121.76, 110.12, 109.91, 107.17, 107.15, 107.05, 106.96, 106.75, 106.49, 106.17, 69.65, 69.61, 69.57, 69.42, 69.23, 69.16, 69.05, 67.36, 67.33, 57.25, 53.31, 48.27, 33.81, 31.73, 31.66, 31.61, 29.50, 29.44, 29.25, 29.20, 25.94, 25.89, 25.80, 25.76, 24.06, 22.69, 22.65, 11.10. MALDI-MS: calculated for C232H328N6O36 [M + Na]+, 3799.3985; found: 3799.3978.Results and discussion
Synthesis and characterization
The synthesis of the new H-shaped triphenylene discotic liquid crystal tetramers is outlined in Scheme 1. The click reaction relies on the initial materials comprising a linear internal alkyne and diazide compounds. Initially, 2-hydroxy-3,6,7,10,11-penta(hexyloxy)triphenylene was synthesized in moderate yield from 1,2-dihexyloxybenzene using FeCl3 as the oxidative cycloaddition reagent following a one pot literature procedure; the corresponding carboxylic acid derivative 3 was obtained after a Williamson ether synthesis and hydrolysis reaction (the detailed procedure of which are described elsewhere). The dimers 4 was subsequently prepared from the corresponding carboxylic acid derivative 3 and 1,4-butynediol using 4-dimethylaminopyridine (DMAP) as the catalyst and dicyclohexylcarbodiimide (DCC) as the condensation agent. In the click reaction, dimer 4 and corresponding diazide 6 or 10 were dissolved in xylene and heated above 140 °C for 24 h; then, the H-shaped triphenylene tetramers were successively obtained. To study the self-assembly properties, the diazide derivatives were changed from soft alkyl diazides (compounds 6a and 6b) to rigid bibenzene diazide derivatives (compounds 10a and 10b). | ||
| Scheme 1 Synthesis of new H-shaped triphenylene discotic liquid crystal tetramers via copper-free click chemistry. Reagents and conditions: (a) BrCH2COOC2H5, K2CO3, acetone, 24 h, reflux; (b) (1) KOH, ethanol, H2O, overnight, reflux, (2) 2 M HCl; (c) 1,4-butynediol, DCC, DMAP, DCM, 12 h, reflux; (d) NaN3, DMF, 24 h, 85 °C; (e) 6a or 6b, xylene, 140 °C for 24 h; (f) 4-bromobutyric acid or 6-bromohexanoic acid, DCC, DMAP, DCM, 12 h; (g) NaN3, DMF, 24 h, 85 °C; and (h) 10a or 10b, xylene, 140 °C for 24 h. | ||
The structures of all new compounds 7a, 7b, 11a, and 11b were confirmed by NMR spectra and high resolution mass spectrometry. The thermal stability of all the target products was determined using thermal gravimetric analysis (TGA). The results show that the products are thermally stable compounds, and the decomposition temperatures at 5% weight loss are over 338 °C (Fig. S1 in the ESI†). The mesomorphism was fully characterized using a combination of polarizing optical microscopy (POM), differential scanning calorimetry (DSC), and X-ray diffraction (XRD).
Mesomorphic properties
Fig. 1a shows one of the typical POM textures obtained for the compound 7a at 25 °C upon cooling under cross polarization. The texture obvious is broken focal conics with straight linear defects, and it is characteristic of an ordered columnar hexagonal phase (Colho).64 The inset image in Fig. 1a is obtained at the same time as obtained for compound 7a in Fig. 1a using POM with a parallel polarizer. It shows that the black areas in Fig. 1a exhibit dendritic textures and expand from the center to outwards. This suggests that the discotic cores have been self-assembled into an ordered column, and the optical axes are perpendicular to the glass surface.65 A similar texture was also obtained for compound 7b, as shown in Fig. 1b. It also exhibited a typical enantiotropic hexagonal columnar phase (Colh). The compounds 11a and 11b did not show any mesophase, and 11a crystallized at 80 °C upon cooling. After melting, the compound 11b did not crystallize upon cooling, and the obtained soft film remained stable without any crystallization phenomenon under room temperatureconditions. | ||
| Fig. 1 The POM images with cross polarization upon cooling obtained for 7a at 90 °C (a) and 7b at 105 °C (b). The inset in Fig. 1a shows the image obtained at the same time obtained for 7a with a parallel polarizer. | ||
The DSC curves obtained for the compounds 7a, 7b, 11a, and 11b during the first cooling and second heating processes are shown in Fig. 2 (7a and 7b) and Fig. S2 (11a and 11b) in the ESI.† The data of DSC was summarized in Table 1, and the peak temperatures are given in °C. Fig. 2 show that there is only one peak in the heating or cooling curves obtained for compounds 7a and 7b. The peak temperatures were 150 °C and 145.5 °C corresponding to the second heating and first cooling procedure for 7a, respectively. Considering the POM results, the peak in the DSC curve can be assigned to the isotropic point, which corresponds to the Colh-to-isotropic liquid transition. The transition temperature of the liquid crystal phase-to-isotropic liquid was 130.9 °C in the second heating curve obtained for 7b, which was lower than that found for compound 7a at about 20 °C. This difference was attributed to the length of the soft spacer between dimers in 7b being longer than that in 7a; therefore, it induces a relatively weaker effect in the system and which is bearing on the stability of the phase.66 As a result, the discotic cores have much more freedom in the column, and the degree of order in 7b was slightly lower than that in 7a, which displayed a relatively low isotropic point temperature. It should be noted that the compounds 7a and 7b cannot be crystallized even if the temperature was reduced to −50 °C or the heating and cooling procedure was conducted many times. Therefore, the compound 7a and 7b are room-temperature enantiotropic DLC materials and reveal a much wider mesophase range as compared to the previously reported triphenylene tetramers.48,67,68
 | ||
| Fig. 2 The DSC thermograms obtained for 7a and 7b on the second heating and first cooling procedure (scan rate: 10 °C min−1). | ||
| Mesophases, transition temperature and enthalpy changes | ||
|---|---|---|
| Second heating/°C (ΔH, kJ mol−1) | First cooling/°C (ΔH, kJ mol−1) | |
| Abbreviations: Cr = crystalline phase; Colh = hexagonal columnar phase; g = glassy state; Iso = isotropic liquid. | ||
| 7a | Colh 150.0 (41.40) Iso | Iso 145.5 (41.86) Colh |
| 7b | Colh 130.9 (35.89) Iso | Iso 123.5 (31.47) Colh |
| 11a | Cr 118.2 (41.16) Iso | Iso 79.5 (18.53) Cr |
| 11b | Cr 109.4 (50.68) Iso | Iso 38.8 (5.9) g |
The DSC results obtained for 11a and 11b are in good accordance with the POM results, and there is only one phase change to the crystal-to-liquid phase transition. The temperature of melting and crystallization for 11a are 118.2 °C and 79.5 °C, respectively. Compound 11b does not display any exothermic peak upon cooling, and there is only one glass transition at 38.8 °C. The most probable reason for this is that the self-assembly of the triphenylene discs is seriously destroyed by the biphenyl group, and they cannot form a liquid crystal phase.
Furthermore, to probe the precise phase structure and molecular self-assembly properties in the mesophase, the compounds 7a and 7b have been studied using variable-temperature XRD. The XRD experiments of 7a and 7b were performed at 25 °C on cooling; the results are presented in Fig. 3 (7a) and Fig. S3 (ESI†) (7b), and the data are summarized in Table 2. For 7a, the X-ray pattern in the wide angle region exhibits a broad amorphous halo, which is centered around 2θ = 20.52°, and a relatively sharp shoulder peak at 2θ = 25.49°. The broad amorphous halo corresponds to the Bragg spacing of the molten alkyl chains, and the shoulder peak can be indexed as the (001) reflection. The d-spacing of the (001) reflection was 3.49 Å, which resulted from the short-range π–π stacking interactions and is referred to the average distance between discs within a column. Previous reports have shown that a DLC with a much higher hole mobility always exhibits a narrow interlayer spacing of around 3.4 Å.69 In general, these two peaks are characteristic for the discotic liquid crystal mesophase.
 | ||
| Fig. 3 The XRD pattern of 7a at 25 °C upon cooling. | ||
| Compound | 2θ | hkl | hkl (Col) | d exp | d cal | Cell const a (Å) | |
|---|---|---|---|---|---|---|---|
| 7a | 2.84 | 100 | 31.08 | 31.08 | |||
| 4.90 | 1′0′0′ | 18.02 | 18.00 | 20.79 | |||
| 5.60 | 200 | 15.77 | 15.54 | ||||
| 8.51 | 300 | 1′1′0′ | 10.38 | 10.36 | 10.39 | ||
| 20.52 | Broad | 4.32 | |||||
| 25.49 | 001 | 3.49 | |||||
| 7b | 2.89 | 100 | 30.56 | 30.55 | |||
| 4.94 | 1′0′0′ | 17.84 | 17.94 | 20.72 | |||
| 5.64 | 200 | 15.56 | 15.28 | ||||
| 8.50 | 300 | 1′1′0′ | 10.40 | 10.18 | 10.36 | ||
| 20.15 | Broad | 4.42 | |||||
| 24.76 | 001 | 3.59 | |||||
There are four reflection peaks at 2θ = 2.84, 4.90, 5.60, and 8.51° in the small-angle region, and their reciprocal spacings are 31.08 Å, 18.02 Å, 15.77 Å, and 10.38 Å, respectively. For ease of description, the four peaks are referred as a, b, c, and d. According to these precise calculations, we believed that the mesophase of 7a was an unusual mesophase of the lamellar–hexagonal columnar phase (Colhl).70 In the Colhl mesophase, the molecules of 7a are arranged in layers similar to calamitic mesogens in smectic phases, and in every layer, the molecules of 7a self-assemble into a hexagonal columnar phase due to the π–π interactions and the effect of the spacers. In Fig. 3, the peaks a, c, and d can be indexed to the (100), (200), and (300) reflections in the layered structure, and the ratio of the these spacings are 1:1/2:1/3, respectively. Therefore, the interlayer spacing was about 3.1 nm according to the (100) reflection. On the other hand, the d-spacing of the strong b and d peaks show a ratio of 1:1/√3, and it is the fundamental reflection of a hexagonal columnar phase. Thus, it can be assigned to the (100) and (110) reflections in a hexagonal columnar phase.
The intercolumnar distance (a) in Colhl was calculated to be 2.08 nm using the following equation: a = 2 〈d1′0′0′〉/√3, 〈d1′0′0′〉 = (d1′0′0′ + √3d1′1′0′)/2. The number (n) of molecules per unit cell in a hexagonal lattice is estimated to be 0.68 from n =√3NAa2hρ/2M, where NA is the Avogadro's number (6.02 × 1023 mol−1), M is the molecular weight (3450.27 g mol−1), and ρ is the density of the material, which is approximately equal to 1 g cm−3 at 20 °C.71 The value of n equal to 0.68 suggests that a 2D hexagonal structure cell cannot be formed in a monomeric fashion, which allows the Colhl of discotic mesophase 7a to be schematically drawn, as shown in Fig. 4.
 | ||
| Fig. 4 A schematic of the lamellar–hexagonal columnar phase of the H-shaped DLC compounds 7a and 7b in LC. | ||
The XRD results obtained for compound 7b show a conclusion similar to those of 7a. The mesophase of 7b was also a Colhl, and the intercolumnar distance (a) was calculated to be 20.72 Å. The average distance between disc–disc within a column was 3.59 Å, which suggested that the degree of order of 7b in the column was weaker than that in 7a due to the weaker restriction of the longer spacer.
Photophysical properties
To examine the photophysical and electrochemical properties of the H-shaped DLC compounds, the UV-vis absorption spectra, photoluminescence spectra (PL) (Fig. 5), and cyclic voltammograms (CVs) were obtained. The experimental values of the maximum of absorption (λmax), optical band gap (Eoptg), and HOMO/LUMO energy level are summarized in Table 3. The HOMO and LUMO energy levels were calculated according to the following empirical equations: EHOMO = [−(Eoxp − 0.14) − 4.8] eV and ELUMO = [−(Eredp − 0.14) − 4.8] eV, in which Eoxp/Eredp is the peak potential of chemically irreversible oxidation/reduction, 0.14 V is the potential of ferrocene vs. Ag/AgNO3, and 4.8 eV is the energy level of ferrocene vs. vacuum energy level.72 The optical band gap was calculated using the equation: Eoptg = hc/λonset. | ||
| Fig. 5 (a) The UV-vis absorption spectra (solid line) and PL spectra of all the H-shaped DLC compounds obtained in DCM. (b) The PL spectra of all the H-shaped DLC compounds obtained as a thin film. | ||
| Compound | λ abs | λ onset (nm) | λ PL. sol. | λ PL. film | E optg (eV) | E g (eV) | E HOMO (eV) | E LUMO (eV) |
|---|---|---|---|---|---|---|---|---|
| The optical band gap (Eoptg) was calculated from the low energy absorption onset in the UV-vis absorption spectra according to the equation, Eoptg = hc/λonset, where h is the Planck constant and c is the speed of light. λonset = 357 nm; cyclic voltammograms measured in CH2Cl2 with 0.1 M [(n-C4H9)4N]PF6 at a scanning rate of 0.1 V s−1. | ||||||||
| 7a | 280 | 357 | 387 | 389, 405 | 3.47 | 2.29 | −5.37 | −3.08 |
| 7b | 280 | 357 | 387 | 389, 405 | 3.47 | 2.29 | −5.37 | −3.08 |
| 11a | 280 | 357 | 387 | 389, 405 | 3.47 | 2.27 | −5.39 | −3.12 |
| 11b | 280 | 357 | 387 | 389, 405 | 3.47 | 2.27 | −5.40 | −3.13 |
The maximum absorption wavelength in dry dichloromethane (DCM) was 280 nm, and this was attributed to the π–π* transition of the triphenylene portion. A broad emission band in the range from 350 to 470 nm was observed in the PL of the DCM solution, and λmax = 387 nm. They also showed a strong fluorescence in the solid phase, exhibiting an emission maxima at a wavelength of 389 nm with a very small red-shift as compared to that observed in solution. In addition, it should be noted that the broad emission band at the longer wavelength was caused by the triphenylene excimer, and it implied that there was strong interaction between the adjacent triphenylenes in favor of molecular self-assembly.
The CV was obtained in DCM and tetra-n-butylammonium hexafluorophosphate (n-Bu4NPF6) as the supporting electrolyte. All the typical CVs curves are shown in Fig. 6. All the compounds show three oxidation potentials and one reduction potential in the CV curves. The first oxidation potentials are 0.71, 0.71, 0.73, and 0.74 V corresponding to the compounds 7a, 7b, 11a, and 11b, which can be related to the HOMO energy level. The HOMO energy levels of the H-shaped DLC molecules are well below −5.0 eV; this indicates good air-stability for the photoelectrical devices. In addition, the band gaps obtained from the CV results are in the range of 2.27–2.29 eV, which are significantly different from the optical band gap of 3.47 eV. The deviation of about 1.0 eV was possibly caused by the different aggregation states in the different solutions.
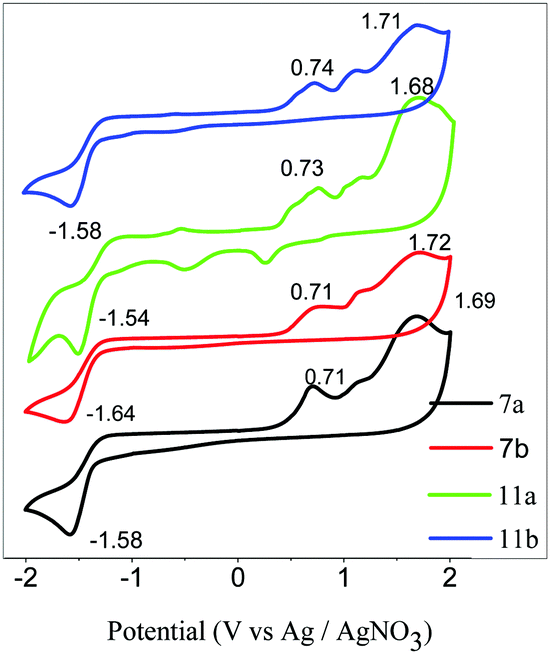 | ||
| Fig. 6 The cyclic voltammograms obtained for 7a, 7b, 11a, and 11b. | ||
Conclusions
A series of new H-shaped triphenylene discotic liquid crystal tetramers has been designed and synthesized using copper-free click chemistry between diazide and a symmetrical triphenylene dimer in which the carbon–carbon triple bond acts as the internal soft linkage. Their mesomorphism and photophysical properties were fully investigated. The compounds 11a and 11b do not display any mesomorphism, whereas the compounds 7a and 7b exhibit a typical enantiotropic liquid crystal phase, and their clearing point is as high as 150.0 °C and 130.9 °C, respectively. The XRD results reveal that 7a and 7b exhibit an unusual mesophase of the lamellar–hexagonal columnar phase (Colhl). In the Colhl mesophase, the molecules of 7a and 7b are arranged in layers similar to calamitic mesogens in smectic phases, and in every layer, the molecules of 7a and 7b are self-assembled into a hexagonal columnar phase. The maximum absorption wavelength of all the newly H-shaped DLC molecules in dry dichloromethane (DCM) was 280 nm, and a strong photoluminescence band at around 390 nm was detected in the film. A new emission band was found around 505 nm, which was attributed to the triphenylene excimer.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the National Natural Science Foundation of China (51073112, 51443004), the Youth Foundation of Sichuan Educational Committee (15ZB0037), and the Foundation of Sichuan Normal University (SYJS2017008) for providing the financial support.Notes and references
- S. Chandrasekhar, B. K. Sadashiva and K. A. Suresh, Pramana, 1977, 9, 471–480 CrossRef CAS.
- T. Woehrle, I. Wurzbach and J. Kirres, Chem. Rev., 2016, 116, 1139–1241 CrossRef CAS PubMed.
- H. K. Bisoyi and Q. Li, Chem. Rev., 2016, 116, 15089–15166 CrossRef CAS PubMed.
- E. K. Fleischmann and R. Zentel, Angew. Chem., Int. Ed., 2013, 52, 8810–8827 CrossRef CAS PubMed.
- X. W. Peng, H. F. Gao, Y. L. Xiao, H. F. Cheng, F. R. Huang and X. H. Cheng, New J. Chem., 2017, 41, 2004–2012 RSC.
- S. Kumar, Chem. Soc. Rev., 2006, 35, 83–109 RSC.
- N. M. Boshta, M. Bomkamp, G. Schnakenburg and S. R. Waldvogel, Eur. J. Org. Chem., 2011, 1985–1992 CrossRef CAS.
- A. Schultz, S. Laschat, S. Diele and M. Nimtz, Eur. J. Org. Chem., 2003, 2829–2839 CrossRef CAS.
- B. Alameddine, R. S. Anju, S. Shetty, N. Baig, F. Al-Sagheer, S. Al-Mousawi and T. A. Jenny, New J. Chem., 2017, 41, 6025–6032 RSC.
- P. H. J. Kouwer and G. H. Mehl, Angew. Chem., Int. Ed., 2003, 42, 6015–6018 CrossRef CAS PubMed.
- L. Y. Zhang, J. L. Li, S. H. Qiu, X. B. Huang and Z. B. Zeng, New J. Chem., 2017, 41, 3260–3264 RSC.
- D. Adam, P. Schuhmacher, J. Simmerer, L. Häussling, K. Siemensmeyer, K. H. Etzbach, H. Ringsdorf and D. Haarer, Nature, 1994, 371, 141–143 CrossRef CAS.
- A. Concellón, M. Marcos, P. Romero, J. L. Serrano, R. Termine and A. Golemme, Angew. Chem., Int. Ed., 2017, 56, 1259–1263 CrossRef PubMed.
- B. M. Zhao, B. Liu, R. Q. Png, K. Zhang, K. A. Lim, J. Luo, J. J. Shao, P. K. H. Ho, C. Y. Chi and J. S. Wu, Chem. Mater., 2010, 22, 435–449 CrossRef CAS.
- S. Tanaka, T. Sakurai, Y. Honsho, A. Saeki, S. Seki, K. Kato, M. Takata, A. Osuka and T. Aida, Chem. – Eur. J., 2012, 18, 10554–10561 CrossRef CAS PubMed.
- M. Yasutake, T. Fujihara, A. Nagasawa, K. Moriya and T. Hirose, Eur. J. Org. Chem., 2008, 4120–4125 CrossRef CAS.
- M. H. Ryu, J. W. Choi, H. J. Kim, N. Park and B. K. Cho, Angew. Chem., Int. Ed., 2011, 50, 5737–5740 CrossRef CAS PubMed.
- T. Kato, N. Mizoshita and K. Kishimoto, Angew. Chem., Int. Ed., 2006, 45, 38–68 CrossRef CAS PubMed.
- J. Pansanel, A. Jouaiti, S. Ferlay, M. W. Hosseini, J. M. Planeix and N. Kyritsakas, New J. Chem., 2006, 30, 71–76 RSC.
- M. M. J. Smulders, P. J. M. Stals, T. Mes, T. F. E. Paffen, A. P. H. J. Schenning, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2010, 132, 620–626 CrossRef CAS PubMed.
- D. Braga, L. Maini, F. Grepioni, A. D. Cian, O. Félix, J. Fischer and M. W. Hosseini, New J. Chem., 2000, 24, 547–553 RSC.
- T. T. Bui, O. Thiebaut, E. Grelet, M. F. Achard, B. G. Bonneval and K. I. M. C. Ching, Eur. J. Inorg. Chem., 2011, 2663–2676 CrossRef CAS.
- A. Głębowska, P. Przybylski, M. Winek, P. Krzyczkowska, A. Krówczyński, J. Szyd1owska, D. Pociecha and E. Górecka, J. Mater. Chem., 2009, 19, 1395–1398 RSC.
- H. Suzuki, K. Kawano, K. Ohta, Y. Shimizu, N. Kobayashi and M. Kimura, ChemistryOpen, 2016, 5, 150–156 CrossRef CAS PubMed.
- Y. Yamauchi, Y. Hanaoka, M. Yoshizawa, M. Akita, T. Ichikawa, M. Yoshio, T. Kato and M. Fujita, J. Am. Chem. Soc., 2010, 132, 9555–9557 CrossRef CAS PubMed.
- G. B. Li, Q. Y. Yang, R. K. Pan, S. G. Liu and Y. W. Xu, New J. Chem., 2017, 41, 6160–6166 RSC.
- N. Terasawa, H. Monobe, K. Kiyohara and Y. Shimizu, Chem. Commun., 2003, 1678–1679 RSC.
- M. H. C. J. V. Houtem, F. Benaskar, C. F. C. Fitié, R. Martín-Rapún, J. A. J. M. Vekemans and E. W. Meijer, Org. Biomol. Chem., 2012, 10, 5898–5908 Search PubMed.
- L. Sosa-Vargas, F. Nekelson, D. Okuda, M. Takahashi, Y. Matsuda, Q. D. Dao, Y. Hiroyuki, A. Fujii, M. Ozaki and Y. Shimizu, J. Mater. Chem. C, 2015, 3, 1757–1765 RSC.
- V. Percec, G. Johansson, G. Ungar and J. P. Zhou, J. Am. Chem. Soc., 1996, 118, 9855–9866 CrossRef CAS.
- G. Johansson, V. Percec, G. Ungar and J. P. Zhou, Macromolecules, 1996, 29, 646–660 CrossRef CAS.
- Z. H. A. Lawati, B. Alkhairalla, J. P. Bramble, J. R. Henderson, R. J. Bushby and S. D. Evans, J. Phys. Chem. C, 2012, 116, 12627–12635 Search PubMed.
- L. D. Campo, T. Varslot, M. J. Moghaddam, J. J. K. Kirkensgaard, K. Mortensen and S. T. Hyde, Phys. Chem. Chem. Phys., 2011, 13, 3139–3152 RSC.
- A. Suemasu, K. Ikegami, N. Maji and T. Tanaka, J. Soc. Inf. Disp., 2005, 13, 599–603 CrossRef CAS.
- I. Bury, B. Heinrich, C. Bourgogne, D. Guillon and B. Donnio, Chem. – Eur. J., 2006, 12, 8396–8413 CrossRef CAS PubMed.
- F. Fernandez-Palacio, M. Poutanen, M. Saccone, A. Siiskonen, G. Terraneo, G. Resnati, O. Ikkala, P. Metrangolo and A. Priimagi, Chem. Mater., 2016, 28, 8314–8321 CrossRef CAS PubMed.
- D. Demus, J. Goodby, G. W. Gray, H. W. Spiess and V. Vill, Handbook of Liquid Crystals: Low Molecular Weight Liquid Crystals II, Liquid Crystal Dimers and Oligomers Liquid Crystal Dimers and Oligomers, 2008, vol. 2B, ch. X, pp. 801–834 Search PubMed.
- S. Kumar, Liq. Cryst., 2005, 32, 1089–1113 CrossRef CAS.
- Y. F. Bai, K. Q. Zhao, P. Hu, B. Q. Wang and C. Redshaw, Curr. Org. Chem., 2013, 17, 871–885 CrossRef CAS.
- C. W. Ong, Y. C. Chan, M. C. Yeh, H. Y. Lin and H. F. Hsu, RSC Adv., 2013, 3, 8657–8659 RSC.
- A. Pegenau, T. Hegmann, C. Tschierske and S. Diele, Chem. – Eur. J., 1999, 5, 1643–1670 CrossRef CAS.
- C. Tschierske, Angew. Chem., Int. Ed., 2013, 52, 8828–8878 CrossRef CAS PubMed.
- C. Y. Bao, R. Lu, M. Jin, P. C. Xue, C. H. Tan, T. H. Xu, G. F. Liu and Y. Y. Zhao, Chem. – Eur. J., 2006, 12, 3287–3294 CrossRef CAS PubMed.
- A. Timme, R. Kress, R. Q. Albuquerque and H. W. Schmidt, Chem. – Eur. J., 2012, 18, 8329–8339 CrossRef CAS PubMed.
- B. Roy, N. De and K. C. Majumdar, Chem. – Eur. J., 2012, 18, 14560–14588 CrossRef CAS PubMed.
- E. Beltran, M. Garzoni, B. Feringan, A. Vancheri, J. Barbera, J. L. Serrano, M. P. Giovanni and S. Teresa, Chem. Commun., 2015, 51, 1811–1814 RSC.
- C. P. Umesh, A. T. M. Marcelis and H. Zuilhof, Liq. Cryst., 2015, 42, 1450–1459 CrossRef CAS.
- A. Zelcer, B. Donnio, C. Bourgogne, F. D. Cukiernik and D. Guillon, Chem. Mater., 2007, 19, 1992–2006 CrossRef CAS.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS PubMed.
- C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef.
- Applications of click chemistry themed issue in Chem. Soc. Rev. such as: C. L. Droumaguet, C. Wang and Q. Wang, Chem. Soc. Rev., 2010, 39, 1233–1239 RSC; Y. Hua and A. H. Flood, Chem. Soc. Rev., 2010, 39, 1262–1271 RSC and so on.
- J. N. Brantley, K. M. Wiggins and C. W. Bielawski, Science, 2011, 333, 1606–1609 CrossRef CAS PubMed.
- B. Pradhan, N. Chakraborty, R. K. Gupta, G. Shanker and A. S. Achalkumar, New J. Chem., 2017, 41, 879–888 RSC.
- B. S. Sumerlin and A. P. Vogt, Macromolecules, 2010, 43, 1–13 CrossRef CAS.
- Y. Angell and K. Burgess, Angew. Chem., Int. Ed., 2007, 46, 3649–3651 CrossRef CAS PubMed.
- L. Ye, L. Q. Wan and F. R. Huang, New J. Chem., 2017, 41, 4424–4430 RSC.
- V. Saravanan, A. Kannan and P. Rajakumar, New J. Chem., 2017, 41, 1714–1722 RSC.
- A. S. Kumar, N. Kommu, V. D. Ghule and A. K. Sahoo, J. Mater. Chem. A, 2014, 2, 7917–7926 CAS.
- D. C. Kennedy, C. S. McKay, M. C. B. Legault, D. C. Danielson, J. A. Blake, A. F. Pegoraro, A. Stolow, Z. Mester and J. P. Pezacki, J. Am. Chem. Soc., 2011, 133, 17993–18001 CrossRef CAS PubMed.
- S. E. Hook, H. L. Osborn, L. A. Golding, D. A. Spadaro and S. L. Simpson, Environ. Sci. Technol., 2014, 48, 3504–3512 CrossRef CAS PubMed.
- M. Rózga and W. Bal, Chem. Res. Toxicol., 2010, 23, 298–308 CrossRef PubMed.
- Y. F. Bai, L. Bao, P. Hu, B. Q. Wang, C. Redshaw and K. Q. Zhao, Liq. Cryst., 2013, 40, 97–105 CrossRef CAS.
- K. Q. Zhao, Y. F. Bai, P. Hu, B. Q. Wang and Y. Shimizu, Mol. Cryst. Liq. Cryst., 2009, 509, 77–88 Search PubMed.
- S. Laschat, A. Baro, N. Steinke, F. Giesselmann, C. Hägele, G. Scalia, R. Judele, E. Kapatsina, S. Sauer, A. Schreivogel and M. Tosoni, Angew. Chem., Int. Ed., 2007, 46, 4832–4887 CrossRef CAS PubMed.
- K. M. Psutka, J. Williams, J. A. Paquette, O. Calderon, K. J. A. Bozek, V. E. Williams and K. E. Maly, Eur. J. Org. Chem., 2015, 1456–1463 CrossRef CAS.
- N. Boden, R. J. Bushby, A. N. Cammidge, A. El-Mansoury, P. S. Martina and Z. Lu, J. Mater. Chem., 1999, 9, 1391–1402 RSC.
- R. Zniber, R. Achour, M. Z. Cherkaoui, B. Donnio, L. Gehringer and D. Guillon, J. Mater. Chem., 2002, 12, 2208–2213 RSC.
- J. L. Schulte, S. Laschat, V. Vill, E. Nishikawa, H. Finkelmann and M. Nimtz, Eur. J. Org. Chem., 1998, 2499–2506 CrossRef CAS.
- E. M. Garća-Frutos, U. K. Pandey, R. Termine, A. Omenat, J. Barberá, J. L. Serrano, A. Golemme and B. Gómez-Lor, Angew. Chem., Int. Ed., 2011, 50, 7399–7402 CrossRef PubMed.
- H. Sakashita, A. Nishitani, Y. Sumiya, H. Terauchi, K. Ohta and I. Yamamoto, Mol. Cryst. Liq. Cryst., 1988, 163, 211–219 CrossRef CAS.
- T. Kushida, A. Shuto, M. Yoshio, T. Kato and S. Yamaguchi, Angew. Chem., Int. Ed., 2015, 54, 6922–6925 CrossRef CAS PubMed.
- J. Heinze, Angew. Chem., Int. Ed. Engl., 1984, 23, 831–847 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7nj02695a |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2018 |