
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Substituent effects on the basicity (pKa) of aryl guanidines and 2-(arylimino)imidazolidines: correlations of pH-metric and UV-metric values with predictions from gas-phase ab initio bond lengths†
Christophe
Dardonville
 *a,
Beth A.
Caine
bc,
Marta
Navarro de la Fuente
a,
Guillermo
Martín Herranz
a,
Beatriz
Corrales Mariblanca
a and
Paul L. A.
Popelier
bc
*a,
Beth A.
Caine
bc,
Marta
Navarro de la Fuente
a,
Guillermo
Martín Herranz
a,
Beatriz
Corrales Mariblanca
a and
Paul L. A.
Popelier
bc
aInstituto de Química Médica, IQM-CSIC, Juan de la Cierva 3, E-28006 Madrid, Spain. E-mail: dardonville@iqm.csic.es; Fax: +34 915644853; Tel: +34 912587490
bManchester Institute of Biotechnology (MIB), 131 Princess Street, Manchester, M1 7DN, UK
cSchool of Chemistry, Univ. of Manchester, Oxford Road, Manchester M13 9PL, UK
First published on 30th August 2017
Abstract
The dissociation constants of two related series of 2-(arylimino)imidazolidine and aryl guanidine α2-adrenoceptor antagonists (35 compounds in total) were measured by potentiometric titrations and by UV-spectrophotometry using the 96-well microtitre plate method. The experimental values obtained using both methods were quite consistent and showed a very good agreement with the pKa values calculated using the AIBLHiCoS methodology, which uses only a single bond length obtained using ab initio calculations at a low level of theory. The prediction power of the imidazolidine and guanidine set of compounds was very good with deviations typically <0.30 and <0.24 pKa units, and a mean absolute error (MAE) of 0.23 and 0.29, respectively. The study of the quantitative effect of diverse substituents on the basicity of aryl guanidine and 2-(arylimino)imidazolidine derivatives is useful for medicinal chemists working with biologically relevant guanidine-containing molecules.
1. Introduction
The guanidine group of the amino acid arginine is ubiquitous in Nature. In its protonated state, several resonance forms that delocalize the cationic charge of the guanidinium cation over the entire functional group contribute to the high basicity of guanidine (pKaH = 13.6 in water).1 The H-bonding ability of guanidine makes this group a valuable tool for the design of compounds for different pharmacological applications such as DNA binding drugs (e.g. anticancer and antiprotozoal agents),2–11 analgesic compounds,12–14 α1-noradrenaline receptors (e.g. α1-AR antagonists for the treatment of benign prostate hyperplasia),15 or α2-AR modulators for neuropsychiatric therapies.16–19Centrally acting α2-adrenoceptor antagonists are potentially useful pharmacological tools for the treatment of depression and schizophrenia.16 Among these, arylguanidines and 2-(arylimino)imidazolidines represent an important family of α-adrenoceptor modulators.16–20 The prototypical example of a (phenylimino)imidazolidine drug is α2-AR and imidazoline receptor agonist clonidine (i.e. N-(2,6-dichlorophenyl)-4,5-dihydro-1H-imidazol-2-amine) used, among other applications, as an antihypertensive drug and for controlling neuropathic pain (Fig. 1).21 As both the guanidine and 2-aminoimidazoline groups are strong bases, they are protonated at physiological pH. Thus, this physicochemical property may impair the absorption and/or limit the uptake of guanidine-containing molecules through biological membranes, including the blood–brain barrier,22,23 a drawback that is highly relevant for centrally acting drugs.
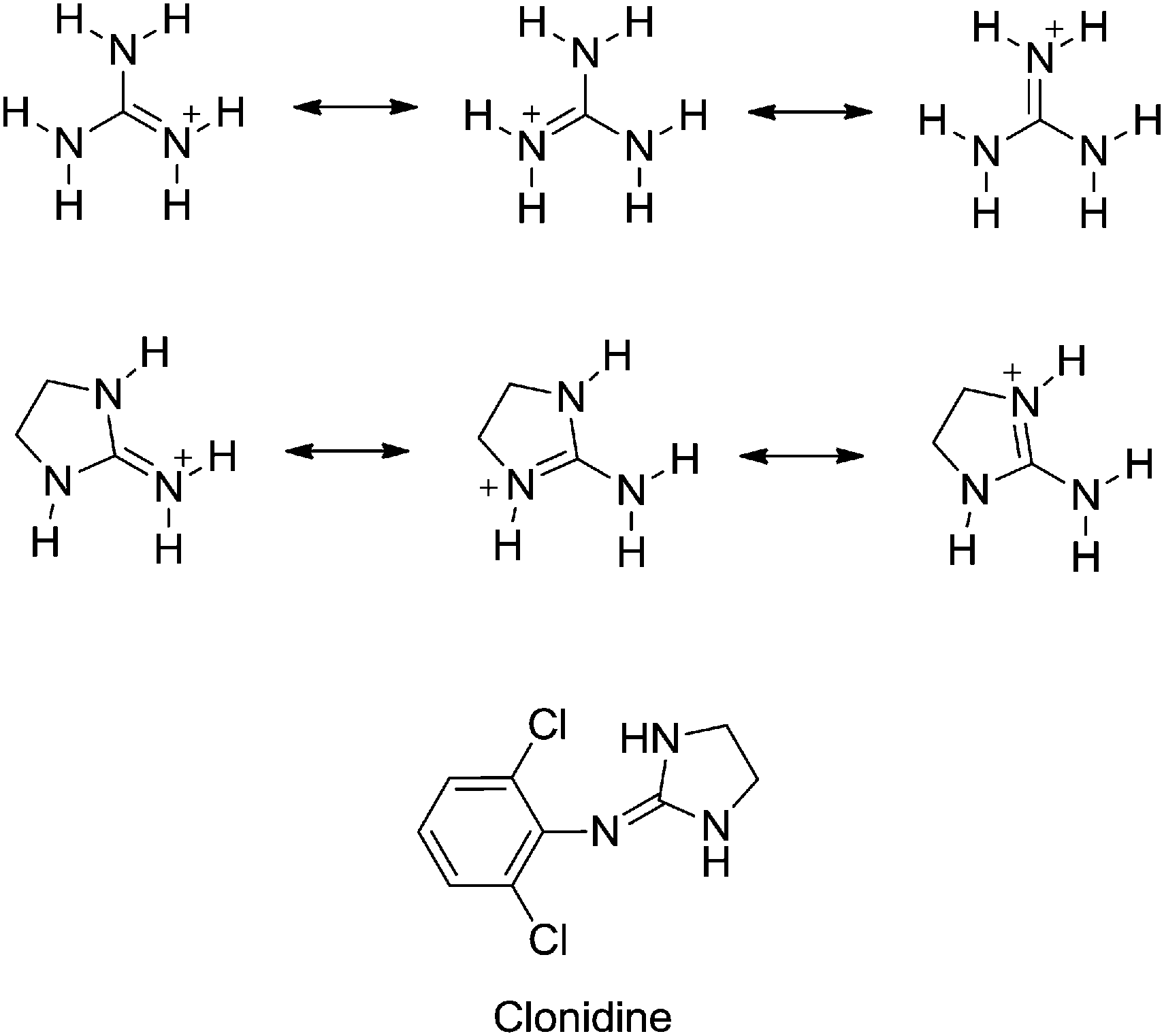 | ||
| Fig. 1 Resonance forms of the guanidinium and 2-iminoimidazolidinium cations responsible for the high basicity of these functional groups. Structure of the archetypal 2-(arylimino)imidazolidine drug clonidine. | ||
The protonation state of a drug molecule determines its lipophilicity, solubility in biological fluids, ability to cross biological membranes, degree of protein binding, and its capacity to bind to biological targets. Hence, knowing the dissociation constants (pKa) of drugs is crucial because this parameter affects the key pharmacokinetic properties such as administration, distribution, metabolism and excretion (ADME).24
One of the objectives of this work is to understand the quantitative effect of diverse substituents on the aqueous basicity of pharmacologically important families of compounds: aryl guanidines and 2-(arylimino)imidazolidines. This knowledge is useful to assist in the design of new biologically relevant guanidine-containing molecules with improved pharmacokinetic properties.22 Another objective is to compare the experimental pKa values of these series with those obtained by ab initio calculations using the AIBLHiCoS method.25,26 The Ab Initio Bond Lengths High Correlation Subsets protocol works on the basis that, for a series of electronically congeneric compounds, chemical space may be partitioned according to a linear free energy relationship (LFER) between the calculated gas-phase equilibrium bond lengths and aqueous pKa values. The equations resulting from highly correlated, local linear relationships for structurally similar molecules are then used as predictive models for calculating the pKa values of other compounds belonging to the appropriate High Correlation Subset (HiCoS). In this work, using a specific bond length and tautomeric form in the neutral state, we have constructed one predictive model with a training set of 13 guanidines and a second model with a training set of 23 2-(phenylimino)imidazolidines. This method is a valuable tool that has already proven its power for the accurate determination of pKas of other guanidine-containing drugs in addition to a number of other functional groups.25,27–29 However, this method has not been tested, until now, with the medically relevant 2-(phenylimino)imidazolidine scaffold. The predictive models built with these series show that the AIBLHiCoS method is an accurate pKa prediction tool with this pharmacologically important family of compounds.
2. Results and discussion
2.1. Synthesis
Trifluoroacetate salts of phenylguanidines (1b, 2b, 3b, 6b, 7b, and 24–27b) and 2-(phenylimino)imidazolidines (2a–10a, and 23a) were synthesized from their Boc-protected precursors using CF3CO2H/CH2Cl2 as reported (Scheme 1).7 These Boc-protected precursors were obtained by a reaction of the corresponding anilines with N,N′-bis-(tert-butoxycarbonyl)imidazolidin-2-thione or N,N′-bis(tert-butoxycarbonyl)thiourea and HgCl2/Et3N/DMF.7 The pyridino derivatives 17–22 were synthesized as hydrochloride salts by Rozas and co-workers following a procedure similar to that shown in Scheme 1.30 | ||
| Scheme 1 Synthesis of 2a–10a, 23a, 1b–3b, 6b, 7b, and 24b–27b. | ||
Compounds 1a and 11a–15a were synthesized as shown in Scheme 2 following the procedure of Mundla et al.31
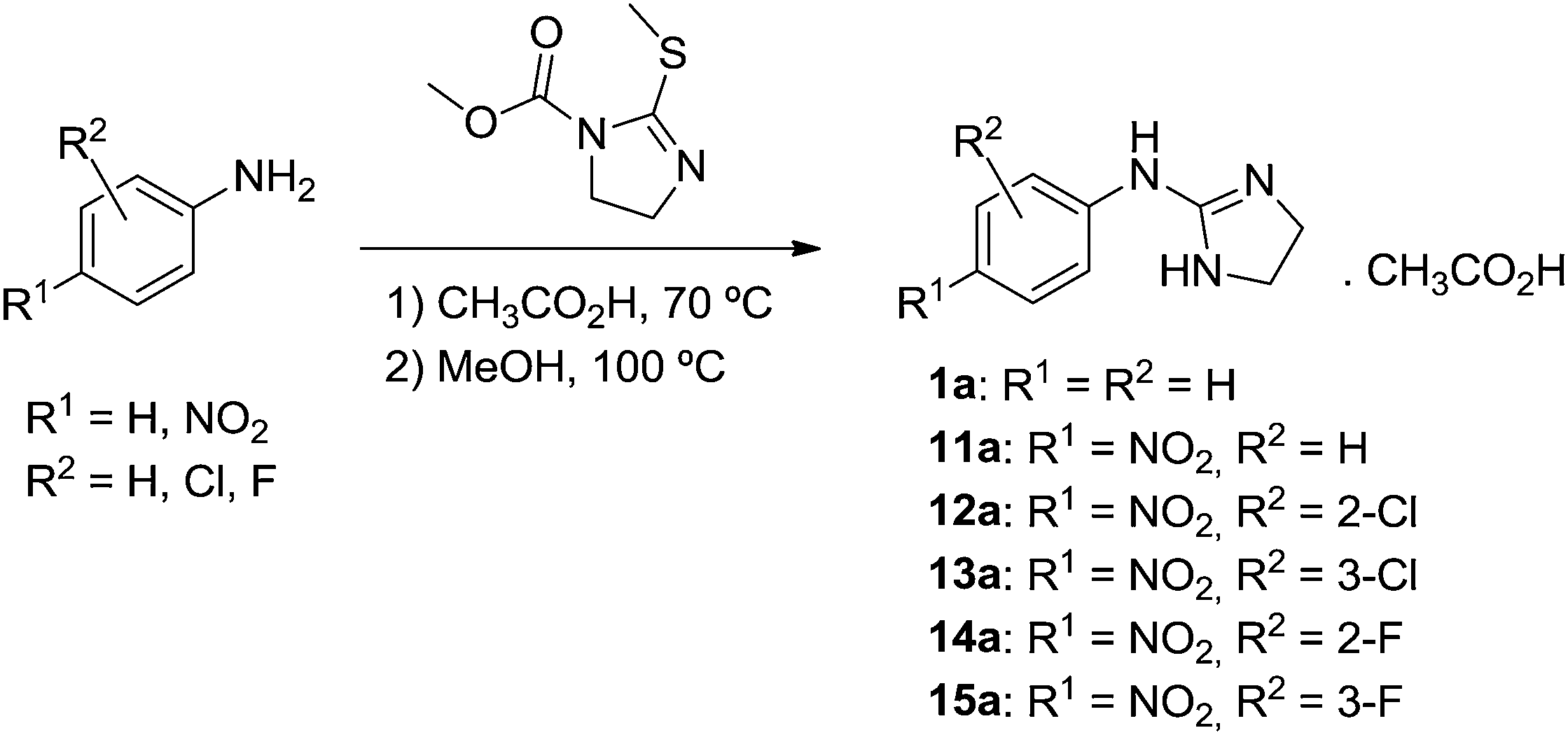 | ||
| Scheme 2 Synthesis of 1a and 11a–15a. | ||
2.2. Correlation between experimental pH-metric, UV-metric, and calculated pKa
The pKa values of two sets of 23 2-(arylimino)imidazolidines (1a–23a) and 12 arylguanidines (1b, 2b, 3b, 6b, 7b, 17b, 18b, 22b–27b), most of them developed by Rozas and co-workers as α2-adrenoceptor ligands,16–20 were measured by potentiometric titration (i.e. pH-metric) at 25 °C using the Sirius T3 apparatus, and by spectrophotometry (i.e. UV-metric) using the 96-well microtitre plate method developed in our group.32 The pKa values determined with the T3 apparatus were highly consistent (SD < 0.03, n = 3) whereas the 96-well microtitre plate method gave somewhat less precise results (SD = 0.02–0.45, n ≥ 3) (Table 1).| Cmpd | Structure | Predicted pKa C–N(im)a pKa N–C(Ph)d | pH-metric pKab | UV-metric pKac |
|---|---|---|---|---|
| a Calculated using the AIBLHiCos approach with the C–N(im) bond model of the imino tautomer. b pH-metric determination at 25 °C using Sirius T3 apparatus. Mean of 3 titrations ± SD. c Determined by UV-spectrophotometry at 30 °C using the 96-well microtiter plate method.32 Mean of ≥ 3 titrations ± SD. d Bond length model built from the same imidazolidine training set with compounds in the imino tautomeric form, but with the C–N(Ph) bond length connecting 2-iminoimidazolidine to benzene. e psKa measured with MeOH as a co-solvent (Yasuda–Shedlowsky extrapolation in H2O). f pKa value of the pyridin-3-yl nitrogen. g The pKa value of the pyridin-2-yl nitrogen could not be worked out. h Literature: pKa = 10.77.33 | ||||
| 1a |
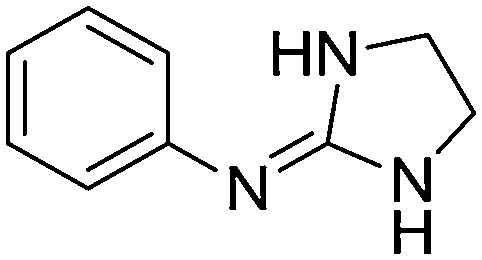
|
10.54
9.90 |
10.24 ± 0.001 | 10.91 ± 0.10 |
| 2a |

|
10.34
9.89 |
10.49 ± 0.01 | — |
| 3a |

|
10.16
9.78 |
10.29 ± 0.01 | 10.40 ± 0.13 |
| 4a |
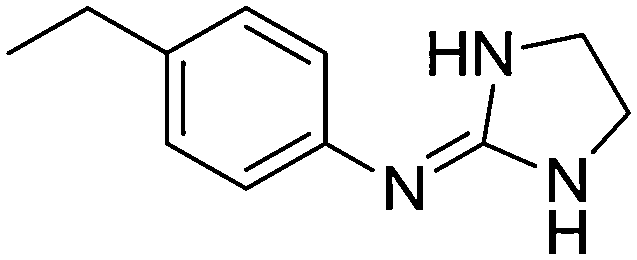
|
10.33
9.89 |
10.42 ± 0.001 | 10.79 ± 0.10 |
| 5a |

|
10.49
9.94 |
10.50 ± 0.01 | 11.07 ± 0.15 |
| 6a |
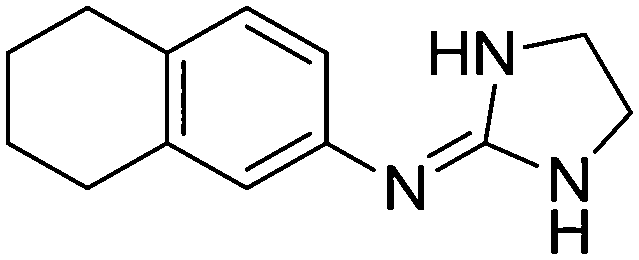
|
10.54
9.97 |
10.44 ± 0.01 | 10.27 ± 0.17 |
| 7a |
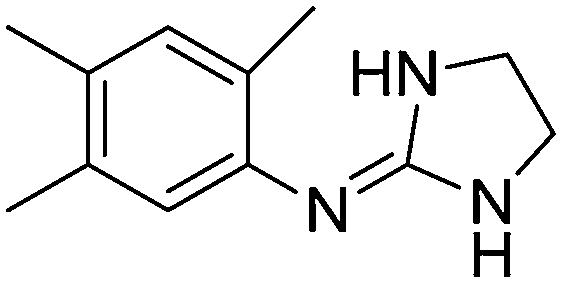
|
10.76
10.26 |
10.78 ± 0.01 | 10.91 ± 0.09 |
| 8a |
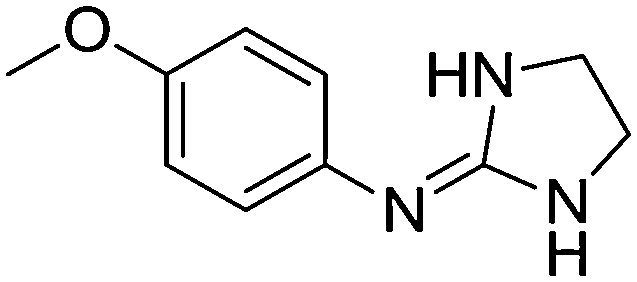
|
10.67
10.16 |
10.62 ± 0.01 | 10.80 ± 0.13 |
| 9a |

|
10.16
9.73 |
10.17 ± 0.01 | 10.28 ± 0.45 |
| 10a |

|
8.90
8.79 |
9.11 ± 0.001 | 9.42 ± 0.02 |
| 11a |

|
8.54
8.26 |
8.52 ± 0.001 | 8.37 ± 0.06 |
| 12a |
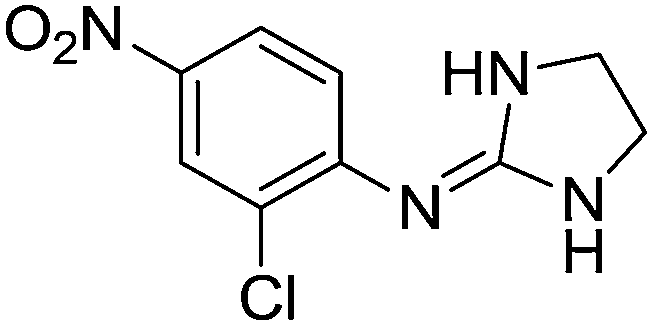
|
7.28
7.48 |
7.29 ± 0.07e | 7.33 ± 0.03 |
| 13a |

|
7.28
8.01 |
8.07 ± 0.001 | 8.06 ± 0.07 |
| 14a |

|
7.60
7.32 |
7.41 ± 0.001 | 7.41 ± 0.02 |
| 15a |

|
7.18
7.89 |
7.80 ± 0.001 | 7.69 ± 0.06 |
| 16a |
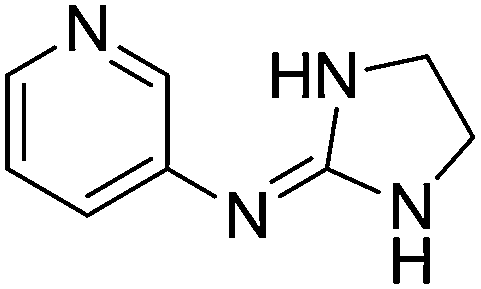
|
9.50
9.30 |
8.99 ± 0.001
3.05 ± 0.01f |
8.99 ± 0.24
3.11 ± 0.17 |
| 17a |
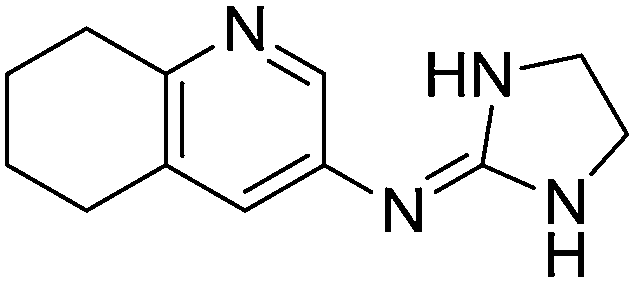
|
9.95
9.54 |
9.37 ± 0.001
4.36 ± 0.001f |
9.57 ± 0.32
4.37 ± 0.19 |
| 18a |

|
10.63
10.02 |
10.33 ± 0.001
4.97 ± 0.001f |
10.35 ± 0.42
4.96 ± 0.08 |
| 19a |
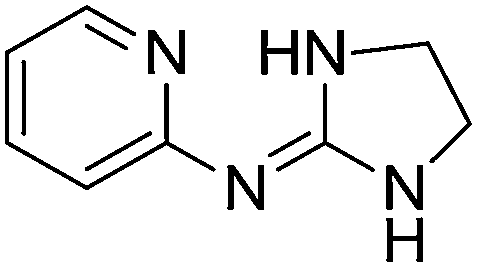
|
9.17
7.65 |
9.45 ± 0.001g | 9.60 ± 0.06 |
| 20a |

|
9.46
7.81 |
9.47 ± 0.01g | 9.98 ± 0.05 |
| 21a |

|
9.59
7.91 |
9.68 ± 0.001g | 10.15 ± 0.15 |
| 22a |

|
8.51
7.30 |
8.97 ± 0.001g | 9.10 ± 0.09 |
| 23a |

|
8.83
9.14 |
9.08 ± 0.01e | 9.26 ± 0.06 |
| 1b |
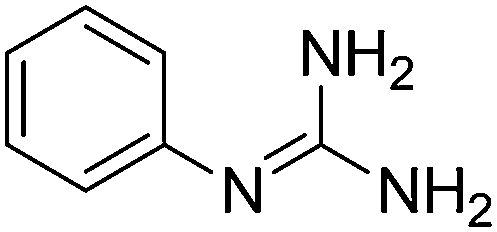
|
11.01 | 10.90 ± 0.001h | — |
| 2b |

|
11.22 | 11.36 ± 0.01 | — |
| 3b |
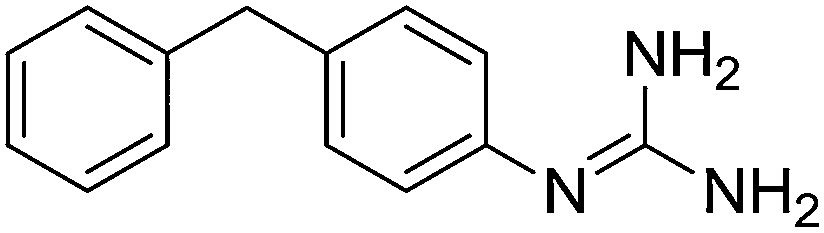
|
11.10 | 10.98 ± 0.001 | 10.95 ± 0.25 |
| 6b |
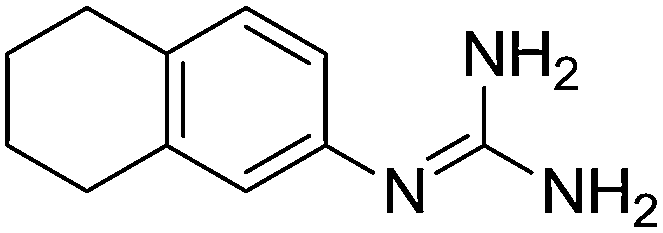
|
11.15 | 10.98 ± 0.001 | — |
| 7b |

|
11.24 | 11.35 ± 0.01 | — |
| 17b |

|
10.60 |
10.05 ± 0.001
4.44 ± 0.001f |
N/A
4.13 ± 0.35 |
| 18b |

|
10.77 |
11.01 ± 0.01
4.94 ± 0.01f |
N/A
4.98 ± 0.09 |
| 22b |

|
10.52 | 10.47 ± 0.001g | 10.88 ± 0.08 |
| 24b |
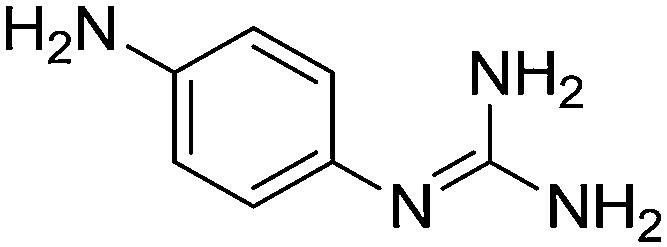
|
11.31 |
12.01 ± 0.03
3.63 ± 0.001 |
— |
| 25b |

|
11.79 | 11.68 ± 0.01 | 11.70 ± 0.09 |
| 26b |

|
10.89 | 11.03 ± 0.01 | 11.23 ± 0.15 |
| 27b |

|
11.03 | 11.01 ± 0.01 | 11.12 ± 0.15 |
A good correlation (r2 = 0.993) was observed between the pKa values measured using spectrophotometry and potentiometry (Fig. 2A). The pKa differences observed between both experimental techniques were generally ≤0.3 pKa units except for compounds 1a (+0.67), 4a (+0.37), 5a (+0.57), 20a (+0.51), 21a (+0.47), and 22b (+0.41).
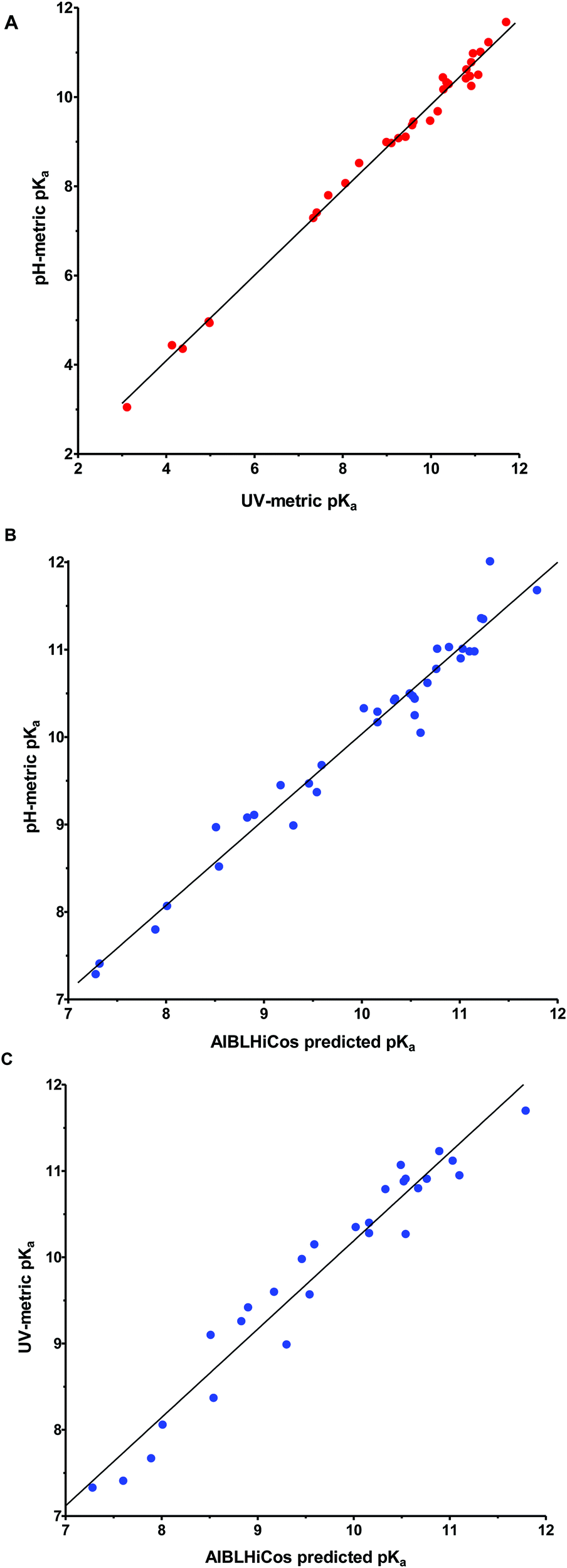 | ||
| Fig. 2 (A) Correlation (r2 = 0.993) between experimental pH-metric and UV-metric pKa values for compounds 3a–23a, 3b, 17b, 18b, 22b, 26b, and 27b. (B) Correlation (r2 = 0.965) between pH-metric pKas and AIBLHiCoS predicted pKa values for compounds 1a–23a and 1b–27b. (C) Correlation (r2 = 0.947) between UV-metric pKas and AIBLHiCoS predicted pKa values for compounds 3a–23a, 3b, 17b, 18b, 22b, 26b, and 27b. | ||
It is known that low aqueous solubility and chromophore strength can affect the accuracy of experimental pKa determination for certain bases.34 As all the compounds with greater discrepancies between UV-metric and pH-metric pKa values have weak chromophores, this could explain the lack of accuracy with these compounds. In contrast, if we take the nitro derivatives 11a–15a as an example of compounds with a strong chromophore and a smooth transition from the protonated (λmax ≈ 298 nm at pH 3) to the neutral form (λmax ≈ 364 nm at pH 12), we observe an excellent correlation between the UV-metric and pH-metric values (Table 1).
The pKa values calculated using the AIBLHiCoS methodology correlated well (r2 = 0.965) with the experimental results obtained using pH-metric titrations (see Fig. 2B) with deviations <0.30 pKa units for the phenyl-imidazolidine derivatives. However, some deterioration was observed for pyridinyl compounds (<0.58) and two compounds containing nitro groups, 13a (−0.79) and 15a (−0.62), were also poorly predicted and are discussed in the next section. The mean absolute error (MAE) was found to be 0.23 with a standard deviation of 0.22 for the error values using the model which returned the highest validation statistics during calibration (C–N(im), Fig. S1, ESI†). This may be compared to the MAE value for ChemAxon predictions using the imino tautomer, which was found to be 0.77, with a standard deviation of 0.47 (Table S1 in the ESI†). The prediction power with the guanidine set of compounds was slightly lower with an MAE of 0.29, and most deviations ≤0.24 pKa units except for compounds 17b (−0.55), 24b (+0.70) and one very high error for 22b (+1.05), which will also be discussed in the next section (Table S2, ESI†).
Finally, with respect to the UV-metric pKa values, deviations ≤0.52 pKa units were observed against the predictions of AIBLHiCoS (and r2 = 0.947) with the exception of compounds 5a (+0.58), 13a (−0.94), 15a (−0.58), 21a (+0.56) and 22a (−0.59) (Fig. 2C).
2.3. Validation of the AIBLHiCoS prediction method applied to arylguanidines and 2-(arylimino)imidazolidines
The bond length vs. pKa model used for most of the imidazolidine predictions in this work was chosen on the basis of its superior r2, leave-one-seventh-out-q2 and Root Mean Error of Estimation (RMSEE) values compared to a number of other candidate bond length models. For the 22 compounds of the training set (listed 1i–22i in Table S3, ESI†), the highest validation statistics correspond to a model constructed using a C–N bond length of the imidazolidine ring, when compounds were represented as the imino tautomer “T3”. This model is labelled C–N(im) in Fig. S1 (ESI†) and will be referred to as such throughout the following discussion. The second best validation statistics were observed for the N–C(Ph) bond, which is also labelled as such in Fig. S1 (ESI†). Predictions using both models are also listed in Table 1.For guanidines, superior validation statistics for the training set (listed g1–g13 in Table S4, ESI†) were obtained using the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond length with compounds represented as amino tautomer “A”, as illustrated in Fig. S2 (ESI†). All predictions listed in Table 1 and Table S2 (ESI†) were obtained using this model.
N bond length with compounds represented as amino tautomer “A”, as illustrated in Fig. S2 (ESI†). All predictions listed in Table 1 and Table S2 (ESI†) were obtained using this model.
A high degree of prediction accuracy in modern terms is considered to be within 1 pKa unit per prediction, with a mean absolute error (MAE) for a test set of less than 0.5 units.35 As the training sets consist of pKa values taken from various sources, and therefore contain measurements obtained using different techniques, noise within the experimental datasets is to be expected. Small variations in conditions are also possible, i.e. solvent purity, sample purity and lab temperature, which can also have an effect on the values measured. According to the above conditions, the error statistics for both test sets show that both the guanidine and imidazolidine AIBLHiCoS models perform well. On only one occasion does any error exceed 1 unit, and the MAE values for both test sets are well below 0.5 (0.23 for imidazolidines and 0.29 for guanidines). For predictions made using the pKa predictor plug-in within the Marvin Suite by ChemAxon,36 with the imino tautomer as the input structure (Tables S1 and S2, ESI†), nine prediction errors exceed 1.0 pKa units and the MAE values are in excess of 0.5 units for both imidazolidines and guanidines (0.77 and 0.54). AIBLHiCoS therefore finds its place here as a useful tool in instances where the empirical tools with a wider applicability radius struggle, as is the case for these guanidine-type ionisable groups.
However, two of the largest prediction errors of the imidazolidine test set correspond to those for 13a and 15a, 2-(3-chloro-4-nitro-phenylamino) and 2-(3-fluoro-4-nitro-phenylamino)imidazolidine. Errors for the pH-metric measurements are −0.79 and −0.62, and compared to the UV-metric values they also give errors of −0.94 and −0.58. The resonance-induced strong electron-withdrawing effect of nitro groups is known to perturb active bond lengths to such an extent that compounds containing these groups often form their own separate HiCoS.28,29 Contrary to the previous results, the partitioning of the imidazolidine set into more structurally specific subsets may not be defined in this case by the presence of a nitro group at the para-position. This is explained by the fact that compound 7i, (2-(2,6-Cl2,4-NO2-phenylimino)imidazolidine), is present within the training set used to form the predictive HiCoS, and 11a, 12a and 14a of the test set fall in the expected region of the trend line for the LFER, as illustrated by their low prediction errors. According to the LFER exhibited between the C–N(im) bond length and pKa, a shorter equilibrium bond length simply corresponds to a lower pKa. In the case of 13a and 15a, we therefore observe an anomalously short C–N(im) bond, resulting in lower pKa values than we would expect according to the LFER exhibited by most other congeners.
Looking to the optimised structures for 13a and 15a to explain these short bond lengths reveals the presence of an interaction between the O− atom of the p-nitro group, and the m-Cl (13a) and m-F (15a) atoms. As a result of this interaction, the nitro group is seen to adopt a geometry whereby it is no longer co-planar with benzene. It may be asserted that the presence of this substituent interaction and deviation in nitro geometry, which is absent in all remaining p-nitro compounds, may be the cause of the anomalously short C–N(im) bond lengths.
The internal and external validation statistics for the chosen model (C–N(im)) are superior to any other bond length vs. pKa plot (r2 = 0.97, q2 = 0.96 and RMSEE = 0.23). The next best model according to these validation statistics is observed for the same imidazolidine training set with compounds in the imino tautomeric form, but for the C–N bond length connecting benzene to 2-iminoimidazolidine (marked as N–C(Ph) within the structure labelled “T3” in Fig. S1, ESI†). This model has an r2 value of 0.96, a q2 value of 0.96 and a RMSEE value of 0.25. When the linear equation for this alternative model is used to predict for the whole test set 1a–23a, the resultant MAE is significantly worse than the original model we chose to implement based on its superior statistics (the MAE is now 0.57 vs. 0.23).
For the 3-pyridyl compounds synthesized in this work, 16a–18a, we observe errors of +0.51, +0.58 and +0.30 for the pH-metric measurements and −0.51, +0.38 and +0.28 for the UV-metric values. Despite the increase in MAE when the whole set is considered, use of the alternative N–C(Ph) model is seen to reduce the errors for 16a and 17a to −0.31 and +0.17. This improvement of predicted values can also be seen for the problematic nitro compounds, 13a and 15a, for which the errors decrease dramatically when predicted using the N–C(Ph) model (−0.26, +0.19, +0.06, −0.09 and +0.09 for 11a–15a).
We now take a moment to briefly introduce the Interacting Quantum Atoms (IQA) energy partitioning scheme in accordance with the Quantum Theory of Atoms in Molecules (QTAIM).37,38 By partitioning the total energy of a system into intra- and interatomic terms, it has been shown that we may derive a quantitative measure of covalent-like interactions between atoms in molecules. This comes in the form of the exchange–correlation potential energy VABxc, which is the sum of the exchange energy VABx and the correlation energy VABc. The former term dominates VABxc, and expresses the Fock–Dirac exchange, a consequence of the Pauli Principle, which describes the ever reducing probability of finding two electrons of the same spin as they approach each other (i.e. the Fermi hole). The latter term VABc is associated with the Coulomb hole, which corresponds to the electrostatic repulsion between electrons. The absolute value of VABxc between two atoms can be taken as the extent of delocalization of electrons between them, a factor that also determines the bond distance.
For the training set compounds, for C–N(im) and N–C(Ph), the VCNxc values (Table S5, ESI†) for the bonded atoms are found to have r2 values of 0.990 and 0.992 with the corresponding bond lengths (Fig. S3(a) and (d), ESI†). However, the analogous plots for other bonds (marked “b” and “c” in Fig. S1 and (b and c) in Fig. S3, ESI†) have weaker correlations (r2 = 0.939 and 0.897). Therefore, we can conclude that the bond lengths that correlate most highly with aqueous pKa are those which are also most accurately reflecting the extent of delocalization between the two bonded atoms. Including the VCNxcvs. bond lengths for our outliers, 13a, 15a, 16a and 17a, shows that they do not diverge substantially from the highly correlated lines of the best fit for either C–N(im) or N–C(Ph). Correspondingly, these four compounds remain as outliers for the plot of VCNxc values vs. pKa for C–N(im), and as observed for the bond lengths, fall within the expected region of the plot for VCNxc values vs. pKa for N–C(Ph) (Fig. S4, ESI†).
On the basis of previous work, we suspect that for compounds with m-halogen and p-nitro substituents (13a and 15a), the halo-nitro interaction perturbs the charge distribution of the guanidine fragment to such a degree that the relationship between C–N(im) and pKa is no longer reflected in the general trend observed for most other congeners. If more pKa data were available, it is proposed that we could form a new HiCoS consisting of only compounds of this type. Currently, in the absence of an adequate quantity of pKa data, a separate predictive HiCoS using the C–N(im) bond lengths cannot be constructed. Given that the N–C(Ph) bond lengths and corresponding VCNxc values do not diverge from the general trend as a result of the halo-nitro interaction, the C–N(Ph) model should be used to predict compounds of this type. For two of three 3-pyridyl compounds (16a–18a), an explanation for their >0.5 prediction errors is not immediately accessible in terms of their structure. This is due to the fact that one 3-pyridyl containing compound, 18a, has a relatively low error of +0.30 predicted using the C–N(im) model. We can therefore assert that the presence of the heteroatom at the 3-position is not the sole cause of the higher pKa values we predict, based on their longer C–N(im) bond lengths. Once again, in the absence of an adequate amount of experimental data for like compounds at the time of writing, we cannot prove indefinitely that these compounds would form their own subset. We therefore suggest that future predictions for all 3-pyridyl compounds be made via implementation of the N–C(Ph) model equation.
N model. pKa data for 1-(2-pyridinyl)guanidine and 1-(6-methyl-2-pyridinyl)guanidine are available, and when the bond lengths of their optimised structures are included in our CN vs. pKa plot using tautomer A (shown in Fig. S2 of the ESI,† bond labelled ii), they also appear below the line of the best fit, near the data point for 22b. When predicted using the model equation, errors of +1.51 and +1.22 units are observed. It appears that the presence of the heteroatom at the 2-position of the aryl group causes further partitioning of the set once again. A new HiCoS cannot be constructed using only two data points, and so a predictive equation which would better describe the LFER for these compounds cannot be formulated until more pKa data become available.
Boltzmann weighting of tautomers according to either internal or Gibbs energies at the level of theory used to obtain the bond lengths for the AIBLHiCoS protocol reveals a general prevalence of the “imino” tautomer for both sets of guanidines and imidazolidines. This tautomer is characterised by a CN double bond between the central carbon atom of the Y-shaped guanidine fragment and the nitrogen adjacent to the phenyl group. For imidazolidines, the optimal model emerges from one of the C–N bond lengths of the 5-membered ring of this most stable tautomer. The emergence of the optimal model from the most stable tautomer is therefore congruent with it being the dominant species in solution, confirmed by the energetics. In terms of the ChemAxon predictions, using the most stable tautomer as an input structure allows for better prediction accuracy for both the imidazolidines and guanidines. However, for AIBLHiCoS the best internal validation statistics for guanidines are revealed using equilibrium bond lengths from a specific conformation of a higher energy tautomeric form, which is not in agreement with the energy rankings. Regardless of that these results corroborate those of our previous work,27 which states that in the case of guanidines conformational commonality undermines stability; that is to say, as long as the compound of interest containing the guanidine group is optimised from this specific form, a very high degree of prediction accuracy may be obtained.
2.4. Quantitative effect of aryl substituents on the pKa of aryl guanidines and 2-(arylimino)imidazolidines
According to a survey by Manallack on the pKa distribution of drugs,24 no CNS drug has a pKa of above 10.5. Thus, phenylguanidine (1b: pKa = 10.90), the reference compound in our study, is unlikely to have good CNS activity. Replacing the guanidine group of 1b by a 2-iminoimidazolidine group (1a: pKa = 10.24) leads to a drop in the basicity of the molecule (ΔpKa = −0.66). Changing the phenyl ring of 1a for a 2-pyridinyl (19a) or a 3-pyridinyl (16a) group has a strong effect on the pKa decrement (ΔpKa = −0.79 and −1.25, respectively). The effect of electron-donating and electron-withdrawing groups on the pKa of 2-(arylimino)imidazolidines and arylguanidines is summarized in Fig. 3 and Table S6 (ESI†). As expected, these effects depend mainly on the capacity of these substituents to donate or withdraw electrons from the aryl ring, resulting in higher or lower electron density on the 2-imino nitrogen, respectively. | ||
| Fig. 3 Substituent effects on the pKa of 2-(arylimino)imidazolidines and arylguanidines. The pKa values were determined using potentiometric titrations at 25 °C. ΔpKa is the difference in pKa of the substituted compound with respect to the unsubstituted compound (1a, 1b, 19a, and 16a, respectively). | ||
A linear relationship was found between the pKa values and the Hammett σ values of meta and para substituents as shown in Fig. 4.39,40 For para-amino substituents conjugated with the imidazolidine nitrogen (2a, 2b, 24b), σp− values were used to get a good correlation. In contrast, compounds with an amide group (9a) in para position correlated poorly with the Hammett constant (red points in Fig. 4).
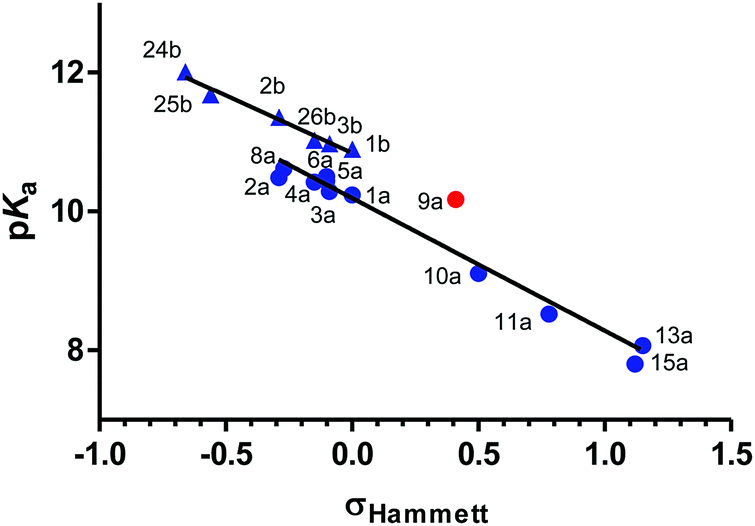 | ||
| Fig. 4 Relationship between experimental pKa (determined by potentiometric titrations at 25 °C) and Σ Hammett σ constants39 for meta and para substituents of 2-(arylimino)imidazolidines (●) and arylguanidines (▲). Compounds that deviate significantly from the bottom (right) linear relationship appear in red (9a). | ||
Amino substituents (18a, 18b, 24b) in the para-position to the guanidine/imidazoline group yield the strongest pKa increase (ΔpKa = +1.11 to 1.34), with the exception of the aminophenyl group (2a, 2b), the electron donating capacity of which is much reduced by the presence of the phenyl ring (ΔpKa = +0.25 to 0.46). Expectedly, the pKa of the pyridine nitrogen is influenced to a larger extent by the presence of amino and alkyl electron-donating substituents (ΔpKa = +1.31 and +1.92 for 17a and 18a, respectively). Alkoxy substituents (8a, 26b, 27b) have a smaller effect on the pKa increase (ΔpKa = +0.06 to 0.38) with respect to the phenoxy group (25b: ΔpKa = +0.78). For electron-withdrawing substituents, the pKa decrement follows this order: 4-PhNHCO < 4-Ac < 4-NO2 < (3-Cl,4-NO2) < (3-F,4-NO2) < (2-F,4-NO2) < (2Cl,4-NO2). The addition of one halogen atom on the aryl ring results in pKa decrements of 0.45 (3-Cl), 0.72 (3-F), 1.11 (2-F), and 1.23 (2-Cl). These values are consistent with the pKa decrements measured for a series of chloro- and fluoro-substituted bis-2-(arylimino)imidazolidines reported previously.22
Timmermans et al. have shown that the presence of two chlorine atoms in the position ortho to the 2-imino nitrogen reduces the pKa value of the molecule by 1.66 units (e.g. 4-nitroclonidine: pKa = 6.86 at 20 °C).41 According to the pKa values reported by Rozas and co-workers for a series of symmetric and unsymmetrical bisguanidines and bis-2-(phenylimino)imidazolidines,42,43 the addition of a 4-imidazoline or 4-guanidine group on the phenyl substituent of 2a–b and 3a–b results in a pKa decrement of approximately −0.50 units with respect to 2a–b and 3a–b. The values of ΔpKa with respect to 1a–b are shown in Fig. 5.
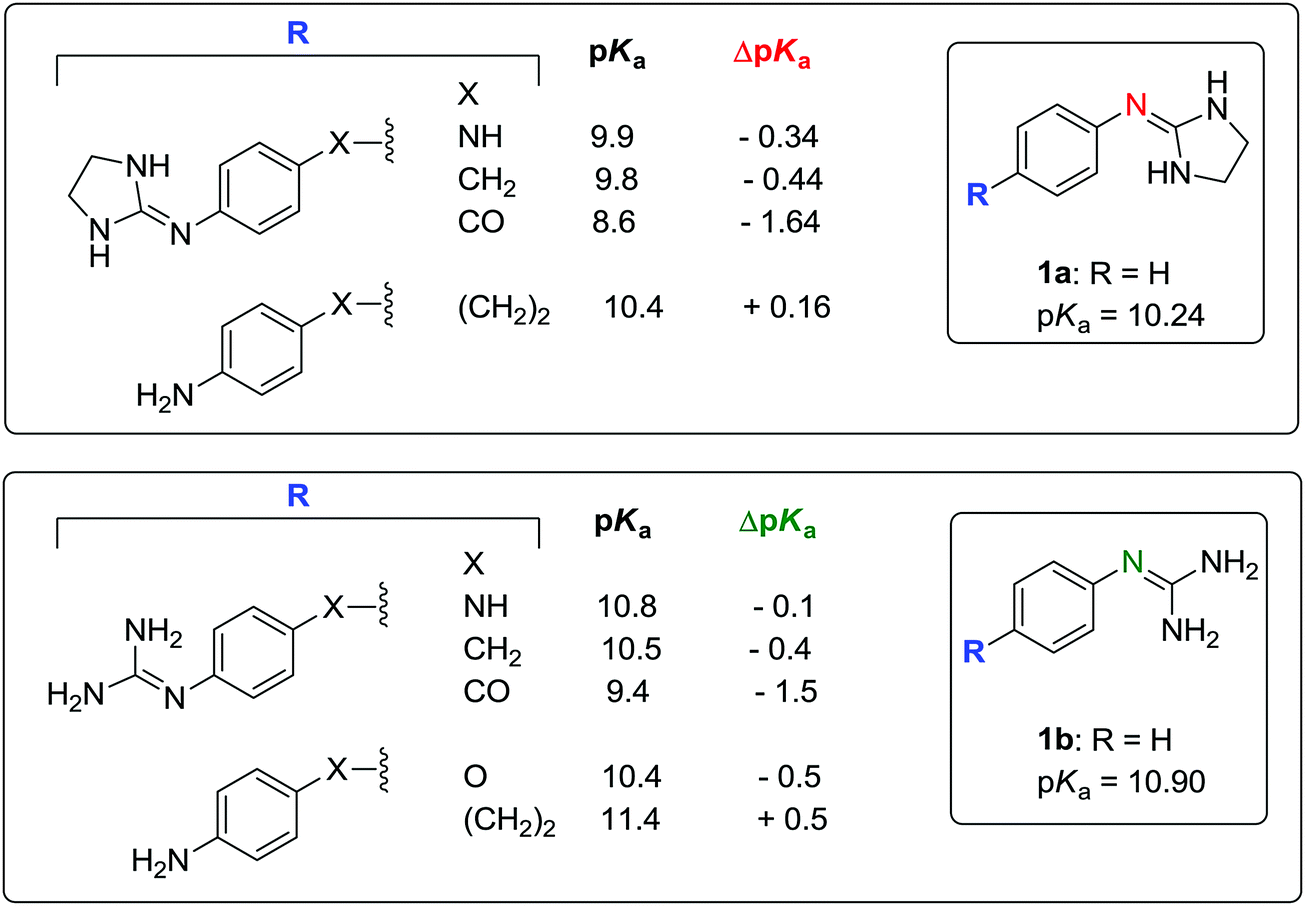 | ||
| Fig. 5 Substituent effects on the pKa of bis(2-(phenylimino)imidazolidines) and bisguanidines. Reported pKa values determined using potentiometric42 or UV-spectrophotometric43 titrations at 25 °C. ΔpKa is the difference in pKa of the 4-substituted compound with respect to 1a or 1b. | ||
3. Conclusions
Arylguanidines and 2-(arylimino)imidazolidines whose pKa values are reported here belong to a pharmacologically important class of molecules with promising applications as centrally active α2-AR modulators. As the ionisation constant is a key physicochemical parameter that governs the membrane permeability of drugs (e.g. the BBB permeability in the case of CNS active compounds), the data of substituent effects on the basicity of arylguanidines and 2-(arylimino)imidazolidines can be useful for the design of new molecules of this class.The dissociation constants of 23 arylguanidines and 12 2-(arylimino)imidazolidines measured using the spectrophotometry method were consistent with those measured using potentiometric titrations, further validating the 96-well microtitre plate method32 as a useful tool for the medium throughput determination of pKa of series of molecules.
Finally, we have shown that the AIBLHiCoS method is useful to predict the pKa values of aryl guanidines and 2-(arylimino)imidazolidines with low RMSD values, using only a single bond length obtained at a low level of theory. It must be noted that compound 12a is an excellent example of the predictive power of the AIBLHiCoS model. The initial (pH-metric) pKa value we were working with was 8.13, meaning that our prediction of 7.28 was 0.85 units out, whilst for 14a, the 2-fluoro analogue, the prediction error was only 0.19. Re-measurement of the pKa value for 12a then revealed the first to be erroneous, for reasons that are not clear, and the new pKa value was observed to be 7.29, only 0.01 units out from the prediction. This value was also in excellent agreement with the UV-metric pKa value (7.33).
4. Experimental
Chemistry
Melting points were measured in open capillary tubes using a Stuart Scientific SMP3 apparatus or a Mettler Toledo MP70 melting point system and are uncorrected. LC-MS spectra were recorded on a WATERS apparatus integrated with a HPLC separation module (2695), a PDA detector (2996) and a Micromass ZQ spectrometer. Analytical HPLC was performed with a SunFire C18–3.5 μm column (4.6 mm × 50 mm). Mobile phase A: CH3CN + 0.08% formic acid and B: H2O + 0.05% formic acid. UV detection was carried over 190 to 440 nm. Accurate mass was measured with an Agilent Technologies Q-TOF 6520 spectrometer using electrospray ionisation. 1H NMR and 13C NMR spectra were registered on Bruker Avance-300, Varian Inova-400, and Varian-system-500 spectrometers. Chemical shifts of the 1H NMR spectra were referenced to the residual proton resonance of the deuterated solvents: D2O (δ 4.79) and CD3OD (δ 3.31). Chemical shifts of the 13C NMR spectra were internally referenced for D2O and CD3OD (δ 49.0). Coupling constants J are given in hertz (Hz).![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MeOH (10:1) (compounds 1a, 11a, 13a, 15a) or CH2Cl2:MeOH (15:1 → 10:1) (compounds 12a and 14a).
:MeOH (10:1) (compounds 1a, 11a, 13a, 15a) or CH2Cl2:MeOH (15:1 → 10:1) (compounds 12a and 14a).
The raw UV data were processed using the Excel program (see the Excel sheet template in the ESI†) and the pKa values were determined by linear regression from the total absorbance vs. pH curve as reported.32 The pKa values are the mean of 3 or 4 experiments (expressed as Ka values) ± SD.
Gas-phase ab initio calculations
The initial structures of each member of the respective training sets (guanidines and 2-(phenylamino)imidazolidines) were built using the program GaussView in each available tautomeric form. This equates to three tautomers for the imidazolidines, labelled T1–T3 in Fig. S1 (ESI†) and five “tautomers” for guanidines, labelled A–E in Fig. S2 (ESI†). For compounds with higher degrees of conformational freedom, several starting structures were generated using the “Conformers” plug-in within Marvin Sketch by ChemAxon.35 Geometry optimization in the gas-phase was then performed for each conformer of each compound in each of the tautomeric forms using the GAUSSIAN09 program.44 Calculations were initially carried out at the B3LYP/6-31G(d,p) level but the basis set was changed to 6-311G(d,p) for reasons that will be explained below. For biguanide derivatives included in the guanidine training set, various tautomeric forms of the guanidine fragment present within the R group were considered, whilst maintaining the specific tautomeric form of the guanidine fragment under consideration. Frequency calculations were carried out on optimised structures again using GAUSSIAN09, at the same level of theory. The output files were then inspected to confirm the absence of negative eigenvalues after diagonalization of the Hessian matrix, so that geometries are confirmed as true energy minima. Where a number of starting conformers of a tautomer were generated, the most stable conformer was chosen by comparing the total energies of each optimised structure. At this point we noticed that the chosen basis set was causing an issue in terms of ranking conformer stability between 2-(2-halogen-phenylimino)imidazolidine geometries with and without IHBs. These IHBs are seen to exist between an N–H group on imidazolidine and the o-halogen atom on benzene, and are ubiquitous throughout the most stable conformations of all compounds containing this substituent for tautomer T3, with the exception of 2-(2,4-Cl-phenylimino) and 2-(2,5-Cl-phenylimino)imidazolidine. This observation prompted the re-optimisation of all T3 training set compounds at the B3LYP/6-311G(d,p) level of theory. Using the valence triple zeta basis set revealed a consistent picture, where the presence of an IHB is a stabilising feature for all compounds. At this point all other calculations for T1 and T2 and tautomers A–E of the guanidine training set were also repeated for consistency. All analysis corresponds to the results of the calculations using the 6-311G(d,p) basis set.Seven bond lengths were extracted from each imidazolidine compound, corresponding to each of the three CN bonds of the guanidine fragment, in addition to the N–C(Ph) bond connecting imidazolidine to benzene and the N–C, C–C and C–N bonds of the imidazolidine ring. These are labelled a–g in Fig. S1 of the ESI.† Five bond lengths from within the guanidine moiety were also extracted, which correspond to the two CN single and one CN double bond of the guanidine group, the N–H bond attached to the imine nitrogen in A, B, D and E, and one N–H bond of a primary amine group. These are labelled i–v, in Fig. S2 (ESI†). The five bond lengths i–v within tautomers A–E were then regressed against the pKa values for the set of guanidines. The seven bond lengths a–g of the three tautomers T1–T3 were also regressed against their pKa values for the set of 2-(phenylimino)imidazolidines. The squared correlation coefficient (r2), Root-Mean-Squared Error of Estimation (RMSEE) and leave-one-seventh-out q2 values were obtained using the program SIMCA-P 10.0.45
By a comparison of internal and external validation metrics obtained for each bond length model, an optimal linear equation was chosen. For guanidines, the model was constructed using the CN bond lengths of training set compounds as tautomer A, labelled ii in Fig. S2 (ESI†). For imidazolidines, the model was constructed using an endocyclic C–N bond length of the imino tautomer T3, labelled “a” in Fig. S1 (ESI†). The predictions for test set compounds 1a–23a (2-(phenylimino)imidazolidines) and 1b–27b (aryl guanidines) were obtained by energy minimization, (via a conformational search using Marvin followed by geometry optimisation), frequency calculations, and insertion of the appropriate bond length into the linear equation for the optimal bond length vs. pKa model. The test compounds were constructed in the same tautomeric form as those of the training set used to construct the model.
The program AIMAll (version 14.04.17)46 was used to carry out QTAIM analysis and the IQA partitioning calculations to obtain Vxc values for the atomic interactions corresponding to the C–N(im) and N–C(Ph) bonds. Calculations were carried out on B3LYP/6-311G(d,p) wavefunctions of the following species: the five nitro compounds (11a–15a), compound 11 of the training set and five compounds for which no pKa data exists, 2-(3-Cl-phenylimino), 2-(3-F-phenylimino), 2-(3,5-Cl2-4-NO2-phenylimino), 2-(3-Br-4-NO2-phenylimino) and 2-(3-F,5-Cl-4-NO2-phenylimino)imidazolidine. For the five additional compounds, optimisation and frequency calculations were also performed at the B3LYP/6-311G(d,p) level before AIMAll analysis and the relevant bond lengths were extracted.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the Spanish Ministerio de Economia y Competitividad (Grant SAF2015-66690-R). B. A. Caine thanks BBSRC and Syngenta Ltd for PhD funding. P. L. A. P. acknowledges the EPSRC for funding through the award of an Established Career Fellowship (grant EP/K005472). We acknowledge the assistance of G. Romero and F. Peréz Gordillo (Instrumental Analysis Department at IQM) with the potentiometric pKa measurements. We thank Chansèle Jourdan for her assistance with the UV-metric pKa measurements. Dr Rozas is gratefully acknowledged for the gift of the pyridino derivatives 16–22 and the Boc-protected precursors of compounds 1–10 and 23–27.References
- J. W. Shaw, D. H. Grayson and I. Rozas, Topics in Heterocyclic Chemistry, Springer Berlin Heidelberg, Berlin, Heidelberg, 2015, pp. 1–51, DOI:10.1007/7081_2015_174.
- P. O'Sullivan and I. Rozas, ChemMedChem, 2014, 9, 2065–2073 CrossRef PubMed.
- P. S. Nagle, C. McKeever, F. Rodriguez, B. Nguyen, W. D. Wilson and I. Rozas, J. Med. Chem., 2014, 57, 7663–7672 CrossRef CAS PubMed.
- P. S. Nagle, F. Rodriguez, B. Nguyen, W. D. Wilson and I. Rozas, J. Med. Chem., 2012, 55, 4397–4406 CrossRef CAS PubMed.
- P. S. Nagle, S. J. Quinn, J. M. Kelly, D. H. O'Donovan, A. R. Khan, F. Rodriguez, B. Nguyen, W. D. Wilson and I. Rozas, Org. Biomol. Chem., 2010, 8, 5558–5567 CAS.
- P. S. Nagle, F. Rodriguez, A. Kahvedzic, S. J. Quinn and I. Rozas, J. Med. Chem., 2009, 52, 7113–7121 CrossRef CAS PubMed.
- F. Rodríguez, I. Rozas, M. Kaiser, R. Brun, B. Nguyen, W. D. Wilson, R. N. García and C. Dardonville, J. Med. Chem., 2008, 51, 909–923 CrossRef PubMed.
- C. Dardonville, M. P. Barrett, R. Brun, M. Kaiser, F. Tanious and W. D. Wilson, J. Med. Chem., 2006, 49, 3748–3752 CrossRef CAS PubMed.
- C. Dardonville and R. Brun, J. Med. Chem., 2004, 47, 2296–2307 CrossRef CAS PubMed.
- C. Dardonville and J. J. Nué Martínez, Curr. Med. Chem., 2017 DOI:10.2174/0929867324666170623091522.
- C. Millan, F. Acosta-Reyes, L. Lagartera, G. Ebiloma, L. Lemgruber, J. J. Nué Martinez, N. Saperas, C. Dardonville, H. de Koning and J. L. Campos, Nucleic Acid Res., 2017, 45, 8378–8391 CrossRef PubMed.
- C. Dardonville, C. Fernandez-Fernandez, S. L. Gibbons, G. J. Ryan, N. Jagerovic, A. M. Gabilondo, J. J. Meana and L. F. Callado, Bioorg. Med. Chem., 2006, 14, 6570–6580 CrossRef CAS PubMed.
- C. Dardonville, N. Jagerovic, L. F. Callado and J. J. Meana, Bioorg. Med. Chem. Lett., 2004, 14, 491–493 CrossRef CAS PubMed.
- C. Dardonville, I. Rozas, P. Goya, R. Giron, C. Goicoechea and M. I. Martin, Bioorg. Med. Chem., 2003, 11, 1283–1291 CrossRef CAS PubMed.
- A. Alsasua, M. J. Borrego, M. T. Dannert, C. Dardonville, M. Rozas and M. I. Martin, Fundam. Clin. Pharmacol., 2001, 15, 84 Search PubMed.
- B. Kelly, M. McMullan, C. Muguruza, J. E. Ortega, J. J. Meana, L. F. Callado and I. Rozas, J. Med. Chem., 2015, 58, 963–977 CrossRef CAS PubMed.
- C. Muguruza, F. Rodriguez, I. Rozas, J. J. Meana, L. Uriguen and L. F. Callado, Neuropharmacology, 2013, 65, 13–19 CrossRef CAS PubMed.
- F. Rodríguez, I. Rozas, J. E. Ortega, A. M. Erdozain, J. J. Meana and L. F. Callado, J. Med. Chem., 2008, 51, 3304–3312 CrossRef PubMed.
- F. Rodriguez, I. Rozas, J. E. Ortega, J. J. Meana and L. F. Callado, J. Med. Chem., 2007, 50, 4516–4527 CrossRef CAS PubMed.
- F. Rodriguez, I. Rozas, J. E. Ortega, A. M. Erdozain, J. J. Meana and L. F. Callado, J. Med. Chem., 2009, 52, 601–609 CrossRef CAS PubMed.
- C. Dardonville and I. Rozas, Med. Res. Rev., 2004, 24, 639–661 CrossRef CAS PubMed.
- C. H. Rios Martinez, J. J. Nue Martinez, G. U. Ebiloma, H. P. de Koning, I. Alkorta and C. Dardonville, Eur. J. Med. Chem., 2015, 101, 806–817 CrossRef CAS PubMed.
- C. Ríos Martínez, F. Miller, K. Ganeshamoorthy, F. Glacial, M. Kaiser, H. de Koning, A. Eze, L. Lagartera, T. Herraiz and C. Dardonville, Antimicrob. Agents Chemother., 2015, 59, 890–904 CrossRef PubMed.
- D. T. Manallack, Perspect. Med. Chem., 2007, 1, 25–38 Search PubMed.
- C. Anstöter, B. A. Caine and P. L. A. Popelier, J. Chem. Inf. Model., 2016, 56, 471–483 CrossRef PubMed.
- I. Alkorta and P. L. A. Popelier, ChemPhysChem, 2015, 16, 465–469 CrossRef CAS PubMed.
- M. Z. Griffiths, I. Alkorta and P. L. A. Popelier, Mol. Inf., 2013, 32, 363–376 CrossRef CAS PubMed.
- A. P. Harding and P. L. A. Popelier, Phys. Chem. Chem. Phys., 2011, 13, 11283–11293 RSC.
- A. P. Harding and P. L. A. Popelier, Phys. Chem. Chem. Phys., 2011, 13, 11264–11282 RSC.
- B. Kelly, M. McMullan, C. Muguruza, J. E. Ortega, J. J. Meana, L. F. Callado and I. Rozas, J. Med. Chem., 2015, 58, 963–977 CrossRef CAS PubMed.
- S. R. Mundla, L. J. Wilson, S. R. Klopfenstein, W. L. Seibel and N. N. Nikolaides, Tetrahedron Lett., 2000, 41, 6563–6566 CrossRef CAS.
- C. H. Ríos Martínez and C. Dardonville, ACS Med. Chem. Lett., 2013, 4, 142–145 CrossRef PubMed.
- T. L. Davis and R. C. Elderfield, J. Am. Chem. Soc., 1932, 54, 1499–1503 CrossRef CAS.
- L. Settimo, K. Bellman and R. M. A. Knegtel, Pharm. Res., 2014, 31, 1082–1095 CrossRef CAS PubMed.
- A. D. Bochevarov, M. A. Watson, J. R. Greenwood and D. M. Philipp, J. Chem. Theory Comput., 2016, 12, 6001–6019 CrossRef CAS PubMed.
- MarvinSketch, version 6.2.2, calculation module developed by ChemAxon, http://www.chemaxon.com/products/marvin/marvinsketch/, 2014.
- P. L. A. Popelier, Atoms in Molecules. An Introduction, Pearson Education, London, UK, 2000 Search PubMed.
- R. F. W. Bader, Atoms in Molecules. A Quantum Theory, Oxford Univ. Press, Oxford, UK, 1990 Search PubMed.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- L. P. Hammett, J. Am. Chem. Soc., 1937, 59, 96–103 CrossRef CAS.
- P. B. Timmermans and P. A. van Zwieten, Arzneimittelforschung, 1978, 28, 1676–1681 CAS.
- G. K. Kinsella, F. Rodriguez, G. W. Watson and I. Rozas, Bioorg. Med. Chem., 2007, 15, 2850–2855 CrossRef CAS PubMed.
- P. Nagle, A. Kahvedžić, T. McCabe and I. Rozas, Struct. Chem., 2011, 1–9 Search PubMed.
- M. Frisch, G. Trucks, H. B. Schegel, G. Scuseria, M. Robb, J. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. Petersson, et al., GAUSSIAN 09, 2009 Search PubMed.
- UMETRICS, in SIMCA-P 10.0, http://www.umetrics.com, Umeå, Sweden, 2002.
- T. A. Keith, AIMAll; TK Gristmill Software, Overland Park, KS, 2016, available at: http://aim.tkgristmill.com, accessed 1 May 2017.
Footnote |
| † Electronic supplementary information (ESI) available: Excel sheet template for the data analysis of UV-metric pKa determination. Tables S1–S6 and Fig. S1–S4. 1H and 13C NMR spectra of compounds 1a and 11a–15a. See DOI: 10.1039/c7nj02497e |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2017 |