
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tuning diastereoisomerism in platinum(II) phosphino- and aminothiolato hydrido complexes†
A.
Polo
 *a,
J.
Duran
a,
R.
Juanola
a,
J.
Real
b,
J.
Benet-Buchholz
c,
M.
Solà
ad and
A.
Poater
*ad
*a,
J.
Duran
a,
R.
Juanola
a,
J.
Real
b,
J.
Benet-Buchholz
c,
M.
Solà
ad and
A.
Poater
*ad
aDepartament de Química, Universitat de Girona, Facultat de Ciències, Ma. Aurèlia Campmany 69, 17003 Girona, Catalonia, Spain. E-mail: alfons.polo@udg.edu; crous.juanola@gmail.com
bDepartament de Química, Universitat Autònoma de Barcelona, 08193 Bellaterra, Spain
cInstitute of Chemical Research of Catalonia (ICIQ), Av. Països Catalans 16, 43007 Tarragona, Spain
dInstitut de Química Computacional i Catàlisi, Universitat de Girona, Campus de Montilivi, 17003 Girona, Catalonia, Spain. E-mail: albert.poater@udg.edu
First published on 6th March 2017
Abstract
Chelate assisted oxidative addition of one equivalent of 2-phosphinothiols (2-(diphenylphosphino)ethanethiol 1, 1-(benzyloxy)-2-(diphenylphosphino)ethanethiol 2 and 2-(diphenylphosphino)-cyclohexanethiol 3) to tetrakis(triphenylphosphine)platinum(0) gives the corresponding hydrido[2-(phosphino-κP)thiolato-κS]triphenylphosphineplatinum(II) complexes 8–10. Temperature variable NMR studies show that these complexes display a chemical equilibrium between the cis-P,P and trans-P,P geometries, strongly displaced toward the trans-P,P configuration (70–90%). XRD studies carried out on crystals of 8 indicate that although the two geometric isomers are present in solution, only the trans-P,P is obtained in the solid state. These results differ from the cis-P,N geometry observed for the related hydrido[2-(amino-κN)thiolato-κS]triphenylphosphineplatinum(II) complexes 5–7 in solution. The crystal structures obtained for these aminothiolate hydrides show that the cis-P,N configuration is the only one observed in the solid state. Chelate assisted oxidative addition of one equivalent of 3-(diphenylphosphino)propanethiol 4 to tetrakis(triphenylphosphine)platinum(0) gives the resultant hydrido[3-(phosphino-κP)thiolato-κS]triphenylphosphineplatinum(II) complex 11. This 3-phosphinothiolate hydride shows in solution only a trans-P,P geometry but presents two chelate ring conformational isomers. Density functional theory calculations have been used to explore the ligand-based stereoelectronic effects that are determinant in the different diastereoisomerism observed in these platinum(II) hydrides (5–11).
Introduction
Chelating diphosphines are the prevalent chiral bidentate ligands for catalytic asymmetric transformations. Despite their characteristic chirality, many of these diphosphines have two donor atoms with similar electronic and steric properties (e.g. DIOP, BINAP). To decrease the symmetry of the bidentate ligand, diphosphines with different substituents at each phosphorus have been synthesized (e.g. DIPAMP), and some chemical changes in the phosphorus group have been introduced (e.g. BINAPHOS). Larger differentiation between the coordinating groups can be achieved by using mixed-donor ligands, such as P–O, P–N, and P–S donors.1 In this context, amino- and phosphinothiolates are suitable candidates because they are asymmetric chelating ligands with substantial electronic and steric differences between the two donor atoms.2 Thus, sulfur supports a formal negative charge and, once bound to the metal, can act as a π donor owing to the two nonbonding electron pairs on sulfur.3 Moreover, the thiolate group has only one substituent, the chain that connects it to the amine or phosphine group. Conversely, nitrogen and phosphorus are neutral and, once bound to the metal, do not support any nonbonding electron pairs, acting as classical σ donor and σ donor/π acceptor, respectively.Low symmetry hydrido[2-(amino-κN)ethanethiolato-κS]triphenylphosphineplatinum(II) complexes (5–7) with potential applications in selective catalysis could be obtained by oxidative addition of 2-aminoethanethiols, some of them straight from the chiral pool (cysteamine, methyl and ethyl ester-protected natural L-cysteine derivatives) to a platinum(0) precursor.4 These complexes show four different donor atoms around metal and become asymmetric for the natural chiral ligands because the symmetry plane of the square-planar coordination is broken by the asymmetric carbon of the aminoacid. A similar synthetic strategy has been used to obtain low symmetry hydrido(phosphinothiolate) complexes such as the hydrido[2-(diphenylphosphino-κP)thiophenolate-κS] triphenylphosphineplatinum(II)5 and the hydrido[1-(diphenylphosphino-κP)butane-2-thiolato-κS]triphenylphosphineplatinum(II)6 complexes. The last one was obtained in a mixture with triphenylphosphine. Despite the promising properties of these complexes, no more low symmetry platinum(II) hydrides with amino- or phosphinothiolate chelating ligands have been described. The reduced attention given to this class of complexes can be due, besides other reasons, to the lack of knowledge on the ligand-based stereoelectronic effects that determine the stereochemistry of these complexes. In this context, the diastereochemistry of these complexes has particular importance, as different diastereomers should present different activity, regioselectivity, and enantioselectivity in a given catalytic process.7
As part of a project on the synthesis of new amino- and phosphinothiolato complexes of the group 10 metals and on the analysis of the ligand-based stereoelectronic effects that are determinants in the coordination conformations around the metal,3,8–13 we now report the preparation of new platinum(II) hydride complexes (8–11) with 2-phosphinoethanethiolate ligands (2-(diphenylphosphino)ethanethiolate dppet 8, 1-(benzyloxy)-2-(diphenylphosphino)ethanethiolate bdppet 9, and 2-(diphenylphosphino)cyclohexanethiolate dppcyt 10) and a 3-phosphinothiolate ligand (3-(diphenylphosphino)propanethiolate dpppt 11). We also present, for the first time to our knowledge, the crystal structure determined by the X-ray analysis of a platinum(II) hydride complex with 2-phosphinoalkylthiolate ligand (8) and with 2-aminothiolate ligands: cysteamine (5) and natural chiral ligands methyl (cysMe) and ethyl (cysEt) ester-protected natural L-cysteine derivatives (6–7). Moreover, DFT calculations are carried out to rationalize the diastereoisomerism observed in platinum(II) hydrides with amino- and phosphinothiolate ligands.
Results and discussion
The hydrido[(2-amino-κN)ethanethiolato-κS]triphenylphosphineplatinum(II) complexes [Pt(H)(SCH2CH2NH2)(PPh3)] (5), [Pt(H)((L)-SCH2CH(COOCH3)NH2)(PPh3)] (6), and [Pt(H)((L)-SCH2CH(COOCH2CH3)NH2)(PPh3)] (7) were prepared as previously reported4 and XRD studies were carried out on crystals obtained from CH2Cl2–hexane (Fig. 1). Crystal data for the structures are given in Table S1 (ESI†). Selected bond lengths and bond angles are listed in Table 1.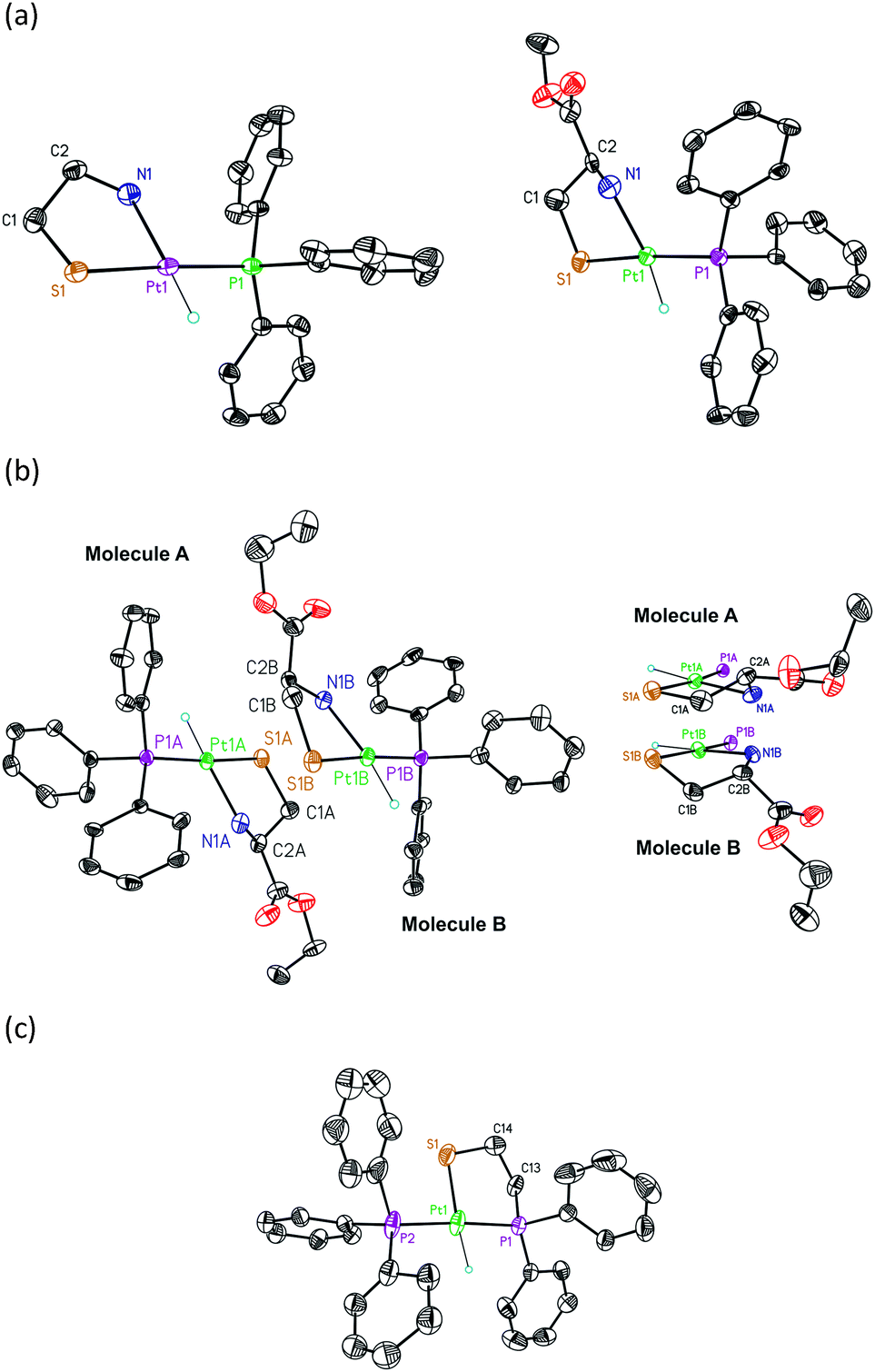 | ||
| Fig. 1 ORTEP view and atom-numbering scheme for (a) 5 (left) and 6 (right), (b) the two diastereomeric molecules (molecules A and B) observed for 7 (left) and their chelate rings (right), and (c) 8. Thermal ellipsoids are drawn at the 50% probability level. Non-relevant hydrogen atoms are omitted to better appreciate the geometry of the complexes. | ||
| Species | 5 | 6 | 7A/7B | 8 |
|---|---|---|---|---|
| Pt–S | 2.301(3) | 2.306(2) | 2.328(3)/2.291(4) | 2.355(2) |
| Pt–N | 2.153(4) | 2.129(8) | 2.142(11)/2.176(10) | |
| Pt–P(1) | 2.2406(13) | |||
| Pt–P(2) | 2.215(2) | 2.213(2) | 2.204(3)/2.228(3) | 2.2757(14) |
| S–C(1) | 1.825(5) | 1.807(11) | 1.827(12)/1.836(12) | 1.832(6) |
| C(1)–C(2) | 1.506(7) | 1.53(2) | 1.510(13)1.501(12) | 1.527(9) |
| C(2)–N | 1.473(7) | 1.52(2) | 1.481(11)/1.489(11) | |
| C(2)–P(1) | 1.832(6) | |||
| S–Pt–N | 85.37(12) | 84.0(2) | 84.4(3)/84.0(3) | |
| S–Pt–P(1) | 86.64(5) | |||
| S–Pt–P(2) | 175.41(4) | 174.58(10) | 173.6(2)/174.17(14) | 97.89(6) |
| P(2)–Pt–N | 99.01(12) | 101.4(2) | 101.7(3)/101.9(3) | |
| P(2)–Pt–P(1) | 175.35(5) | |||
| Pt–S–C(1) | 98.16(17) | 99.3 (3) | 98.8 (4)/98.6(3) | 105.9(2) |
| Pt–N–C(2) | 113.2(3) | 106.6(8) | 108.3(6)/115.3(6) | |
| Pt–P(1)–C(2) | 108.3(2) | |||
| S–C(1)–C(2) | 110.7(4) | 113.3(9) | 110.3(7)/111.9(6) | 112.6(4) |
| C(1)–C(2)–N | 110.4(4) | 111.6(12) | 108.4(7)/106.3(8) | |
| C(1)–C(2)–P(1) | 110.9(4) |
The crystal structures reveal mononuclear square-planar complexes with an SP-4-2 configuration (cis-N,P arrangements, from this point forward cis) for all aminothiolate complexes 5–7 (Fig. 1(a) and (b)). This geometry is also the only one observed in solution. This result differs from the trans-N,P geometry (but also SP-4-2 configuration, from now on trans) observed for the related chloride complexes (Table 2, entries 4 and 5).14 The square-planar geometry is slightly distorted with an S–Pt–N angle somewhat reduced from the idealized value of 90°. Quite similar distortions from the idealized geometry are commonly observed in platinum(II) complexes with chelated 2-aminoethanethiolate ligands (Table 2, entries 4 and 6–8).14a,15 The Pt–S distances are in the range observed for mononuclear [2-(diphenylphosphino-κP)ethanethiolato-κS]triphenylphosphine16 and [ethane-1,2-dithiolato-κ2S1,S2]triphenylphosphine17 platinum(II) complexes with a trans-P,S arrangement, but the Pt–P distances are substantially lower (Table 2, entries 18–23), indicating a smaller trans influence, and then a smaller chelate effect, of the thiolate group of 2-aminothiolate ligands compared to those of 2-phosphinothiolate or 2-dithiolate ligands.
| Entry | Complex | Pt–S | Pt–N | Pt–P(1) | Pt–P(2) | Chelate angle | Ref. |
|---|---|---|---|---|---|---|---|
a L1 = 3-methylisoxazole-4,5-dione-4-iximato.
b L2 = cis-[Pt(C6F5)2(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C-2-C5H3N-κC)(PPh3)].
c L3 = (1R,2S,5R)-1-((diphenylphosphino)methyl)-2-isopropyl-5-methylcyclohexanethiolate.
d L4 = 1-cyclohexenyl. C-2-C5H3N-κC)(PPh3)].
c L3 = (1R,2S,5R)-1-((diphenylphosphino)methyl)-2-isopropyl-5-methylcyclohexanethiolate.
d L4 = 1-cyclohexenyl.
|
|||||||
| 1 | cis-[PtH(SCH2CH2NH2)(PPh3)] (5) | 2.301(3) | 2.153(4) | 2.215(2) | 85.37(12) | This work | |
| 2 | cis-[PtH(SCH2CH(CO2Me)NH2)(PPh3)] (6) | 2.307(2) | 2.114(8) | 2.213(2) | 83.6(2) | This work | |
| 3 | cis-[PtH(SCH2CH(CO2Et)NH2)(PPh3)] (7) | 2.328(3) | 2.142(11) | 2.204(3) | 84.4(3) | This work | |
| 4 | trans-[PtCl(SCH2CH(Me)NMe2)PMe2Ph] | 2.235(2) | 2.130(9) | 2.247(2) | 88.6(2) | 14a | |
| 5 | trans-[PtCl(2-SC5H4N)(PBut3)] | 2.331(2) | 2.094(6) | 2.3146(18) | 69.14(18) | 14b | |
| 6 | [Pt6(SCH2CH2NH2)8]Br4 | 2.276(5) | 2.08(2) | 86.5(5) | 15a | ||
| 2.278(5) | 2.08(2) | 87.5(5) | |||||
| 2.293(5) | 2.08(1) | 85.8(4) | |||||
| 2.305(5) | 2.10(1) | 84.7(5) | |||||
| 7 | trans-[PtCl(SCH2CH(CO2H)NH2)]3 | 2.279(2) | 2.071(6) | 87.5(2) | 15b | ||
| 8 | trans-[Pt(SC(Me)2CH(CO2H)NH2)2] | 2.298 | 2.043 | 85.96 | 15c | ||
| 9 | [PtH(C3F7)(Me2NCH2CH2NMe2)] | 2.155(10) | trans H | 82.4(3) | 18 | ||
| 10 | trans-[PtH(PhHNNC3H6)(PPh3)2]BF4 | 2.166(6) | 19a | ||||
| 11 | trans-[PtH(L1-κN)(PPh3)2]a | 2.148 | 19b | ||||
| 12 | trans-[PtH(L2-κN) (PPh3)2]b | 2.120(16) | 19c | ||||
| 13 | trans-[PtH(MeCN) (PCy3)2]BPh4 | 2.079 | 20a | ||||
| 14 | trans-[PtH(N(C(CN)2)2)(PPh3)2] | 2.066 | 20b | ||||
| 15 | trans-[PtH(SCH2CH2PPh2)(PPh3)] (8) | 2.355(2) | 2.240(2) | 2.275(3) | 86.63(5) | This work | |
| 16 | trans-[PtH(SC6H4-2-PPh2)(PPh3)] | 2.357(3) | 2.243(3) | 2.278(3) | 86.9(1) | 5 | |
| 17 | trans-[Pt(L3)2]c | 2.3138(9) | 2.2620(9) | 86.37(3) | 10 | ||
| 3.3386(9) | 2.2787(9) | 85.94(3) | |||||
| 18 | cis-[PtCl(SCH2CH2PPh2)(PPh3)] | 2.319(3) | 2.215(4) | 2.281(4) | 86.34 | 16 | |
| 19 | [Pt(SCH2CH2S)(PPh3)2] | 2.327 | 2.280 | 87.71 | 17a | ||
| 2.312 | 2.295 | ||||||
| 20 | [Pt(cyclohepta-3,5-diene-1,2-dithiolate)(PPh3)2] | 2.339(4) | 2.275(3) | 88.96(13) | 17b | ||
| 2.308(4) | 2.296(3) | ||||||
| 21 | [Pt(cis-cyclooctane-1,2-dithiolato)(PPh3)2] | 2.3136(9) | 2.2826(9) | 86.63(10) | 17c | ||
| 2.309(5) | 2.2750(9) | ||||||
| 22 | [Pt(trans-cyclooctane-1,2-dithiolato)(PPh3)2] | 2.3025(10) | 2.2888(10) | 87.39(3) | 17c | ||
| 2.3185(10) | 2.2748(10) | ||||||
| 23 | [Pt(SCH(CH2OH)CH2S)(PPh3)2] | 2.385 | 2.272 | 85.77 | 17d | ||
| 2.350 | 2.293 | ||||||
| 24 | trans-[PtH(HCS2)(PCy3)2] | 2.401 | 2.306 | 26 | |||
| 2.313 | |||||||
| 25 | trans-[PtH((PhCH2)2NCS2)(PCy3)2] | 2.403 | 2.288 | 27 | |||
| 2.308 | |||||||
| 26 | [Pt3H3(μ-CH3C(CH2S)3)(PPh3)3] | 2.382 | trans H | 2.221 | 28 | ||
| 2.386 | trans H | 2.231 | |||||
| 2.387 | trans H | 2.240 | |||||
| 27 | [Pt(L4)(Cy2PCH2CH2PCy2)(PPh3)]HCO3d | 2.313 | 2.331 | 85.2 | 29a | ||
| 2.356 | |||||||
| 28 | [Pt(CHNMe2-κC)(Ph2PCH2CH2Ph2)(PPh3)]PF6 | 2.3164(12) | 2.3503 | 84.09(5) | 29b | ||
| 2.3303(13) | |||||||
| 29 | [PtCl(7,8-(PPh2)2-7,8C2B9H10)(PPh3)] | 2.287(2) | 2.345(2) | 85.55(7) | 29c | ||
| 2.237(2) | |||||||
The Pt–N distances are similar to those observed for complexes with trans H–Nsp318 and trans H–Nsp219 arrangements but larger than those obtained for complexes with trans H–Nsp20 geometries (Table 2, entries 9–14). For complex 7, the unit cell comprises two diastereomeric molecules (A and B, Fig. 1(b) left). The two molecules are different in the conformation adopted by the chelate ring. In molecule A, this conformation places the carboxylate substituent of the asymmetric carbon above the molecular plane. In molecule B, the conformation of the chelate ring locates this substituent below the molecular plane (Fig. 1(b) right). As a result, the configuration of the platinum atom is different in each molecule: molecule A can be designated as (RPt,RC) and molecule B as (SPt,RC) according to the nomenclature developed for square-planar π-allyl palladium complexes and also useful for these complexes.21 The difference observed in the solid state between these two molecules disappears in solution owing to the R configuration of the asymmetric carbon that forces the chelate ring to adopt a classic λ conformation in order to locate the carboxylate substituent in an equatorial position. The presence of the two diastereomeric molecules in the solid state can also be observed for complex 6 if the structure is refined in the chiral P1 space group. However, the crystal structure of this complex refines better using a disordered model in the centrosymmetric P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group.
space group.
Chelate assisted oxidative addition4,13,22 of one equivalent of 2-(diphenylphosphino)ethanethiol (Hdppet, 1) to tetrakis(triphenylphosphine)platinum(0) gives the corresponding hydrido[2-(diphenylphosphino-κP)ethanethiolato-κS]triphenylphosphineplatinum(II) complex ([Pt(H)(dppet)(PPh3)], 8, Scheme 1). Complex 8 is isolated as a white solid in 68% yield and purified by recrystallization in dichloromethane/ether. A critical point of this reaction is the stoichiometry. If Hdppet is added to default, a mixture of 8 and the reactant complex is obtained making impossible further separations. On the other hand, if an excess of Hdppet is used, a mixture of 8 and the bischelate [Pt(dppet)2] is obtained,13,23 which is also difficult to be separated especially if the bischelate complex is present in high quantities. Complex 8 presents a satisfactory EA and has been characterized by IR and NMR spectroscopy, as well as XRD.
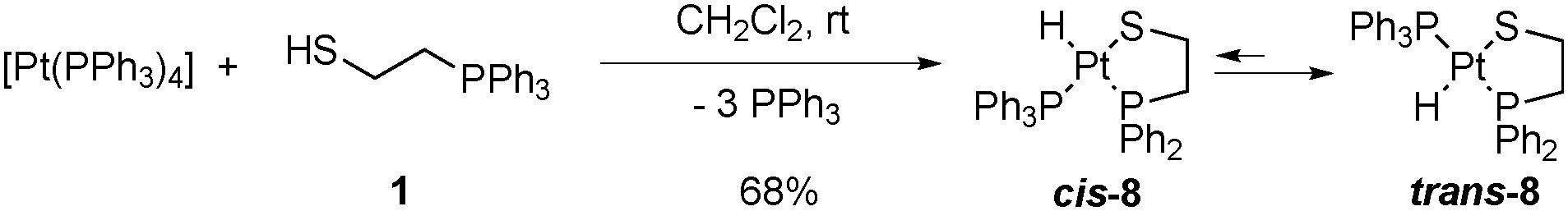 | ||
| Scheme 1 Hydrido[2-(diphenylphosphino-κP)ethanethiolato-κS]triphenylphosphineplatinum(II) (8) by chelate assisted oxidative addition of one equivalent of 2-(diphenylphosphino)ethanethiol (Hdppet, 1) to a platinum(0) complex. | ||
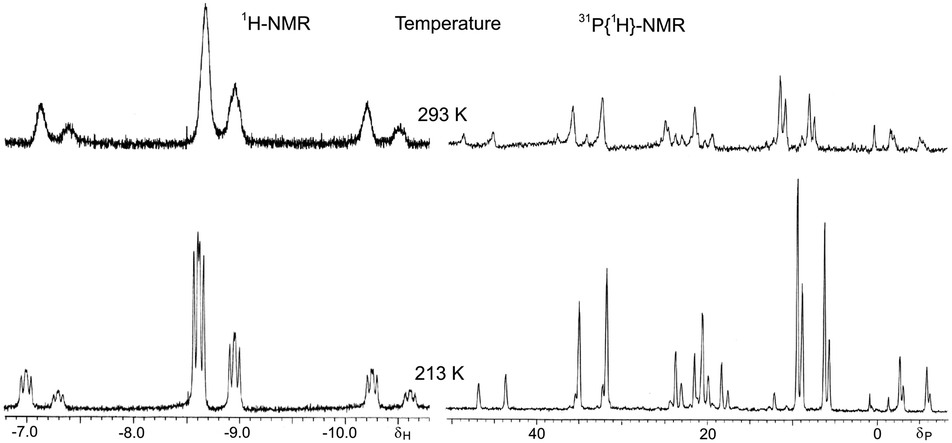
The IR spectrum of complex 8 exhibits an intense ν(PtH) vibration at 2082 cm−1. In solution, complex 8 shows a chemical equilibrium between the cis-P,P (SP-4-2-configuration, hereafter named cis-8) and trans-P,P (SP-4-4 configuration, hereafter named trans-8) geometries strongly displaced toward the trans-8 form (ca. 80%) as can be seen in the NMR spectra (Fig. 2). Equilibrium in solution between cis and trans geometries in Pt(II) (phosphino)thiolato complexes are profusely described, including hydrido(phosphino)thiolato complexes.5,6 The trans nature of the major isomer agrees with the more thermodynamically stable trans form in [PtH(SAr)(PPh3)2] complexes.24 The major isomer, trans-8, shows a temperature dependent 1H NMR. At room temperature, the hydride resonance appears as a broad doublet at δ −7.31 (JH–P ≈ 9 Hz) with 195Pt satellites (JPt–H = 969.7 Hz). When the sample is cooled to −60 °C, the broad signal resolves into a doublet of doublets with characteristic cis H–P coupling constants (2JH–P = 16.8 and 9.7 Hz, JPt–H = 977.9 Hz). In contrast, the 1H NMR of cis-8 does not display the same temperature dependence, showing at room and at low temperatures the hydride resonance as a resolved doublet of doublets, between δ −4.15 and −4.43, with a trans and a cis typical H–P coupling constants (2JH–P = 186.2–182.8 and 22.9–22.0 Hz respectively) and 195Pt satellites (JPt–H = 967.0–937.8 Hz). Also the 31P{1H} NMR of isomer trans-8 shows to be dependent on the temperature. At room temperature only two broad doublets at δ ca. 33 and 67 were observed, for PPh3 and PPh2 groups respectively, with a distinctive trans P–P coupling constant at about 393 Hz. At −60 °C, these signals transform into sharp doublets at δ 33.03 and 66.71 (2JP–P = 392.8 Hz) with 195Pt satellites (JP–Pt = 2901.6 and 2953.9 Hz respectively).
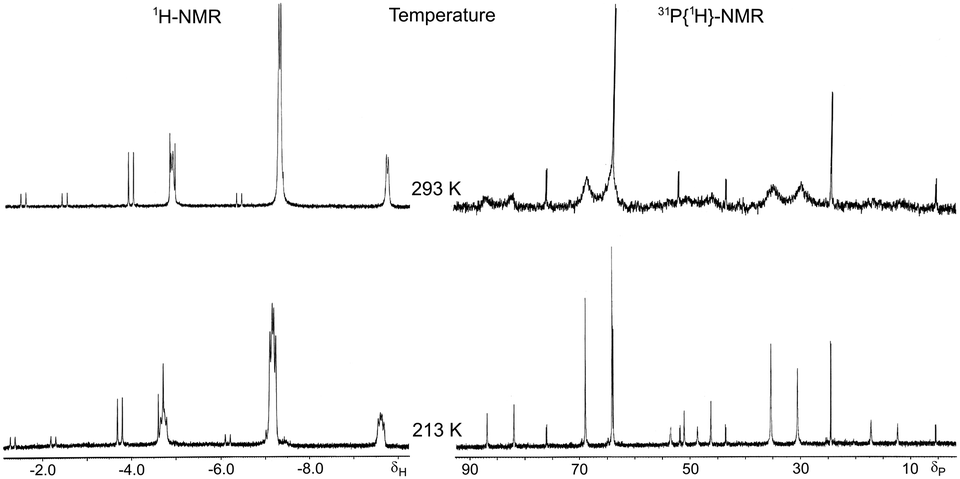 | ||
| Fig. 2 Hydride region of 1H NMR (left) and 31P{1H} NMR (right) of hydrido[2-(diphenylphosphino-κP)ethanethiolato-κS]triphenylphosphineplatinum(II) (8) at room temperature (up) and at −60 °C (down). | ||
XRD studies were carried out on crystals of 8 obtained from CH2Cl2–hexane. Selected bond lengths and bond angles are listed in Table 1. The crystal structure reveals a mononuclear square-planar complex with a trans-P,P arrangement (SP-4-4 configuration, Fig. 1c) that contrasts with the trans-S,P geometry observed for the related chloride complex.16 It should also be considered that the pyridine or triphenylphosphine promoted cis to trans isomerization on [PtH(SAr)(PPh3)2] complexes indicates that the trans isomer is thermodynamically the most stable.25 Although the two geometric isomers of 8 are present in solution, only one of them is observed in the solid state. Presumably, lower solubility and greater concentration cause the crystallization of trans-8 exclusively. In order to confirm this assumption, solid 8 was dissolved at −78 °C, under a nitrogen atmosphere, with precooled dichloromethane, and proton NMR and 31-phosphorus NMR were recorded directly at −60 °C, observing practically single trans-8 in solution. Only traces of cis-8 can be detected. After the temperature increases to room temperature, both cis-8 and trans-8 were perfectly visible in solution (see Fig. S8, ESI†). In solid state, the phenyl groups of the triphenylphosphine ligand appear disordered in two positions each and the square-planar geometry of the complex is somewhat distorted with a chelate angle slightly reduced from the idealized value of 90°. Quite similar distortions from the idealized geometry are also observed in other platinum(II) complexes with bidentate 2-phosphinoethanethiolato10,16 and 2-phosphinophenolato5 ligands (Table 2, entries 16–18). The Pt–S distance is comparable to the related 2-phosphinophenolato complex5 but appreciably longer than those observed for complexes with cyclic alkanethiolate ligands trans to triphenylphosphine16,17 (Table 2, entries 16 and 18–23). Conversely, the Pt–S bond in trans-8 is shorter than those presented by complexes with methanedithioate26 and dibenzylcarbamodithioate27trans to hydride (Table 2, entries 24 and 25). The Pt–S distance is also slightly smaller than those found in complexes with bridging μ-alkanethiolate trans to a hydride ligand (Table 2, entry 26).28 The Pt–PPh3 distance is analogous to that of the hydrido(2-phosphinophenolate)platinum(II) complex5 but smaller than those observed in neutral and cationic complexes with a PPh3 ligand trans to a phosphorus of a chelated 1,2-ethanediphosphine (Table 2, entries 16 and 27–29),29 suggesting a lower chelate effect of the 2-phosphinothiolate ligands compared to these diphosphines. However, also the Pt–PPh2 bond lengths in 2-phosphinothiolate complexes are slightly smaller than those measured for the phosphorous of the chelated ligand in diphosphine complexes (Table 2, entries 16 and 27–29).29
The observed structural preferences, cis for aminothiolato complexes 5–7 and trans for the phosphinothiolato complex 8, could be interpreted, in simple terms, on the basis of the antisymbiosis rule.30 Thus, the ligand with a greater trans influence, the hydride, is placed trans to the one with a smaller trans influence, amino for aminothiolato complexes 5–7 and thiolato for phosphinothiolato complex 8. More detailed information can be extracted from the DFT calculations on complexes 5–8. The optimization of the two geometric isomers of 2-aminothiolate complexes 5–7 gives as a result a favorable energy difference, of at least 3.2 kcal mol−1, for the cis isomer (3.2, 3.5, and 3.4 kcal mol−1 for 5–7 respectively). For 2-phosphinothiolate complex 8 the difference between the two stereoisomers is lower, indeed the trans isomer becomes slightly more stabilized by 1.0 kcal mol−1. For aminothiolato complexes 5–7, covalent bonding interactions (antisymbiotic effects) which promote the cis isomer should be considerable, and predominant over the effect of the maximum hardness principle31 that favors the trans one, although the differences of chemical hardness are small (Δη = 0.9–2.0 kcal mol−1). In these compounds, the geometric isomers seem to not present large steric differences resulting in a total significant energetic preference for the cis isomer. For phosphinothiolato complex 8, both hardness and antisymbiotic effect favor the trans isomer. The effect of the maximum hardness principle is stronger (Δη = 2.5 kcal mol−1), but the trans arrangement of the two strong σ-donor phosphine ligands should lead to a very weak antisymbiotic effect. Moreover, in this case, the steric repulsion between bulky ligands (partially cancelled by dispersion) favors also the trans isomer that becomes somewhat more stable than the cis one. These results are in agreement with an equilibrium between the two geometric isomers displaced towards the cis isomer for 2-aminothiolate complexes 5–7 and towards the trans isomer for phosphinothiolate complex 8. To point out that when the solvent correction was introduced, the energetic preference for the cis arrangement increases by around 1.0–1.1 kcal mol−1, for aminothiolato complexes 5–7, bringing the computational results into much better agreement with the experimental observation: only the cis isomer is present in solution. The large dipole moment of the cis isomer provides larger solvation energy in comparison with the non-polar trans isomer,32 whereas for phosphinothiolato complex 8, the solvation effects reduce the energetic preference for the trans isomer by 0.2 kcal mol−1, allowing the existence of the two geometric isomers in solution. These results absolutely agree with the experimental observations.
To get more insight into the bonding of the ligands in complexes 5 and 8, an energy decomposition analysis (EDA) was carried out. The aim of the EDA is to break the binding energy (BE) into deformation energy (ΔEdef) and interaction energy (ΔEint). This last attractive term can also be split into a Pauli repulsion term (ΔEPauli), an orbital interaction term (ΔEorb), and an electrostatic term (ΔEelstat). ΔEorb is related to the covalent character of the bond, whereas ΔEelect is more connected with the ionic character of the bond. The deformation energy is the difference between the energy of the optimized fragments and the energy of the fragments at the geometry they have in the complex. The EDA of the molecules has been done for three different fragmentation schemes (see Table S3 and ESI† for further details). Table 3 gives the EDA of the binding energy between the fragments H–Pt–PPh3 and the corresponding bidentate thiolato ligand to form complexes 5 and 8 in cis and trans configurations. The results support the experimental observations because cis-5 and trans-8 present higher BE values. The deformation energies are relatively high because the position of the ligands in a free metal fragment has to change significantly to achieve the geometry that the ligands have in the final complex. Furthermore, the electrostatic term is also remarkable, surely due to the charged fragments. The larger stability of the cis-5 as compared to trans-5 is found to be the result of lower deformation energies, whereas the higher stability of trans-8 with respect to cis-8 is due to better orbital interaction and electrostatic stabilizing interactions.
| Complex | ΔEPauli | ΔEoi | ΔEelstat | ΔEint | ΔEdef![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) fr.1 fr.1 |
ΔEdeffr.2 |
ΔEdef | BE |
|---|---|---|---|---|---|---|---|---|
| cis-5 | 287.6 | −202.7 | −341.3 | −256.5 | 3.2 | 23.3 | 26.5 | −230.0 |
| trans-5 | 295.3 | −207.6 | −346.6 | −258.9 | 4.4 | 27.2 | 31.5 | −227.4 |
| cis-8 | 286.0 | −189.8 | −326.8 | −230.6 | 6.8 | 24.2 | 31.0 | −199.6 |
| trans-8 | 290.4 | −206.1 | −331.9 | −247.6 | 6.4 | 39.0 | 45.5 | −202.1 |
Concerning the behavior shown in solution by the two geometric isomers of complexes 5–8, only one of the geometric isomers of 8 shows a temperature dependent NMR spectrum. The temperature dependent NMR presented by some 2-phosphinothiolato platinum(II) hydrides was previously assigned to a rapid exchange between coordinated and free PPh3, present in excess.5,6 After several recrystallization processes of product 8, the solution does not contain free PPh3, although the presence of traces cannot be ruled out. In fact, a fast dissociation equilibrium of PPh3 from trans-[PtH(SAr)(PPh3)2] complexes was observed at room temperature,24 suggesting that similar dissociation equilibrium can be operative in complex trans-8 (up in Scheme 2), being the cause of the temperature dependence of the NMR of this complex. The same dissociation equilibrium for the minor isomer cis-8 should be slower attending to the smaller trans effect of the thiolate ligand relative to the phosphine one, generating a temperature independent NMR spectrum for this isomer. The same reasoning about the trans effect difference between an amine and a phosphine ligand can be used to explain the no observation of the dissociative equilibrium for complexes 5–7.4 The proposed dissociation equilibrium for 8 may end up in the cis/trans isomerization via a dissociative mechanism through a 14 electron intermediate. Although experimental evidence of cis/trans isomerization via dissociative mechanisms for some square planar complexes of Pt(II) exists,33 usually this route is discarded in front of the more widely accepted associative mechanism (down in Scheme 2).34 However, in order to generate the necessary nucleophile (PPh3) for the cis/trans isomerization via the associative mechanism, the dissociation equilibrium should be ever operative. If the PPh3 dissociation does not occur or is too slow, as in 2-aminothiolate complexes 5–7, the isomerization via the associative route is also impossible. In a previous theoretical study on the axial bonding capabilities of square planar d8 complexes, those complexes proved to be able to act as electrophiles because of the presence of an empty d orbital (usually accepted the dz2). The coordination of a fifth ligand with σ-donor properties requires the pyramidalization of the structure to create an empty orbital with a suitable geometry (usually accepted a pz–dz2 hybrid), and the electrophilicity of the complex depends basically on its capacity to distort the initial square planar structure.35 Although the calculated electrophilicities of the cis isomers of 5–8 (129.1, 124.9, 123.0, 136.3 kcal mol−1) are greater than those of the trans ones (110.6, 111.5, 111.6, 120.3 kcal mol−1), the pyramidalization of the structure is sterically favored for trans-8 in comparison to cis-8 with a cis arrangement between the two voluminous phosphine groups. Therefore, an associative equilibrium would also be faster for the trans isomer of 8, and could generate a temperature dependent NMR spectrum for this isomer. Similar behavior differences between cis and trans isomers have been observed for other platinum(II) complexes.36 The association equilibrium for 5–7 is not favored because of their smaller electrophilicities. Thus, the latter complexes have a smaller tendency to coordinate with the fifth ligand necessary to generate the isomer via an associative mechanism. Actually, all attempts to optimize complexes 5–7 with an incoming coordinated fifth ligand, i.e. a triphenylphosphine ligand, failed. For complex 8, the new inserted ligand cannot be coordinated to the metal either, but at about 3.5 Å a sort of adduct is formed (see Fig. S7, ESI†). Anyway, this indicates that the associative mechanism is not easier even for the 2-phosphinothiolate complexes.
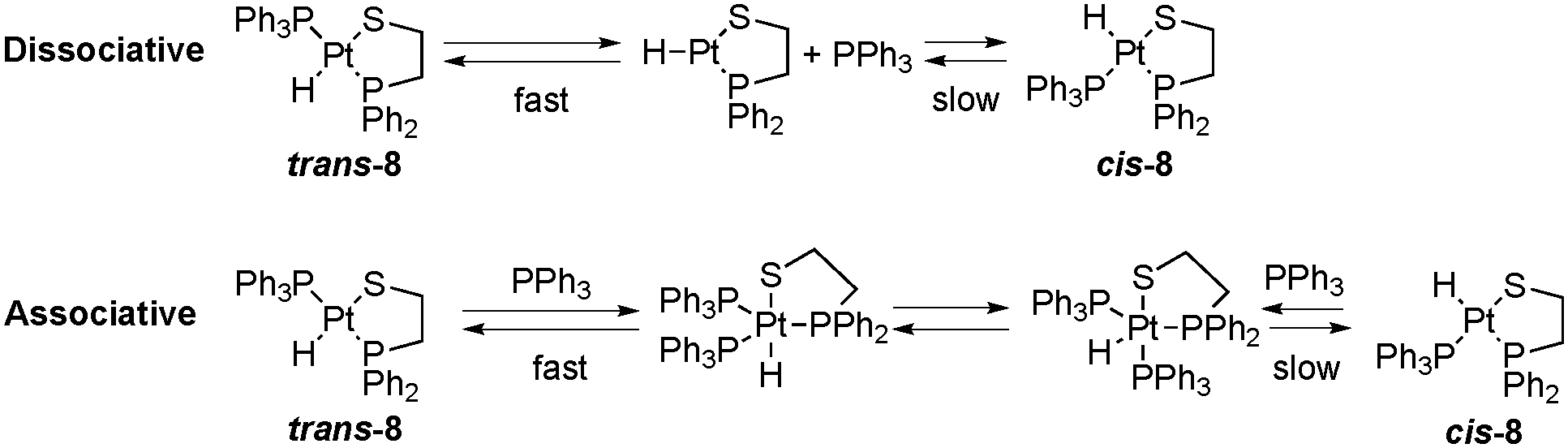 | ||
| Scheme 2 cis/trans equilibrium via dissociative (up) and associative (down) mechanisms for 8. | ||
The geometrical analysis of the isomers of compounds 5–8 shows that, in all cases, the Pt–S bond distance increases when going from the cis to the trans isomer (see Table S6, ESI†), and that for the 2-phosphinothiolate complex 8, this bond is slightly weaker with respect to the same isomer of 2-aminothiolate complexes 5–7. The Pt–H bond distance is similar for all the trans isomers. This distance is longer for the trans isomers of 2-aminothiolate complexes 5–7 and for the cis isomer of 2-phosphinothiolate complex 8. The opposite behavior is observed for the Pt–P(2) bond. On the other hand, the Pt–N or Pt–P(1) bond distances are longer for the cis isomer, regardless of the chelate ligand. All these results are in agreement with the trans influence series: H− > PRPh2 > SR− > NRH2. The Pt–P(1) bond distances in 2-phosphinothiolate complex 8 are much longer than the Pt–N ones in 2-aminothiolato complexes 5–7, providing more space for the two phenyl units attached to the phosphorus atom of the 2-phosphinothiolato ligand. This is especially important for cis-8 where the steric repulsions among the phenyl rings of the two phosphine groups are stronger than those in the trans isomer.37 With regard to the bond angles, the cis isomers present larger chelate angles (S–Pt–N for 5–7 and S–Pt–P(1) for 8), and thus the compact packing of the bidentate ligand is higher for the trans isomers, and this is particularly important for complex 8 because of the steric hindrance of the two phenyl groups attached to the phosphorous of the 2-phosphinothiolato ligand. In this way, the higher packing of the 2-phosphinothiolate ligand in trans-8 provides more space for the other ligands. For example, in the trans isomers, the P(2)–Pt–P(1) angle of 8 is appreciably lower than the P(2)–Pt–N of 5–7, while in the cis isomers these angles are similar. Moreover, the steric maps38,39 in Fig. 3 for both isomers of complex 8 reveal that the trans conformation suffers less sterical hindrance because it avoids the sterical hindrance between both phosphine moieties. The corresponding percentages of buried volume, %VBur, are 49.7 and 50.5% for cis-8 and trans-8 systems,40 respectively. The detailed hindered region defined by the interaction between the two phosphine groups in the cis conformation is clearly demonstrated by the steric maps.
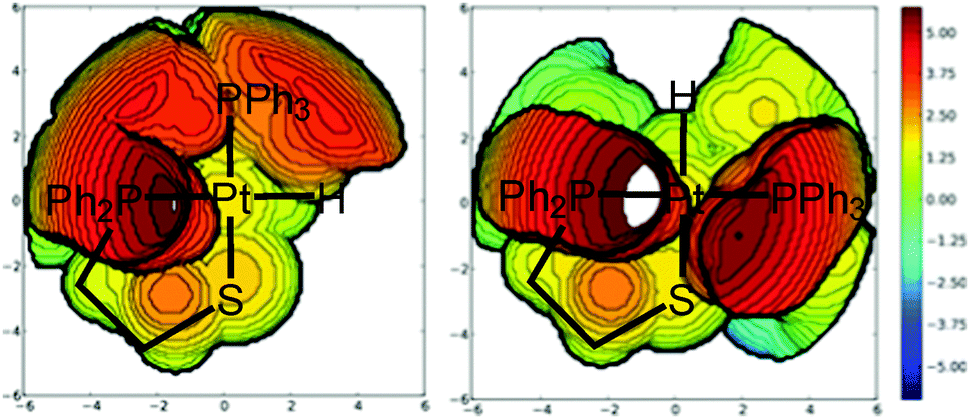 | ||
| Fig. 3 Steric maps for cis-8 (left) and trans-8 (right). The isocontour curves are given in Å. Pt is located in the centre, and all the four ligands in the same plane, the 2-phosphinothiolato ligand is displayed with the left position (P atom) and down position (S atom). The PPh3 group is up for cis-8 and right for trans-8. The systems are oriented with the two Pt free coordination positions on the z axis. The two maps at the top are computed with a radius of the sphere equal to 3.5 Å. | ||
It is well-known that modifications in the chelate ring of the phosphinothiolate ligand can produce significant changes in the coordination conformation around the metal and in the nuclearity of the complexes.10–12,22 Accordingly, three modifications were introduced in the former phosphinothiol ligand intending to explore the parameters that affect the cis/trans equilibrium observed for 8: introduction of a substituent in the chelate ring by using 1-(benzyloxy)-2-(diphenylphosphino)ethanethiol (Hbdppet, 2), inclusion of the chelate ring carbons in a cyclic structure by means of 2-(diphenylphosphino)cyclohexanethiol (Hdppcyt, 3) and enlargement of the chelate ring by using 3-(diphenylphosphino)propanethiol (Hdpppt, 4). Phosphinothiols 211 and 441 were synthetized as previously reported and racemic 3 was prepared from cyclohexene oxide in a low yield two-step synthesis42,43 and has been structurally characterized in solution by NMR spectroscopy (see the Experimental section and the ESI†).
Hydrido[(diphenylphosphino-κP)alkanethiolato-κS]triphenylphosphinoplatinum(II) complexes 9–11 were prepared by chelate assisted oxidative addition4,13,22 of one equivalent of the corresponding phosphinothiol ligand (2–4) to tetrakis(triphenylphosphine)platinum(0) in moderate yields and were characterized by IR and NMR spectroscopy.
For isomer trans-9, the hydride resonance does not resolve at low temperature because of the rapid exchange between coordinated and free PPh3 present in solution. As in complex 8, the minor isomer cis-9 shows a temperature independent NMR spectrum in the whole range studied.
The same cis/trans equilibrium in solution is observed for complex 10 (ca. 70% trans-10). As for the previous phosphinothiolate complexes, the major isomer of 10 shows temperature dependent NMR spectra that resolve at −40 °C (Fig. S5, ESI†). Once more, the minor isomer cis-10 has a different behavior showing, in whole temperature range studied, a temperature independent NMR spectrum.
Complexes 9 and 10 were also analyzed computationally. For complex 9, the use of the two enantiomers of ligand 2 was studied. The optimization of any geometric isomer of 9 results in the adoption of a different chelate ring conformation for each enantiomer of the ligand (λ for the R enantiomer and δ for the S enantiomer). In this way, the axial position of the chelate ring substituent is avoided, and two enantiomers appear for each geometric isomer.10–12 Obviously, the energy of each pair of enantiomers is calculated to be the same and the trans isomer is slightly more stable than the cis one by a difference of only 0.2 kcal mol−1, which confirms the more displaced equilibrium to the trans isomer, observed experimentally. For complex 10, only an enantiomer of the ligand was used for the optimization of the geometric isomers, resulting in a more stable cis form in the gas phase by only 0.3 kcal mol−1. This result is in part in disagreement with the experimental observations that point out that the trans conformation should be favored slightly, but suggests that the isomeric equilibrium must be less displaced to the trans form than in complex 9, as has been experimentally observed.
The NMR analysis of complex 11 also shows the presence of two different molecules in solution, but in this case both display temperature dependent NMR spectra (Fig. 4). In the hydride region of the 1H NMR spectrum at room temperature, two broad bands can be observed at very near chemical displacement δ −8.66 and −8.95 with 195Pt satellites and an integral ratio of ca. 1:2 respectively. When the solution is cooled to −60 °C, the broad signals transform into a doublet of doublets each. All the H–P coupling constants have characteristic values for cis coupling, 15.8 and 10.8 Hz for the major isomer and 14.8 and 11.0 Hz for the minor isomer, suggesting a trans-P,P arrangement (SP-4-4 configuration) for both products in solution. Also the 31P{1H} NMR of compound 11 shows to be dependent on the temperature. At room temperature only broad doublets with 195Pt satellites can be observed for both molecules and both phosphine groups. At −60 °C, these signals resolve in sharper doublets at δ 33.18 (PPh2) and 7.63 (PPh3) for the major isomer and at δ 33.59 (PPh2) and 7.09 (PPh3) for the minor isomer. The P–P coupling constant values of 388.2 and 290.2 Hz confirm the trans-P,P arrangement of both products. The experimental observations are in agreement with the presence in solution of two complexes with the same configuration around the metal: SP-4-4 or trans-P,P geometry and a very similar hydride cis to the two phosphorus donor atoms. As a result, the difference between the two complexes can only reside in the chelate ring conformation.
 | ||
| Fig. 4 Hydride region of 1H NMR (left) and 31P{1H} NMR (right) of hydrido[3-(diphenylphosphino-κP)propanethiolato-κS]triphenylphosphineplatinum(II) (11) at room temperature (up) and at low temperature (down). | ||
It is well-known that the six membered chelate rings can basically adopt three conformations: chair, skew-boat, and boat (Scheme 3). The boat conformation is usually energetically not preferred because its unfavorable torsion arrangement,44 and the chair and skew-boat conformations coexist in complexes with six membered chelated diamines45,46 and diphosphines.47 In the absence of other factors, as chiral carbons in the chelate ring, the chair conformation is normally slightly more stable than the skew-boat one, because the latter has additional torsional strain energy (ca. 1.6 kcal mol−1 in diamines), resulting principally from the flattening of the chelate ring.48 In solution, the two achiral forms possible for a chair conformation of a six membered chelated diamine habitually interconvert quickly through a skew-boat conformation intermediate. This skew-boat conformation exists in two chiral forms and numerous unsymmetrical geometries, and the equilibrium between the different conformations is too quick to allow the observation of the independent conformations by NMR.45 These conformational interconversions in six membered chelated diamines are so fast because the activation energy is very low, lower than that for cyclohexane (5.6 vs. 10.7 kcal mol−1).44 For six membered chelated diphosphines, the conformational interconversions are slower, allowing the observation of the different conformational isomers by temperature variable NMR techniques.49,50
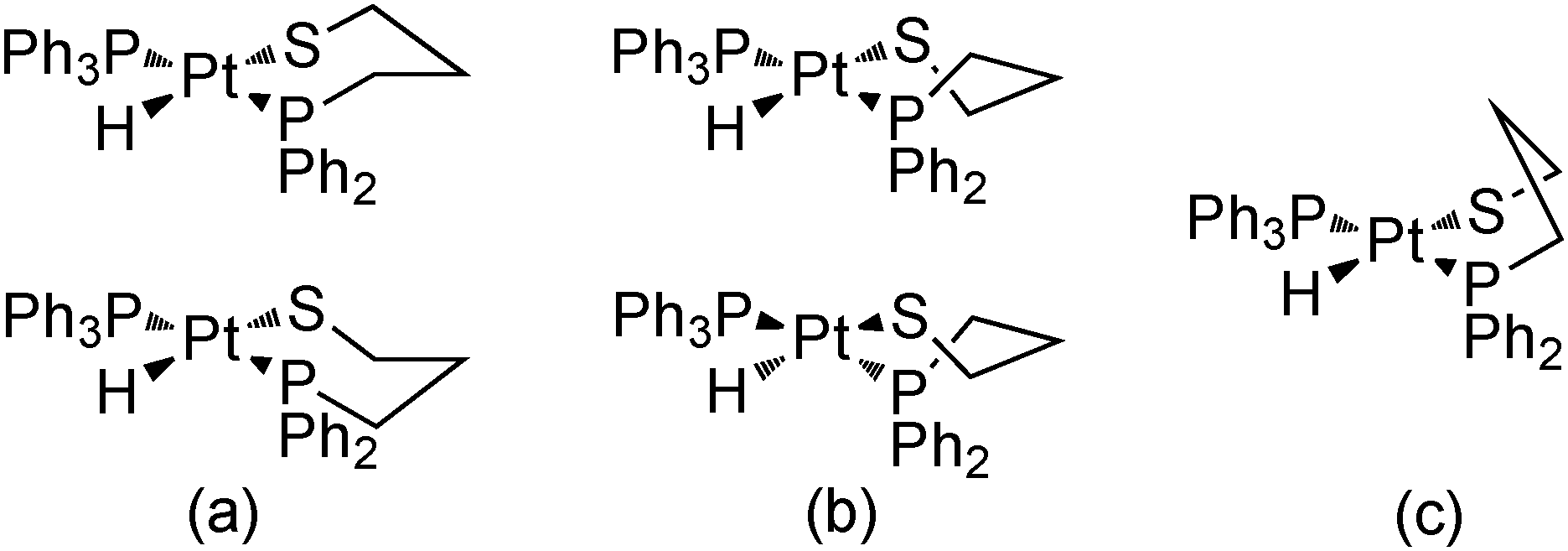 | ||
| Scheme 3 Conformations for six membered chelate rings of complex 11: (a) chair, (b) skew-boat, and (c) boat. | ||
Probably, these differences between diamines and diphosphines are related, among other parameters, to the different bonding to the metal. The ability of the interconversion between the conformers depends on the torsional barrier around the M-donor atom bond.44 Therefore, the torsional barrier about the M–P bond, with some partial double bond character, must be greater than that of the single M–N bond. In this context, also the M–S bond can show some partial double bond character because of the additional π-donor ability of the thiolate ligand with two lone pairs in the donor atom. With the aim of eliminating this π-donor capacity of the thiolate moiety, two equivalents of a Lewis acid, SnCl2, were added to an NMR solution of complex 11 at room temperature. The replacement of the two formerly broad bands in the hydride region by a single and resolved hydride resonance at δ 9.77 with only cis-H–P coupling constants was observed (Fig. 5). Accordingly, and taking into account that the two chair conformations of complex 11 are enantiomers due to the four different ligands around the metal atom, the two isomers observed in its solutions could be assigned each one to a chair and a skew-boat conformation of the chelate ring (Scheme 3).
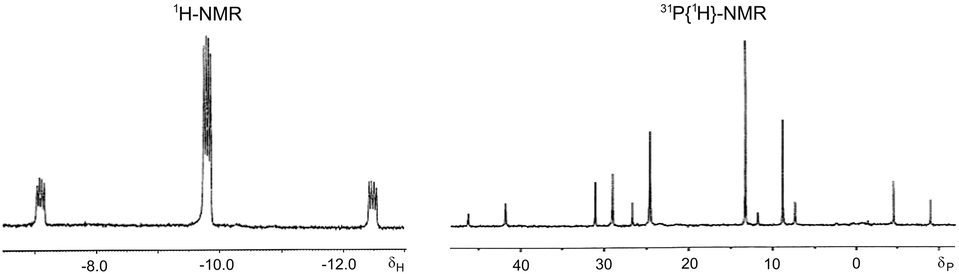 | ||
| Fig. 5 Hydride region of 1H NMR of hydrido[3-(diphenylphosphino-κP)propanethiolato-κS]triphenylphosphineplatinum(II) (11) at room temperature after the addition of two equivalents of SnCl2. | ||
To better understand these experimental results, complex 11 was analyzed computationally. Given that the cis isomer is at roughly 3–4 kcal mol−1 less favored, the calculation of the different conformations was done only for the trans isomer (Fig. 6). For this isomer, the three potential forms coexist with a preference for the chair and skew-boat conformations. From the skew-boat, there is a barrier of 2.1 kcal mol−1 that links this isomer to the chair isomer, whereas the barrier to reach the boat from the skew-boat is 3.2 kcal mol−1. Even though the latter barrier is relatively low, the thermodynamics will not favor the boat conformation that is 2.2 kcal mol−1 higher in energy than the chair conformation.
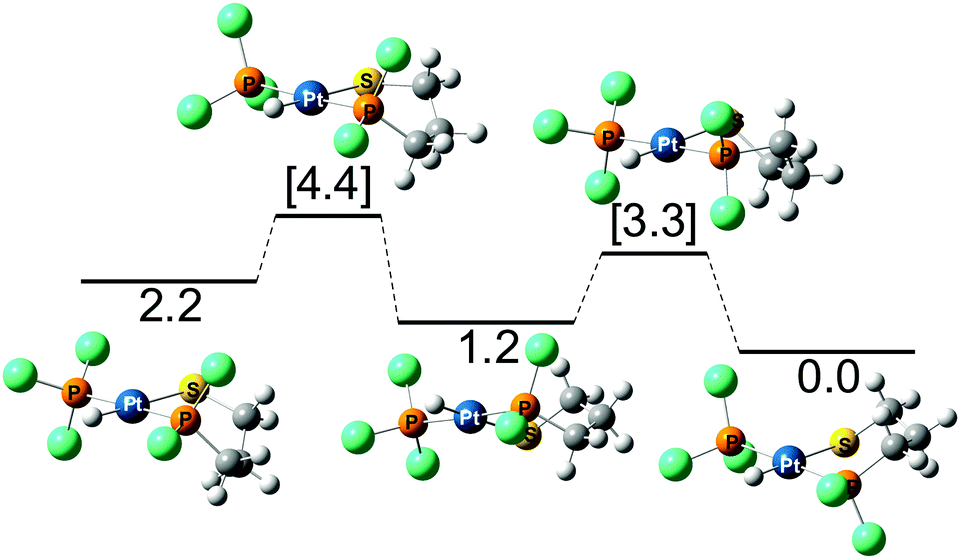 | ||
| Fig. 6 Interconversion between the boat (left), skew-boat (middle), and chair (right) conformations of trans-11 (energies in kcal mol−1, phenyl groups are substituted by green balls for the sake of clarity). | ||
Conclusions
A new family of potentially catalytic platinum(II) hydrides bearing phosphinothiolato ligands (8–11) were synthetized. 2-Phosphinothiolato complexes (8–10) display a chemical equilibrium between the cis-P,P and trans-P,P geometries, strongly displaced towards the trans-P,P configuration, whereas the corresponding complexes with aminothiolato ligands (5–7) showed the inverse stability trend, with the absence of trans isomers. The X-ray characterization of a platinum(II) hydride complex with 2-phosphinothiolate ligand (8) and also with 2-aminothiolate ligands (5–7) is reported for the first time. Density functional theory was used to calculate the energy differences between the cis–trans diastereomers of the above Pt(II) complexes, and the results were in agreement with the experimentally observed cis/trans ratios. We found that the preference for the cis or trans isomer is the result of a delicate balance between higher steric repulsions in the cis isomers as indicated by steric maps and electronic effects, especially those due to trans substituents. DFT calculations are also applied to understand the different behaviour in solution shown between the cis and trans diastereomers of the phosphinothiolato platinum(II) hydrides and between the phosphinothiolato and aminothiolato complexes. 3-Phosphinothiolato platinum(II)hydride (11) appeared as a single geometric isomer, trans, but as two conformational diastereomers because of the six membered chelate ring. Also this surprising result was confirmed by DFT.Experimental section
General considerations
Reactions were performed under nitrogen using standard techniques. Solvents were dried, distilled under nitrogen, kept over molecular sieves, and deaerated prior to use. Pt(PPh3)4 was prepared by established procedures.51 Aminothiolate complexes [Pt(H)(SCH2CH2NH2)(PPh3)] (5), [Pt(H)((L)-SCH2CH(COOCH3)NH2)(PPh3)] (6) and [Pt(H)((L)-SCH2CH(COOCH2CH3)NH2)(PPh3)] (7) were prepared as previously reported and recrystallized from CH2Cl2/hexane.4 The syntheses of 2-(diphenylphosphino)ethanethiol (Hdppet, 1),52 1-(benzyloxy)-2-(diphenylphosphino)ethanethiol (Hbdppet, 2)11 and 3-diphenylphosphinoropanethiol (Hdpppt, 4) have been previously described. Microdistillation procedures were performed on a Buchi GK-R-51 instrument. The C, H, and S analyses were carried out using a Carlo Erba microanalyzer. Infrared spectra (range 4000–400 cm−1) were recorded using KBr pellets on a Nicolet 205 spectrophotometer. Deuterated solvents for NMR measurements were dried over molecular sieves. NMR spectra were obtained on a Bruker DPX-200 operating at 200.13 MHz for 1H, 50.32 MHz for 13C and 80.01 MHz for 31P, or on a Varian Gemini 300 operating at 300.00 MHz for 1H, 75.43 MHz for 13C and 121.44 MHz for 31P. NMR data are given in the δ scale and are referred to internal TMS for 1H and 13C and to external H3PO4 (85%) for 31P.Preparation of 2-(diphenylphosphino)cyclohexanethiol (Hdppcyt, 3)
Step 1. Thiourea (6.222 g, 81.7 mmol) was suspended in a 5/1 mixture of ethanol/deionized water (32 mL) and commercial cyclohexene oxide (2.06 mL, 20.4 mmol) was added with stirring. The mixture was stirred at room temperature for 90 h. Deionized water (15 mL) and ether (15 mL) were added to the reaction mixture. The aqueous phase was extracted with diethyl ether (3 × 10 mL). The organic phase was washed with deionized water (15 mL), dried with magnesium sulfate, filtered, and evaporated to dryness at low temperature. Purification of the crude product by distillation under vacuum (125–130 °C, 0.1 Torr) afforded 477 mg (21%) of cyclohexene sulfide of purity >97% by NMR. Data for cyclohexene sulfide: 1H NMR (CDCl3, 200.13 MHz, 293 K): 1.26 (2H, m, H3 axial); 1.51 (2H, m, H3 equatorial); 2.14 (4H, m, H2); 3.22 (2H, m, H1). 13C{1H} NMR (CDCl3, 50.32 MHz, 293 K): 19.34 (CH2, C3); 25.73 (CH2, C2); 36.93(CH, C1).Step 2. A solution of cyclohexene sulfide (390 mg, 3.4 mmol) in THF (10 mL) was slowly added to an ice-cold solution of commercial potassium diphenylphosphide (0.5 M in tetrahydrofuran, 6.9 mL, 3.5 mmol) with stirring. The reaction mixture was then vigorously stirred at room temperature. After 30 min, deoxygenated methanol (1 mL), deoxygenated saturated ammonium chloride aqueous solution (10 mL), deoxygenated deionized water (15 mL) and ether (15 mL) were consecutively added to the reaction mixture. The aqueous phase was extracted with diethyl ether (3 × 15 mL). The organic phase was washed with water (15 mL), dried with magnesium sulfate, filtered and evaporated to dryness. Purification of the crude product by distillation under vacuum (215–220 °C, 0.1 Torr) afforded 200 mg (21%) of 2-(diphenylphosphino)cyclohexanethiol. Data for Hdppcyt (3): IR: 3053, 2926, 2854, 2538, 1480, 745, 697. 1H NMR (CDCl3, 200.13 MHz, 293 K): 0.7–2.0 (7H, m), 2.18 (1H, d, J = 5.3 Hz, SH); 2.25 (1H, m, H2); 2.40 (1H, td, J = 8.4 Hz, J = 8.4 Hz, J = 3.5 Hz, H6 axial); 2.88 (1H, m, H1); 7–8 (10H, PPh2). 13C{1H} NMR (CDCl3, 50.32 MHz, 293 K): 24.49 (s, CH2, C5); 24.98 (ps. d, JC–P < 3 Hz, CH2, C4); 26.36 (s, CH2, C3); 35.89 (d, JC–P = 6.8 Hz, CH2, C6); 40.68 (d, JC–P = 19.1 Hz, CH, C2); 43.16 (d, JC–P = 14.4 Hz, CH, C1); 128–135 (PPh2). 31P{1H} NMR (CD2Cl2, 81.01 MHz, 293 K): 31P{1H} NMR (CDCl3, 81.01 MHz, 293 K): −5.75 (PPh2).
The yields were low, but as the reagents are easily available, the procedure is still convenient.
Preparation of [Pt(H)(dppet)(PPh3)] (8), hydrido[2-(diphenylphosphino-κP)ethanethiolato-κS]triphenylphosphineplatinum(II)
The platinum(0) complex Pt(PPh3)4 (350 mg, 0.28 mmol) was dissolved in dichloromethane (ca. 10 mL) to give a clear yellow solution. Upon slow addition of Hdppet (2-(diphenylphosphino)ethanethiol, 76 mg, 0.28 mmol, neat) to the stirred solution, its color turned lighter. The mixture was allowed to react for 15 min, the solvent was evaporated in vacuo down to 1–2 mL, and ethylic ether (1–2 mL) was slowly added to precipitate a white microcrystalline solid. The product was separated by filtration, washed with Et2O and dried under a current of nitrogen and under vacuum. Yield: 132 mg (68%). Completely dry complex 8 was stable in air, soluble in dichloromethane and only sparingly soluble in chloroform, and insoluble in ether and hexane. Data for 8: elem. anal. for C32H30P2PtS, found (calcd), C, 53.8 (54.6); H, 4.3 (4.3); S, 4.8 (4.6). IR: 3047, 3006, 2924, 2852, 2082, 1430, 1099, 747, 700, 510. 1H NMR (CD2Cl2, 200.13 MHz, 293 K): cis-8: −4.43 (1H, dd, JH–P = 186.2 Hz, JH–P = 22.9 Hz, JH–Pt = 937.8 Hz); 2.2–3.0 (4H, m); 7–8 (25H, m). trans-8: −7.31 (1H, br d, JH–P = 8.8 Hz, JH–Pt = 969.7 Hz); 2.2–3.0 (4H, m); 7–8 (25H, m). 1H NMR (CD2Cl2, 200.13 MHz, 213 K): cis-8: −4.15 (1H, dd, JH–P = 182.8 Hz, JH–P = 22.0 Hz, JH–Pt = 967.0 Hz); 2.1–3.0 (4H, m); 7–8 (25H, m). trans-8: −7.31 (1H, dd, JH–P = 16.8 Hz, JH–P = 9.7 Hz, JH–Pt = 977.9 Hz); 2.1–3.0 (4H, m); 7–8 (25H, m). 13C{1H} NMR (CD2Cl2, 50.32 MHz, 293 K): 25.44 (d, CH2S, JC–P = 10.1 Hz); 41.47 (d, CH2P, JC–P = 39.3 Hz); 126–136 (PPh2R, PPh3). 31P{1H} NMR (CD2Cl2, 81.01 MHz, 293 K): cis-8: 24.47 (d, PPh3, JP–P = 10.1 Hz, JP–Pt = 3097.9 Hz); 64.22 (d, PPh2R, JP–P = 10.1 Hz, JP–Pt = 1945.9 Hz). trans-8: ca. 33 (br d, PPh3, JP–P = 392.8 Hz); ca. 67 (br d, PPh2R, JP–P = 392.8 Hz). 31P{1H} NMR (CD2Cl2, 81.01 MHz, 213 K): cis-8: 24.53 (d, PPh3, JP–P = 10.3 Hz, JP–Pt = 3100.1 Hz); 64.12 (d, PPh2R, JP–P = 10.3 Hz, JP–Pt = 1964.9 Hz). trans-8: 33.03 (d, PPh3, JP–P = 392.8 Hz, JP–Pt = 2953.9 Hz); 66.71 (d, PPh2R, JP–P = 392.8 Hz, JP–Pt = 2901.6 Hz). After several recrystallization processes product 8 showed the same spectroscopic properties.Preparation of [Pt(H)(bdppet)(PPh3)] (9), hydrido[1-(benzyloxy)-2-(diphenylphosphino-κP)ethanethiolato-κS]triphenylphosphinoplatinum(II)
To a solution of Pt(PPh3)4 (200 mg, 0.16 mmol) in dichloromethane (5 mL) a solution of Hbdppet (1-(benzyloxy)-2-(diphenylphosphino)ethanethiol, 59 mg, 0.16 mmol) in 2 ml of dichloromethane was slowly added with stirring. The mixture was allowed to react for 15 min, the solvent was evaporated in a vacuum down to 1 mL, and ethylic ether (1–2 mL) was slowly added to precipitate a white microcrystalline solid. The product was separated by filtration, washed with Et2O and dried under a current of nitrogen and under vacuum. Yield: 102 mg of a mixture of complex 9 with free PPh3. Completely dry complex 9 was stable in air, soluble in dichloromethane and chloroform, and insoluble in ether and hexane. Data for 9: IR: 2059. 1H NMR (CD2Cl2, 200.13 MHz, 300 K): cis-9: −4.45 (dd, JH–P = 186.1 Hz, JH–P = 22.0 Hz, JH–Pt = 964.6 Hz). trans-9: −7.46 (s, JH–Pt = 978.6 Hz). 1H NMR (CD2Cl2, 200.13 MHz, 213 K): cis-9: −4.21 (dd, JH–P = 184.1 Hz, JH–P = 22.0 Hz, JH–Pt = 956.6 Hz). trans-9: −7.33 (br d, JH–P = 6.0 Hz, JH–Pt = 988.6 Hz). 31P{1H} NMR (CD2Cl2, 81.01 MHz, 300 K): cis-9: 23.93 (d, PPh3, JP–P = 10.8 Hz, JP–Pt = 3120.8 Hz); 56.53 (d, PPh2R, JP–P = 10.8 Hz, JP–Pt = 1891.2 Hz). trans-9: ca. 59 (br, PPh2R, JP–Pt ≈ 2845 Hz). 31P{1H} NMR (CD2Cl2, 81.01 MHz, 193 K): cis-9: 23.88 (d, PPh3, JP–P = 10.7 Hz, JP–Pt = 3125.6 Hz); ca. 59 (PPh2R, JP–Pt ≈ 2051 Hz). trans-9: 32.89 (d, PPh3, JP–P = 384.2 Hz, JP–Pt = 2889.9 Hz); 60.46 (d, PPh2R, JP–P = 384.1 Hz, JP–Pt = 2819.5 Hz).Preparation of [Pt(H)(dppcyt)(PPh3)] (10), hydrido[2-(diphenylphosphino-κP)cyclohexanethiolato-κS]triphenylphosphineplatinum(II)
Complex 10 was prepared as described for complex 9 using 48 mg (0.16 mmol) of Hdppcyt (2-(diphenylphosphino)cyclohexanethiol). Yield: 78 mg (64%). Completely dry complex 10 was stable in air, sparingly soluble in dichloromethane, slightly soluble in chloroform, and insoluble in ether and hexane. Data for 10: elem. anal. for C36H36P2PtS, found (calcd), C, 56.8 (57.1); H, 4.5 (4.8); S, 4.4 (4.2). IR: 3045, 2941, 2850, 2080, 1431, 752, 695. 1H NMR (CD2Cl2, 200.13 MHz, 293 K): cis-10: −4.88 (1H, dd, JH–P = 190.3 Hz, JH–P = 23.8 Hz, JH–Pt = 977.2 Hz); 0.5–2.5 (10H, m); 6.5–8.0 (25H, m). trans-10: −7.67 (1H, br d, JH–P = 9.4 Hz, JH–Pt = 986.2 Hz); 0.5–2.5 (10H, m); 6.5–8.0 (25H, m). 1H NMR (CD2Cl2, 200.13 MHz, 233 K): cis-10: −4.84 (1H, dd, JH–P = 180.7 Hz, JH–P = 23.2 Hz, JH–Pt = 970.2 Hz); 0.5–2.5 (10H, m); 6.5–8.0 (25H, m). trans-10: −7.66 (1H, dd, JH–P = 18.0 Hz, JH–P = 10.2 Hz, JH–Pt = 998.6 Hz); 0.5–2.5 (10H, m); 6.5–8.0 (25H, m). 31P{1H} NMR (CD2Cl2, 81.01 MHz, 293 K): cis-10: 26.27 (d, PPh3, JP–P = 10.8 Hz, JP–Pt = 3106.3 Hz); 61.73 (d, PPh2R, JP–P = 10.8 Hz, JP–Pt = 1908.1 Hz). trans-10: ca. 33 (br d, PPh3, JP–P ≈ 383 Hz); ca. 60 (br d, PPh2R, JP–P ≈ 383 Hz). 31P{1H} NMR (CD2Cl2, 81.01 MHz, 233 K): cis-10: 26.31 (d, PPh3, JP–P = 11.0 Hz, JP–Pt = 3108.4 Hz); 61.43 (d, PPh2R, JP–P = 11.0 Hz, JP–Pt = 1923.9 Hz). trans-10: 33.84 (d, PPh3, JP–P = 385.0 Hz, JP–Pt = 2877.1 Hz); 59.92 (d, PPh2R, JP–P = 385.0 Hz, JP–Pt = 2783.5 Hz).Preparation of [Pt(H)(dpppt)(PPh3)] (11), Hydrido[3-(diphenylphosphino-κP)propanethiolato-κS]triphenylphosphineplatinum(II)
Complex 11 is prepared as described for complex 8 using 73 mg (0.16 mmol) of Hdpppt (3-(diphenylphosphino)propanethiol). Yield: 143 mg (72%). Completely dry complex 10 was stable in air, sparingly soluble in dichloromethane, slightly soluble in chloroform, and insoluble in ether and hexane. Data for 11: elem. anal. for C33H32P2PtS, found (calcd), C, 54.6 (55.2); H, 4.2 (4.5); S, 5.3 (4.5). IR: 3048, 2901, 2078, 2054, 1434, 1099, 692, 519. 1H NMR (CD2Cl2, 300.00 MHz, 293 K): major isomer: −8.66 (1H, br, JH–Pt ≈ 925 Hz); 1–3 (6H, m); 7–8. (25H, m). Minor isomer: −8.95 (1H, br, JH–Pt ≈ 945 Hz); 1–3 (6H, m); 7–8 (25H, m). 1H NMR (CD2Cl2, 300.00 MHz, 213 K): major isomer: −8.58 (1H, dd, JH–P = 15.8 Hz, JH–P = 10.8 Hz, JH–Pt = 930.0 Hz); 1.2–2.8 (6H, m); 7–8 (25H, m). minor isomer: −8.90 (1H, dd, JH–P = 14.8 Hz, JH–P = 11.0 Hz, JH–Pt = 950 Hz); 1.2–2.8 (6H, m); 7–8 (25H, m). 31P{1H} NMR (CD2Cl2, 121.44 MHz, 293 K): major isomer: ca. 9.4 (br d, PPh3, JP–P ≈ 396 Hz, JP–Pt ≈ 3010 Hz); ca. 32.1 (br d, PPh2R, JP–P ≈ 397 Hz, JP–Pt ≈ 2923 Hz). Minor isomer: ca. 8.8 (br d, PPh3, JP–P ≈ 396 Hz); ca. 33.8 (br d, PPh2R, JP–P ≈ 395 Hz). 31P{1H} NMR (CD2Cl2, 121.44 MHz, 213 K): major isomer: 7.63 (d, PPh3, JP–P = 388.4 Hz, JP–Pt = 2937.0 Hz); 33.18 (d, PPh2R, JP–P = 388.4 Hz, JP–Pt = 2885.0 Hz). Minor isomer: 7.09 (d, PPh3, JP–P = 390.3 Hz, JP–Pt = 2871.0 Hz); 33.59 (d, PPh2R, JP–P = 390.3 Hz, JP–Pt = 2796.9 Hz).Single crystal X-ray diffraction
White crystals of complexes 5–8 were obtained after different trials by slow evaporation of CH2Cl2–hexane solutions. Crystal structure determinations were carried out using a Siemens P4 diffractometer equipped with a SMARTCCD-1000 area detector, a MACScience Co rotating anode with Mo Kα radiation, a graphite monochromator, and a Siemens LT2 low-temperature device (T = −120 °C). The measurements were made in the range 2.14–31.54° (5), 1.58–31.56° (6), 1.57–31.51° (7) and 2.22–31.53° (8) for θ. Full-sphere data collection was carried out using ω and φ scans. Programs used: Data collection using Smart,53 data reduction using Saint54 and absorption correction using SADABS.55 Crystal structure solution for 5–8 was achieved using direct methods, as implemented in SHELXTL56 and visualized using the XP program. Missing atoms were subsequently located from difference Fourier syntheses and added to the atom list. Least squares refinement on F2 using all measured intensities was carried out using the program SHELXTL.57 All non-hydrogen atoms were refined including anisotropic displacement parameters. Hydrogen atoms were invariably placed in geometrically optimized positions and forced to ride on the atom to which they are attached. Crystallographic data and structure refinement parameters are presented in Table S1 (ESI†), and selected distances and angles are presented in Table 1.:48. The structure shows pseudo-symmetry and was refined in the space groups P1 and P. Refinement in the space group P applying a disorder model in the chiral atom (C2) was preferred. The R1 value was lowered by doing this from 7.39% to 6.06%. If the platinum atom is considered as chiral, the two disordered orientations would correspond to diastereoisomers. Structure 7 shows a similar behavior to structure 6 but in this case, although pseudo-symmetry is observed, the space group P1 was preferred. The asymmetric unit contains two independent molecules which would correspond to diastereoisomers. In structure 8 a high electron density was localized at 1.44 Å from the platinum atom and 0.95 Å from the sulfur atom which could not be refined properly since it does not correspond to logical localization. Attempts to refine a disordered model were not successful. Taking into account that in general high electron densities in the surrounding of heavy atoms are common it was assumed that the presence of this electron density was reinforced by problems in the crystal quality (maybe a second small crystal present) and with difficulties during the absorption correction.
Theoretical background
To analyze the electrophilicity of the optimized complexes, we have calculated the electrophilicity index defined by Parr et al. as:58where μ and η are the chemical potential and the molecular hardness, respectively. In the framework of the conceptual Density Functional Theory (DFT), the chemical potential (μ) and molecular hardness (η) can be numerically calculated as:
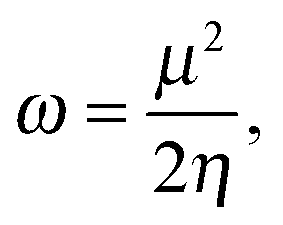
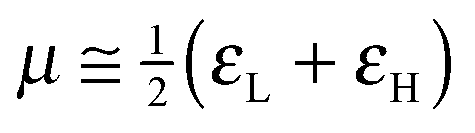
where εH and εL are the energies of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO), respectively.
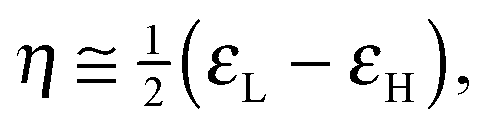
Computational details
All DFT static calculations were performed using the Gaussian set of programs.59 The electronic configuration of the molecular systems was described with the standard split-valence basis set with a polarization function of Ahlrichs and co-workers for C, N, P, O, S and H (SVP keyword in Gaussian09).60 For Pt we used the small-core, quasi-relativistic Stuttgart/Dresden effective core potential, with an associated valence basis set contracted (standard SDD keywords in Gaussian09).61 The geometry optimizations were carried out without symmetry constraints, and the characterization of the located stationary points was performed by analytical frequency calculations. The B3LYP-D3 functional was employed in geometry optimizations and frequency calculations.62,63 Gibbs energies, ΔG, were built through single point energy calculations on the B3LYP-D3/SVP geometries using the M06 functional64 and the triple-ζ valence plus polarization on main group atoms (TZVP keyword in Gaussian). Solvent effects were included with the PCM model using CH2Cl2 as the solvent.65 To these M06/TZVP electronic energies in solvent, zero point energy and thermal and entropic corrections were included from the gas phase frequency calculations at the B3LYP-D3/SVP level of theory.To validate the computational method a geometry optimization of the X-ray data of the complex cis-7 was made. The results show a good agreement between the experimental and theoretical data as can be seen in Table S2 (ESI†). The experimental and theoretical bond lengths and angles differ by less than 0.069 Å and 2.5°, respectively. And the standard deviation for the distances is 0.038 Å, and for the angles 1.2°, which confirms the validity of the chosen computational method. The optimized geometries of the stereoisomers of complexes 5–11 can be seen in Fig. S6 (other methods from a benchmark study are also included in the ESI†).66
We have also carried out energy decomposition analyses (EDAs).67 In these EDAs, the total binding energy (BE) has been divided into deformation energy and interaction energy (BE = ΔEdef + ΔEint). The deformation energy (ΔEdef) is the energy needed to modify the geometry of the ground state free fragments to attain the geometry that they have in the intermediate. The interaction energy (ΔEint) is the energy released when the two singlet free deformed fragments are brought to the position that they have in the intermediate, and it has been, in turn, split into electrostatic, Pauli repulsion, and orbital interaction terms (ΔEint = ΔEelstat + ΔEPauli + ΔEoi). The term ΔEelstat corresponds to the classical electrostatic interaction between the unperturbed charge distributions of the prepared fragments and is usually attractive. The Pauli repulsion term, ΔEPauli, comprises the destabilizing interactions between occupied orbitals and is responsible for the steric repulsion. The orbital interaction term, ΔEoi, accounts for bond pair formation (interaction between single occupied orbitals in the two fragments), charge transfer (interaction between occupied orbitals on one moiety with unoccupied orbitals of the other, including the HOMO–LUMO interactions) and polarization (empty occupied orbital mixing on one fragment due to the presence of another fragment).68 The reported EDA calculations have been carried out by using the Amsterdam density functional (ADF) package developed by Baerends and coworkers69 and vectorized by Ravenek.70 The numerical integration scheme employed has been that of te Velde and Baerends.71 An uncontracted triple-ς basis set has been used for describing the orbitals of platinum. For C, N, P, O, S and H atoms double-ς basis sets have been employed. Both basis sets have been augmented by an extra polarization function.72 A set of auxiliary s, p, d, f, and g functions, centered in all nuclei, has been introduced in order to fit the molecular density and Coulomb potential accurately in each SCF cycle.73 Relativistic effects have been included in the energy calculations and geometry optimizations using the ZORA method.74 Geometries and energies have been evaluated using a generalized gradient approximation (GGA) that includes the GGA exchange correction of Becke75 and the GGA correlation correction of Perdew.76 The 2014 release of the ADF package has been used for all EDA calculations.77
Experimentally, it is found that mononuclear species 5–11 are diamagnetic. For this reason, optimizations were carried out for neutral closed-shell singlet ground state structures. However, since it has been found that the triplet state is close in energy to the singlet or even it can be more stable we have checked for all compounds the relative stability of both states by optimizing the triplet state. In all cases, the triplet state geometry presents a higher energy, indicating that the singlet state is the most favorable. The difference between these two states is never lower than 8 kcal mol−1.
%VBur calculations
The buried volume calculations were performed using the SambVca package developed by Cavallo et al.38,40 The radius of the sphere around the metal center was set to 3.5 Å, while for the atoms we adopted the Bondi radii scaled by 1.17, and a mesh of 0.1 Å was used to scan the sphere for buried voxels. The steric maps were evaluated using a development version of the SambVca package.38Acknowledgements
We thank the Spanish Ministerio de Economía y Competitividad (MINECO) for projects BQU2002-04070-C02-01, CTQ2014-54306-P and CTQ2014-59832-JIN. This work has also been supported by the Generalitat de Catalunya (project number 2014SGR931, Xarxa de Referència en Química Teòrica i Computacional, and ICREA Academia 2014 prize for M. S.).Notes and references
- F. Agbossou-Niedercorn, in Applied Homogeneous Catalysis with Organometallic Compounds, ed. R. Comils and W. A. Herrmann, VCH, Weinheim, 2002, vol. 2, pp. 1014–1033 Search PubMed.
- (a) N. Salvarese, A. Dolmella, F. Refosco and C. Bolzati, Inorg. Chem., 2015, 54, 1634–1644 CrossRef CAS PubMed; (b) N. Salvarese, N. Morellato, A. Venzo, F. Refosco, A. Dolmella and C. Bozati, Inorg. Chem., 2013, 52, 6365–6377 CrossRef CAS PubMed; (c) C. Bolzati, N. Salvarese, D. Carta, F. Refosco, A. Dolmella, H. J. Pietzsch, R. Bergmann and G. Bandoli, JBIC, J. Biol. Inorg. Chem., 2011, 16, 137–155 CrossRef CAS PubMed; (d) P. Perez-Lourido, J. Romero, L. Rodriguez, J. A. Garcia-Vazquez, J. Castro, A. Sousa, J. R. Dilworth and O. R. Nascimento, Inorg. Chem. Commun., 2002, 5, 337–339 CrossRef CAS; (e) K. A. Giffin, I. Korobkov and R. T. Baker, Dalton Trans., 2015, 44, 19587–19596 RSC.
- E. J. Fernandez, A. Laguna, J. M. Lopez-de-Luzuriaga, M. Monge and E. Sanchez-Forcada, Dalton Trans., 2011, 40, 3287–3294 RSC.
- J. Real, A. Polo and J. Duran, Inorg. Chem. Commun., 1998, 1, 457–459 CrossRef CAS.
- J. Real, E. Prat-Gil, M. Pagès-Barenys, A. Polo, J. F. Piniella and A. Álvarez-Larena, Dalton Trans., 2016, 45, 3964–3973 RSC.
- A. Dervisi, R. L. Jenkins, K. M. A. Malik, M. B. Hursthouse and S. Coles, Dalton Trans., 2003, 1133–1142 RSC.
- P. W. N. M. Van Leeuwen, P. C. J. Kamer, J. N. H. Reek and P. Dierkes, Chem. Rev., 2000, 100, 2741–2769 CrossRef CAS PubMed.
- J. Real, M. Pagès, A. Polo, J. F. Piniella and A. Álvarez-Larena, Chem. Commun., 1999, 277–278 RSC.
- J. Real, E. Prat, A. Polo, A. Álvarez-Laren and J. F. Piniella, Inorg. Chem. Commun., 2000, 3, 221–223 CrossRef CAS.
- J. Duran, N. Brugat, A. Polo, J. Real, X. Fontrodona and J. Benet-Buchholz, Organometallics, 2003, 22, 3432–3438 CrossRef CAS.
- N. Brugat, J. Duran, A. Polo, J. Real, A. Álvarez-Larena and J. F. Piniella, Tetrahedron: Asymmetry, 2002, 13, 569–577 CrossRef CAS.
- J. Duran, A. Polo, J. Real, J. Benet-Buchholz, A. Poater and M. Solà, Eur. J. Inorg. Chem., 2003, 4147–4151 CrossRef CAS.
- J. Duran, A. Polo, J. Real, J. Benet-Buchholz, A. Poater and M. Solà, ChemistryOpen, 2016, 5, 51–59 CrossRef CAS PubMed.
- (a) S. Dey, L. B. Kumbhare, V. K. Jain, T. Schurr, W. Kaim, A. Klein and F. Belaj, Eur. J. Inorg. Chem., 2004, 4510–4520 CrossRef CAS; (b) N. Ghavale, A. Wadawale, S. Dey and V. K. Jain, J. Organomet. Chem., 2010, 695, 2296–2304 CrossRef CAS.
- (a) D. Gibson and S. Lippard, J. Inorg. Chem., 1986, 25, 219–222 CAS; (b) P. P. Corbi, E. E. Castellano, F. Cagnin and A. C. Massabni, J. Chem. Crystallogr., 2007, 37, 91–95 CrossRef CAS; (c) N. Yoshinari, Y. Hashimoto, A. Igashira-Kamiyama and T. Konno, Bull. Chem. Soc. Jpn., 2011, 84, 623–625 CrossRef CAS.
- P. V. Rao, B. Sumit, J. Jiang, D. Hong and R. H. Holm, J. Am. Chem. Soc., 2005, 127, 1933–1945 CrossRef CAS PubMed.
- (a) S. A. Bryan and D. M. Roundill, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1983, C39, 184–186 CrossRef CAS; (b) A. Ishii, S. Kashiura, Y. Hayashi and W. Weigand, Chem. – Eur. J., 2007, 13, 4326–4333 CrossRef CAS PubMed; (c) N. Nakata, M. Sakashita, C. Komatsubara and A. Ishii, Eur. J. Inorg. Chem., 2010, 447–453 CrossRef CAS; (d) C. Mügge, C. Rothenburger, A. Beyer, H. Görls, C. Gabbiani, A. Casini, E. Michelucci, I. Landini, S. Nobili, E. Mini, L. Messori and W. Weigand, Dalton Trans., 2011, 40, 2006–2016 RSC.
- R. P. Hughes, J. T. Sweetser, M. D. Tawa, A. Williamson, C. D. Incarvito, B. Rhatigan, A. L. Rheingold and G. Rossi, Organometallics, 2001, 20, 3800–3810 CrossRef CAS.
- (a) S. Krogsrud, L. Toniolo, U. Croatto and J. A. Ibers, J. Am. Chem. Soc., 1977, 99, 5277–5284 CrossRef CAS; (b) E. Leidl, U. Nagel and W. Beck, Chem. Ber., 1983, 116, 1370–1376 CrossRef CAS; (c) J. R. Berenguer, E. Eguizábal, L. R. Falvello, J. Forniés, E. Lalinde and A. Martín, Organometallics, 1999, 18, 1653–1662 CrossRef CAS.
- (a) R. H. Heyn, S. D. Grumbine and T. D. Tilley, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1991, 47, 313–315 CrossRef; (b) L. Jager, C. Tretner, H. Hartung and M. Biedermann, Chem. Ber., 1997, 130, 1007–1012 CrossRef CAS.
- J. W. Faller, H. L. Stokes-Huby and M. A. Albrizzio, Helv. Chim. Acta, 2001, 84, 3031–3042 CrossRef CAS.
- N. Brugat, A. Polo, A. Álvarez-Larena, J. P. Piniella and J. Real, Inorg. Chem., 1999, 38, 4829–4837 CrossRef CAS PubMed.
- M. Kita, T. Yamamoto, K. Kashiwabara and J. Fujita, Bull. Chem. Soc. Jpn., 1992, 65, 2272–2274 CrossRef CAS.
- (a) R. Ugo, G. La Monica, S. Cenini, A. Segre and F. Conti, J. Chem. Soc. A, 1971, 522–528 RSC; (b) A. E. Keskinen and C. V. Senoff, J. Organomet. Chem., 1972, 37, 201–208 CrossRef CAS.
- F. Yamashita, H. Kuniyasu, J. Terao and N. Kambe, Inorg. Chem., 2006, 45, 1399–1404 CrossRef CAS PubMed.
- A. Albinati, A. Musco, G. Carturan and G. Strukul, Inorg. Chim. Acta, 1976, 18, 219–223 CrossRef CAS.
- C. H. Wei, G. B. Cheng, H. F. Guo, S. F. Lu and Q. J. Wu, Chin. J. Struct. Chem., 1991, 10, 180–182 CAS.
- C. A. Ghilardi, S. Midollini, A. Orlandini, G. Scapacci and A. Vacca, J. Organomet. Chem., 1993, 461, C4–C6 CrossRef CAS.
- (a) M. A. Bennett, G. B. Robertson and P. N. Kapoor, Aust. J. Chem., 1999, 52, 949–954 CrossRef CAS; (b) G. Ferguson, Y. Li, A. J. McAlees, R. McCrindle and E. Zang, J. Organomet. Chem., 2001, 617–618, 671–680 CrossRef CAS; (c) D. Zhang, J. Dou, D. Li and D. Wang, Inorg. Chim. Acta, 2006, 359, 4243–4249 CrossRef CAS.
- R. G. Pearson, Inorg. Chem., 1973, 12, 712–713 CrossRef CAS.
- (a) R. G. Pearson, Acc. Chem. Res., 1993, 26, 250–255 CrossRef CAS; (b) R. G. Pearson, J. Chem. Educ., 1999, 76, 267–275 CrossRef CAS.
- J. N. Harvey, K. M. Heslop, A. G. Orpen and P. G. Pringle, Chem. Commun., 2003, 278–279 RSC.
- (a) G. Faraone, V. Ricevuto, R. Romeo and M. Trozzi, J. Chem. Soc. A, 1971, 1877–1881 RSC; (b) R. J. Cross, Adv. Inorg. Chem., 1989, 34, 219–292 CrossRef CAS; (c) R. Romeo, Comments Inorg. Chem., 1990, 11, 21–57 CrossRef CAS.
- F. Basolo, Coord. Chem. Rev., 1996, 154, 151–161 CrossRef CAS.
- G. Aullón and S. Álvarez, Inorg. Chem., 1996, 35, 3137–3144 CrossRef.
- R. Ros, R. A. Michelin, R. Bataillard and R. Roulet, J. Organomet. Chem., 1979, 165, 107–116 CrossRef CAS.
- L. P. Wolters, R. Koekkoek and F. M. Bickelhaupt, ACS Catal., 2015, 5, 5766–5775 CrossRef CAS.
- L. Falivene, R. Credendino, A. Poater, A. Petta, L. Serra, R. Oliva, V. Scarano and L. Cavallo, Organometallics, 2016, 35, 2286–2293 CrossRef CAS.
- (a) L. Cavallo, A. Correa, C. Costabile and H. Jacobsen, J. Organomet. Chem., 2005, 690, 5407–5413 CrossRef CAS; (b) A. Poater, F. Ragone, S. Giudice, C. Costabile, R. Dorta, S. P. Nolan and L. Cavallo, Organometallics, 2008, 27, 2679–2681 CrossRef CAS; (c) S. M. Ahmed, A. Poater, M. I. Childers, P. C. B. Widger, A. M. LaPointe, E. B. Lobkovsky, G. W. Coates and L. Cavallo, J. Am. Chem. Soc., 2013, 135, 18901–18911 CrossRef CAS PubMed; (d) J. P. Martínez, S. V. C. Martínez, L. Falivene, S. P. Nolan, L. Cavallo, M. Solà and A. Poater, Chem. – Eur. J., 2016, 22, 6617–6623 CrossRef PubMed.
- A. Poater, B. Cosenza, A. Correa, S. Giudice, F. Ragone, V. Scarano and L. Cavallo, Eur. J. Inorg. Chem., 2009, 1759–1766 CrossRef CAS.
- G. S. White and D. W. Stephan, Organometallics, 1987, 6, 2169–2175 CrossRef CAS.
- H. Bouda, M. E. Borredon, M. Delmas and A. Gaset, Synth. Commun., 1987, 17, 943–951 CrossRef CAS.
- P. H. Leung, A. C. Willis and S. B. Wild, Inorg. Chem., 1992, 31, 1406–1410 CrossRef CAS.
- J. R. Gollogly and C. J. Hawkins, Inorg. Chem., 1972, 11, 156–161 CrossRef CAS.
- C. J. Hawkins and J. A. Palmer, Coord. Chem. Rev., 1982, 44, 1–60 CrossRef CAS.
- (a) A. K. Mukherjee, M. Mukherjee, S. Ray, A. Ghosh and N. Ray Chaudhuri, J. Chem. Soc., Dalton Trans., 1990, 2347–2350 RSC; (b) P. C. Christidis, C. A. Bolos, G. Bauer, G. Will, N. S. Trendafilova and G. St. Nikolov, Inorg. Chim. Acta, 1995, 228, 173–185 CrossRef CAS; (c) M. F. DaCruz and M. Zimmer, Inorg. Chem., 1996, 35, 2872–2877 CrossRef CAS; (d) L. Cerasino, K. M. Williams, F. P. Intini, R. Cini, L. G. Marzilli and G. Natile, Inorg. Chem., 1997, 36, 6070–6079 CrossRef CAS PubMed; (e) M. F. DaCruz and M. Zimmer, Inorg. Chem., 1998, 37, 366–368 CrossRef CAS.
- V. Schulz, A. Frick and G. Huttner, Eur. J. Inorg. Chem., 2002, 3111–3128 CrossRef CAS.
- G. R. Brubaker and D. W. Johnson, Coord. Chem. Rev., 1984, 53, 1–60 CrossRef CAS.
- J. Karas, G. Huttner, K. Heinze, P. Rutsch and L. Zsolnai, Eur. J. Inorg. Chem., 1999, 405–420 CrossRef CAS.
- A. Frick, V. Schulz and G. Huttner, Eur. J. Inorg. Chem., 2002, 3129–3147 CrossRef CAS.
- R. Ugo, F. Cariati and G. La Monica, Inorg. Synth., 1968, 11, 105–108 CrossRef CAS.
- J. Chatt, J. R. Dilworth, J. A. Schmutz and J. A. Zubieta, J. Chem. Soc., Dalton Trans., 1979, 1595–1599 RSC.
- Data collection: SMART software; version 5.060, Bruker AXS 1999 Search PubMed.
- Data processing: Saint +; version 6.02, Bruker AXS 1999 Search PubMed.
- Absorption correction: SABADS, Bruker AXS 1999 Search PubMed.
- G. M. Sheldrick, Structure solution: XS implemented in SHEXLTL version 5.10, Universtität Göttingen, Göttingen, Germany, 1998 Search PubMed.
- G. M. Sheldrick, Structure refinement: XL implemented in SHEXLTL version 5.10, Universtität Göttingen, Göttingen, Germany, 1998 Search PubMed.
- R. G. Parr, L. von Szentpály and S. Liu, J. Am. Chem. Soc., 1999, 121, 1922–1924 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- A. Schaefer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef CAS.
- (a) U. Haeusermann, M. Dolg, H. Stoll and H. Preuss, Mol. Phys., 1993, 78, 1211–1224 CrossRef; (b) W. Kuechle, M. Dolg, H. Stoll and H. Preuss, J. Chem. Phys., 1994, 100, 7535–7542 CrossRef CAS; (c) T. Leininger, A. Nicklass, H. Stoll, M. Dolg and P. Schwerdtfeger, J. Chem. Phys., 1996, 105, 1052–1059 CrossRef CAS.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS; (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS; (c) P. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef CAS.
- (a) V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS; (b) J. Tomasi and M. Persico, Chem. Rev., 1994, 94, 2027–2094 CrossRef CAS.
- A. Poater, E. Pump, S. V. C. Vummaleti and L. Cavallo, J. Chem. Theory Comput., 2014, 10, 4442–4448 CrossRef CAS PubMed.
- (a) F. M. Bickelhaupt, N. M. Nibbering, E. M. van Wezenbeek and E. J. Baerends, J. Phys. Chem., 1992, 96, 4864–4873 CrossRef CAS; (b) T. Ziegler and A. Rauk, Inorg. Chem., 1979, 18, 1558–1565 CrossRef CAS; (c) T. Ziegler and A. Rauk, Inorg. Chem., 1979, 18, 1755–1759 CrossRef CAS; (d) T. Ziegler and A. Rauk, Theor. Chim. Acta, 1977, 46, 1–10 CrossRef CAS; (e) K. Kitaura and K. Morokuma, Int. J. Quantum Chem., 1976, 10, 325–331 CrossRef CAS.
- (a) C. Fonseca Guerra and F. M. Bickelhaupt, Angew. Chem., Int. Ed., 1999, 38, 2942–2945 CrossRef CAS; (b) C. Fonseca Guerra, F. M. Bickelhaupt, J. G. Snijders and E. J. Baerends, Chem. – Eur. J., 1999, 5, 3581–3595 CrossRef; (c) C. Fonseca Guerra and F. M. Bickelhaupt, Angew. Chem., Int. Ed., 2002, 41, 2092–2095 CrossRef; (d) J. Poater, X. Fradera, M. Solà, M. Duran and S. Simon, Chem. Phys. Lett., 2003, 369, 248–255 CrossRef CAS.
- (a) E. J. Baerends, D. E. Ellis and P. Ros, Chem. Phys., 1973, 2, 41–51 CrossRef CAS; (b) C. Fonseca Guerra, O. Visser, J. G. Snijders, G. te Velde and E. J. Baerends, Methods and Techniques for Computational Chemistry, STEF, Cagliari, 1995, p. 305 Search PubMed; (c) G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. Fonseca Guerra, S. J. A. van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931–967 CrossRef CAS.
- W. Ravenek, Algorithms and Applications on Vector and Parallel Computers, Elsevier, Amsterdam, 1987 Search PubMed.
- G. te Velde and E. J. Baerends, J. Comput. Phys., 1992, 99, 84–98 CrossRef CAS.
- (a) J. G. Snijders, E. J. Baerends and P. Vernooijs, At. Data Nucl. Data Tables, 1982, 26, 483–509 CrossRef; (b) P. Vernooijs and E. J. Baerends, Slater Type Basis Functions for the Whole Periodic System. Internal Report, Vrije Universiteit of Amsterdam, Amsterdam, 1981.
- J. B. Krijn and E. J. Baerends, Fit Functions in the HFS Method. Internal Report, Vrije Universiteit of Amsterdam, Amsterdam, 1984 Search PubMed.
- (a) E. van Lenthe, A. E. Ehlers and E. J. Baerends, J. Chem. Phys., 1999, 110, 8943–8953 CrossRef CAS; (b) E. van Lenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys., 1993, 99, 4597–4610 CrossRef CAS; (c) E. van Lenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys., 1994, 101, 9783–9792 CrossRef CAS; (d) E. van Lenthe, J. G. Snijders and E. J. Baerends, J. Chem. Phys., 1996, 105, 6505–6516 CrossRef CAS; (e) E. van Lenthe, E. J. Baerends and J. G. Snijders, Int. J. Quantum Chem., 1996, 57, 281–293 CrossRef CAS.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS.
- J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822–8824 CrossRef.
- (a) G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. Fonseca Guerra, S. J. A. van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931–967 CrossRef CAS; (b) C. Fonseca Guerra, J. G. Snijders, G. te Velde and E. J. Baerends, Theor. Chem. Acc., 1998, 99, 391–403 Search PubMed; (c) E. J. Baerends, T. Ziegler, J. Autschbach, D. Bashford, A. Bérces, F. M. Bickelhaupt, C. Bo, P. M. Boerrigter, L. Cavallo, D. P. Chong, L. Deng, R. M. Dickson, D. E. Ellis, M. van Faassen, L. Fan, T. H. Fischer, C. Fonseca Guerra, M. Franchini, A. Ghysels, A. Giammona, S. J. A. van Gisbergen, A. W. Götz, J. A. Groeneveld, O. V. Gritsenko, M. Grüning, S. Gusarov, F. E. Harris, P. van den Hoek, C. R. Jacob, H. Jacobsen, L. Jensen, J. W. Kaminski, G. van Kessel, F. Kootstra, A. Kovalenko, M. V. Krykunov, E. van Lenthe, D. A. McCormack, A. Michalak, M. Mitoraj, S. M. Morton, J. Neugebauer, V. P. Nicu, L. Noodleman, V. P. Osinga, S. Patchkovskii, M. Pavanello, P. H. T. Philipsen, D. Post, C. C. Pye, W. Ravenek, J. I. Rodríguez, P. Ros, P. R. T. Schipper, H. van Schoot, G. Schreckenbach, J. S. Seldenthuis, M. Seth, J. G. Snijders, M. Solà, M. Swart, D. Swerhone, G. te Velde, P. Vernooijs, L. Versluis, L. Visscher, O. Visser, F. Wang, T. A. Wesolowski, E. M. van Wezenbeek, G. Wiesenekker, S. K. Wolff, T. K. Woo and A. L. Yakovlev, ADF2014, SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR data for ligand 3 and complexes 9 and 10. Computational details, cartesian coordinates, and crystal data for the XRD structures. CCDC 1501904–1501907. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6nj04042j |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2017 |