
A metal–semiconductor nanocomposite as an efficient oxygen-independent photosensitizer for photodynamic tumor therapy†
Jin-Xuan
Fan‡
,
Miao-Deng
Liu‡
,
Chu-Xin
Li
,
Sheng
Hong
,
Di-Wei
Zheng
,
Xin-Hua
Liu
,
Si
Chen
,
Hong
Cheng
and
Xian-Zheng
Zhang
 *
*
Key Laboratory of Biomedical Polymers of Ministry of Education & Department of Chemistry, Wuhan University, Wuhan 430072, China. E-mail: xz-zhang@whu.edu.cn
First published on 12th July 2017
Abstract
Photodynamic therapy (PDT) is regarded as one of the most promising cancer treatments, and oxygen-independent photosensitizers have been intensively explored for advancing the development of PDT. Here, we reported on a superior hybrid nanocomposite (HNC) consisting of a metal (Au deposition) and a semiconductor (CdSe-seeded/CdS nanorods) as a photosensitizer. Under visible light, the photogenerated holes were three-dimensionally confined to the CdSe quantum dots and the delocalized electrons were transferred to the Au tips, which provided hydrogen and oxygen evolution sites for water splitting to generate reactive oxygen species (ROS) with no need for oxygen participation. Compared with semiconductors without deposited metal (i.e. raw CdSe-seeded/CdS nanorods (NRs)) under a normoxic or hypoxic environment, the HNCs exhibited substantially enhanced light-triggered ROS generation in vitro. After being modified with an Arg-Gly-Asp (RGD) peptide sequence, the nanocomposite was deemed as a tumor-targeting, long-lived and oxygen-independent photosensitizer with promoted PDT efficiency for in vivo anti-tumor therapy. This oxygen-independent nanocomposite successfully overcame the hypoxia-related PDT resistance by water splitting, which opened a window to develop conventional semiconductors as photosensitizers for effective PDT.
Conceptual insightsMetal deposition on semiconductors as photocatalysts has been a promising method to improve their catalytic efficiencies in many fields, nevertheless rarely in biomedicine. Here, we innovatively employed CdSe-seeded/CdS nanorods as a representative semiconductor for PDT tumor therapy, and verified that after deposition of Au on top of the nanorods, the catalytic efficiency increased substantially to generate plenty of ROS for cell death. In addition, the overall process of ROS generation utilized water splitting without oxygen participation, which provided a novel oxygen-independent photosensitizer to overcome the hypoxic microenvironment of tumors. |
1. Introduction
Photodynamic therapy (PDT) is considered as a promising cancer treatment due to its minimal invasiveness, low side effects and ideal site-specific controllability.1–4 In the traditional PDT process, a photosensitizer (PS) is activated upon light irradiation and then it transfers energy to molecular oxygen,5,6 thus generating cytotoxic reactive oxygen species (ROS) such as singlet oxygen (1O2) and hydroxyl radicals (˙OH).7–9 However, it is well-known that hypoxia (pO2 ≤ 2.5 mmHg) is a common feature of most solid tumors and could severely blunt the therapeutic effect of PDT.10 To achieve efficient PDT, various strategies have been proposed, such as subcellular organelle targeting PDT for irreversible cell damage, utilizing perfluorohexane for intratumoral oxygen enrichment and producing oxygen in situ through H2O2 decomposition for tumor hypoxia alleviation.11 Self-evidently, the concept of oxygen-independent photodynamic therapy is quite attractive, and an oxygen-independent photosensitizer with high-efficacy is the key to overcome the hypoxia-related PDT resistance.It is well-known that semiconductor materials have been widely used for sensing management, energy catalysis and electrochemistry applications owing to their excellent electron conduction ability and broadband light response from the UV to visible region. Moreover, numerous semiconductor materials such as titania (TiO2),12–14 carbon nitride (C3N4),15–17 cadmium sulfide (CdS)18–20 and tin tungstate (α-SnWO4)21 universally possess appropriate band gaps, which are suitable to promote charge separation of photogenerated holes and electrons under visible light irradiation, and are used for photocatalytic water splitting.22,23 In general, the processes of water splitting can be divided into down-hill reactions and up-hill reactions.24 In our previous study, we utilized C3N4 to achieve an up-hill reaction based on water splitting to produce oxygen in vivo for enhanced PDT,25 but the integrated ROS producing efficiency was not satisfactory. It is worth noting that heterogeneous photocatalysis based on semiconductor materials tends to undergo a down-hill reaction during water splitting, and thus directly produces reactive substances including 1O2 and ˙OH. This specific property makes it possible to exploit new semiconductor materials for oxygen-independent PDT treatment.
Conventional semiconductor materials are usually accompanied by a low quantum efficiency (no more than 7%)26 and limited utilization of solar light (the excitation wavelength of TiO2 is <380 nm).27,28 As mentioned above, it is quite necessary to improve the photocatalytic efficiency as well as the long-term photosensitivity of traditional semiconductor nanomaterials.29 Having attracted extensive interest, semiconductor doping and surface modification are promising strategies for performance improvement of semiconductor nanomaterials.30–33 Semiconductor nanoparticles doped using metallic tips, which could serve as anchor points for the retention of photoexcited electrons, could significantly promote the separation of photogenerated electrons and holes,34 thus enhancing the photocatalytic efficiency.35–37 For example, after the growth of Pt,38 Au,39 and CuS40 on CdSe-seeded/CdS semiconductor nanorods, the efficiency of the hybrid nanocomposite (HNC) for water splitting is significantly enhanced.
Here, we report on a superior hybrid nanocomposite, which consists of a metal (Au deposition) and a semiconductor (CdSe-seeded/CdS nanorods). In consideration of tumor targeting, the Arg–Gly–Asp (RGD) peptide sequence, which can specifically bind tumors with a high level of integrin αvβ3 expressed in most of the solid tumors, was further introduced to construct a PDT photosensitizer as illustrated in Scheme 1A.41 Under visible light irradiation, this nanocomposite was able to promote charge separation of photogenerated holes and electrons.42 The separated holes were three-dimensionally confined to the CdSe quantum dots, while the delocalized electrons were transferred to the Au tips.43,44 The aforesaid process provided reaction sites for high-efficiency water splitting to generate plenty of ROS with no need for oxygen participation. In vitro studies demonstrated that the hybrid nanocomposite had a high efficiency for ROS generation upon visible light irradiation under both aerobic and anaerobic conditions. In vivo studies based on an orthotopic breast cancer BALB/c mouse model also showed remarkable tumor inhibition ability after a biodistribution study, a tumor suppression study and evaluation of the side effects following treatment with the HNCs.
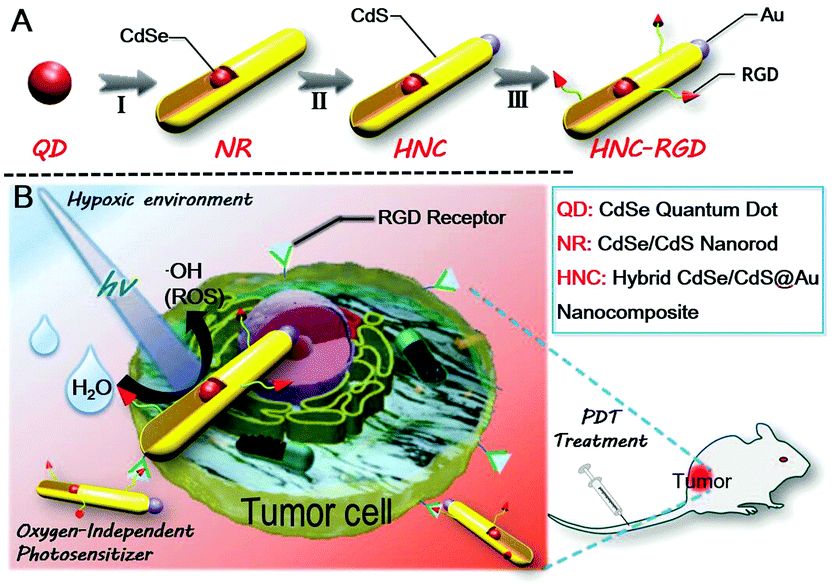 | ||
| Scheme 1 (A) Structure of HNCs: (I) anisotropic growth of NRs; (II) gold deposition on top of NRs; (III) RGD modification of HNCs; (B) schematic diagram of visible light driven water splitting to generate ROS for PDT treatment. | ||
2. Results and discussion
2.1 Synthesis and characterization
The structural composition and schematic process of the HNCs are displayed in Fig. 1A. Firstly, the CdSe quantum dots (QDs) that served as the seeds were synthesized, and the mixture of precursors for the growth of NRs was added into the QD solution. The length of the NRs was controlled by maintaining a specific process temperature and amount of raw materials. Subsequently, gold growth proceeded in an inert atmosphere at room temperature to form HNCs (see the methods section for details). TEM images of the different phases of the nanocomposites are displayed in Fig. 1B–E. As the seeds of the composites, the QDs showed a homogeneous size of around 5 nm (Fig. 1B), and after polarized growth, the NRs had an average length of around 60 nm (Fig. 1C). As expected, high-resolution transmission electron microscopy (HRTEM) of the NRs loaded with Au tips showed non-epitaxial growth at one side of the CdS shell (Fig. 1D and E), and their homogeneous nanoscale size (50–150 nm) was considered to be beneficial for circulation in the body as testified in previous research.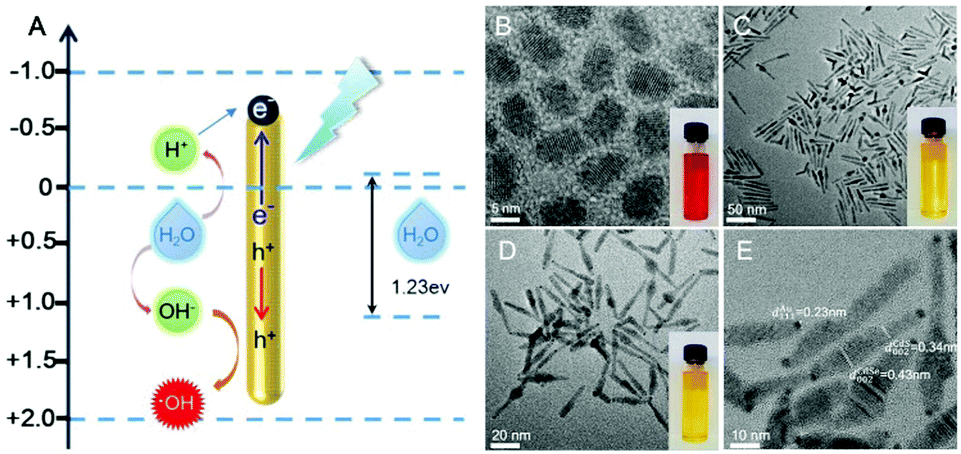 | ||
| Fig. 1 (A) The structural composition and schematic process of HNCs; (B) TEM image of cadmium selenide quantum dots (QDs) (scale bar: 5 nm); (C) TEM image of CdSe/CdS nanorods (NRs) (scale bar: 50 nm); (D) TEM image of CdSe/CdS–Au hybrid nanocomposites (HNCs) (scale bar: 20 nm); (E) high resolution TEM image of HNCs (scale bar: 10 nm), the measured d-spacing values from the visible lattice fringes 0.23 nm, 0.34 nm and 0.43 nm are assigned to Au (111), CdS (002) and CdSe (002), respectively. | ||
Moreover, the chemical constitution of the HNCs was evaluated subsequently. Intuitively, the distribution of different elements such as cadmium, selenium and aurum in the nanocomposite was shown by elemental mapping (Fig. 2A). As expected, Cd was uniformly dispersed throughout the particles, and Se was located on the end of the rods as well as Au. Next, we quantified the doping concentrations of S, Cd, Au, and Se (Fig. 2B) in the nanocomposites by energy-dispersive X-ray spectroscopy (EDX). X-ray photoelectron spectra (XPS) were characterized with a monochromatic Al Ka X-ray source, and the peaks correspond to the binding energies of Cd3d, S2p and Au4f, respectively, which demonstrated the chemical states of Cd–S, metal sulfide and Au–S bonds (Fig. 2C and Fig. S1, ESI†). Furthermore, as shown in Fig. 2D, the HNCs showed an obvious UV-vis absorption at around 460 nm and a strong fluorescence at 600 nm, which possibly resulted from the CdSe quantum dots. The cadmium selenide quantum dots (QDs) show a fluorescent signal mainly at 580 nm, and the nanorods may induce a shift in the signal to 600 nm (Fig. S2, ESI†). All of the aforesaid results implied that the HNCs were designed as expected.
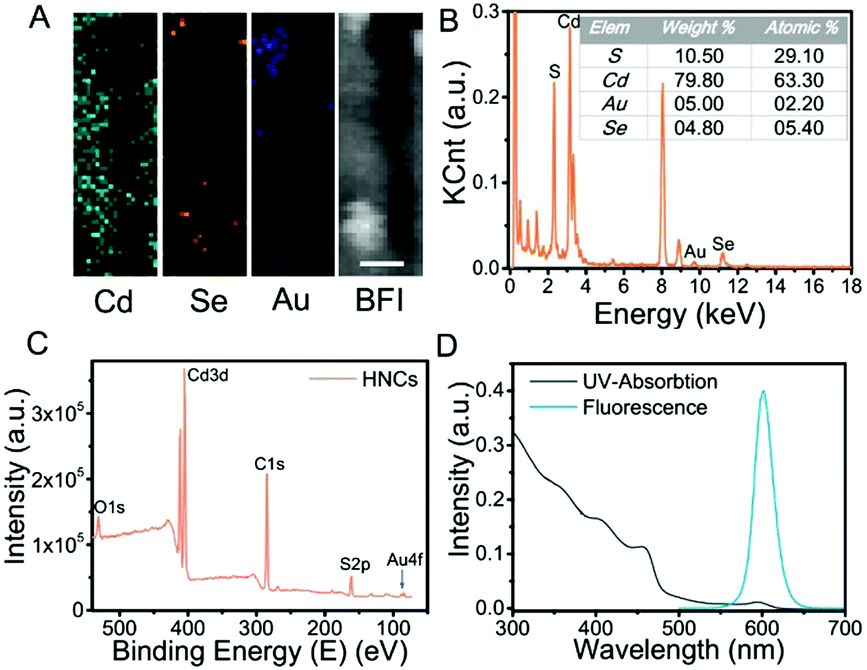 | ||
| Fig. 2 Chemical constitution and optical properties of HNCs. (A) Elemental mapping of HNCs (green: Cd, yellow: Se, blue: Au and white: bright field) (scale bar: 5 nm); (B) energy dispersive X-ray (EDX) of HNCs; (C) X-ray photoelectron spectra (XPS) of HNCs; (D) UV-vis absorption spectra and fluorescence of HNCs. | ||
2.2 In vitro detection of ROS generation and PDT enhancement
In vitro detection of ROS generation was determined by the decrease of UV absorbance of the disodium salt of p-nitroso-N,N-dimethylaniline (RNO).45 As shown in Fig. 3A and B, after LED irradiation, the UV-vis absorption of RNO was reduced to some extent with the same material concentrations of NRs and HNCs. However, whether the surrounding was hypoxic (1% O2) or not (21% O2), the ROS generation produced by the HNCs is much larger than that produced by the NRs. This indicated that the Au tips loaded on the CdSe-seeded/CdS nanorods could serve as anchor points and provide sites for the retention of photoexcited electrons, which ensured higher efficiency in splitting water into ROS than raw NRs. In the meantime, both the HNCs and NRs showed a relatively constant efficiency in producing ROS despite hypoxia or normoxia. This result indicated that the ROS generated by HNCs and NRs were dependent on a light-triggered water-splitting reaction instead of directly activating molecular oxygen. Overall, we demonstrated the high efficiency in oxygen-independent ROS generation of the HNCs.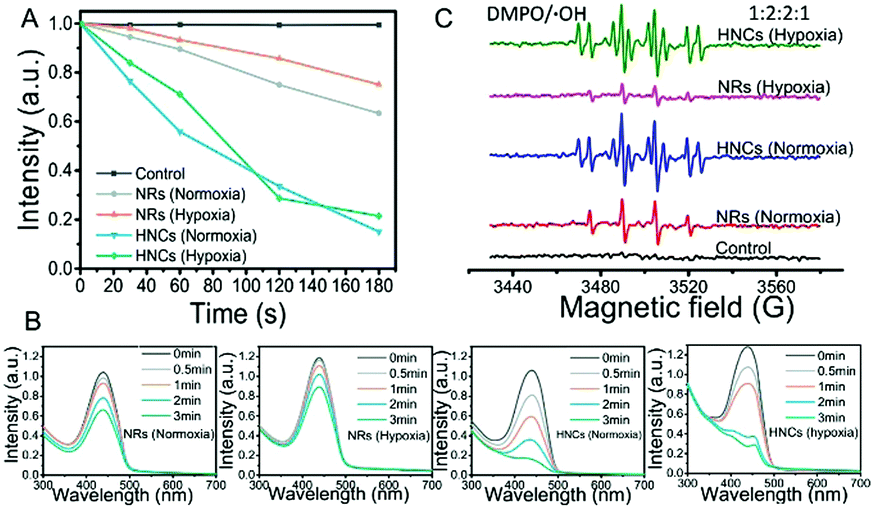 | ||
| Fig. 3 In vitro evaluation of ROS generation. (A and B) Absorbance spectra of RNO solution containing HNCs and NRs treated under hypoxia and normoxia with blue LED light irradiation; (C) significant enhancement effect of HNCs for promoting generation of ROS under simulated sunlight. The ESR spectra obtained from the solutions containing DMPO and HNCs or NRs treated under hypoxia and normoxia after irradiation for five minutes at ambient temperature. | ||
Furthermore, ESR spin trapping is a recognized technique for further detection of short-lived free radicals and paramagnetic species.46 Here we utilized 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) as a spin trap for the detection of ROS, especially hydroxyl radicals (˙OH). Fig. 3C shows the ESR spectra obtained from solutions containing DMPO and NRs or HNCs after irradiation for five minutes. Compared with the control group, the NR and HNC treated groups showed strong signals of DMPO–OH with hyperfine coupling values of AN = 14.9 G and AH = 14.9 G, demonstrating the significant production of hydroxyl radicals triggered by light excitation. Besides, the signal intensity of the HNC treated group showed no significant changes despite oxygen depletion and was much stronger than that of the NR treated group. This result is in accordance with the RNO assay and further demonstrates that HNCs possess a highly efficient and stable ROS producing capacity with no need for oxygen.
Subsequently, confocal laser scanning microscopy (CLSM) indicated the cellular uptake behavior of the HNCs, and COS-7 cells and HeLa cells were incubated with HNCs for 4 h. As shown in Fig. S3 (ESI†), compared with control groups, the CLSM images of both the COS-7 cells and HeLa cells presented distinct autofluorescence of the HNCs. Next, as a common intracellular ROS sensor, 2′,7′-dichlorofluorescin diacetate (DCFH-DA) was used for intracellular ROS detection with the HNCs. The nonfluorescent DCFH-DA could permeate into live cells freely. After being rapidly hydrolyzed by esterase, DCFH-DA could be oxidized by ROS and thereby emit bright green fluorescence, and the fluorescence intensity depended on ROS generation. As shown in Fig. 4A and B, after being irradiated by a blue LED lamp for 10 min (wavelength: 450 nm; 500 mW cm−2), 4T1 cells were incubated with 50 μg mL−1 and 25 μg mL−1 HNCs for 24 h. Under a normoxic environment (21% O2), the CLSM images in Fig. 4A1 showed strong fluorescence within the 4T1 cells that were incubated with 50 μg mL−1 HNCs under 450 nm LED irradiation, and after the decrease of the HNC concentration to 25 μg mL−1, the fluorescence intensity decreased obviously (Fig. 4B1). In contrast, 50 μg mL−1 HNCs could not trigger any fluorescence in cells without irradiation (Fig. 4C1). This result indicated that the HNCs could fundamentally increase the intracellular ROS generation upon visible light. Furthermore, irradiation alone and the negative control could not cause an increase in the intracellular ROS level (Fig. 4D1 and E1). As shown in Fig. 4A2–E2, under the hypoxic environment (1% O2), the HNCs showed the same trend in accordance with the previous results, and there was no apparent decrease in fluorescence intensity. This demonstrated that the HNCs have potential applications for improving PDT efficacy, especially for overcoming hypoxia in cancer therapy.
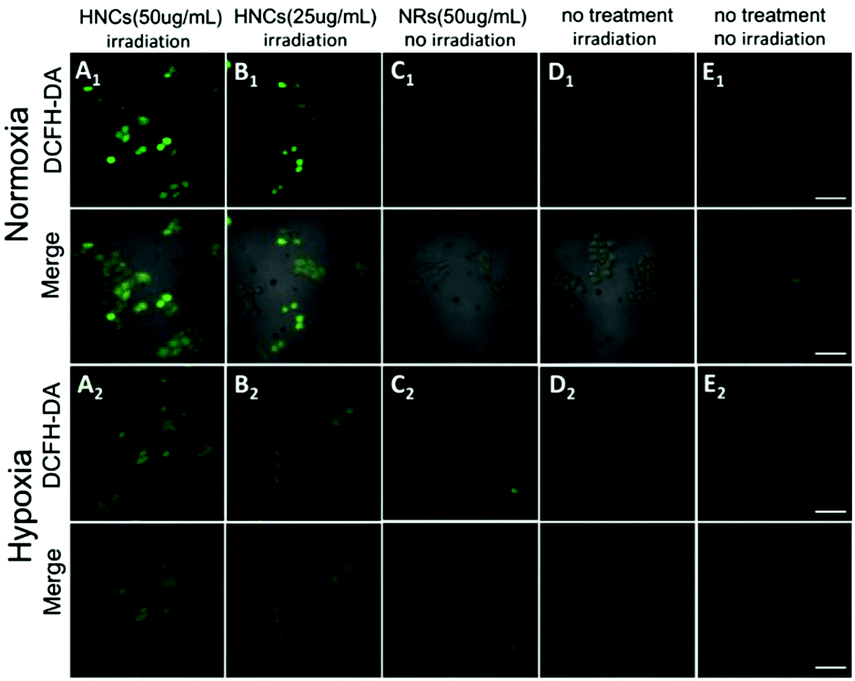 | ||
| Fig. 4 Intracellular ROS detection by confocal fluorescence imaging after 4T1 cells were treated with (A) HNCs (50 μg mL−1) and irradiation; (B) HNCs (25 μg mL−1) and irradiation; (C) HNCs (50 μg mL−1) without irradiation; (D) irradiation; (E) no treatment under normoxia and hypoxia environments (scale bar: 20 μm). | ||
2.3 In vitro cytotoxicity assay
The cytotoxicity and toxicity mechanism of the HNCs were evaluated in vitro. In order to prove that the cytotoxicity of the HNCs was dependent on the irradiation time, mouse 4T1 breast cancer cells were incubated with 50 μg mL−1 HNCs and under the same irradiation by a blue LED lamp for 1 min, 2 min and 3 min, respectively. As shown in Fig. S4 (ESI†), CLSM images revealed that along with the extension of the irradiation time, the intracellular fluorescence intensity increased visibly, accompanied by a rapid decline in the amount of live cells.An MTT assay was used to evaluate the cytotoxicity of both the HNCs and NRs under hypoxic and normoxic environments in 4T1 cells. As shown in Fig. 5A, the cell viability of the HNCs in the dark was always maintained at more than 90%, which showed no significant cytotoxicity from the HNCs. In the meantime, under blue LED lamp irradiation, the HNCs showed a much more toxic effect than the NRs accompanied by an increase in the concentration. Whether in hypoxia (1% O2) or normoxia (21% O2), the viability of the 4T1 cells incubated with 25 μg mL−1 HNCs was less than 20%, while more than 80% cells were alive with an equal concentration of NRs. This result demonstrated that both HNCs and NRs showed phototoxicity independent of oxygen, while the HNCs had a much higher efficiency for water-splitting to produce ROS than NRs. In this case, the metal–semiconductor nanocomposite showed promising application potential to accelerate ROS generation in hypoxic surroundings for oxy-PDT treatment.
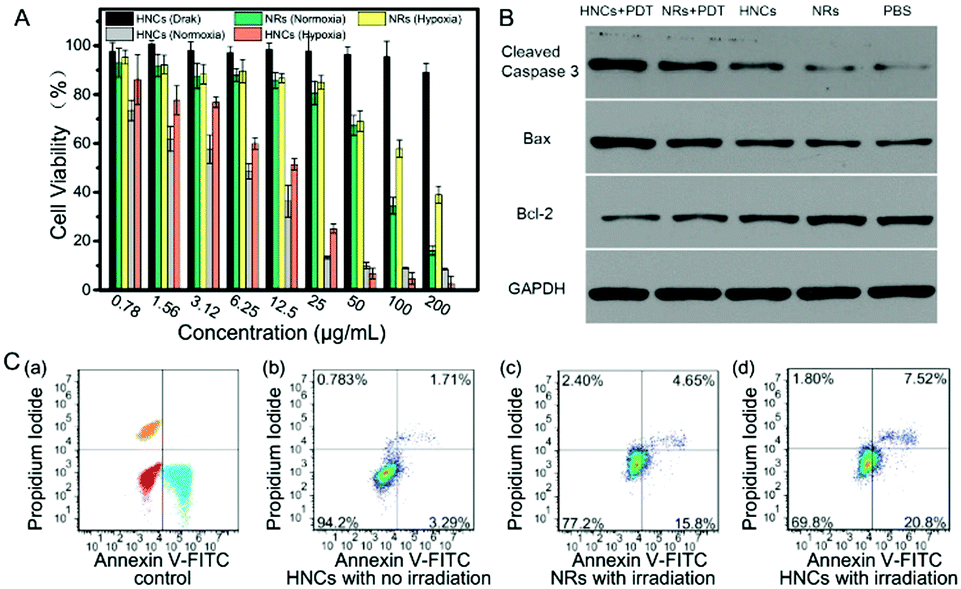 | ||
| Fig. 5 (A) Cytotoxicity of 4T1 cells after incubation with HNCs at various concentrations and treated under normoxia or hypoxia environments; (B) western blot analysis of cleaved Caspase-3, Bax, and Bcl-2 protein expression after various treatments. (C) Flow cytometric analysis of 4T1 cells for apoptosis and necrosis after different treatments. | ||
Next, to further verify the mechanism of cell death caused by HNCs under irradiation, the cell apoptosis and necrosis analyses of 4T1 cells were conducted by staining with annexin V-FITC and propidium iodide. As presented in Fig. 5C, after incubation with 4T1 cells without any irradiation, the HNCs did not induce remarkable cell death (<5%), mainly due to the significant biocompatibility of the HNCs at low concentration, as well as extremely low ROS generation in the dark. Impressively, after incubation under blue LED lamp irradiation for 10 min (500 mW cm−2), both the NRs and HNCs triggered apoptosis (Fig. 5C(c) and (d)), while the apoptotic percentage of cells treated by the HNCs (≈27.8%) was apparently higher than that of cells treated by the NRs (≈18%). Moreover, neither the HNCs nor NRs caused cell necrosis, which demonstrated that the simplex toxicity source was ROS generation from water-splitting.
Furthermore, to validate the apoptosis-promoting effect of the HNCs in PDT treatment, the expression of the cleaved cysteine protease Caspase-3, B-cell lymphoma 2 (Bcl-2) and Bcl-2-associated X protein (Bax) was examined by western blot analysis in 4T1 cells, and GAPDH was used as the internal control. Cleaved Caspase-3 plays a key role in the execution phase of cell apoptosis, and Bax functions as an apoptotic activator which has an antagonistic effect toward Bcl-2. As shown in Fig. 5B, compared with the no treatment group and no irradiation groups, the cells incubated with the HNCs under LED irradiation remarkably expressed the highest levels of cleaved Caspase-3 and Bax, and meanwhile the lowest level of Bcl-2. In addition, the same trend could be observed in the NR mediated PDT group, though it was less prominent than in the HNC mediated PDT group. This result demonstrated that both the HNCs and NRs could promote cell apoptosis under irradiation, and the HNCs showed higher phototoxicity than the NRs.
2.4 In vivo biodistribution study
In consideration of tumor targeting, an RGD peptide sequence, which can specifically bind tumors with a high level of integrin αvβ3 expressed in most of the solid tumors, was further introduced to construct HNCs for tumor-specific accumulation in vivo. The hydrophilic integrin receptor PEG-RGD was synthesized via solid-phase synthesis and its molecular weight was determined to be 873 by electrospray ionization mass spectrometry (ESI-MS; Fig. S5, ESI†), and it was purified by HPLC (>94%; Fig. S6, ESI†). Moreover, the peptide was linked to the surface of the HNCs via ligand exchange confirmed by Infrared Spectroscopy (IR) as shown in Fig. S7 (ESI†). In the meantime, after C-PEG8-RGD modification, the HNCs can disperse into water stably and metal sulfides can be easily coated onto the HNCs using chemical substances containing sulfydryl groups due to the good hydrophilicity of PEG-RGD.After being linked with the RGD sequence, the ex vivo bio-distribution of the HNCs-RGD was evaluated using a 4T1 cell-bearing balb/c mouse model. Due to the autofluorescence of HNCs-RGD in the visible spectrum, a small animal fluorescence imaging system (IVIS) was used to detect the accumulation of HNCs-RGD in different organs after the mice were sacrificed. As shown in Fig. 6A, 6 hours after the intravenous tail injection of HNCs-RGD, owing to the RES clearance of the NPs, the hepatic tissue displayed high fluorescence levels, and a large amount of fluorescence was also found in the tumor tissues, while the other organs such as the heart, spleen, liver and kidneys displayed limited fluorescence. Furthermore, 24 hours after the intravenous tail injection of HNCs-RGD, when the fluorescence intensity in other organs began to decrease, the tumor tissues still displayed high fluorescence levels. Fig. 6B shows the changes in the mean fluorescence intensity in different organs at 6 hours and 24 hours after injection. The aforesaid result demonstrated that HNCs-RGD not only had the ability for tumor-specific accumulation, but could also reside in tumor tissues for a long time. The major factor is that the RGD sequence can specifically bind to the tumor with a high level of integrin αvβ3 expressed in most of the solid tumors; in the meantime, a particle size of around 60 nm results in remarkable accumulation ability in tumors due to the enhanced permeability and retention (EPR) effect.
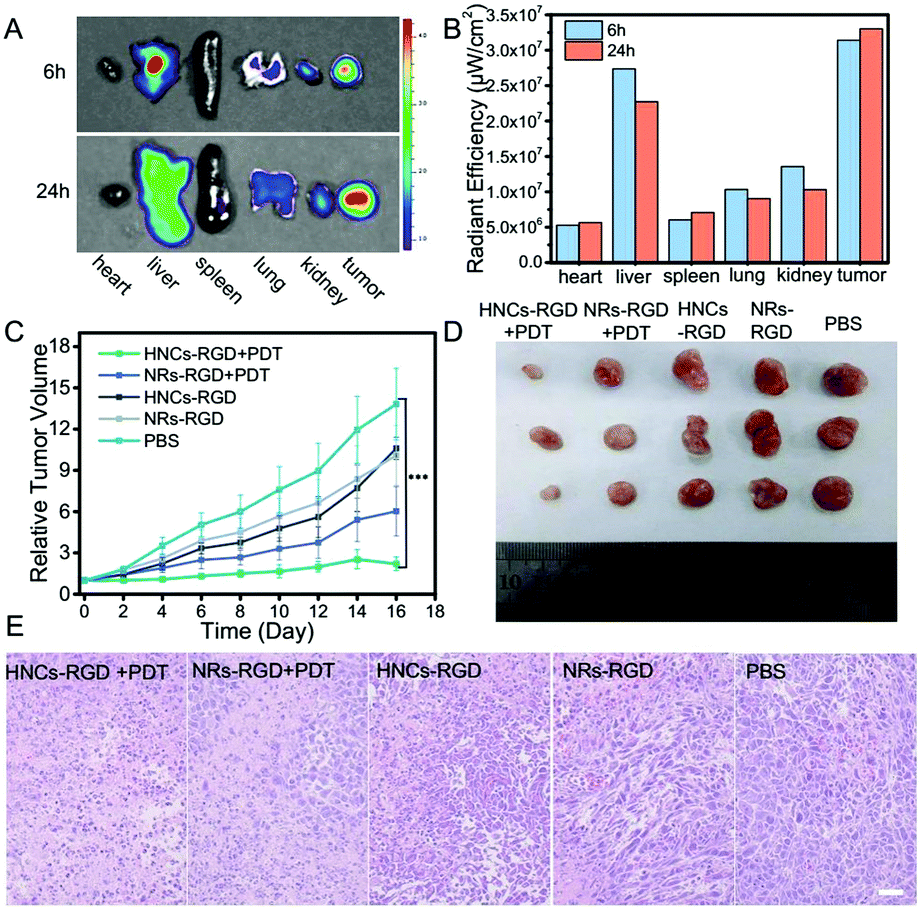 | ||
| Fig. 6 (A) Ex vivo fluorescence images of the major organs harvested from 4T1 tumor-bearing mice at 6 h and 12 h postinjection (from left to right: heart, liver, spleen, lung, kidney and tumor); (B) fluorescence intensity of the major organs; (C) relative tumor volumes from the different treatment groups; (D) representative tumor digital photos after various treatments for 16 days; (E) H&E staining of tumor tissues harvested from 4T1 tumor-bearing mice in the different groups at the end of the treatment (scale bar: 20 μm). | ||
2.5 Tumor suppression study and evaluation of side effects
Subsequently, an orthotopic tumor mouse model was established using the murine 4T1 breast-adenocarcinoma mouse cell line, which closely mimics human breast cancer. Female balb/c mice were subcutaneously hypodermically injected with 4T1 cells (5 × 106 cells per mouse) into the hind flank. The tumor volume reached approximately 100 mm3 after a week, and the tumor bearing mice were randomly divided into five groups with six mice each to obtain statistical data. Subsequently, after being treated with PBS, NRs-RGD, NRs-RGD under irradiation, HNCs-RGD and HNCs-RGD with irradiation, the tumor volumes were recorded every day. As recognized, a smaller relative tumor volume represented a higher antitumor therapeutic efficacy. As shown in Fig. 6C, a remarkable tumor growth inhibition effect was observed after treatment with HNCs-RGD under irradiation. As a comparison, the group treated with NRs-RGD under irradiation showed limited tumor inhibition, which confirmed that the metal–semiconductor nanocomposite showed a higher treatment efficiency than the raw nanorods as reflected in antitumor therapy. The control groups which were treated with PBS or without irradiation hardly showed any antitumor effect, and two weeks after injection, the tumor volumes of the negative control reached more than ten times compared with the effective treatment group. This result was also confirmed by representative tumor images (Fig. 6D and Fig. S8, ESI†) at the 16th day.In order to further evaluate the therapeutic efficacy as well as side effects, the organs of the treated mice were separated for H&E staining at the 16th day. As shown in Fig. 6E, entire cells in the view were observed to be dead in tumor tissues treated with HNCs-RGD under irradiation, while a certain number of dead cells were observed in the NRs-RGD group. In contrast, dead cells were hardly observed within the tumor tissues of the control groups. The result indicated that HNCs-RGD under irradiation showed the highest antitumor ability for PDT treatment. In the meantime, H&E staining of other organs such as the heart, liver, spleen, lungs and kidneys showed that HNCs-RGD and NRs-RGD exhibited negligible systemic toxicity to normal tissues (Fig. S9, ESI†). In addition, the changes in body weight were recorded to assess the side effects (Fig. S10, ESI†). The average body weight of all groups exhibited a similar stabilization tendency, suggesting that all treatments affected the heath condition negligibly.
In consideration of the possible hematotoxicity caused by Cd and Se, blood biochemical indices and blood routine indices were tested in vivo respectively. Typically, a blood assay can reflect the physical condition and the function of organs. For instance, the levels of ALT, AST and GGT signify the running condition of the liver. Herein, we could evaluate the side effects for different organs by blood assay. As shown in Tables S1 and S2 (ESI†), compared with the control group, there were no statistically significant fluctuations in hematology and clinical chemistry parameters in all groups. And the blood biochemical indices and hematological indices were all in the normal range.
3. Conclusion
In summary, by using semiconductor nanoparticles for splitting water to generate ROS without oxygen participation, we engineered a remarkable hybrid metal–semiconductor nanocomposite (HNC) based on CdSe-seeded/CdS nanorods (NRs) and Au tips as an efficient photosensitizer for oxygen-independent photodynamic tumor therapy. It was found that after metal deposition on one end of the rods, the HNCs showed a higher efficiency in ROS generation than NRs in vitro under visible light irradiation. Furthermore, due to the EPR effect and after being modified with the RGD ligand, HNCs-RGD showed excellent accumulation in tumor tissues, and the preferable therapeutic effect of HNCs-RGD was also confirmed in vivo. This hybrid nanocomposite could bypass tumor hypoxia and improve the therapeutic effect of PDT, which might find great potential in tumor therapy.Acknowledgements
This work was supported by the National Natural Science Foundation of China (51690152 and 51233003).References
- S. Li, X. Wang, R. Hu, H. Chen, M. Li, J. Wang, Y. Wang, L. Liu, F. Lv, X.-J. Liang and S. Wang, Chem. Mater., 2016, 28, 8669–8675 CrossRef CAS.
- L.-H. Liu, W.-X. Qiu, B. Li, C. Zhang, L.-F. Sun, S.-S. Wan, L. Rong and X.-Z. Zhang, Adv. Funct. Mater., 2016, 26, 6257–6269 CrossRef CAS.
- Z. Yu, Q. Sun, W. Pan, N. Li and B. Tang, ACS Nano, 2015, 9, 11064–11074 CrossRef CAS PubMed.
- J.-J. Hu, Q. Lei, M.-Y. Peng, D.-W. Zheng, Y.-X. Chen and X.-Z. Zhang, Biomaterials, 2017, 128, 136–146 CrossRef CAS PubMed.
- X. Duan, C. Chan, N. Guo, W. Han, R.-R. Weichselbaum and W. Lin, J. Am. Chem. Soc., 2016, 138, 16686–16695 CrossRef CAS PubMed.
- H. Fan, G. Yan, Z. Zhao, X. Hu, W. Zhang, H. Liu, X. Fu, T. Fu, X.-B. Zhang and W. Tan, Angew. Chem., Int. Ed., 2016, 55, 5477–5482 CrossRef CAS PubMed.
- Z. Yu, W. Pan, N. Li and B. Tang, Chem. Sci., 2016, 7, 4237–4244 RSC.
- D.-W. Zheng, Q. Lei, J.-Y. Zhu, J.-X. Fan, C.-X. Li, C. Li, Z. Xu, S.-X. Cheng and X.-Z. Zhang, Nano Lett., 2017, 17, 284–291 CrossRef CAS PubMed.
- C. M. Courtney, S. M. Goodman, J. A. McDaniel, N. E. Madinger, A. Chatterjee and P. Nagpal, Nat. Mater., 2016, 15, 529–534 CrossRef CAS PubMed.
- D. M. Gilkes, G. L. Semenza and D. Wirtz, Nat. Rev. Cancer, 2014, 14, 430–439 CrossRef CAS PubMed.
- H. Chen, J. Tian, W. He and Z. Guo, J. Am. Chem. Soc., 2015, 137, 1539–1547 CrossRef CAS PubMed.
- Z. Hou, Y. Zhang, K. Deng, Y. Chen, X. Li, X. Deng, Z. Cheng, H. Lian, C. Li and J. Lin, ACS Nano, 2015, 9, 2584–2599 CrossRef CAS PubMed.
- S. S. Lucky, N. Muhammad Idris, Z. Li, K. Huang, K. C. Soo and Y. Zhang, ACS Nano, 2015, 9, 191–205 CrossRef CAS PubMed.
- N. Kotagiri, G. P. Sudlow, W. J. Akers and S. Achilefu, Nat. Nanotechnol., 2015, 10, 370–379 CrossRef CAS PubMed.
- L. Feng, F. He, B. Liu, G. Yang, S. Gai, P. Yang, C. Li, Y. Dai, R. Lv and J. Lin, Chem. Mater., 2016, 28, 7935–7946 CrossRef CAS.
- E. Ju, K. Dong, Z. Chen, Z. Liu, C. Liu, Y. Huang, Z. Wang, F. Pu, J. Ren and X. Qu, Angew. Chem., Int. Ed., 2016, 55, 11467–11471 CrossRef CAS PubMed.
- X. Zhang, X. Xie, H. Wang, J. Zhang, B. Pan and Y. Xie, J. Am. Chem. Soc., 2013, 135, 18–21 CrossRef CAS PubMed.
- S. Chakrabortty, J. A. Yang, Y. M. Tan, N. Mishra and Y. Chan, Angew. Chem., Int. Ed., 2010, 49, 2888–2892 CrossRef CAS PubMed.
- K. A. Brown, D. F. Harris, M. B. Wilker, A. Rasmussen, N. Khadka, H. Hamby, S. Keable, G. Dukovic, J. W. Peters and L. C. Seefeldt, Science, 2016, 352, 448–450 CrossRef CAS PubMed.
- L. Liu, M. Sun, H. Zhang, Q. Yu, M. Li, Y. Qi, C. Zhang, G. Gao, Y. Yuan, H. Zhai, W. Chen and P. J. Alvarez, Nano Lett., 2016, 16, 688–694 CrossRef CAS PubMed.
- C. Seidl, J. Ungelenk, E. Zittel, T. Bergfeldt, J. P. Sleeman, U. Schepers and C. Feldmann, ACS Nano, 2016, 10, 3149–3157 CrossRef CAS PubMed.
- S. Wang, X. Li, Y. Chen, X. Cai, H. Yao, W. Gao, Y. Zheng, X. An, J. Shi and H. Chen, Adv. Mater., 2015, 27, 2775–2782 CrossRef CAS PubMed.
- H. W. Tseng, M. B. Wilker, N. H. Damrauer and G. Dukovic, J. Am. Chem. Soc., 2013, 135, 3383–3386 CrossRef CAS PubMed.
- T. Abe, M. Okumura, Y. Kikuchi, T. Itoh and K. Nagai, J. Mater. Chem. A, 2017, 5, 7445–7450 CAS.
- D.-W. Zheng, B. Li, C.-X. Li, J.-X. Fan, Q. Lei, C. Li, Z. Xu and X.-Z. Zhang, ACS Nano, 2016, 10, 8715–8722 CrossRef CAS PubMed.
- Y. Tachibana, L. Vayssieres and J. R. Durrant, Nat. Photonics, 2012, 6, 511–518 CrossRef CAS.
- J. J. Choi, X. Yang, Z. M. Norman, S. J. Billinge and J. S. Owen, Nano Lett., 2014, 14, 127–133 CrossRef CAS PubMed.
- L. Zeng, W. Ren, L. Xiang, J. Zheng, B. Chen and A. Wu, Nanoscale, 2013, 5, 2107–2113 RSC.
- K. K. Sakimoto, A. B. Wong and P. Yang, Science, 2016, 351, 74–77 CrossRef CAS PubMed.
- H. Gong, Y. Chao, J. Xiang, X. Han, G. Song, L. Feng, J. Liu, G. Yang, Q. Chen and Z. Liu, Nano Lett., 2016, 16, 2512–2521 CrossRef CAS PubMed.
- S. Bai, J. Jiang, Q. Zhang and Y. Xiong, Chem. Soc. Rev., 2015, 44, 2893–2939 RSC.
- Y. Ben-Shahar, F. Scotognella, I. Kriegel, L. Moretti, G. Cerullo, E. Rabani and U. Banin, Nat. Commun., 2016, 7, 10413 CrossRef CAS PubMed.
- S. Chen, Q. Lei, W.-X. Qiu, L.-H. Liu, D.-W. Zheng, J.-X. Fan, L. Rong, Y.-X. Sun and X.-Z. Zhang, Biomaterials, 2017, 117, 92–104 CrossRef CAS PubMed.
- Y. Nakibli, P. Kalisman and L. Amirav, J. Phys. Chem. Lett., 2015, 6, 2265–2268 CrossRef CAS PubMed.
- Y. Kim, D. Shin, W.-J. Chang, H.-L. Jang, C.-W. Lee, H.-E. Lee and K.-T. Nam, Adv. Funct. Mater., 2015, 25, 2369–2377 CrossRef CAS.
- H. Li, Y. Gao, Y. Zhou, F. Fan, Q. Han, Q. Xu, X. Wang, M. Xiao, C. Li and Z. Zou, Nano Lett., 2016, 16, 5547–5552 CrossRef CAS PubMed.
- Z. Shi, W. Ren, A. Gong, X. Zhao, Y. Zou, E. M. Brown, X. Chen and A. Wu, Biomaterials, 2014, 35, 7058–7067 CrossRef CAS PubMed.
- C. Caddeo, V. Calzia, L. Bagolini, M. T. Lusk and A. Mattoni, J. Phys. Chem. C, 2015, 119, 22663–22668 CAS.
- W. He, H. K. Kim, W. G. Wamer, D. Melka, J. H. Callahan and J. J. Yin, J. Am. Chem. Soc., 2014, 136, 750–757 CrossRef CAS PubMed.
- I. Jen-La Plante, A. Teitelboim, I. Pinkas, D. Oron and T. Mokari, J. Phys. Chem. Lett., 2014, 5, 590–596 CrossRef CAS PubMed.
- J. Zhang, Z.-F. Yuan, Y. Wang, W.-H. Chen, G.-F. Luo, S.-X. Cheng, R.-X. Zhuo and X.-Z. Zhang, J. Am. Chem. Soc., 2013, 135, 5068–5073 CrossRef CAS PubMed.
- C. M. Courtney, S. M. Goodman, J. A. McDaniel, N. E. Madinger, A. Chatterjee and P. Nagpal, Nat. Mater., 2016, 15, 529–534 CrossRef CAS PubMed.
- P. Kalisman, Y. Nakibli and L. Amirav, Nano Lett., 2016, 16, 1776–1781 CrossRef CAS PubMed.
- N. Waiskopf, Y. Ben-Shahar, M. Galchenko, I. Carmel, G. Moshitzky, H. Soreq and U. Banin, Nano Lett., 2016, 16, 4266–4273 CrossRef CAS PubMed.
- G. Lv, W. Guo, W. Zhang, T. Zhang, S. Li, S. Chen, A. S. Eltahan, D. Wang, Y. Wang, J. Zhang, P.-C. Wang, J. Chang and X.-J. Liang, ACS Nano, 2016, 10, 9637–9645 CrossRef CAS PubMed.
- F. Xu, J. Li, T.-T. Zhu, S.-S. Yu, C. Zuo, R.-S. Yao and H.-S. Qian, RSC Adv., 2016, 6, 102647–102656 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7nh00087a |
| ‡ J.-X. F. and M.-D. L. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |