
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fully degradable protein nanocarriers by orthogonal photoclick tetrazole–ene chemistry for the encapsulation and release†
Keti
Piradashvili
a,
Johanna
Simon
ab,
David
Paßlick
ab,
Julian R.
Höhner
a,
Volker
Mailänder
ab,
Frederik R.
Wurm
 *a and
Katharina
Landfester
*a
*a and
Katharina
Landfester
*a
aMax-Planck-Institut für Polymerforschung, Ackermannweg 10, Mainz 55128, Germany. E-mail: wurm@mpip-mainz.mpg.de; landfester@mpip-mainz.mpg.de
bUniversity Medical Center, Dermatology Clinic, Johannes Gutenberg University, Langenbeckstr. 1, 55131 Mainz, Germany
First published on 19th June 2017
Abstract
The encapsulation of sensitive drugs into nanocarriers retaining their bioactivity and achieving selective release is a challenging topic in current drug delivery design. Established protocols rely on metal-catalyzed or unspecific reactions to build the (mostly synthetic) vehicles which may inhibit the drug's function. Triggered by light, the mild tetrazole–ene cycloaddition enables us to prepare protein nanocarriers (PNCs) preserving at the same time the bioactivity of the sensitive antitumor and antiviral cargo Resiquimod (R848). This catalyst-free reaction was designed to take place at the interface of aqueous nanodroplets in miniemulsion to produce core–shell PNCs with over 90% encapsulation efficiency and no unwanted drug release over storage for several months. Albumins used herein are major constituents of blood and thus ideal biodegradable natural polymers for the production of such nanocarriers. These protein carriers were taken up by dendritic cells and the intracellular drug release by enzymatic degradation of the protein shell material was proven. Together with the thorough colloidal analysis of the PNCs, their stability in human blood plasma and the detailed protein corona composition, these results underline the high potential of such naturally derived drug delivery vehicles.
Conceptual insightsNanocarrier-mediated drug delivery is an important platform for future medicine. However, the preparation of fully degradable nanocarriers that can be loaded with sensitive cargo and that guarantee controlled release inside of cells is challenging. Mild, orthogonal, and metal-free chemistries with efficient drug loading, release and no deactivation of the cargo remains demanded. Here, we achieved such demands for protein nanocarriers (PNCs) prepared by a miniemulsion polymerization. With the light-triggered and catalyst-free tetrazole–ene cycloaddition (TET-click) degradable PNCs loaded with the sensitive antitumor and antiviral cargo Resiquimod (R848) were prepared. For the first time, the TET-click was adjusted to take place at the interface of aqueous nanodroplets in miniemulsion to produce PNCs with high encapsulation. The PNCs were colloidally stable in human blood plasma and the drug-loaded PNCs were taken up by dendritic cells of the immune system. They released their cargo intracellularly by enzymatic degradation of the protein shell more efficiently compared to conventional (i.e. isocyanate) chemistry. These results underline the high potential of nanocarriers prepared by the orthogonal TET-click polycondensation for modern immunotherapy. In addition, the synthetic concept can be expanded to other materials besides proteins in order to prepare well-defined nanocarriers for applications other than nanomedicine. |
Introduction
Bioorthogonal chemistry is an efficient strategy for selective protein labeling, polymer conjugation, and surface modification.1–3 In this study, bioorthogonal 1,3-dipolar tetrazole–ene cycloaddition chemistry was used for the first time as an interfacial cross-linking reaction to synthesize biodegradable nanocarriers out of naturally occurring proteins for drug delivery.For the application of nanocarriers for biomedical purposes it is essential that the shell material is biocompatible and degradable within the body without producing toxic side products. Many polymers used for this purpose fulfill the first aspect but often fail for the latter.4,5 Therefore, proteins from the abundant albumin family can be advantageous over synthetic materials, since the body is acquainted with processing these substances, which make them ideal for degradable drug delivery vehicles.6–8
Hollow nanocarriers have furthermore the advantage to protect the payload from the outer environment, and they can be designed to release their cargo with certain stimuli, such as pH, temperature, enzymes, or redox potential.9–14 Hollow nanocarriers are most frequently synthesized by forced polymerization at the droplet's interface in miniemulsion.15 Successful nanocarrier formation in inverse miniemulsion has been reported by reactive, but rather unselective compounds such as isocyanates (e.g. toluene diisocyanate, TDI)16,17 making the encapsulation of biopolymers, due to unwanted side reactions, difficult.
Thus, these kinds of reactions do not allow the encapsulation of sensitive biological cargoes since the electrophilic isocyanates will react with the payload and eventually deactivate it partially or completely. An attractive strategy for successful encapsulation of biomolecules and therapeutic substances is the application of bioorthogonal chemistry.1,18,19 Ruthenium-catalyzed olefin cross-metathesis20,21 and copper-catalyzed azide–alkyne interfacial click reaction22 have been reported as bioorthogonal and mild alternatives to isocyanates for the preparation of nanocarriers. Nevertheless, these reactions depend on the use of potentially toxic transition metal catalysts.
In the class of bioorthogonal reactions, the tetrazole–ene 1,3 dipolar cycloaddition between a diphenyltetrazole and a dipolarophile, reported first by Huisgen in 1967,23 stands out. In recent years, this method was established as an efficient bioorthogonal ligation technique by Lin and coworkers and termed “photoclick-reaction”, since it is induced by UV-light.24–26 Irradiation with a low-powered UV-lamp is sufficient for the diaryltetrazole to undergo a cycloreversion, releasing nitrogen and generating a nitrile imine dipole, which readily reacts with an olefin as dipolarophile.24 The tetrazole–ene cycloaddition is claimed to be one of the fastest bioorthogonal reactions, which is an ideal prerequisite for an interfacial reaction.27 In addition, it proceeds at ambient conditions (in water, at physiological pH, at room temperature), does not require metal catalysts and yields a fluorescent product.28,29
Herein, we adjusted the TET-click reaction to take place selectively at the interface of nanodroplets in miniemulsion to generate nanocarriers with a cross-linked protein shell. This is the first report on bio-orthogonal chemistry to prepare fully biodegradable protein nanocarriers based on different proteins that can be loaded with functional drugs and exhibit a high loading capacity and density. They further are nontoxic and can be uptaken by cells and release their cargo intracellularly. In contrast to simple protein ligation, the tetrazole chemistry and the proteins ovalbumin (OVA) and human serum albumin (HSA) were exploited for the preparation of hollow nanocarriers. The proteins were modified with tetrazole groups (acting as An-type monomer) and for cross-linking, a difunctional oil-soluble norbornene derivative was chosen as the B2-type monomer, due to the high reactivity of the strained olefins.30 The encapsulation of a hydrophilic compound into the non-toxic protein nanocarriers (PNCs) was proven to be efficient (encapsulation efficiency 91%). Furthermore, the PNC interaction with human blood plasma and the proteolytic degradation are shown. To verify that the PNCs are taken up by dendritic cells and degraded intracellularly, resiquimod (R848) was encapsulated. R848 is an immunostimulant which has antitumor and antiviral properties (structure cf. Fig. S1, ESI†).31 As the receptor for R848 is located in the endosomes of dendritic cells, an upregulation of the surface proteins, like CD86, after stimulation with the R848-loaded nanocarriers indicates their successful uptake and proteolytic degradation to release R848 with a preserved functionality. Control experiments with toluene diisocyanate (TDI) as crosslinker showed a significantly lower activity of R848 caused by an inactivation through reaction. The results for the survival and intracellular release of an active drug makes the PNCs synthesized by the light-triggered tetrazole-chemistry a general platform for the design of new drug delivery vehicles without interference of the drug throughout the formation process.
Results and discussion
Upon irradiation with UV-light, tetrazole derivatives release nitrogen and then react with olefins.32,33 We chose to synthesize 4-(2-phenyl-2H-tetrazol-5-yl)benzoic acid (TET) which carries a carboxyl group and thereby allows us to attach it to proteins or other polymeric compounds. TET was attached to HSA and OVA by Steglich amidation. The degree of functionalization was determined by UV/vis measurements (Table S1 and Fig. S2, ESI†). By this method, a TET/protein ratio of 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 for OVA and of 12:1 for HSA was determined. Due to the decrease in hydrophilicity for the proteins after TET-conjugation, we decided not to increase the degree of modification further. Since the modified proteins were used as nanocarrier material not aiming to preserve the native state, a detailed analysis of protein secondary and ternary structures was not performed.
:1 for OVA and of 12:1 for HSA was determined. Due to the decrease in hydrophilicity for the proteins after TET-conjugation, we decided not to increase the degree of modification further. Since the modified proteins were used as nanocarrier material not aiming to preserve the native state, a detailed analysis of protein secondary and ternary structures was not performed.
Recently, the bioorthogonality of the tetrazole–ene cycloaddition has been doubted, since the nitrile imine intermediate can also react with strong nucleophiles under certain conditions.34–37 Nevertheless, the use of highly reactive dipolarophiles such as strained norbornene derivatives and the confinement of the reaction at the interface of two liquids can decrease the probability for cross-reactivity. Since tetrazoles react readily with strained double bonds,30 we prepared a difunctional norbornene cross-linker (DN1, Fig. 1a). Norbornenes are advantageous over other strained ring systems such as cyclooctynes which are laborious to synthesize.
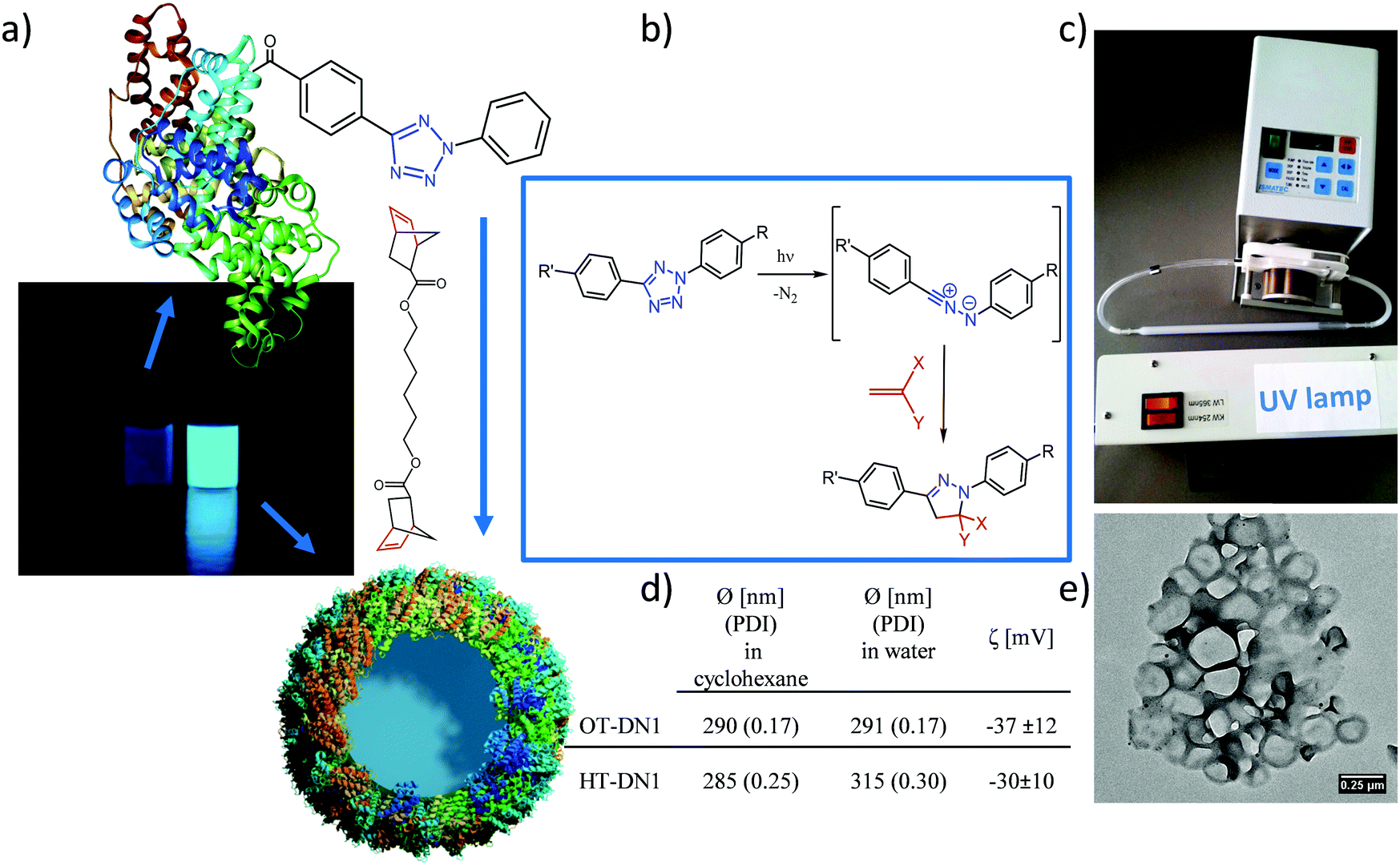 | ||
| Fig. 1 Preparation of protein nanocarriers (PNCs). (a) Non-fluorescent protein–TET conjugates were cross-linked by dinorbornene in inverse miniemulsion to obtain self-fluorescent protein nanocarriers; (b) reaction mechanism of the bioorthogonal UV-light induced 1,3 dipolar tetrazole–ene cycloaddition; (c) experimental setup with a peristaltic pump pumping the emulsion through a quartz cuvette with the UV-lamp placed in front; (d) average size of the PNCs determined by DLS and their zeta-potential (OT-DN1 for OVA-PNCs, HT-DN1 for HSA-PNCs); (e) TEM image of the PNCs. | ||
The protein nanocarriers (PNCs) were prepared by an interfacial cross-linking reaction in a water-in-oil miniemulsion. The TET-conjugated proteins (OT stands for ovalbumin–TET conjugates, HT for HSA-TET) were dissolved in chloride-free aqueous buffer (due to the inhibitory effect of chloride on the cycloaddition reaction).38 Cyclohexane, containing the surfactant P((E/B)-b-EO) was used as the organic phase and the mixture was homogenized by ultrasound to obtain stable aqueous nanodroplets. By addition of dinorbornene cross-linker to the miniemulsion and irradiation of the mixture with UV light (254 nm), the reaction at the oil/water interface between the TET moieties and the cross-linker was triggered leading to the formation of a core–shell morphology (Fig. 1e). Upon reaction, the appearance of fluorescence was observed (Fig. 1a), which is also typical for the formed pyrazoline cycloadduct.26 As comparison, also non orthogonal isocyanate-crosslinked nanocarriers (with toluene diisocyanate) were prepared.
After transfer of the PNCs into water, their diameter from DLS was about 300 nm with a zeta potential of about −30 mV (cf.Fig. 1d). The core–shell morphology was confirmed by transmission electron microscopy (TEM & SEM, Fig. 1e and Fig. S3, ESI†). The PNCs appear smaller compared to the values from DLS, which can be attributed to their soft shell, leading to shrinkage upon drying and under high-vacuum conditions. Also trehalose-embedded samples were visualized by TEM, in which individual nanocarriers with core–shell morphology are clearly visible (Fig. S3, ESI†).
The PNCs were loaded with the hydrophilic and low molecular weight sulforhodamine SR101 as model drug (theoretical loading 4.3 wt% SR101:protein) to assess the encapsulation efficiency of the PNCs for their potential application as drug carriers. After preparation of the PNCs and redispersion in water, the amount of SR101 in the continuous phase was determined, this is the amount of non-encapsulated cargo. By comparing the measured value with the amount of used initially, an encapsulation efficiency of over 91% (i.e. ca. 4 wt% compared to protein amount) was established. Leakage of the dye over a time period of up to 78 days was recorded by taking samples and measuring the fluorescence intensity of the aqueous supernatant after removal of the PNCs by centrifugation, proving no leakage over the investigated time (Fig. S4, ESI†).
A major feature of protein nanocarriers is their potential to (bio-)degrade into low molecular weight fragments which can be cleared from the body by using naturally occurring enzymes. However, also a certain stability of protein nanocarriers is essential, in order to deliver their cargo to the place of action.39,40 HSA and OVA have been widely studied for the preparation of protein nanoparticles by different formulations,6 that is why we decided also to use these proteins as shell material for the preparation of our PNCs via interfacial TET-click.
To prove that the cross-linked PNCs can be cleaved by proteases, DQ-ovalbumin was encapsulated in OT-DN1 and HT-DN1 PNCs. DQ-ovalbumin is a commercial, self-quenched fluorescent marker, consisting of ovalbumin conjugated with several BODIPY molecules. Upon proteolytic degradation, green fluorescence (λem = 515 nm) can be detected. The DQ-ovalbumin-loaded PNC dispersions in PBS buffer (at pH = 7.4) do not show any change in fluorescence over several hours. After the addition of an excess of the serine protease trypsin (20 µL of trypsin with a concentration of 0.5 mg mL−1 in PBS buffer were added to 100 µL PNC dispersion, i.e. a ca. 300× higher trypsin concentration compared to normal serum values41) an fast increase in fluorescence intensity can be detected (Fig. 2a). The fluorescence increases over time as trypsin degrades the PNCs and thus the cargo is released. Similarly, the proteolytic degradation was also verified by encapsulation and release of CdTe quantum dots, proving the versatility of the process to encapsulate various cargo molecules and particles (Fig. S5, ESI†). These experiments prove the general protease-lability of the PNCs in vitro; to monitor the degradation in situ via fluorescence, a high excess of the enzyme was used. Under normal blood concentrations of trypsin, other protein nanocarriers were reported to be stable over several hours to days.6,39
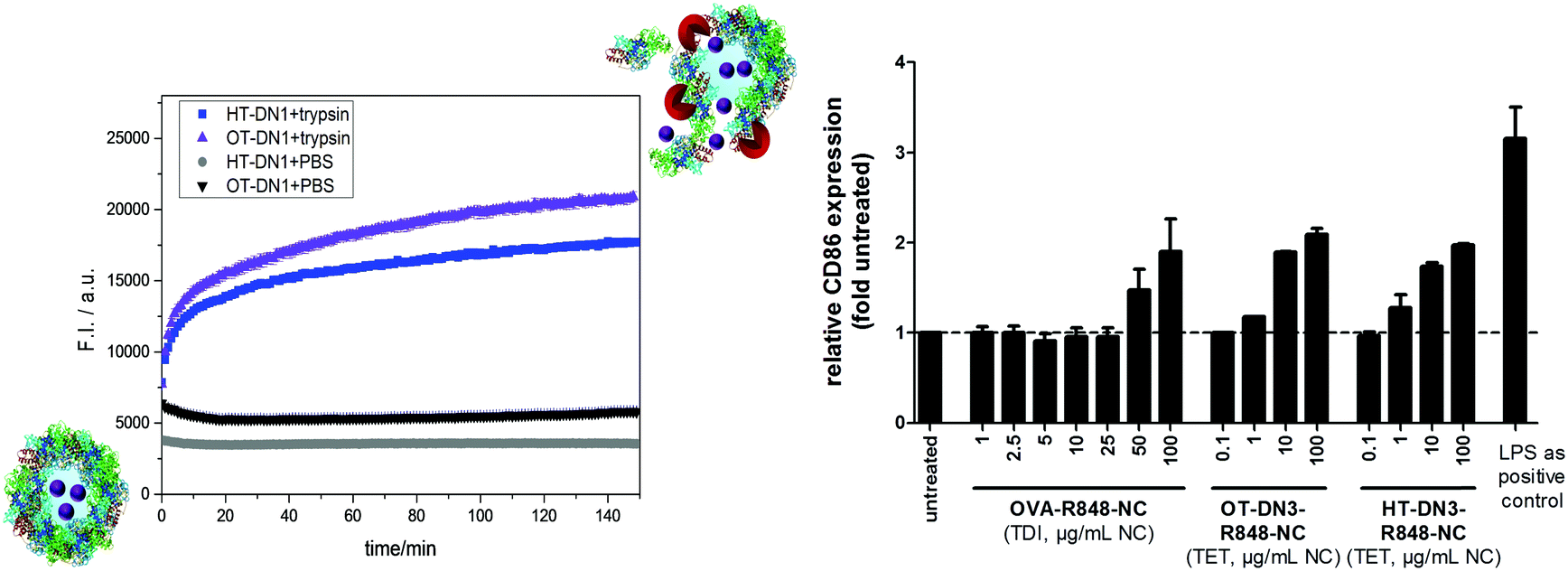 | ||
| Fig. 2 (left) Biodegradability of the nanocarriers: change of fluorescence intensity over time of OT-DN1 and HT-DN1 PNCs containing DQ-ovalbumin and incubated with trypsin or PBS buffer at pH 7.4, 37 °C for 2.5 h. (right) Upregulation of CD86 expression by bone marrow derived dendritic cells (BMDCs) upon treatment with PNCs, crosslinked via TDI or TET, loaded with R848 indicating a successful intracellular cleavage of the PNCs. | ||
The encapsulation of the immune-stimulant R848 into the PNCs offers the opportunity to prove the cellular uptake of PNC and the fast intracellular degradation and release of the cargo in a functional manner. R848 is recognized by the highly specific toll-like receptor 7, localized in the endosomes, and induces an upregulation of specific surface proteins on the corresponding cells of the immune system, such as dendritic cells.31,42 These proteins are necessary and essential for the activation of T cells and therefore for all adaptive immune reactions against tumors and pathogens.43 Thus, to develop its stimulatory potential, R848 has to reach the endosomally localized receptor. For this reason, unstimulated bone marrow derived dendritic cells (BMDCs) were treated with different doses of TET-crosslinked HT-DN1 and OT-DN1 (0.1–100 µg mL−1) as well as TDI-crosslinked OVA-NC (1–100 µg mL−1) as control, both loaded with comparable amounts of R848, for 24 h. Lipopolysaccharide (LPS), an immunostimulant, whose receptor is located on the dendritic cell surface, was used as the common immunological positive control. Afterwards, BMDCs were harvested and the CD86 expression was measured by flow cytometry.
OT-DN1-R848 and HT-DN1-R848, respectively, induced a dose dependent upregulation of the dendritic cell surface protein CD86 (Fig. 3). Compared to the untreated samples, the CD86 expression was strongly enhanced by these nanocarriers, especially with 10–100 µg mL−1 and started as early as 1 µg mL−1, while the stimulatory effect of the TDI-crosslinked OVA-NC started only at 50 µg mL−1. This is due to the fact that R848 has partly reacted with TDI and therefore is not active any more. The dose dependent stimulatory effect of the PNCs illustrates that the TET-click chemistry does not restrict R848 in its biological activity and an fast intracellular degradation of the PNCs is feasible, whereas TDI inactivates R848 and only 10% are biologically active. Notably, the supernatant of the nanocarriers did not show any biological effect, proving the high loading efficiency of the TET-click reaction. In addition after ca. 8 months of storage, no leakage of the R848 was detected and the nanocarriers kept their performance to the freshly prepared dispersion (i.e. no aggregation, degradation occurs under these conditions, cf. ESI†).
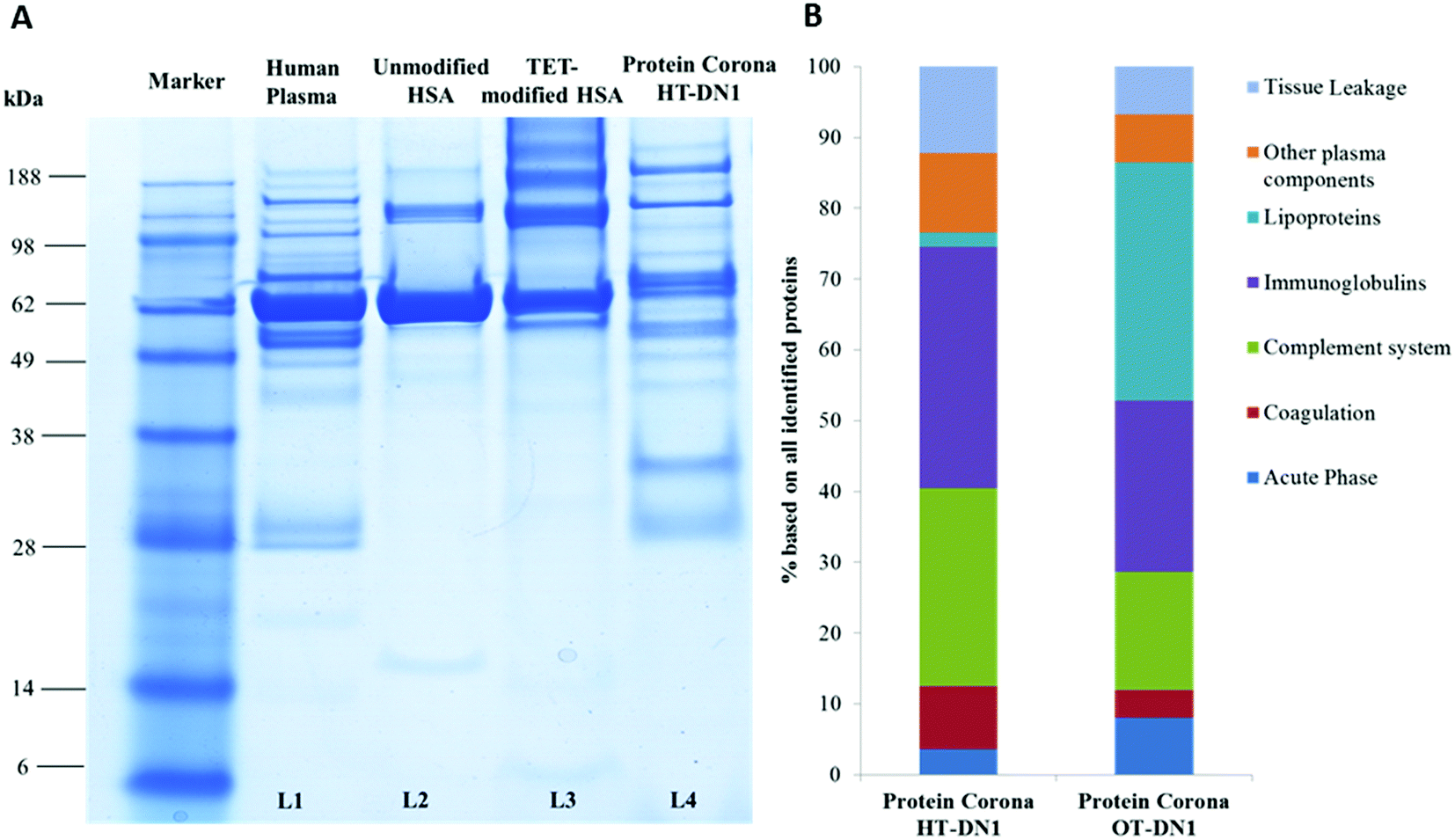 | ||
| Fig. 3 Interaction of PNCs with human blood plasma. Protein nanocarriers were incubated with human plasma for 1 h at 37 °C. To remove unbound and loosely bound proteins, PNCs were centrifuged (20000g, 30 min) and the remaining supernatant was discharged. This procedure was repeated three times. Bound proteins, constituting the protein corona, were eluted from nanocarriers with lysis buffer and analyzed by SDS-PAGE (A) and LC-MS/MS (n =4) (B). Human blood plasma is applied to lane L1, unmodified HSA = L2, TET-modified HSA = L3 and eluted corona proteins bound to HT-DN1 nanocarriers = L4. | ||
For the successful biomedical application of nanocarriers, it is essential to understand their behavior in physiological fluids such as blood plasma. Interactions of blood proteins with nanocarriers can dramatically alter their physicochemical properties and highly influence the further biological fate. It is crucial that the drug delivery vehicles are non-toxic and, for intravenous administration, do not aggregate in blood. Human monocyte derived dendritic cells (moDCs) were incubated with 1 to 100 µg L−1 of OT-DN1 and HT-DN1 nanocarriers for 24 h. Propidium iodide staining did not reveal any increase in cytotoxicity even at the highest concentration of PNCs (Fig. S6, ESI†).
To understand the behavior of nanocarriers once introduced to blood and exclude potential side effects (e.g. aggregation formation),44 we investigated their interaction with human blood plasma. With dynamic light scattering measurements we proved that PNCs remained stable in human blood plasma and do not aggregate (Fig. S7, ESI†). In addition, incubation with human blood plasma altered the zeta-potential of PNCs, indicating protein adsorption and protein corona formation (Table S2, ESI†). As the surface of PNCs is covered with proteins, this additional protein layer mediates the further biological response of PNCs such as their interaction with specific cells and surface receptors.45–47 For qualitative and quantitative characterization of the protein corona of the nanocarriers in human blood plasma, we applied SDS-PAGE and liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS). First, we found the binding affinity of distinct proteins towards PNCs did not correlate with their native abundance in human blood plasma (exemplary shown for PNCs prepared from human serum albumin, Fig. 3A. L1: Human Plasma vs. L4: Protein Corona from the nanocarrier). This is in agreement with other studies.48,49 Corona proteins associated with PNCs prepared from HSA (HT-DN1) or ovalbumin (OVA-DN1) were identified by LC-MS and further classified into six major classes: Acute Phase, coagulation, complement system, immunoglobulins, lipoproteins and tissue leakage. PNCs prepared from HSA carry a protein corona strongly enriched with immunoglobulins and proteins from the complement system (Fig. 3B). The enrichment of these proteins on the surface of PNCs may trigger interactions with immune cells, causing a clearance of PNCs by mononuclear phagocytic cells (e.g. macrophages) and affecting their half-life and bio distribution in vivo.50 In contrast, a great amount of lipoproteins (e.g. clusterin, ApoA IV) adsorbed on PNCs prepared from ovalbumin (OT-DN1-PNCs) which are of importance to prolong blood circulation and obtain a stealth behavior in vivo.51,52
This further renders the use of proteins as the building material for nanocarriers interesting, as it allows the tailoring of the protein corona in blood and should enable us to control selective targeting in vivo, eventually. A detailed overview of all identified proteins is summarized in the ESI† (Table S3).
Conclusion
In summary, we developed a robust and successful synthesis method for the preparation of biodegradable protein nanocarriers that are taken up by dendritic cells and release an active drug intracellularly. The bioorthogonal 1,3-dipolar tetrazole–ene cycloaddition was adjusted precisely at the interface of nanodroplets in miniemulsion to generate crosslinked protein nanocarriers by low-intensity UV-light in 30 min at ambient conditions. A high drug payload of more than 90% was achieved. The non-toxic PNCs were stable in blood plasma and were enzymatically degradable. They released their intact cargo only after proteolytic cleavage of the shell which was shown by encapsulation of the immunostimulant R848 into the PNCs. Proteins are ideal non-toxic and biodegradable building blocks to be combined with the bioorthogonal TET chemistry. This unique combination, suitable for a great variety of proteins, offers us a diverse tool to design drug delivery vehicles for the encapsulation of sensitive drugs.Acknowledgements
We thank Dr Anette Pietrzak-Nguyen (Uniclinics Mainz) for toxicity analysis, Christine Rosenauer (MPIP) for DLS in plasma measurements, Dr Maria Kokkinopoulou (MPIP) and Dr Ingo Lieberwirth (MPIP) for TEM measurements, and the Max Planck Graduate Center and DFG (SFB 1066) for financial support. Open Access funding provided by the Max Planck Society.References
- E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974–6998 CrossRef CAS PubMed.
- K. Lang and J. W. Chin, Chem. Rev., 2014, 114, 4764–4806 CrossRef CAS PubMed.
- G. Delaittre, A. S. Goldmann, J. O. Mueller and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2015, 54, 11388–11403 CrossRef CAS PubMed.
- A. S. Hoffman, Adv. Drug Delivery Rev., 2013, 65, 10–16 CrossRef CAS PubMed.
- P. Couvreur, Adv. Drug Delivery Rev., 2013, 65, 21–23 CrossRef CAS PubMed.
- A. O. Elzoghby, W. M. Samy and N. A. Elgindy, J. Controlled Release, 2012, 161, 38–49 CrossRef CAS PubMed.
- J. Ge, E. Neofytou, J. Lei, R. E. Beygui and R. N. Zare, Small, 2012, 8, 3573–3578 CrossRef CAS PubMed.
- J.-W. Yoo, D. J. Irvine, D. E. Discher and S. Mitragotri, Nat. Rev. Drug Discovery, 2011, 10, 521–535 CrossRef CAS PubMed.
- Z. Y. Zhang, Y. D. Xu, Y. Y. Ma, L. L. Qiu, Y. Wang, J. L. Kong and H. M. Xiong, Angew. Chem., Int. Ed., 2013, 52, 4127–4131 CrossRef CAS PubMed.
- E. Fleige, M. A. Quadir and R. Haag, Adv. Drug Delivery Rev., 2012, 64, 866–884 CrossRef CAS PubMed.
- Y.-J. Pan, Y.-Y. Chen, D.-R. Wang, C. Wei, J. Guo, D.-R. Lu, C.-C. Chu and C.-C. Wang, Biomaterials, 2012, 33, 6570–6579 CrossRef CAS PubMed.
- S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991–1003 CrossRef CAS PubMed.
- K. Landfester, A. Musyanovych and V. Mailänder, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 493–515 CrossRef CAS.
- J. Nicolas, S. Mura, D. Brambilla, N. Mackiewicz and P. Couvreur, Chem. Soc. Rev., 2013, 42, 1147–1235 RSC.
- K. Piradashvili, E. M. Alexandrino, F. R. Wurm and K. Landfester, Chem. Rev., 2015, 116, 2141–2169 CrossRef PubMed.
- S. Taheri, G. Baier, P. Majewski, M. Barton, R. Forch, K. Landfester and K. Vasilev, J. Mater. Chem. B, 2014, 2, 1838–1845 RSC.
- D. Crespy, M. Stark, C. Hoffmann-Richter, U. Ziener and K. Landfester, Macromolecules, 2007, 40, 3122–3135 CrossRef CAS.
- W. R. Algar, D. E. Prasuhn, M. H. Stewart, T. L. Jennings, J. B. Blanco-Canosa, P. E. Dawson and I. L. Medintz, Bioconjugate Chem., 2011, 22, 825–858 CrossRef CAS PubMed.
- Y. Jiang, J. Chen, C. Deng, E. J. Suuronen and Z. Zhong, Biomaterials, 2014, 35, 4969–4985 CrossRef CAS PubMed.
- K. Malzahn, F. Marsico, K. Koynov, K. Landfester, C. K. Weiss and F. R. Wurm, ACS Macro Lett., 2013, 3, 40–43 CrossRef.
- K. Breitenkamp and T. Emrick, J. Am. Chem. Soc., 2003, 125, 12070–12071 CrossRef CAS PubMed.
- R. Roux, L. Sallet, P. Alcouffe, S. Chambert, N. Sintes-Zydowicz, E. Fleury and J. Bernard, ACS Macro Lett., 2012, 1, 1074–1078 Search PubMed.
- J. S. Clovis, A. Eckell, R. Huisgen and R. Sustmann, Chem. Ber., 1967, 100, 60–70 CrossRef CAS.
- W. Song, Y. Wang, J. Qu, M. M. Madden and Q. Lin, Angew. Chem., 2008, 120, 2874–2877 CrossRef.
- Y. Wang, C. I. Rivera Vera and Q. Lin, Org. Lett., 2007, 9, 4155–4158 CrossRef CAS PubMed.
- R. K. Lim and Q. Lin, Chem. Commun., 2010, 46, 1589–1600 RSC.
- C. P. Ramil and Q. Lin, Curr. Opin. Chem. Biol., 2014, 21, 89–95 CrossRef CAS PubMed.
- Y. Fan, C. Deng, R. Cheng, F. Meng and Z. Zhong, Biomacromolecules, 2013, 14, 2814–2821 CrossRef CAS PubMed.
- J. O. Mueller, D. Voll, F. G. Schmidt, G. Delaittre and C. Barner-Kowollik, Chem. Commun., 2014, 50, 15681–15684 RSC.
- Z. Yu, R. K. Lim and Q. Lin, Chem. – Eur. J., 2010, 16, 13325–13329 CrossRef CAS PubMed.
- H. Hemmi, T. Kaisho, O. Takeuchi, S. Sato, H. Sanjo, K. Hoshino, T. Horiuchi, H. Tomizawa, K. Takeda and S. Akira, Nat. Immunol., 2002, 3, 196–200 CrossRef CAS PubMed.
- W. Song, Y. Wang, J. Qu and Q. Lin, J. Am. Chem. Soc., 2008, 130, 9654–9655 CrossRef CAS PubMed.
- Z. Yu, T. Y. Ohulchanskyy, P. An, P. N. Prasad and Q. Lin, J. Am. Chem. Soc., 2013, 135, 16766–16769 CrossRef CAS PubMed.
- Z. Li, L. Qian, L. Li, J. C. Bernhammer, H. V. Huynh, J. S. Lee and S. Q. Yao, Angew. Chem., Int. Ed., 2015, 128, 2042–2046 CrossRef.
- Y. Zhang, W. Liu and Z. K. Zhao, Molecules, 2013, 19, 306–315 CrossRef PubMed.
- W. Siti, A. K. Khan, H.-P. M. de Hoog, B. Liedberg and M. Nallani, Org. Biomol. Chem., 2015, 13, 3202–3206 CAS.
- W. Feng, L. Li, C. Yang, A. Welle, O. Trapp and P. A. Levkin, Angew. Chem., Int. Ed., 2015, 54, 8732–8735 CrossRef CAS PubMed.
- X. S. Wang, Y.-J. Lee and W. R. Liu, Chem. Commun., 2014, 50, 3176–3179 RSC.
- W. Lohcharoenkal, L. Wang, Y. C. Chen and Y. Rojanasakul, BioMed Res. Int., 2014, 2014, 12 Search PubMed.
- F. R. Wurm and C. K. Weiss, Front. Chem., 2014, 2 DOI:10.3389/fchem.2014.00049.
- J. M. Artigas, M. E. Garcia, M. R. Faure and A. M. Gimeno, Postgrad. Med. J., 1981, 57, 219–222 CrossRef CAS PubMed.
- M. Jurk, F. Heil, J. Vollmer, C. Schetter, A. M. Krieg, H. Wagner, G. Lipford and S. Bauer, Nat. Immunol., 2002, 3, 499 CrossRef CAS PubMed.
- A. Iwasaki and R. Medzhitov, Science, 2010, 327, 291–295 CrossRef CAS PubMed.
- K. Mohr, M. Sommer, G. Baier, S. Schöttler, P. Okwieka, S. Tenzer, K. Landfester, V. Mailänder, M. Schmidt and R. G. Meyer, J. Nanomed. Nanotechnol., 2014, 5, 193 Search PubMed.
- D. Walczyk, F. B. Bombelli, M. P. Monopoli, I. Lynch and K. A. Dawson, J. Am. Chem. Soc., 2010, 132, 5761–5768 CrossRef CAS PubMed.
- M. P. Monopoli, D. Walczyk, A. Campbell, G. Elia, I. Lynch, F. B. Bombelli and K. A. Dawson, J. Am. Chem. Soc., 2011, 133, 2525–2534 CrossRef CAS PubMed.
- S. Tenzer, D. Docter, S. Rosfa, A. Wlodarski, J. Kuharev, A. Rekik, S. K. Knauer, C. Bantz, T. Nawroth, C. Bier, J. Sirirattanapan, W. Mann, L. Treuel, R. Zellner, M. Maskos, H. Schild and R. H. Stauber, ACS Nano, 2011, 5, 7155–7167 CrossRef CAS PubMed.
- M. Lundqvist, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 14265–14270 CrossRef CAS PubMed.
- T. Cedervall, I. Lynch, M. Foy, T. Berggad, S. C. Donnelly, G. Cagney, S. Linse and K. A. Dawson, Angew. Chem., Int. Ed., 2007, 46, 5754–5756 CrossRef CAS PubMed.
- O. Lunov, T. Syrovets, C. Loos, J. Beil, M. Delacher, K. Tron, G. Nienhaus, A. Musyanovych, V. Mailänder, K. Landfester and T. Simmett, ACS Nano, 2011, 5, 1657–1669 CrossRef CAS PubMed.
- S. Schöttler, G. Becker, S. Winzen, T. Steinbach, K. Mohr, K. Landfester, V. Mailänder and F. R. Wurm, Nat. Nanotechnol., 2016, 11, 372–377 CrossRef PubMed.
- S. Ritz, S. Schöttler, N. Kotman, G. Baier, A. Musyanovych, J. R. Kuharev, K. Landfester, H. R. Schild, O. Jahn and S. Tenzer, Biomacromolecules, 2015, 16, 1311–1321 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7nh00062f |
| This journal is © The Royal Society of Chemistry 2017 |