
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tyr25, Tyr58 and Trp133 of Escherichia coli bacterioferritin transfer electrons between iron in the central cavity and the ferroxidase centre†
Justin M.
Bradley
a,
Dimitri A.
Svistunenko
b,
Geoffrey R.
Moore
a and
Nick E.
Le Brun
 *a
*a
aCentre for Molecular and Structural Biochemistry, School of Chemistry, University of East Anglia, Norwich Research Park, Norwich, NR4 7TJ, UK. E-mail: n.le-brun@uea.ac.uk; Fax: +44 (0)1603 592003; Tel: +44 (0)1603 592699
bSchool of Biological Sciences, University of Essex, Wivenhoe Park, Colchester CO4 3SQ, UK
First published on 6th September 2017
Abstract
Ferritins are 24meric proteins that overcome problems of toxicity, insolubility and poor bioavailability of iron in all types of cells by storing it in the form of a ferric mineral within their central cavities. In the bacterioferritin (BFR) from Escherichia coli iron mineralization kinetics have been shown to be dependent on an intra-subunit catalytic diiron cofactor site (the ferroxidase centre), three closely located aromatic residues and an inner surface iron site. One of the aromatic residues, Tyr25, is the site of formation of a transient radical, but the roles of the other two residues, Tyr58 and Trp133, are unknown. Here we show that these residues are important for the rates of formation and decay of the Tyr25 radical and decay of a secondary radical observed during Tyr25 radical decay. The data support a mechanism in which these aromatic residues function in electron transfer from the inner surface site to the ferroxidase centre.
Significance to metallomicsThe iron-storage/detoxification protein bacterioferritin (BFR) can acquire several thousand Fe2+ ions per protein molecule, which are stored in a central cavity as a polynuclear oxy-hydroxide Fe3+ mineral. Thus the process of mineral-formation involves the oxidation of Fe2+ ions. Recently it has been reported that BFR from Escherichia coli employs a tyrosyl radical during the mineralization process to transmit electrons from Fe2+ oxidation in the cavity to dinuclear iron sites within each of the 24 protein subunits where O2 is reduced. This paper identifies additional aromatic amino acids that are involved in the mineralization electron-transfer pathway. |
Introduction
Bacterioferritins are members of the ferritin super-family of proteins,1–3 named after the Latin word ferratus (furnished with iron) because of their high iron content when isolated as native proteins.4,5 They are homopolymers of 24 subunits that pack together to create an approximately spherical molecule with a central cavity of ∼80 Å diameter (Fig. 1A), in which a polynuclear iron mineral can form. They are unique amongst ferritins in that they contain up to 12 hemes per protein, located at inter-subunit binding sites.6 Their ability to lay down an iron-rich mineral inside a protein shell was one of the first indicators that ferritins had a role in iron storage,4 though it is now not certain that is the primary function of all bacterioferritins. While bacterioferritin (BFR) in Pseudomonas aeruginosa does act as the general housekeeping store for iron,7 in Escherichia coli it does not.8 Both P. aeruginosa and E. coli contain another ferritin in addition to BFR, prokaryotic ferritin (Ftn), which is also a homopolymer of 24 subunits,9 and it is Ftn that functions as the general housekeeping store for iron in E. coli.8 In order for either BFR or Ftn to lay down an iron-containing mineral in their central cavities, the protein takes up Fe2+ and catalyses its oxidation to Fe3+ by O2 (or H2O2).2,3,10 Thus, oxidation of Fe2+ is a key feature of the iron storage role of some ferritins, and it also seems to be important in dealing with oxidative stress in bacteria. Such stress occurs when the cell becomes burdened with too great a concentration of reactive oxygen species (ROS). Without a defence mechanism, the ROS would kill the cell through uncontrolled reactions with organic material. Provided there is Fe2+ available, BFR and Ftn can reduce the level of ROS while building up their mineral cores.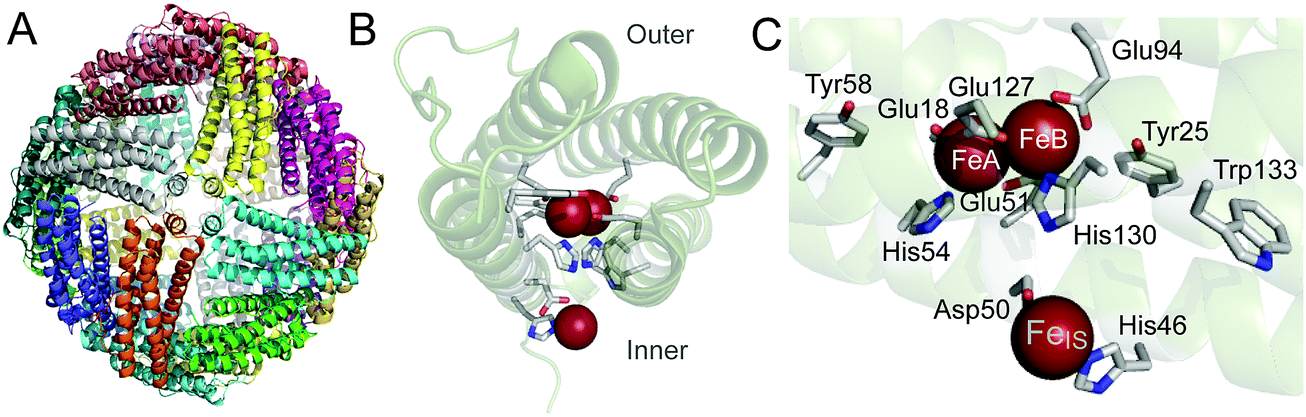 | ||
| Fig. 1 Structural features of E. coli BFR. (A) Ribbon cartoon representation of the 24 meric ferritin rhombic dodecahedron viewed along one of six four-fold axes. Each of the subunits are coloured differently. (B) A single subunit of the protein shell, viewing the helical bundle along the long axis, with the iron ions of the ferroxidase centre (centre of the bundle) and inner surface site (below the bundle) depicted as red spheres. (C) View of the catalytic site of one subunit. The diiron ferroxidase centre (FeA and FeB) and inner surface iron site (FeIS) are shown with coordinating residues. The nearby aromatic residues (Tyr25, Tyr58 and Trp133) are also shown. Residues are shown in stick representation with carbon in grey, nitrogen in blue and oxygen in red. Generated using PyMol with PDB file 3E1M.6 | ||
Much of the oxidation of Fe2+ in ferritins involves dinuclear iron centres located in the middle of the subunits.2,3,10 These centres, which are commonly referred to as ferroxidase centres, are present in most types of ferritin subunit, but not all.11 Ferritins containing subunits where they are absent are heteropolyomers with other subunits having a ferroxidase centre so that the molecule is still able to rapidly oxidise Fe2+.2,11 In BFRs, the ferroxidase centres play a central role in Fe2+ oxidation, while the heme groups do not; instead they function in iron release from BFRs.12,13 The ferroxidase centre of E. coli BFR (Fig. 1B and C) has both iron ions ligated by terminal glutamate and histidine residues and by two bridging glutamates.14 This ligation scheme is highly symmetric, which is a major difference when compared with other ferritin ferroxidase centres,10,15 and similar to the dinuclear iron centres of enzymes that transform organic substrates, such as ribonucleotide reductase.16 Whilst the ferroxidase centres of some ferritins act as transit points for the movement of iron into the cavity,17–19 the E. coli BFR ferroxidase centre acts as a true cofactor rather than as a substrate binding site,14,20–22 continually cycling between its oxidized (bridged di-Fe3+) and reduced (di-Fe2+) states. The catalytic cycling of the ferroxidase centre is driven by the oxidation of Fe2+ ions in the central cavity, with the electrons resulting from this process channeled to the ferroxidase centre and reducing it to the di-Fe2+ form, which is then primed to react again with O2 (or H2O2).21,23 Hydrolysis of the accumulating hydrated Fe3+ in the cavity leads to mineral formation.
About 9 Å from the ferroxidase centre of E. coli BFR is a mononuclear Fe2+ site, referred to as the inner surface site FeIS (Fig. 1B and C). The Fe2+ ion at this site is coordinated by Asp50 and His46 and also by three water molecules. Studies of Asp50Ala and His46Ala variants of E. coli BFR indicated that this site plays a role in the transmission of electrons from the growing mineral in the cavity to the ferroxidase centre, and may also have a role in moving iron into the cavity.14 Though the 24 FeIS sites per 24-mer are important, by themselves they are not sufficient to efficiently transfer electrons resulting from Fe2+ oxidation at the site or in the cavity to effect reduction of their neighbouring ferroxidase centres. The need for a more extensive electron transfer pathway was recognized at the time the FeIS sites were discovered and a variety of methods applied to E. coli BFR in attempts to identify it. A combination of data from electron paramagnetic resonance spectroscopy (EPR), site-directed mutagenesis and kinetic studies of Fe2+ oxidation demonstrated that three aromatic residues are required for electron transfer during mineralization.24 These residues are Tyr25, Tyr58 and Trp133, all of which are within 10 Å of the ferroxidase centre of the same subunit (Fig. 1C). While Bradley et al.24 established that the role of Tyr25 included it becoming a free-radical as the di-Fe3+ ferroxidase centre is reduced to the di-Fe2+ form, the precise roles of Tyr58 and Trp133 were not established.
Starting with iron-free (apo) BFR, three kinetic phases have been detected by UV-visible spectroscopy in the process leading to mineral formation in the central cavity.20 Phase 1 is the reversible binding of Fe2+ to the ferroxidase centres, phase 2 is the oxidation of these Fe2+ ions at the ferroxidase centres to generate di-Fe3+ centres, and phase 3 is the actual formation of the mineral in the cavity. The rate at which the phase 2 reaction occurs is much faster than the rate of the phase 3 reaction, and it requires rapid reaction procedures to monitor it accurately. Whilst the phase 2 reaction is only observed for iron additions to apo-BFR, the centre is required for the phase 3 reaction to proceed with the rate observed for the wild-type protein since variants of E. coli BFR with an impaired ferroxidase centre have much lower rates of mineralization than the wild-type protein.25 Wild-type protein in the presence of Zn2+, which inhibits ferroxidase activity by directly binding the centre and effectively blocking it, is also unable to mineralize iron rapidly.23 In the earlier work by Bradley et al.24 reporting that Tyr25, Tyr58 and Trp133 were important for mineralization, it was shown that the Tyr25Phe, Tyr58Phe and Trp133Phe variants had unaltered rates of the phase 2 reaction but considerably diminished rates of the phase 3 reaction. This is consistent with a role for Tyr25, Tyr58 and Trp133 in the reductive half of the ferroxidase centre reaction cycle; transmitting electrons from the Fe2+ oxidation occurring in the cavity to reduce the di-Fe3+ ferroxidase centres, prior to their re-oxidation by O2 (or H2O2). Here we present further kinetic and EPR studies of wild type E. coli BFR and its Tyr25Phe, Tyr58Phe and Trp133Phe variants that show Tyr58 and Trp133 play important roles in the formation and decay of the Tyr25 radical.
Experimental
Protein purification
Wild type, Y25F, Y58F and W133F BFR proteins were prepared as previously described,24,26 with expression induced with 10 μM IPTG. Non-heme iron was removed by treatment with sodium dithionite and bipyridyl, as previously described.27 The concentrations of BFR proteins were determined using per subunit ε280nm values: 33![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 (wild type);14,21 23375 (W133F);26 25585 (Y25F)24 and 24600 (Y58F)24 all in units of M−1 cm−1. Heme content of proteins was determined following non-heme iron removal using the heme Soret absorbance intensity (ε418nm = 107000 M−1 cm−1),28 and found to be 1.0–1.5 heme/BFR for all variants. Note that the lack of heme does not significantly affect the rate of mineralization at low to mid-iron loadings.29,30
000 (wild type);14,21 23375 (W133F);26 25585 (Y25F)24 and 24600 (Y58F)24 all in units of M−1 cm−1. Heme content of proteins was determined following non-heme iron removal using the heme Soret absorbance intensity (ε418nm = 107000 M−1 cm−1),28 and found to be 1.0–1.5 heme/BFR for all variants. Note that the lack of heme does not significantly affect the rate of mineralization at low to mid-iron loadings.29,30
Absorbance monitored Fe2+ oxidation
The kinetics of iron oxidation by apo and Zn2+-incubated wild type and variant BFRs was determined according to the method of Baaghil et al.23 Oxidation of iron to the ferric state results in the appearance of broad features in the absorbance spectrum at approximately 340 nm due to charge transfer transitions between oxygenic ligands and iron that are absent for the ferrous state. Solutions of 0.5 μM protein (1.6 mL volume in 1 cm pathlength cuvettes) were mixed with 400 equivalents (200 μM) Fe2+, added as 6.4 μL of a 50 mM ferrous ammonium sulfate solution (Sigma), and the rate of iron oxidation deduced from the increase in absorbance at 340 nm as a function of time. Data were recorded on a Hitachi U2900 spectrophotometer. The final absorbance of the Zn2+ free samples was used to deduce an extinction coefficient for mineralized iron and these values (1750 M−1 cm−1 for wild type and 1950 M−1 cm−1 for variants) used to calculate initial rates of iron oxidation (μM min−1) from the initial slope of traces of absorbance as a function of time.Zn2+ incubated samples were prepared by replacing an appropriate volume of buffer with 1 mM stock ZnCl2 (Sigma) solution, such that in each case protein concentration was 0.5 μM and sample volume was 1.6 mL, as with the apo-protein samples, prior to the addition of Fe2+.
EPR spectroscopy and analysis
Spectra were recorded at 10 K on a Bruker EMX (X-band) EPR spectrometer equipped with an Oxford Instruments liquid helium system and a spherical high-quality ER 4122 SP 9703 Bruker resonator. Spectra were recorded using field modulation of frequency 100 kHz and amplitude 3.0 G in all cases, and incident microwave power of 3.18 mW (high power) or 0.05 mW (low power). Protein samples in EPR tubes were mixed with the appropriate volume of a freshly prepared 25 mM stock Fe2+ solution and frozen at the appropriate time thereafter by plunging the tubes into methanol cooled with solid CO2. The final protein concentration was 8.33 μM (200 μM in monomer) in all cases.Kinetic dependences of the Tyr radical and of the secondary isotropic signal are expressed in absolute units of concentration (Fig. 3C and 4D), where the radical concentration was determined by reference to a Cu2+ concentration standard (80 μM Cu2+ with excess EDTA in 50 mM phosphate buffer, pH 7.0). Both the protein radical EPR spectra and the spectra of the standard were analysed for their saturation behaviour upon increasing microwave power. The second integrals of the EPR signals for the non-saturating conditions were compared. EPR lineshapes of each species were obtained as described in Fig. S1, ESI.† The fact that the overall line shape of the free radical EPR spectrum changes with reaction time was used in the deconvolution of overlapping EPR signals. Spectra subtraction with variable coefficient,31 applied to the spectra measured at two different microwave power values, allowed extraction of the two line shapes that have been further used in the quantitation.
Experimentally measured kinetics of the free radical species formation and decay were fitted by computer generated (Origin 8, Origin Labs) time dependences of radical B˙(t) in the simple two step kinetic scheme A → B˙ → C, involving two first order reaction rate constants for formation (rate constant kAB) and subsequent decay (rate constant kBC) of the radical B˙. An offset parameter was also included to account for non-zero plateauing of the intermediate species. Although this scheme does not describe the full mechanism by which the radicals are formed and decay (which is undoubtedly more complex), it does allow ready quantitative comparison of the differing rates of radical formation and decay (when Fe2+ is exhausted) in the proteins studied, something that is also apparent from visual inspection of the data.
Results and discussion
Tyr25, Trp58 and Trp133 have an indirect effect on the role of the ferroxidase centres in mineralization
Bradley et al established that the rate of the phase 2 reactions in the Tyr25Phe, Tyr58Phe and Trp133Phe variants of BFR were the same as the wild type protein but that of the phase 3 reaction was severely decreased.24 However, because the phase 2 reaction only reports on the oxidative half of the ferroxidase centre reaction cycle (in which the di-Fe2+ centre is oxidized) and not on the reductive half (in which the di-Fe3+ centre is reduced back to the di-Fe2+ form) there remained a possibility that the substitutions directly affected the latter rather than just the rate at which electrons arrive from the cavity. In order to investigate this further we studied the effect of added Zn2+ on the rates of the phase 3 reactions. Zn2+ inhibits mineralization by binding more tightly to the ferroxidase centre residues than does Fe2+, thereby completely inhibiting ferroxidase centre reactivity at a ratio of 48 Zn2+ per 24-mer.14,21 Monitoring the phase 3 reaction of wild type BFR and its Tyr25Phe, Tyr58Phe and Trp133Phe variants with variable amounts of Zn2+ present, as described in the Experimental section, revealed that though the rates of the phase 3 reactions differed, the profiles of the effect of Zn2+ on them are similar (Fig. 2A–D). This is most clearly seen in the summary plot with normalised activities (Fig. 2E). Firstly, since iron oxidation can be completely eliminated by inhibiting the ferroxidase center with zinc, this plot demonstrates that the previously reported24,26 residual activity of the variants results from ferroxidase centre-mediated oxidation and is not due to adventitious Fe2+ oxidation occurring elsewhere. Secondly it highlights the similar effect of Zn2+ on all of the variants, which shows that the mutations have no direct effect on the activities of the ferroxidase centres throughout the phase 3 reactions. This is consistent with the high resolution crystal structures of the variants, which showed that changes were confined to the substituted side chains.24,26,32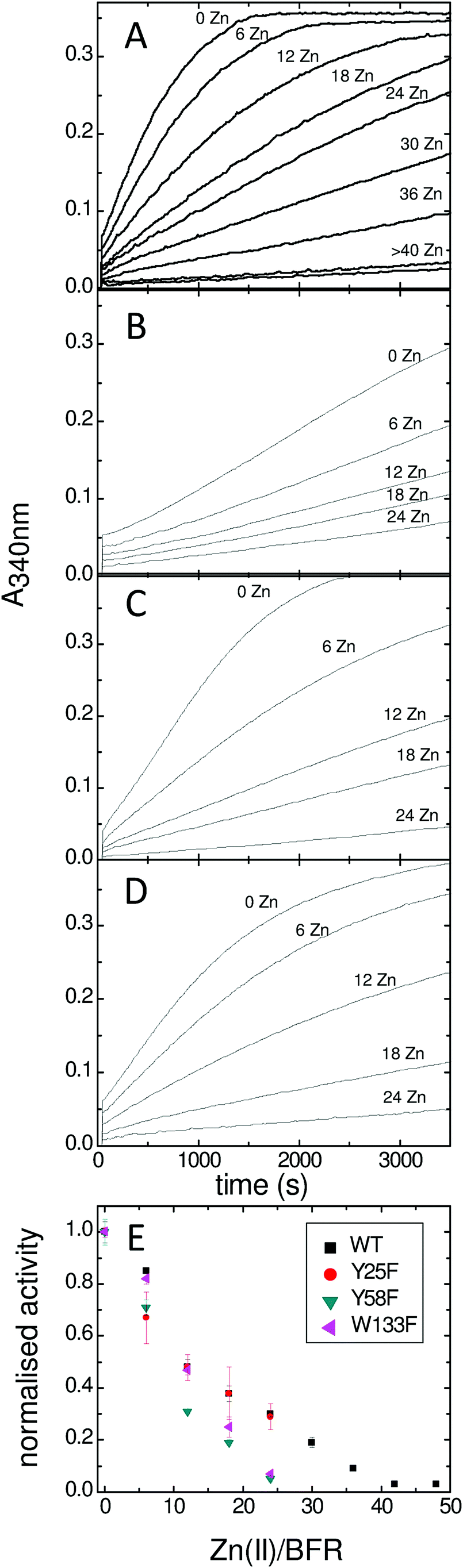 | ||
| Fig. 2 Effect of Zn2+ on the rate of E. coli BFR mineralisation (phase 3). (A) Wild type BFR, (B) Tyr25Phe BFR, (C) Tyr58Phe BFR, (D) Trp133Phe BFR, (E) Summary plot of the normalised rates versus the Zn(II)/BFR ratios. E. coli BFR (0.5 μM) was in 100 mM MES, pH 6.5. | ||
EPR studies reveal that Tyr58 and Trp133 influence the Tyr25 radical during mineralization
Having demonstrated that Tyr25, Tyr58 and Trp133 are needed for mineralization and that their replacement by phenylalanine does not directly perturb the ferroxidase centres, we turned to the issue of their roles in electron transfer from Fe2+ at the FeIS site, or Fe2+ in the cavity, to the ferroxidase centres. As Tyr25 formed a free-radical it seemed its role is directly related to this, especially since phenylalanine, which cannot form a similar radical, could not sustain activity in its place.24 As neither Tyr58 nor Trp133 form a detectable radical during the reaction, and both the Tyr58Phe and Trp133Phe variants formed Tyr25 radicals,24 it seemed plausible that one route by which Tyr58 and Trp133 could influence mineralization is by perturbing the properties of the functionally important Tyr25 radical. This was investigated using EPR spectroscopy.We previously reported a time course of Tyr25 radical in the wild type BFR during iron mineralization over a range of reaction time from 0.3 s to 16 min.24 Fitting this time dependence (Fig. 3D in ref. 24) of the Tyr25˙ radical to a two-step kinetic scheme, 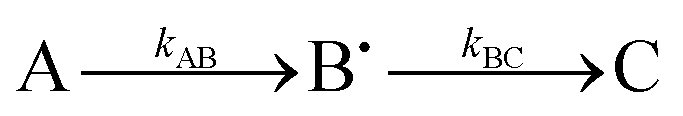 , as described in the Experimental section, gave the values kAB = 1.2 ± 0.2 s−1 and kBC = 0.03 ± 0.006 s−1 at 4 °C (Fig. 3C, blue curve, and Table 1). Here, species A is the bridged di-ferric species formed at completion of the phase 2 reaction, B˙ is formed by transfer of one electron from Fe2+ bound at FeIS and one from Tyr25 to the di-ferric ferric ferroxidase centre (generating the Tyr radical) and C is the product formed upon dissipation of the Tyr radical. It is important to note that during mineralization, the Tyr radical is rapidly quenched by oxidation of Fe2+ most likely located within the cavity. The slow decay process observed here is that which occurs when all of the Fe2+ is exhausted. Kinetic dependences of the Tyr25˙ radical of the Tyr58Phe and Trp133Phe variants over a mineralization reaction time range of 15 s–16 min (Fig. 3A and B) were obtained in a similar fashion. The contribution of the Tyr25˙ radical EPR signal to the spectra was determined by applying the methodology of spectra subtraction with variable coefficient,31 and the kinetic dependences for the radical in the two variants were fitted with the same
, as described in the Experimental section, gave the values kAB = 1.2 ± 0.2 s−1 and kBC = 0.03 ± 0.006 s−1 at 4 °C (Fig. 3C, blue curve, and Table 1). Here, species A is the bridged di-ferric species formed at completion of the phase 2 reaction, B˙ is formed by transfer of one electron from Fe2+ bound at FeIS and one from Tyr25 to the di-ferric ferric ferroxidase centre (generating the Tyr radical) and C is the product formed upon dissipation of the Tyr radical. It is important to note that during mineralization, the Tyr radical is rapidly quenched by oxidation of Fe2+ most likely located within the cavity. The slow decay process observed here is that which occurs when all of the Fe2+ is exhausted. Kinetic dependences of the Tyr25˙ radical of the Tyr58Phe and Trp133Phe variants over a mineralization reaction time range of 15 s–16 min (Fig. 3A and B) were obtained in a similar fashion. The contribution of the Tyr25˙ radical EPR signal to the spectra was determined by applying the methodology of spectra subtraction with variable coefficient,31 and the kinetic dependences for the radical in the two variants were fitted with the same 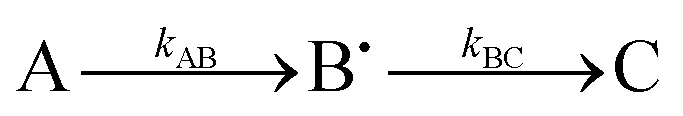 kinetic scheme (Fig. 3C) with the rate constants reported in Table 1. An immediate conclusion from the three kinetic dependences in Fig. 3 is that the Tyr25˙ radical in wild type BFR both forms and decays significantly faster than in the two variants (see Table 1 for a comparison of the apparent rate constants). This strongly correlates with the importance of both Trp133 and Tyr58 for iron mineralization. It thus appears that the affected mineralization in both the Tyr58Phe and Trp133Phe variants might follow directly from the necessity of these residues for cycling of Tyr25 through its radical state during the mineralization process.
kinetic scheme (Fig. 3C) with the rate constants reported in Table 1. An immediate conclusion from the three kinetic dependences in Fig. 3 is that the Tyr25˙ radical in wild type BFR both forms and decays significantly faster than in the two variants (see Table 1 for a comparison of the apparent rate constants). This strongly correlates with the importance of both Trp133 and Tyr58 for iron mineralization. It thus appears that the affected mineralization in both the Tyr58Phe and Trp133Phe variants might follow directly from the necessity of these residues for cycling of Tyr25 through its radical state during the mineralization process.
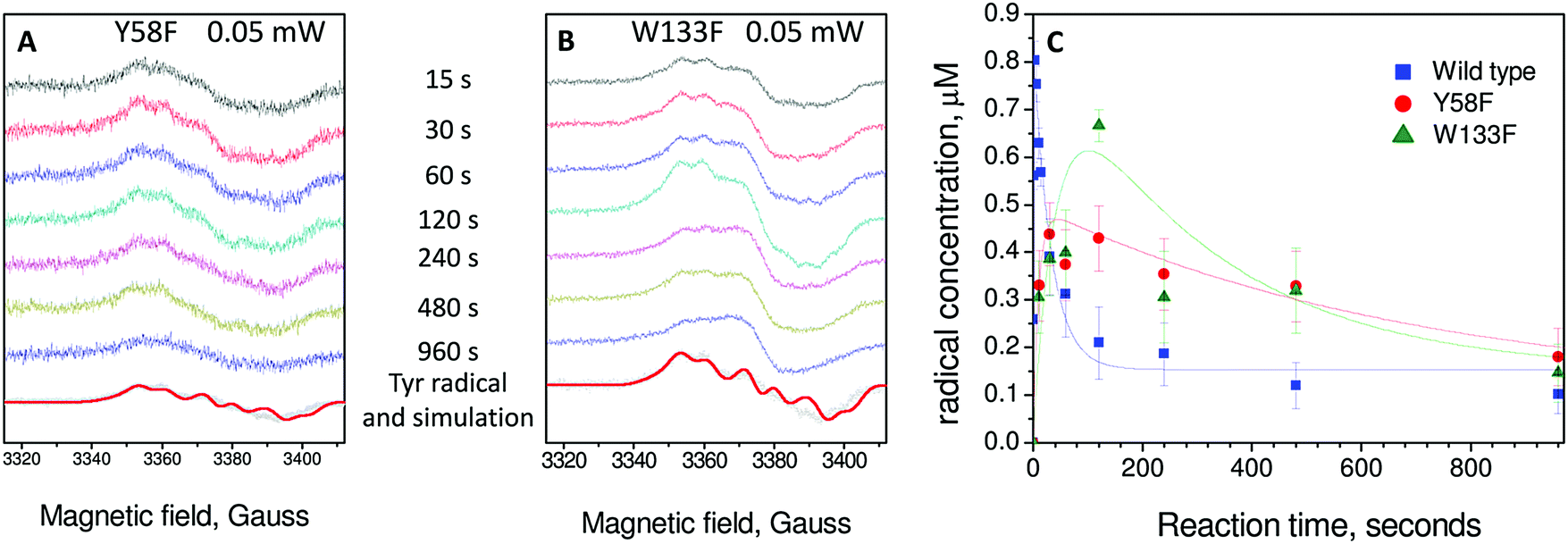 | ||
| Fig. 3 EPR analysis of the formation and decay of the Tyr25 radical in wild type and variant BFR proteins. (A) Tyr58Phe BFR and (B) Trp133Phe BFR. Samples were 8.3 μM in 100 mM MES pH 6.5 frozen at variable times (as indicated) after addition of 72 Fe2+ per 24-mer. The tyrosyl radical spectrum obtained using the methodology of spectral subtraction as described in ref. 31 is shown in grey along with that simulated for the reported sidechain conformation of Tyr25, in each case the spectra are scaled according to the intensity observed 120 seconds after the addition of Fe2+. Spectra were obtained at 10 K with the following instrumental conditions: microwave frequency = 9.467 GHz, microwave power = 0.05 mW, modulation frequency = 100 kHz, modulation amplitude = 3 G, scan rate = 0.596 G s−1, time constant τ = 82 ms. (C) Plots of tyrosyl radical concentrations versus time following addition of 72 Fe2+ per BFR 24-mer. The error bars principally represent uncertainties resulting from the method of spectral subtraction with variable coefficient used to deconvolute each spectrum into two spectral components and to determine their respective concentrations.23 The data for the wild-type protein are from ref. 24. Data for the radicals are fitted to an A → B˙ → C kinetic scheme as discussed in the text. | ||
| Protein | Tyr25 radical | Secondary isotropic radical | ||
|---|---|---|---|---|
| Apparent rate constantsa (s−1) | ||||
| Formation | Decayb | Formation | Decay | |
| a Rate constants were obtained from fits using a A → B˙ → C kinetic scheme as described in the experimental. For the Tyr25 radical, details of the proposed mechanism are given in the text. For the secondary isotropic radical, B˙ is the secondary radical itself, while A and C refer to the pre-formation and post-decay diamagnetic states, respectively. This is a simplification of the likely true mechanism because B˙ is not a single species but a collection of radical species, as described in the text. b Apparent decay rate constant is associated with the remaining radical intensity when all added Fe2+ has been oxidised, meaning that quenching by electron transfer from Fe2+ within the cavity can no longer occur. Standard errors associated with the fits are shown. | ||||
| Wild type BFR | 1.2 (±0.2) | 3 (±0.6) × 10−2 | 2 (±0.5) × 10−2 | 7 (±3) × 10−3 |
| W133F BFR | 0.025 (±0.01) | 3 (±1) × 10−3 | 1.7 (±0.5) × 10−2 | 2 (±0.5) × 10−3 |
| Y58F BFR | 0.1 (±0.03) | 1.4 (±0.7) × 10−3 | 3.3 (±1.5) × 10−2 | 1.4 (±0.6) × 10−3 |
Decay of the Tyr25 radical involves a second free-radical
An important parameter in EPR spectroscopy is the power of the applied microwaves. Varying the power while monitoring signal intensity provides information on the relaxation characteristics of the signal, and these in turn can provide mechanistic insights. The spectra reported previously for the wild type protein and in Fig. 3 for the variants were acquired at low power. At high power the Tyr25 signal is reduced in intensity relative to a second transient, low intensity radical signal that did not saturate with microwave power as readily as that of Tyr25 (Fig. 4A). This second radical gives rise to a ∼14–16 G wide, apparently isotropic, EPR signal at g = 2.004. Its intensity was measured as a function of time and expressed in concentration units of μM in the wild type and the Tyr58Phe and Trp133Phe variants (Fig. 4D). Rate constants for the formation and decay of this paramagnetic species, resulting from fits of the kinetic data as described for the Tyr25 radical signal, are reported in Table 1.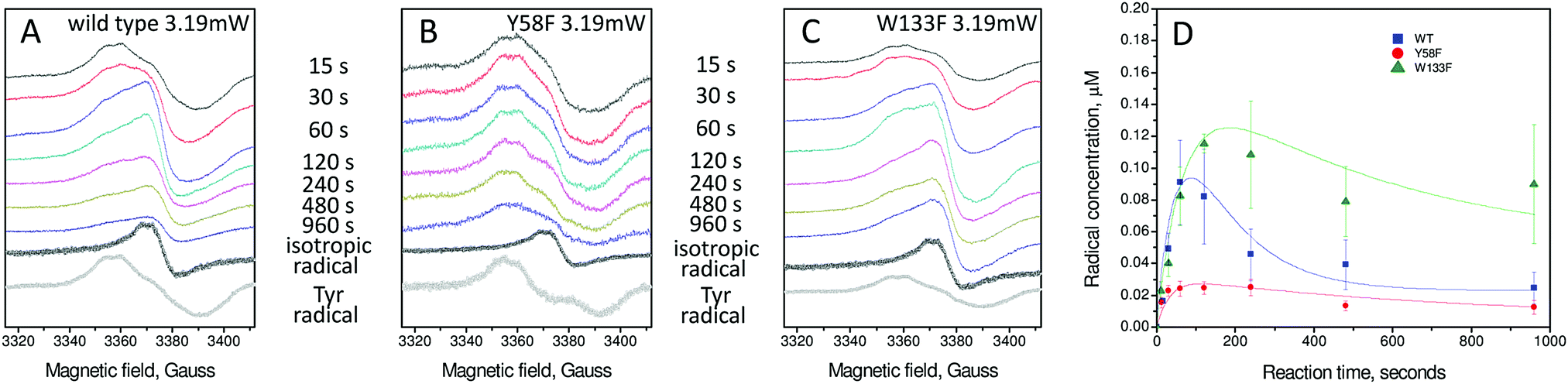 | ||
| Fig. 4 EPR analysis of the formation and decay of the apparently isotropic radical signal of wild type and variant BFR proteins. (A) wild-type BFR, (B) Tyr58Phe BFR and (C) Trp133Phe BFR. Samples were 8.3 μM in 100 mM MES pH 6.5 frozen at variable times (as indicated) after addition of 72 Fe2+ per BFR 24-mer. The spectra of the isotropic and tyrosyl radical obtained using spectral subtraction methodology31 are shown at the bottom of each panel in black and grey, respectively. Spectra were recorded at 10 K with the following instrumental conditions: microwave frequency = 9.467 GHz, microwave power = 3.19 mW, modulation frequency = 100 kHz, modulation amplitude = 3 G, scan rate = 0.596 G s−1, time constant τ = 82 ms. (D) Plots of the radical concentrations versus time following addition of 72 Fe2+ per BFR 24-mer. The error bars principally represent uncertainties resulting from the method of spectral subtraction with variable coefficient used to deconvolute each spectrum into two spectral components and to determine their respective concentrations.29 Data for the radicals are fitted to an A → B˙ → C kinetic scheme as discussed in the text. | ||
Similar isotropic EPR signals have been observed in other proteins. In most cases, their time dependent appearance in the spectra follows the disappearance of a different, often better-resolved, EPR signal. This has been rationalized as a radical dissipation process that occurs when a distinct protein radical species with a multicomponent EPR signal is transferred non-specifically to one of several possible new locations, and the process repeats until two different radicals combine. Thus, when a protein radical at a specific site decays, not only does the radical EPR signal intensity decrease, its line shape loses hyperfine structure, turning into a symmetric 14–17 G wide isotropic signal, which is the same irrespective of the protein concerned.33–35 It appears that the isotropic EPR signal detected in BFR is yet another example of this type of protein radical kinetic behaviour.
Tyr58 and Trp133 modulate the characteristics of the Tyr25 radical
The EPR data presented above strongly indicate that Tyr58 and Trp133 are both required for the rapid turnover of Tyr25 through its radical form to sustain mineralization. If either of the two residues, Tyr58 or Trp133, is missing, the radical on Tyr25 is both formed and quenched more slowly than in the wild type protein (Table 1), thus mirroring the residues’ replacement effect on the mineralization rate. Therefore we suggest that the electron transfer pathway (or pathways) from Fe2+ in the cavity to the O2 bound to the ferroxidase centre includes these two residues, in addition to Tyr25. The fact that the free radical states of these residues have not been detected in this work is not at variance with this hypothesis, but indicates that transient concentration of such paramagnetic species are below the detection limit.According to the reaction scheme proposed by Bradley et al.,24 the Tyr25 radical is largely quenched through the oxidation of an Fe2+ ion that is probably located in the cavity. However, the observation of the isotropic unresolved free radical EPR signal, which likely arises from the radical dissipation initiated by the Tyr25 radical decay, indicates that not all the Tyr25 radicals are consumed by the quenching through Fe2+ oxidation. This is consistent with some Tyr25 radical remaining when no further Fe2+ is available to quench it (due to its oxidation to Fe3+).
While replacing Trp133 and Tyr58 by phenylalanine residues drastically decreases both formation and decay rates for the Tyr25 radical, the effect on the suggested multi-radical state with isotropic EPR line is different: the formation rate is not affected while the decay rate is decreased in both variants (Table 1). This means that these two residues not only ensure a fast turnover of the Tyr25 radical, they also both, and possibly in tandem, participate in radical dissipation through the multi-radical mechanism making it effectively faster (as replacement of either of them makes the isotropic isotropic EPR signal disappear 3–5 times more slowly, Table 1).
Interestingly, the Tyr58Phe protein shows a significantly decreased yield of the species with isotropic resonance (Fig. 4D). Considering the same apparent formation rate constant and 5-time slower apparent decay rate constant, this can only be possible if the mechanism of this species formation and decay is more complex than 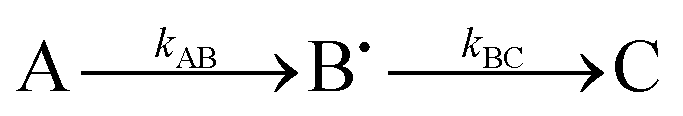 . It is likely therefore that other residues in addition to Trp133 and Tyr58 assist with dissipation of the secondary radicals forming the overall isotropic EPR signal.
. It is likely therefore that other residues in addition to Trp133 and Tyr58 assist with dissipation of the secondary radicals forming the overall isotropic EPR signal.
Comparison of E. coli bacterioferritin with ribonucleotide reductase
The discovery that Tyr58 and Trp133 play important roles in the formation and decay of the Tyr25 radical draws further comparison with the R2 subunit of ribonucleotide reductase (RNR), in which a diiron centre closely related to that of BFR generates a stable radical at the nearby Tyr122 (E. coli R2 numbering). The radical is subsequently shuttled back and forth over >30 Å to the active site of ribonucleotide reduction in the R1 subunit.16 Tyr25 and Trp133 of BFR are in very similar positions to Tyr122 and Trp48 of R2, but on the other side of the diiron site. Tyr58 does not have a direct structural homologue in RNR, but Tyr residues play a key role in shuttling the radical away from (and back to) the diiron site. Thus it is reasonable to propose Tyr58 fulfils a similar shuttling role in BFR. It was recently demonstrated for an RNR R2-inspired maquette that the Tyr and Trp sidechains form a Tyr–Trp dyad in which the phenol group of the tyrosine and the indole group of the tryptophan participate in a dipole–dipole interaction.36 It was proposed that a similar interaction occurs in R2 RNR, accounting for the importance of Trp48 for RNR activity. A similar Tyr–Trp dyad arrangement is found in other Tyr radical forming systems, including photosystem II,37 galactose oxidase38 and GlxA.39 In BFR, we find a similar arrangement, with a distance of ∼6 Å between Tyr25 and Trp133 (Fig. 1C). A dipole–dipole interaction that facilitates radical formation/decay on Tyr25 would account for the effects reported here, and for the importance of Trp133 for BFR mineralisation.Conclusion
The major conclusion of this work is that the E. coli BFR ferroxidase centre should not be considered simply as a diiron site; rather, it should be viewed as a diiron site with a surrounding network of aromatic residues (Tyr25, Tyr58 and Trp133) that function together to facilitate redox cycling of the diiron site during mineralisation. Replacement of any of the aromatic residues does not prevent mineralization from being driven by the ferroxidase centre, but drastically reduces the rate at which it occurs.Abbreviations
| EPR | Electron paramagnetic resonance |
| IPTG | Isopropyl β-D-1-thiogalactopyranoside |
| MES | 2-(N-Morpholino)ethanesulfonic acid |
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the Biotechnology and Biological Sciences Research Council (grant BB/I021884/1). GRM thanks the Leverhulme Trust for an Emeritus Fellowship (EM-2014-088).References
- S. C. Andrews, The ferritin superfamily: structure, function and evolution of the biological iron storeman, Biochim. Biophys. Acta, 2010, 1800, 691–705 CrossRef CAS PubMed.
- E. C. Theil, M. Matzapetakis and X. F. Liu, Ferritins: iron/oxygen biominerals in protein nanocages, J. Biol. Inorg. Chem., 2006, 11, 803–810 CrossRef CAS PubMed.
- J. M. Bradley, N. E. Le Brun and G. R. Moore, Ferritins: furnishing proteins with iron, J. Biol. Inorg. Chem., 2016, 21, 13–28 CrossRef CAS PubMed.
- E. I. Stiefel and G. D. Watt, Azotobacter Cytochrome-B557.5 Is a Bacterioferritin, Nature, 1979, 279, 81–83 CrossRef CAS PubMed.
- V. Laufberger, Sur la cristallisation de la ferritine, Bull. Soc. Chim. Biol., 1937, 19, 1575–1582 CAS.
- N. E. Le Brun, A. Crow, M. E. P. Murphy, A. G. Mauk and G. R. Moore, Iron core mineralisation in prokaryotic ferritins, Biochim. Biophys. Acta, 2010, 1800, 732–744 CrossRef CAS PubMed.
- K. Eshelman, H. Yao, A. N. Punchi Hewage, J. J. Deay, J. R. Chandler and M. Rivera, Inhibiting the BfrB:Bfd interaction in Pseudomonas aeruginosa causes irreversible iron accumulation in bacterioferritin and iron deficiency in the bacterial cytosol, Metallomics, 2017, 9, 646–659 RSC.
- H. Abdul-Tehrani, A. J. Hudson, Y. S. Chang, A. R. Timms, C. Hawkins, J. M. Williams, P. M. Harrison, J. R. Guest and S. C. Andrews, Ferritin mutants of Escherichia coli are iron deficient and growth impaired, and fur mutants are iron deficient, J. Bacteriol., 1999, 181, 1415–1428 CAS.
- A. J. Hudson, S. C. Andrews, C. Hawkins, J. M. Williams, M. Izuhara, F. C. Meldrum, S. Mann, P. M. Harrison and J. R. Guest, Overproduction, Purification and Characterization of the Escherichia coli Ferritin, Eur. J. Biochem., 1993, 218, 985–995 CrossRef CAS PubMed.
- J. M. Bradley, G. R. Moore and N. E. Le Brun, Diversity of Fe2+ entry and oxidation in ferritins, Curr. Opin. Chem. Biol., 2017, 37, 122–128 CrossRef CAS PubMed.
- P. M. Harrison and P. Arosio, Ferritins: molecular properties, iron storage function and cellular regulation, Biochim. Biophys. Acta, 1996, 1275, 161–203 CrossRef.
- S. Yasmin, S. C. Andrews, G. R. Moore and N. E. Le Brun, A new role for heme, facilitating release of iron from the bacterioferritin iron biomineral, J. Biol. Chem., 2011, 286, 3473–3483 CrossRef CAS PubMed.
- S. K. Weeratunga, C. E. Gee, S. Lovell, Y. H. Zeng, C. L. Woodin and M. Rivera, Binding of Pseudomonas aeruginosa apo-bacterioferritin-associated ferredoxin to bacterioferritin B promotes heme mediation of electron delivery and mobilization of core mineral iron, Biochemistry, 2009, 48, 7420–7431 CrossRef CAS PubMed.
- A. Crow, T. L. Lawson, A. Lewin, G. R. Moore and N. E. Le Brun, Structural basis for iron mineralization by bacterioferritin, J. Am. Chem. Soc., 2009, 131, 6808–6813 CrossRef CAS PubMed.
- J. M. Bradley, G. R. Moore and N. E. Le Brun, Mechanisms of iron mineralization in ferritins: one size does not fit all, J. Biol. Inorg. Chem., 2014, 19, 775–785 CrossRef CAS PubMed.
- E. C. Minnihan, D. G. Nocera and J. Stubbe, Reversible, long-range radical transfer in E. coli class Ia ribonucleotide reductase, Acc. Chem. Res., 2013, 46, 2524–2535 CrossRef CAS PubMed.
- F. Bou-Abdallah, G. H. Zhao, H. R. Mayne, P. Arosio and N. D. Chasteen, Origin of the unusual kinetics of iron deposition in human H-chain ferritin, J. Am. Chem. Soc., 2005, 127, 3885–3893 CrossRef CAS PubMed.
- P. Turano, D. Lalli, I. C. Felli, E. C. Theil and I. Bertini, NMR reveals pathway for ferric mineral precursors to the central cavity of ferritin, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 545–550 CrossRef CAS PubMed.
- S. K. Weeratunga, S. Lovell, H. L. Yao, K. P. Battaile, C. J. Fischer, C. E. Gee and M. Rivera, Structural studies of bacterioferritin B from Pseudomonas aeruginosa suggest a gating mechanism for iron uptake via the ferroxidase center, Biochemistry, 2010, 49, 1160–1175 CrossRef CAS PubMed.
- N. E. Le Brun, M. T. Wilson, S. C. Andrews, J. R. Guest, P. M. Harrison, A. J. Thomson and G. R. Moore, Kinetic and structural characterization of an intermediate in the biomineralization of bacterioferritin, FEBS Lett., 1993, 333, 197–202 CrossRef CAS PubMed.
- X. O. Yang, N. E. Le Brun, A. J. Thomson, C. R. Moore and N. D. Chasteen, The iron oxidation and hydrolysis chemistry of Escherichia coli bacterioferritin, Biochemistry, 2000, 39, 4915–4923 CrossRef CAS PubMed.
- Y. Kwak, J. K. Schwartz, V. W. Huang, E. Boice, D. M. Kurtz and E. I. Solomon, CD/MCD/VTVH-MCD studies of Escherichia coli bacterioferritin support a binuclear iron cofactor site, Biochemistry, 2015, 54, 7010–7018 CrossRef CAS PubMed.
- S. Baaghil, A. Lewin, G. R. Moore and N. E. Le Brun, Core formation in Escherichia coli bacterioferritin requires a functional ferroxidase center, Biochemistry, 2003, 42, 14047–14056 CrossRef CAS PubMed.
- J. M. Bradley, D. A. Svistunenko, T. L. Lawson, A. M. Hemmings, G. R. Moore and N. E. Le Brun, Three aromatic residues are required for electron transfer during iron mineralization in bacterioferritin, Angew. Chem., Int. Ed., 2015, 54, 14763–14767 CrossRef CAS PubMed.
- N. E. Le Brun, S. C. Andrews, J. R. Guest, P. M. Harrison, G. R. Moore and A. J. Thomson, Identification of the ferroxidase center of Escherichia coli bacterioferritin, Biochem. J., 1995, 312, 385–392 CrossRef CAS PubMed.
- T. L. Lawson, A. Crow, A. Lewin, S. Yasmin, G. R. Moore and N. E. Le Brun, Monitoring the iron status of the ferroxidase center of Escherichia coli bacterioferritin using fluorescence spectroscopy, Biochemistry, 2009, 48, 9031–9039 CrossRef CAS PubMed.
- E. R. Bauminger, P. M. Harrison, D. Hechel, I. Nowik and A. Treffry, Mössbauer spectroscopic investigation of structure-function relations in ferritins, Biochim. Biophys. Acta, 1991, 1118, 48–58 CrossRef CAS.
- M. R. Cheesman, N. E. Le Brun, F. H. A. Kadir, A. J. Thomson, G. R. Moore, S. C. Andrews, J. R. Guest, P. M. Harrison, J. M. A. Smith and S. J. Yewdall, Heme and non-heme iron sites in Escherichia coli bacterioferritin - spectroscopic and model-building studies, Biochem. J., 1993, 292, 47–56 CrossRef CAS PubMed.
- S. C. Andrews, N. E. Le Brun, V. Barynin, A. J. Thomson, G. R. Moore, J. R. Guest and P. M. Harrison, Site-directed replacement of the coaxial heme ligands of bacterioferritin generates heme-free variants, J. Biol. Chem., 1995, 270, 23268–23274 CrossRef CAS PubMed.
- S. G. Wong, R. Abdulqadir, N. E. Le Brun, G. R. Moore and A. G. Mauk, Fe-haem bound to Escherichia coli bacterioferritin accelerates iron core formation by an electron transfer mechanism, Biochem. J., 2012, 444, 553–560 CrossRef CAS PubMed.
- D. A. Svistunenko, N. Davies, D. Brealey, M. Singer and C. E. Cooper, Mitochondrial dysfunction in patients with severe sepsis: an EPR interrogation of individual respiratory chain components, Biochim. Biophys. Acta, 2006, 1757, 262–272 CrossRef CAS PubMed.
- K. Hingorani, R. Pace, S. Whitney, J. W. Murray, P. Smith, M. H. Cheah, T. Wydrzynski and W. Hillier, Photo-oxidation of tyrosine in a bio-engineered bacterioferritin 'reaction centre'-a protein model for artificial photosynthesis, Biochim. Biophys. Acta, 2014, 1837, 1821–1834 CrossRef CAS PubMed.
- B. S. Rajagopal, A. N. Edzuma, M. A. Hough, K. Blundell, V. E. Kagan, A. A. Kapralov, L. A. Fraser, J. N. Butt, G. G. Silkstone, M. T. Wilson, D. A. Svistunenko and J. A. R. Worrall, The hydrogen-peroxide-induced radical behaviour in human cytochrome c-phospholipid complexes: implications for the enhanced pro-apoptotic activity of the G41S mutant, Biochem. J., 2013, 456, 441–452 CrossRef CAS PubMed.
- B. J. Reeder, D. A. Svistunenko, C. E. Cooper and M. T. Wilson, Engineering tyrosine-based electron flow pathways in proteins: the case of aplysia myoglobin, J. Am. Chem. Soc., 2012, 134, 7741–7749 CrossRef CAS PubMed.
- M. K. Thompson, S. Franzen, R. A. Ghiladi, B. J. Reeder and D. A. Svistunenko, Compound ES of dehaloperoxidase decays via two alternative pathways depending on the conformation of the distal histidine, J. Am. Chem. Soc., 2010, 132, 17501–17510 CrossRef CAS PubMed.
- C. V. Pagba, T. G. McCaslin, G. Veglia, F. Porcelli, J. Yohannan, Z. J. Guo, M. McDaniel and B. A. Barry, A tyrosine-tryptophan dyad and radical-based charge transfer in a ribonucleotide reductase-inspired maquette, Nat. Commun., 2015, 6, 10010 CrossRef CAS PubMed.
- Y. Umena, K. Kawakami, J. R. Shen and N. Kamiya, Crystal structure of oxygen-evolving photosystem II at a resolution of 1.9 Å, Nature, 2011, 473, 55–60 CrossRef CAS PubMed.
- N. Ito, S. E. V. Phillips, C. Stevens, Z. B. Ogel, M. J. McPherson, J. N. Keen, K. D. S. Yadav and P. F. Knowles, Novel thioether bond revealed by a 1.7 Å crystal structure of galactose oxidase, Nature, 1991, 350, 87–90 CrossRef CAS PubMed.
- A. K. Chaplin, M. L. C. Petrus, G. Mangiameli, M. A. Hough, D. A. Svistunenko, P. Nicholls, D. Claessen, E. Vijgenboom and J. A. R. Worrall, GlxA is a new structural member of the radical copper oxidase family and is required for glycan deposition at hyphal tips and morphogenesis of Streptomyces lividans, Biochem. J., 2015, 469, 433–444 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary figure. See DOI: 10.1039/c7mt00187h |
| This journal is © The Royal Society of Chemistry 2017 |