
Phenylarsine oxide (PAO) induces apoptosis in HepG2 cells via ROS-mediated mitochondria and ER-stress dependent signaling pathways
Ping
Huang†
a,
Yu Hua
Zhang†
b,
Xiao Wei
Zheng
a,
Yu Jia
Liu
a,
Hong
Zhang
a,
Luo
Fang
a,
Yi Wen
Zhang
a,
Chang
Yang
cd,
Khairul
Islam
d,
Chao
Wang
d and
Hua
Naranmandura
 *cd
*cd
aDepartment of Pharmacy, Zhejiang Cancer Hospital, Hangzhou, 310022, China
bZhejiang Provincial People's Hospital, Hangzhou 310014, China
cDepartment of Pharmacology, School of Medicine, Zhejiang University, Hangzhou, 310058, China
dDepartment of Toxicology, School of Medicine and Public Health, Zhejiang University, Hangzhou, 310058, China. E-mail: narenman@zju.edu.cn; Fax: +86-571-8820-6736; Tel: +86-571-8820-6736
First published on 3rd August 2017
Abstract
Arsenic trioxide (As2O3) is an old drug that has recently been reintroduced as a therapeutic agent for acute promyelocytic leukemia (APL). Although As2O3 is also applied to treat other types of cancer in vitro and in vivo, it has been reported that single agent As2O3 has poor efficacy against non-hematologic malignant cancers in clinical trials. Recently, a few reports have indicated that organic arsenic compounds can be a possible alternative for the treatment of As2O3-resistant cancers. In this study, we aimed to investigate whether the organic arsenic compound phenylarsine oxide (PAO) has potent cytotoxic effects against human hepatocellular carcinoma (HCC) HepG2 cells. Our results showed that PAO not only had a potent inhibitory effect on the proliferation of HepG2 cells but also activated apoptosis-related proteins (e.g., caspase-3 and -9 and poly-ADP ribose polymerase) in a dose- and time-dependent manner. Furthermore, intracellular ROS were specifically accumulated in the mitochondria and endoplasmic reticulum (ER) after exposure to PAO, implying that they are the target organelles for PAO-induced cytotoxicity. Additionally, when the cells were pretreated with antioxidant N-acetylcysteine (NAC), apoptosis and ER-stress were attenuated significantly, suggesting that induction of apoptosis and cell death probably occurs through the ROS-mediated mitochondria and ER-stress dependent signaling pathways.
Significance to metallomicsMany manufactured compounds containing potentially toxic heavy metals and metalloids (e.g., Hg, Sb, As, etc.) have been used historically for medicinal purposes. Interestingly, As2O3 has been revived again for its therapeutic efficacy against APL that led many scientists towards further research on arsenic compounds and other metalloids (e.g., Sb) in cancer treatment. PAO (organic arsenic derivative) is such a compound that offers a therapeutic approach for resistant cancers like hepatocellular carcinoma (HCC), PML mutated APL, etc. Although metalloids are toxic to normal/healthy cells, through proper formulation and target-based therapy (antibody, aptamers or nanoparticles) PAO can show promise as a new agent for cancer treatment in the future especially for resistant ones. |
Introduction
Hepatocellular carcinoma (HCC) is one of the most common dominant forms of primary liver cancer occurring in patients with chronic liver disease and cirrhosis.1 Among all cancers, liver cancer death rates and incidence rates have increased, and the primary liver cancer is the sixth most common cancer in the world as well as the third leading cause of cancer mortality.2 Although surgery is the optimal treatment for HCC, systemic and partial chemotherapy could be necessary for patients with advanced HCC.3 However, HCC cells showed drug resistance very easily to most common anticancer agents.4The poor response of liver cancer to chemotherapy has whipped up interest in arsenic based therapy, because being an anticancer drug against acute promyelocytic leukemia (APL) arsenic trioxide (As2O3)5,6 has also been proved as an effective compound that can inhibit the growth of some solid cancers such as gastric, colorectal, and pancreatic carcinoma in vitro and in vivo.7,8 Additionally, the liver is well known as a primary site for the biotransformation, accumulation, and excretion of arsenic to its relevant metabolites by the arsenic methyltransferase (AS3MT) enzyme for excretion into bile or to the blood stream.9,10 This evidence suggests that HCC cells could be more likely sensitive as a result of the aggregation of intracellular arsenicals after As2O3 treatment.
However, a phase II clinical trial evaluating the efficacy and toxicity of arsenic trioxide in patients with HCC showed that single-agent arsenic trioxide was not active against advanced HCC.11 Several likely factors could explain the inefficacy of As2O3 against advanced HCC. The appearance of increased drug efflux due to overexpression of transporters from the ATP-binding cassette (ABC) superfamily and acquired resistance might be the important possibilities that contributed to drug resistance. It has already been reported that arsenic resistant HepG2/AS liver cancer cells overexpress the ABCB1 and ABCC1 transporters and p-glycoprotein, which could decrease the intracellular arsenic concentrations by increasing the efflux mechanism.12,13 The limitations of As2O3 against HCC led us to investigate other types of arsenic compounds for use in reversing the chemoresistance that was shown by the excessive efflux mechanism.
Organic arsenic compounds are possible alternatives for the treatment of therapy-resistant HCC, because they have greater affinity toward cells, sub-cellular organelles and biological macromolecules when compared with As2O3.14 Phenylarsine oxide (PAO) is one such organic trivalent arsenic compound that can easily enter into cells by passive diffusion and binds with cysteine residues of intracellular proteins with a higher affinity than As2O3.15 Moreover, PAO is also known as a membrane-permeable protein-tyrosine phosphatase (PTPase) inhibitor that can induce a remarkable increase of phosphotyrosine in many cellular proteins, resulting in altered cell signaling pathways.16 It has also been extensively used as a tool for detecting and blocking thiol-dependent biological processes.17 On the other hand, it has been reported that PAO can also inhibit cell proliferation and induce apoptosis in Bax- and Bak-deficient cancer cells.18 More interestingly, PAO is also found to have the ability to degrade the As2O3-resistant PML mutant proteins.19 All these efficacies of PAO build up potential interest in using it for the treatment of therapy-resistant solid tumors in vitro. We designed this study to investigate the underlying mechanism of the cytotoxic effects of PAO on As2O3-resistant HCC cells.
In the present study, we used human HCC HepG2 cells to determine the cytotoxic effects of PAO on cell viability, generation of ROS, translocation of pro- and anti-apoptotic proteins such as Bax and Bcl-2 in the mitochondria as well as in the cytoplasm, induction of ER-stress and apoptosis. We further demonstrated that PAO-induced cell death predominantly occurs through mitochondrial and ER-stress signaling pathways via increased generation of ROS. These findings suggest that PAO plays an important role because of its ability to induce apoptosis of As2O3-resistant cancer cells, and that this characteristic can be used to develop more efficient anticancer drugs in future.
Methods
Reagents
All reagents were of analytical grade. Milli-Q water (Millipore) was used throughout the experiment. Trizma® HCl and Trizma® base, phenazine methosulfate, decylubiquinone, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), and phenylarsine oxide (PAO) were purchased from Sigma (St. Louis, MO, USA). Sodium arsenite (iAsIII) was purchased from Wako Pure Chemical Industries, Ltd (Osaka, Japan). N-Acetyl-L-cysteine (NAC) was obtained from Beyotime Biotechnology. All the chemicals were dissolved in DMSO and stored at −20 °C. In all experiments, the final DMSO solvent concentration was ≤0.2% (v/v).Antibodies
Antibodies against p-PERK, cyto c, PARP, and cleaved-PARP were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). ATF4, p-EIF2, CHOP, GAPDH, caspase-3, caspase-9, cleaved-caspase-3, cleaved-caspase-9, and β-actin antibodies were purchased from Cell Signaling Technology (Danvers, MA).Cell culture
HepG2 (human HCC) cells were cultured in Dulbecco's modified Eagle's medium (DMEM) plus 10% heat-inactivated FBS. The cells were purchased from Shanghai Institute of Biochemistry and Cell Biology (Shanghai, China), and grown in medium supplemented with 100 units per mL penicillin and 100 mg per mL streptomycin at 37 °C with 5% CO2. After 24 h of seeding, the cultures were washed with PBS, fresh medium was added, and the cells were treated with the indicated doses of PAO for the indicated times.MTT assay
The cells were seeded into 96-well plates and treated with chemicals at various concentrations. After 24 h of incubation, 20 μL of MTT (5 mg mL−1) was added to each well for 4 h of incubation. Then, the supernatant was removed and 150 μL of DMSO was added to solubilize the blue-purple crystals of formazan. Cell viability was measured as the absorbance at 490 nm after shaking for 2 min using a microplate reader (Beckman Coulter, Multimode Detector DTX-880). The cell viability rate was related to the percentage of MTT absorption and was calculated as % survival = (mean experimental absorbance/mean control absorbance) × 100.DAPI staining assay
HepG2 cells were cultured in 6-well plates and treated with PAO for 12 h. The cells were washed twice with PBS and then incubated with 4′,6-diamidino-2-phenylindole (DAPI, 1 mg mL−1) for 30 min. The morphological changes in the nucleus caused by PAO in HepG2 cells were observed using a fluorescence microscope (DMI 4000 B, Leica, Germany). The cells with chromatin condensation and nuclear fragmentation were hallmarks of apoptosis.Annexin V/PI assays for apoptosis
For the Annexin V/PI assays, the cells were dual-stained with Annexin V-FITC and PI and then subsequently evaluated for apoptosis using flow cytometry according to the manufacturer's instructions (BD PharMinutesgen, San Diego, CA, USA). Briefly, 1 × 105 cells were washed twice with D-Hank's solution and stained with 5 μL of Annexin V-FITC and then 5 μL of PI in 500 μL of binding buffer for 5 min in the dark at room temperature. The apoptotic cells were analyzed using BD FACSDiva software (BD Biosciences, Franklin Lakes, NJ). Annexin V staining indicates phosphatidylserine externalization.Mitochondrial membrane potential (MMP) assay
The changes in mitochondrial membrane potential (ΔΨm) were monitored using 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolcarbocyanine iodide dye (JC-1). PAO-exposed HepG2 cells were stained with JC-1 (10 μg mL−1) and kept in the dark at 37 °C for 15 min. Later the cells were washed twice with PBS and then immediately analyzed by flow cytometry (FACS Canto II, Becton Dickinson, USA).Analysis of the sub-cellular distribution of ROS
Immunofluorescence staining was used to analyze the subcellular distribution of ROS in HepG2 cells after exposure to PAO. Cells were cultured in 24-well plates and treated with PAO for 12 h, then washed 3 times with D-Hank's solution. The HepG2 cells were blocked in 3% bovine serum for 30 min and incubated with ER-tracker and MitoTracker and ROS probes for 45 min. After washing (three times) with D-Hank's solution, the subcellular distribution of ROS was determined using a fluorescence microscope (model IX71; Olympus, Tokyo, Japan).Measurement of intracellular ROS
HepG2 cells were seeded in 6-well culture plates and were exposed to various concentrations of PAO with or without NAC for 12 h at 37 °C. Then the cells were re-suspended in fresh culture medium containing 10 μM DCFH-DA. Finally, ROS generation in HepG2 cells was measured by flow cytometry.Western blot analysis
HepG2 cells were lysed in lysis buffer in the presence of protease inhibitors [50 mM Tris–HCl buffer, pH 8.0, 150 mM NaCl, 0.5% Nonidet P-40, and 1 mM phenylmethylsulphonyl fluoride (PMSF)]. The protein concentrations were determined by the Bio-Rad microprotein assay using BSA as the standard. The samples containing whole cell protein were separated on polyacrylamide gels (SDS-PAGE) and blotted onto polyvinylidene fluoride (PVDF) transfer membranes. These membranes were then blocked using 5% non-fat milk, and were then incubated with relevant primary antibodies overnight at 4 °C. Afterwards, these membranes were washed twice with T-TBS followed by incubation with secondary antibodies (Jackson Immunoresearch, West Grove, PA) for 1 h at room temperature. After incubation, using an enhanced chemiluminescence detection kit (Amersham Pharmacia Biotech, Arlington Heights, IL, USA), the protein expression was visualized.Results and discussion
Effects of arsenite on the cell viability of HepG2 cells with or without heat treatment
Arsenite has already been proved as an effective anticancer agent against some solid cancers, e.g., gastric, colorectal, pancreatic carcinoma in vitro and in vivo.7,8 Moreover, its occurrence in the liver during metabolism implies that HCC cells are more likely to be sensitive following As2O3 treatment.9,10 Additionally, the properties of arsenic binding to proteins in cells also influence the cytotoxicity.20 The chemical structure is the determinant for the cytotoxic effects of arsenic; in particular trivalent arsenic compounds have a higher binding affinity to the sulfhydryl groups of proteins or enzymes and inhibit those enzymes, such as glutathione reductase, glutathione peroxidases, thioredoxin peroxidase, and thioredoxin reductase, resulting in induction of the cytotoxicity.21Taken together all these advantages of arsenic, we assessed the effects of iAsIII on the viability of human hepatocellular carcinoma HepG2 cells, as shown in Fig. 1A. However, the HepG2 cells have shown much resistance to iAsIII, and more than 56% of cells were still alive after exposure to iAsIII at higher concentrations (100 μM), implying that iAsIII have less effects on liver cancer cells. Due to the limited effects of iAsIII on the HepG2 cells, we further attempted to apply heat treatment (e.g., 43 °C) to increase the cytotoxic effect of iAsIII, because heat treatment has been reported to induce or accelerate apoptosis in cancer cells. Unexpectedly, there were no significant cytotoxic effects of iAsIII being increased in the HepG2 cells following heat treatment (Fig. 1A).
 | ||
| Fig. 1 Effects of arsenicals on the cell viability of HepG2 cells with or without heat treatment. (A) HepG2 cells were exposed to different concentrations of iAsIII (i.e., 5, 10, 20 and 100 μM) for 24 h (left), or plus two hours of heat treatment at 43 °C (right). Structure of the organic arsenic compound phenylarsine oxide (PAO) presented in this study (B). HepG2 cells were exposed to PAO at different concentrations (1–5 μM) for 24 or 72 h (C). Cell viabilities were determined by MTT assay (C). All data are expressed as mean ± standard deviation (n = 5). | ||
The appearance of excess arsenic transporters (i.e., ABCB1, ABCC1) and p-glycoprotein in liver cells along with acquired resistance might be the possible reasons. It has already been reported that overexpression of human MRP1 decreases the cytotoxicity of iAsIII, AsV and MMAIII in cultured cells; alternatively in Mrp2-deficient rats, accumulation of arsenic increased in the liver, kidney, red blood cells, and plasma, signifying that enterohepatic cycling is self-protection against arsenic, especially to the liver.22
Assessment of the cytotoxic effect of phenylarsine oxide (PAO) on the cell viability of HepG2 cells
Based on the above findings, we observed that HepG2 cells were very resistant to inorganic arsenic as well as to heat shock treatments. Alternative arsenic compounds such as PAO have the ability to overcome these limitations and may be a potential solution for the treatment of therapy-resistant HCC. PAO is a potent trivalent organic arsenic compound that is more cytotoxic than iAsIII, and has anticancer effects on hematological malignancies.15,23–25 The chemical structure of phenylarsine oxide (PAO) is shown in Fig. 1B. However, its anticancer effects on liver cancer cells (e.g., HepG2) have not yet been analyzed. Therefore, we attempted to examine whether PAO has an anticancer effect on cell survival of HepG2 cells.In this manner, the HepG2 cells were exposed to various concentrations of PAO for 24 or 48 h respectively (i.e., in a dose- and time-dependent manner) and the cell viability was determined (Fig. 1C). We found that PAO (at low doses 0.5–1 μM for 24 h) is significantly more cytotoxic than As2O3 with regard to the cell viability, implying that PAO may have potent anticancer effects against liver cancer cells when compared with iAsIII.
Nuclear morphological changes and apoptosis in HCC cells by exposure to PAO
The efficacy of PAO on cell viability makes us enthusiastic to examine the apoptotic effect on HepG2 cells, because induction of apoptosis is a common mechanism for the regulation of tumor growth by chemotherapeutics.26,27 In general, apoptosis is assessed using changes in cell morphology, flow cytometry with Annexin-V staining or Western blot analysis for apoptosis-related proteins (e.g., PARP, caspases etc.).28,29The nuclear morphological changes were assessed by DAPI staining. DAPI permeates the plasma membrane which results in blue chromatin viable cells that display a normal nuclear size and blue fluorescence in the control. Whereas apoptotic-like morphological changes with nuclear condensation were clearly observed in HepG2 cells after PAO treatment, as shown in Fig. 2A. To further assess the effect of PAO on apoptosis, HepG2 cells were treated with PAO, and an Annexin V-FITC assay was performed. Likewise, the increased number of annexin-V-positive cells after PAO treatment further points out the PAO-induced apoptosis of the HepG2 cells (Fig. 2B). Additionally, the apoptotic related proteins such as cleaved PARP and caspase-3 and -9 were significantly increased after being exposed to PAO in a dose- and time-dependent manner (Fig. 2C), suggesting that the apoptosis induced by PAO is associated with such apoptosis cascade mechanisms.
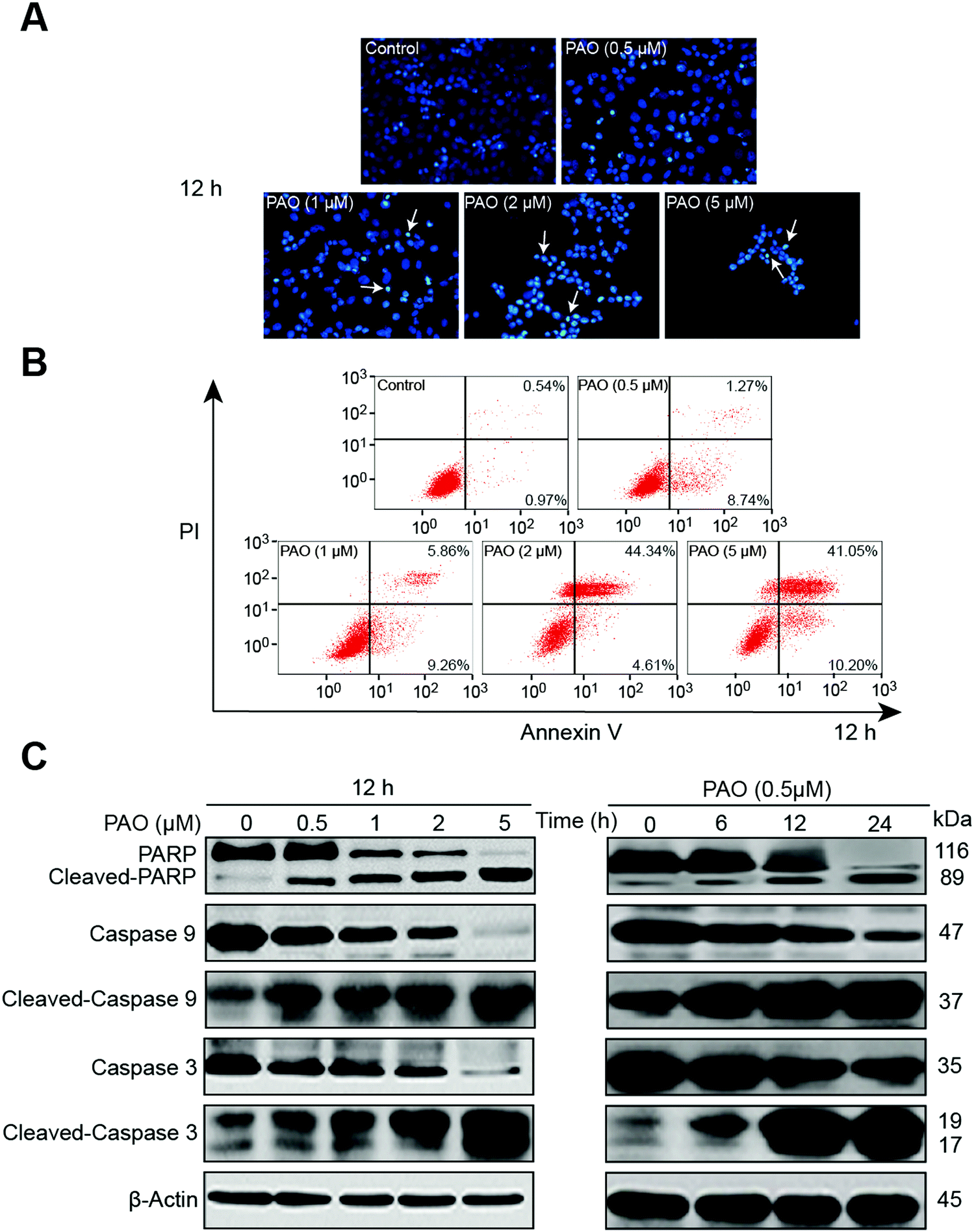 | ||
| Fig. 2 Determination of apoptosis in HepG2 cells after exposure to PAO. Nuclear morphological changes (A) and apoptosis (B) were examined by Wright–Giemsa and flow cytometry respectively after exposure to PAO in a dose-dependent manner. Blue color indicates the cell nucleus, while the arrows indicate chromatin condensation. Apoptotic related proteins poly(ADP-ribose) polymerase (PARP), caspase-3 and -9 and the corresponding cleaved proteins were examined in a dose- and time-dependent manner after exposure to PAO (C). β-Actin was used as a loading control. | ||
Changes in mitochondrial membrane potential and pro- or anti-apoptotic protein expression in HepG2 cells by PAO treatment
Here, we hypothesized that PAO induced cell death may be associated with ROS generation in specific cell organelles. It is well known that the mitochondria play a pivotal role in the signal transduction of apoptosis.30,31 Thus, firstly the changes in mitochondrial membrane potential (ΔΨm) were examined. PAO induced the mitochondrial membrane potential (ΔΨm) loss and increased the pro-apoptotic Bax protein (Fig. 3A), while anti-apoptotic Bcl-2 protein levels were significantly reduced (Fig. 3B), indicating that PAO has a potential effect on mitochondrial dysfunction. | ||
| Fig. 3 Changes in mitochondrial membrane potential, pro- and anti-apoptotic protein expression and ROS generation in HepG2 cells upon PAO treatment. The cells were exposed to PAO at different concentrations (0, 0.5, 1, 2 and 5 μM) for 6 h. After PAO treatment, the mitochondrial membrane potential (ΔΨm) was determined by using the fluorescent dye JC-1 (A). Changes in pro- and anti-apoptotic proteins Bax and Bcl-2 in mitochondrial or cytoplasmic fractions were determined by western blotting (B). ROS generation was examined in PAO-treated HepG2 cells by incubation with 2 μM CM-H2DCFDA (green) and MitoTracker (red) (C). β-Actin was used as a loading control. | ||
Depending on the above findings, we put forward that PAO induced cell death may be associated with ROS generation.32 In general, PAO initiates ROS generation, namely, by NADPH oxidase activation and disruption of the mitochondrial membrane potential. The mitochondrial membrane permeability changes are accelerated by thiol crosslinking (Cys160 and Cys257) in ANT (mitochondrial adenine nucleotide translocase) and therefore lock ANT into an open configuration.21,33 Moreover, PAO activates the cytosolic subunits of the NADPH oxidase complex (e.g., MR-8/MR-14 complex) resulting in the translocation of NADPH oxidase to the membrane and the production of ROS.34 Therefore, in this study, we used MitoTracker and DCFH-DA to establish ROS generation and its localization in cells. As expected, ROS were found to be significantly generated in the mitochondria as shown in Fig. 3C.
Effect of antioxidant N-acetylcysteine on PAO-induced ROS generation and apoptosis in HepG2 cells
To further confirm the involvement of ROS in apoptosis, we determined the generation of ROS in HepG2 cells by exposure to PAO, and we found that ROS were significantly increased. Additionally, antioxidant N-acetylcysteine (2 mM) significantly reduced the PAO induced ROS, as shown in Fig. 4A. On the other hand, the changes in apoptosis-related proteins such as cleaved PARP and caspase-9 were also determined after exposure to PAO with or without NAC treatment in HepG2 cells (Fig. 4B). More intriguingly, NAC extensively prevented the induction of cleaved PARP and caspase-9, explaining our observation that ROS may be the main cause for the induction of apoptosis in HepG2 cells.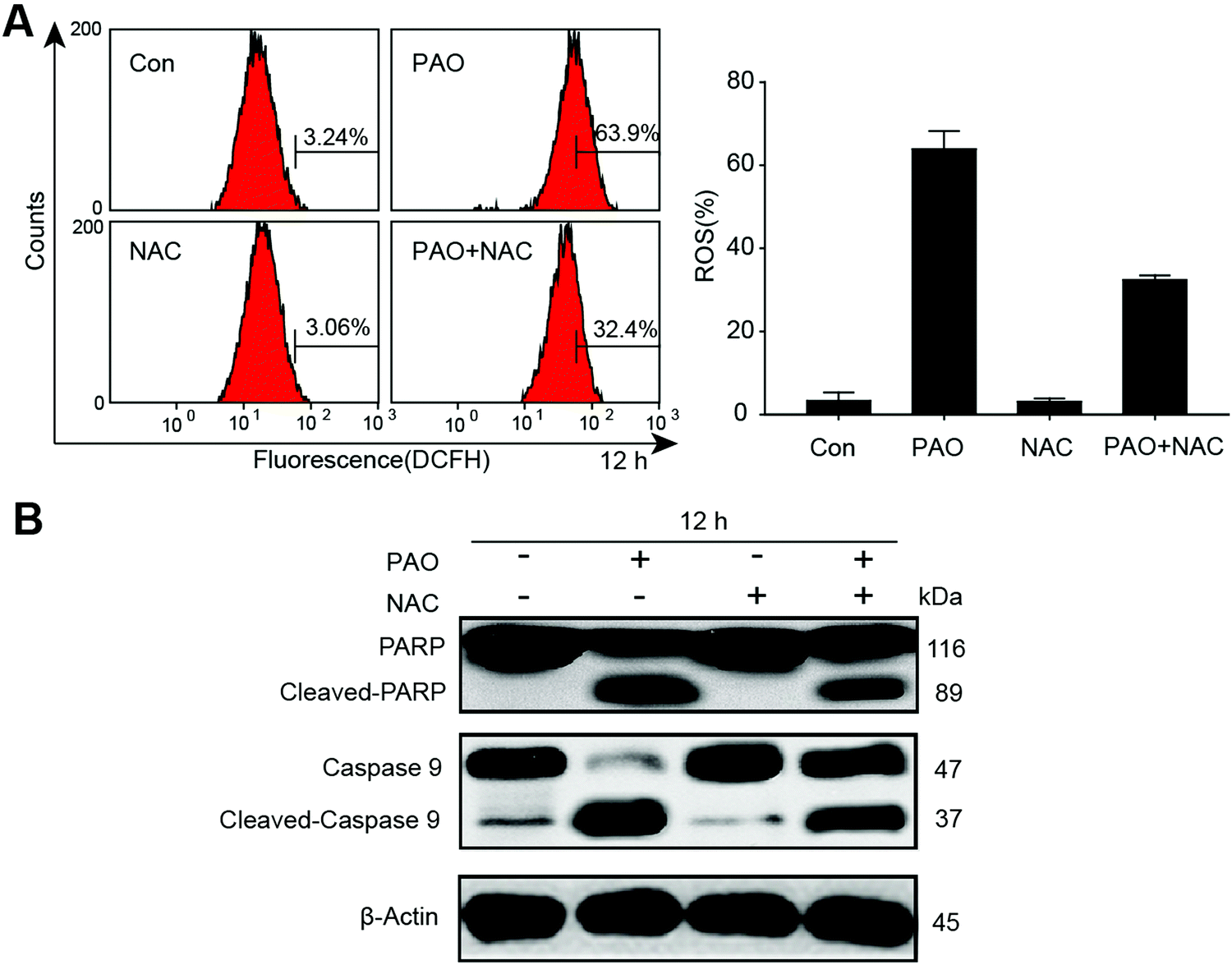 | ||
| Fig. 4 Ameliorating effect of antioxidant N-acetylcysteine (NAC) on PAO-induced ROS generation and apoptosis in HepG2 cells. The cells were treated with 1 μM of PAO with or without antioxidant NAC (2 mM) to determine ROS generation (left) and quantified (right) by flow cytometry (A). Changes in cleaved poly(ADP-ribose) polymerase (PARP) and caspase-9 proteins in HepG2 cells were determined by western blotting after exposure to PAO with or without NAC treatment (B). | ||
Determination of ER-stress in HepG2 cells after exposure to PAO
We hypothesize that apoptosis can also be induced by an ER-dependent signaling pathway following treatment with PAO in HepG2 cells. Therefore, cells were exposed to PAO at a concentration of 1 μM for the indicated time points (i.e., 0, 6, 12 and 24 h) to assess the changes in ER-stress related marker proteins, e.g., p-PERK, ATF4, p-eif2a and Chop, as shown in Fig. 5A. The phosphorylation of PERK can be induced for 6 h, thus decreasing with increasing exposure time up to 24 h (Fig. 5A). Correspondingly, the downstream proteins such as ATF4, phosphorylated eiF2α and Chop were activated significantly, indicating that the PAO is able to induce ER-stress in HepG2 cells. It has already been reported that ROS regulated ER-stress in many different cancer cells (e.g., gastric and lung cancers).35–37 However, it remains unknown whether the ER-stress resulted from the generation of ROS following PAO treatment. Therefore, we try to confirm the involvement of ROS in ER stress after exposure to PAO, and we found that ROS could also be generated in the ER as well as in the mitochondria. Similarly, NAC was used to examine the relationship between the ER-stress and generation of ROS. Interestingly, NAC could inhibit the induction of ER-stress as well as apoptosis, suggesting that the generation of ROS results in inducing cell death after exposure to PAO (Fig. 5B and C). | ||
| Fig. 5 Assessment of ER-stress in HepG2 cells after exposure to PAO. The cells were treated with 1 μM of PAO at different time periods (i.e., 0, 6, 12 and 24 h) to determine the changes in p-PERK, ATF4, p-eif2a and Chop (A) in the absence or presence of NAC (B). Location of ROS generation was further examined by using a confocal microscope in PAO-treated HepG2 cells stained with ER-tracker red (C). | ||
Conclusions
We for the first time showed that the arsenic compound PAO has potent cytotoxic effects compared to As2O3 in inducing apoptosis of HepG2 cells, predominately by ROS-dependent mitochondrial and ER-stress signaling pathways. This study suggests that PAO might offer a novel therapeutic approach for HCC, especially in the case of As2O3-resistant ones.Statement of contributions
Ping Huang and Yu Hua Zhang were responsible for conceiving and designing the experiments. Hong Zhang, Xiao Wei Zheng and Yu Jia Liu took part in performing the experiments. Luo Fang, Yi Wen Zhang, and Chang Yang gave general support for interpretation of the results. Khairul Islam and Chao Wang were involved in manuscript preparation. Hua Naranmandura took the overall responsibility in hypothesis generation, study design and manuscript preparation. All authors read and approved this manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors wish to acknowledge the National Natural Science Foundation of China (81473289 and 81673521ZJ); the Department of Science and Technology of Zhejiang Province (2015C33154 and 2017C33164); the Key Project of Traditional Chinese Medicine Science and Technology of Zhejiang Province (2015ZZ006); Zhejiang Provincial & National Health and Family Planning Commission Project for Medical Sciences (WKJ-ZJ-1504 and WKJ-ZJ-1512), and the Zhejiang Cancer Hospital Program for the Cultivation of 1022 Talents.References
- M. C. Yu, J. M. Yuan and S. C. Lu, Alcohol, cofactors and the genetics of hepatocellular carcinoma, J. Gastroenterol. Hepatol., 2008, 23, S92–S97 CrossRef CAS PubMed.
- R. X. Zhu, W. K. Seto, C. L. Lai and M. F. Yuen, Epidemiology of hepatocellular carcinoma in the Asia-Pacific region, Gut Liver, 2016, 10, 332–339 Search PubMed.
- K. W. Chen, T. M. Ou, C. W. Hsu, C. T. Horng, C. C. Lee, Y. Y. Tsai, C. C. Tsai, Y. S. Liou, C. C. Yang, C. W. Hsueh and W. H. Kuo, Current systemic treatment of hepatocellular carcinoma: A review of the literature, World. J. Hepatol., 2015, 7, 1412–1420 CrossRef PubMed.
- H. Liu, Z. Zhang, X. Chi, Z. Zhao, D. Huang, J. Jin and J. Gao, Arsenite-loaded nanoparticles inhibit PARP-1 to overcome multidrug resistance in hepatocellular carcinoma cells, Sci. Rep., 2016, 6, 31009 CrossRef CAS PubMed.
- V. Mathews, E. Chendamarai, B. George, A. Viswabandya and A. Srivastava, Treatment of Acute Promyelocytic Leukemia with Single-Agent Arsenic Trioxide, Mediterr. J. Hematol. Infect. Dis., 2011, 3, e2011056 CrossRef PubMed.
- B. Chen, Q. Liu, A. Popowich, S. Shen, X. Yan, Q. Zhang, X. F. Li, M. Weinfeld, W. R. Cullen and X. C. Le, Therapeutic and analytical applications of arsenic binding to proteins, Metallomics, 2015, 7, 39–55 RSC.
- Q. S. Shao, Z. Y. Ye, Z. Q. Ling and J. J. Ke, Cell cycle arrest and apoptotic cell death in cultured human gastric carcinoma cells mediated by arsenic trioxide, World J. Gastroenterol., 2005, 11, 3451–3456 CrossRef CAS PubMed.
- P. R. Subbarayan, M. Lima and B. Ardalan, Arsenic trioxide/ascorbic acid therapy in patients with refractory metastatic colorectal carcinoma: a clinical experience, Acta Oncol., 2007, 46, 557–561 CrossRef CAS PubMed.
- A. P. Geubel, M. C. Mairlot, J. P. Buchet and R. Lauwerys, Abnormal methylation capacity in human liver cirrhosis, Int. J. Clin. Pharmacol. Res., 1988, 8, 117–122 CAS.
- E. Marafante, M. Vahter and J. Envall, The role of the methylation in the detoxication of arsenate in the rabbit, Chem.-Biol. Interact., 1985, 56, 225–238 CrossRef CAS PubMed.
- C. C. Lin, C. Hsu, C. H. Hsu, W. L. Hsu, A. L. Cheng and C. H. Yang, Arsenic trioxide in patients with hepatocellular carcinoma: a phase II trial, Invest. New Drugs, 2007, 25, 77–84 CrossRef CAS PubMed.
- T. Zheng, D. Yin, Z. Lu, J. Wang, Y. Li, X. Chen, Y. Liang, X. Song, S. Qi, B. Sun, C. Xie, X. Meng, S. Pan, J. Liu, H. Jiang and L. Liu, Nutlin-3 overcomes arsenic trioxide resistance and tumor metastasis mediated by mutant p53 in Hepatocellular Carcinoma, Mol. Cancer, 2014, 13, 133 CrossRef PubMed.
- X. Chen, M. Zhang and L. X. Liu, The overexpression of multidrug resistance-associated proteins and gankyrin contribute to arsenic trioxide resistance in liver and gastric cancer cells, Oncol. Rep., 2009, 22, 73–80 CrossRef CAS PubMed.
- X. Y. Chen, X. L. Hu, C. F. Xia, C. Q. Qin and Y. Liu, Antibacterial evaluation of novel organoarsenic compounds by the microcalorimetric method, Biol. Trace Elem. Res., 2013, 153, 382–389 CrossRef CAS PubMed.
- Z. Estrov, S. K. Manna, D. Harris, Q. Van, E. H. Estey, H. M. Kantarjian, M. Talpaz and B. B. Aggarwal, Phnylarsine oxide blocks interleukin-1beta-induced activation of the nuclear transcription factor NF-kappaB, inhibits proliferation, and induces apoptosis of acute myelogenous leukemia cells, Blood, 1999, 94, 2844–2853 CAS.
- Y. Zheng, H. Yamaguchi, C. Tian, M. W. Lee, H. Tang, H. G. Wang and Q. Chen, Arsenic trioxide (As(2)O(3)) induces apoptosis through activation of Bax in hematopoietic cells, Oncogene, 2005, 24, 3339–3347 CrossRef CAS PubMed.
- J. Scott, A. Opejin, A. Tidball, N. Stehouwer, J. Rekman and L. L. Louters, Dual action of phenylarsine oxide on the glucose transport activity of GLUT1, Chem.-Biol. Interact., 2009, 182, 199–203 CrossRef CAS PubMed.
- B. Ni, Q. Ma, B. Li, L. Zhao, Y. Liu, Y. Zhu and Q. Chen, Phenylarsine Oxide Induces Apoptosis in Bax- and Bak-Deficient Cells through Upregulation of Bim, Clin. Cancer Res., 2012, 18, 140–151 CrossRef CAS PubMed.
- Y. H. Jiang, Y. J. Chen, C. Wang, Y. F. Lan, C. Yang, Q. Q. Wang, L. Hussain, Y. Maimaitiying, K. Islam and H. Naranmandura, Phenylarsine Oxide Can Induce the Arsenite-Resistance Mutant PML Protein Solubility Changes, Int. J. Mol. Sci., 2017, 25, 18 Search PubMed.
- S. Hirano and Y. Kobayashi, Cytotoxic effects of S-(dimethylarsino)-glutathione: a putative intermediate metabolite of inorganic arsenicals, Toxicology, 2006, 227, 45–52 CrossRef CAS PubMed.
- S. Shengwen, X. F. Li, W. R. Cullen, M. Weinfeld and X. C. Le, Arsenic Binding to Proteins, Chem. Rev., 2013, 113, 7769–7792 CrossRef PubMed.
- B. A. Roggenbeck, M. Banerjee and E. M. Leslie, Cellular arsenic transport pathways in mammals, J. Environ. Sci., 2016, 49, 38–58 CrossRef PubMed.
- B. Moe, H. Peng, X. Lu, B. Chen, L. W. Chen, S. Gabos, X. F. Li and X. C. Le, Comparative cytotoxicity of fourteen trivalent and pentavalent arsenic species determined using real-time cell sensing, J. Environ. Sci., 2016, 49, 113–124 CrossRef PubMed.
- C. Oetken, M. von Willebrand, M. Autero, T. Tutu, L. Andersson and T. Mustelin, Phenylarsine oxide augments tyrosine phosphorylation in hematopoietic cells, Eur. J. Haematol., 1992, 49, 208 CrossRef CAS PubMed.
- C. Oetken, M. von Willebrand, A. Marie-Cardine, T. Pessa-Morikawa, A. Stahls, S. Fisher and T. Mustelin, Induction of hyperphosphorylation and activation of the p56lck protein tyrosine kinase by phenylarsine oxide, a phosphotyrosine phosphatase inhibitor, Mol. Immunol., 1994, 31, 1295 CrossRef CAS PubMed.
- W. Lu, Y. Li, S. Men, J. Y Guo, J. Zhang and W. Zhang, miRNA-296-3p modulates chemosensitivity of lung cancer cells by targeting CX3CR1, Am. J. Transl. Res., 2016, 8, 1848–1856 Search PubMed.
- J. Han, Z. Liu, N. Wang and W. Pan, MicroRNA-874 inhibits growth, induces apoptosis and reverses chemoresistance in colorectal cancer by targeting X-linked inhibitor of apoptosis protein, Oncol. Rep., 2016, 36, 542–550 CAS.
- K. H. Ryu, S. Y. Woo, M. Y. Lee, Y. J. Jung, E. S. Yoo, J. Y. Seoh, J. H. Kie, H. Y. Shin and H. S. Ahn, Morphological and biochemical Changes induced by arsenic trioxide in Neuroblastoma cell lines, Pediatr. Hematol. Oncol., 2005, 22, 609–621 CrossRef CAS PubMed.
- X. Li, X. Ding and T. E. Adrian, Arsenic trioxide causes redistribution of cell cycle, caspase activation, and GADD expression in human colonic, breast, and pancreatic cancer cells, Cancer Invest., 2004, 22, 389–400 CrossRef CAS PubMed.
- S. W. Tait and D. R. Green, Mitochondria and cell death: outer membrane permeabilization and beyond, Nat. Rev. Mol. Cell Biol., 2010, 11, 621–632 CrossRef CAS PubMed.
- S. A. Susin, H. K. Lorenzo, N. Zamzami, I. Marzo, B. E. Snow, G. M. Brothers, J. Mangion, E. Jacotot, P. Costantini, M. Loeffler, N. Larochette, D. R. Goodlett, R. Aebersold, D. P. Siderovski, J. M. Penninger and G. Kroemer, Molecular characterization of mitochondrial apoptosis-inducing factor, Nature, 1999, 4, 441–446 Search PubMed.
- Y. X. Zhang, P. F. Yu, Z. M. Gao, J. Yuan and Z. Zhang, Caffeic acid n-butyl ester-triggered necrosis-like cell death in lung cancer cell line A549 is prompted by ROS mediated alterations in mitochondrial membrane potential, Eur. Rev. Med. Pharmacol. Sci., 2017, 21, 1665–1671 Search PubMed.
- A. J. Kowaltowski, A. E. Vercesi and R. F. Castilho, Mitochondrial membrane protein thiol reactivity with N-ethylmaleimide or mersalyl is modified by Ca2+: correlation with mitochondrial permeability transition, Biochim. Biophys. Acta, 1997, 1318, 395–402 CrossRef CAS.
- J. Doussière, F. Bouzidi and P. V. Vignais, Phenylarsine oxide-binding protein of neutrophil cytosol, which belongs to the S100 family, potentiates NADPH oxidase activation, Biochem. Biophys. Res. Commun., 2001, 285, 1317 CrossRef PubMed.
- X. Huang, L. Li, L. Zhang, Z. Zhang, X. Wang, X. Zhang, L. Hou and K. Wu, Crosstalk between endoplasmic reticulum stress and oxidative stress in apoptosis induced by α-tocopheryl succinate in human gastric carcinoma cells, Br. J. Nutr., 2013, 28, 727–735 CrossRef PubMed.
- J. Ma, J. Liu, C. Lu and D. Cai, Pachymic acid induces apoptosis via activating ROS-dependent JNK and ER stress pathways in lung cancer cells, Cancer Cell Int., 2015, 15, 78 CrossRef PubMed.
- P. Zou, M. Chen, J. Ji, W. Chen, X. Chen, S. Ying, J. Zhang, Z. Zhang, Z. Liu, S. Yang and G. Liang, Auranofin induces apoptosis by ROS-mediated ER stress and mitochondrial dysfunction and displayed synergistic lethality with piperlongumine in gastric cancer, Oncotarget, 2015, 6, 36505–36521 Search PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |