
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recent structural insights into the function of copper nitrite reductases
Sam
Horrell
,
Demet
Kekilli
,
Richard W.
Strange
 and
Michael A.
Hough
*
and
Michael A.
Hough
*
School of Biological Sciences, University of Essex, Wivenhoe Park, Colchester, CO4 3SQ, UK. E-mail: mahough@essex.ac.uk; Tel: +44 1206 873317
First published on 28th June 2017
Abstract
Copper nitrite reductases (CuNiR) carry out the first committed step of the denitrification pathway of the global nitrogen cycle, the reduction of nitrite (NO2−) to nitric oxide (NO). As such, they are of major agronomic and environmental importance. CuNiRs occur primarily in denitrifying soil bacteria which carry out the overall reduction of nitrate to dinitrogen. In this article, we review the insights gained into copper nitrite reductase (CuNiR) function from three dimensional structures. We particularly focus on developments over the last decade, including insights from serial femtosecond crystallography using X-ray free electron lasers (XFELs) and from the recently discovered 3-domain CuNiRs.
Introduction
Copper nitrite reductases (CuNiRs) are key enzymes in the nitrogen cycle, occurring in a wide range of denitrifying bacteria and fungi.1 The denitrification pathway in these organisms couples ATP synthesis with the reduction of nitrate (NO3−) or nitrite (NO2−) to dinitrogen (N2), via nitric oxide (NO) and nitrous oxide (N2O) intermediates.2,3 This pathway is critical in the return of fixed nitrogen to the atmosphere and in controlling the level of biologically available nitrogen in the soil. The extensive use of nitrogen containing fertilisers from the Haber–Bosch process can cause major environmental problems if the denitrification pathway becomes overloaded.2 Moreover, the N2O intermediate in the denitrification pathway is a potent greenhouse gas.4 The roles of different metalloenzymes in denitrification and their environmental and agricultural impacts have been recently reviewed.5The first committed step (i.e. the first effectively irreversible reaction in the pathway that commits the organism to generate the final product) in denitrification is the reduction of nitrite to NO (eqn (1)) and this is carried out either by Fe-containing cytochrome cd1 nitrite reductases, penta- or octa-haem nitrite reductases or by CuNiRs, encoded by the nirk gene.
| NO2− + e− + 2H+ → NO + H2O | (1) |
Overall fold and oligomeric state of CuNiRs
The majority of CuNiRs are trimeric, with each ∼37 kDa monomer comprising two cupredoxin (β-sandwich) domains, Fig. 1A and B.6 Each of these domains has a similar architecture to that of monomeric cupredoxin proteins, such as azurin, pseudoazurin and rusticyanin, that act as electron donor partner proteins to CuNiRs.7 The C-terminal region of each cupredoxin-like monomer forms an extended loop that wraps around an adjacent monomer contributing to the stability of the trimer.6These well-characterised two-domain CuNiRs are assigned as classes I and II, depending on the colour arising from their type 1 Cu (T1Cu) centre, with class I being blue and class II being green. Class III has an additional T1Cu domain with a hexameric oligomer and is greenish-blue due to the presence of two Cu centres, one each of class I and II.8 This class III CuNiR is an example of a recently discovered group of extended (three-domain) CuNiRs,9 that exist with additional fused N- or C-terminal domains containing a T1Cu or cytochrome haem redox centre, respectively (Fig. 1).10 Regardless of such variations, the core structure of T1Cu and type 2 Cu (T2Cu) sites in the trimeric core is largely conserved. We will first discuss the structure and function of the trimeric CuNiRs before highlighting variations in the more recently characterised extended CuNiRs.
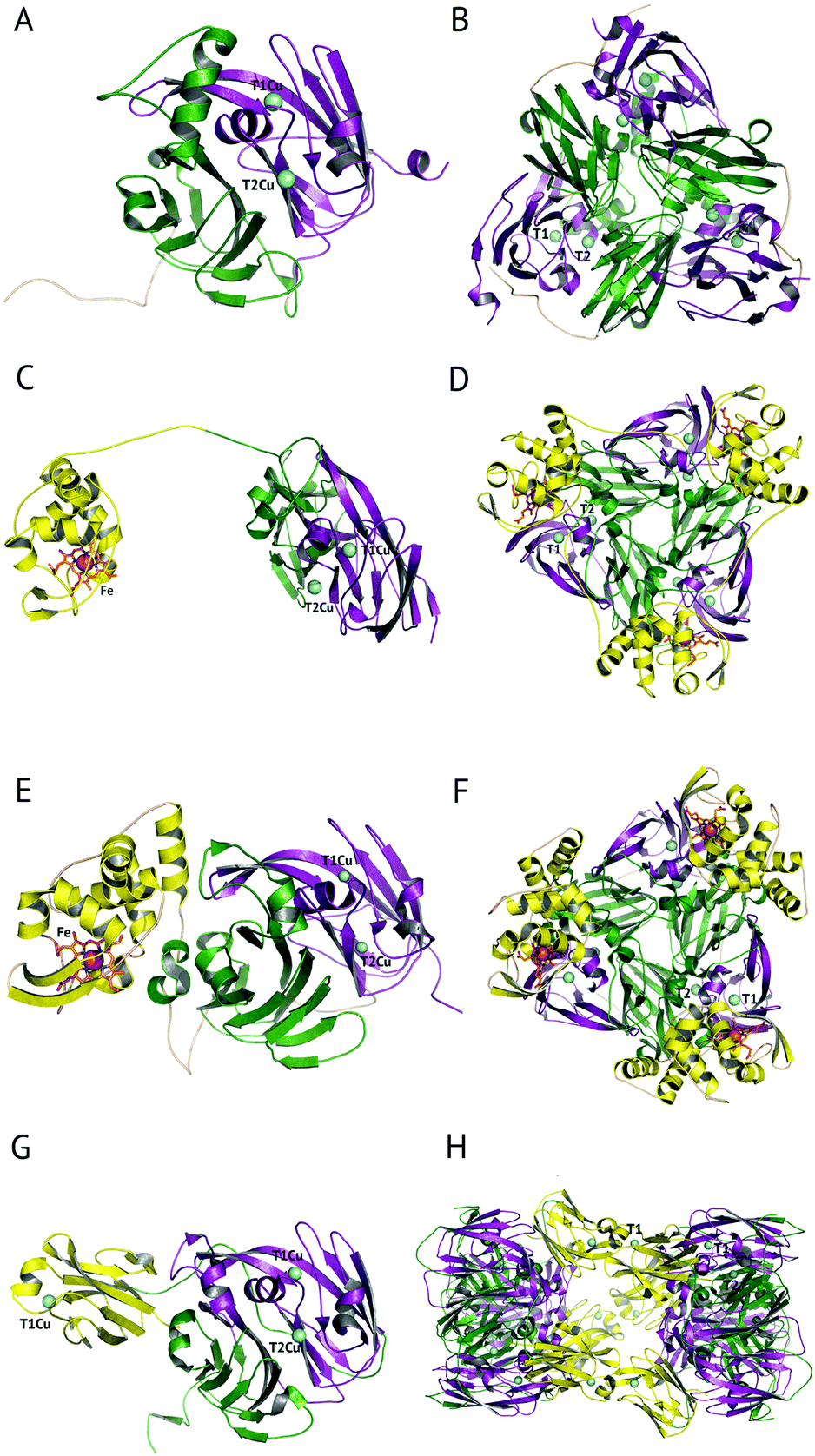 | ||
| Fig. 1 The domain and oligomeric structures of CuNiRs. In class I and II proteins, two cupredoxin domains are present per monomer, here coloured green and purple. Class III CuNiRs have an additional C- or N-terminal domain (yellow) containing T1Cu or haem. The two-domain proteins form a trimeric oligomer, shown in panels A and B. In the three-domain proteins from PhNiR and RpNiR the additional C-terminal cytochrome domain, panels C and E, forms trimeric oligomers, panels D and F, respectively. In the three-domain HdNiR an additional T1Cu N-terminal domain is present, panel G, and the biological unit appears to be a hexameric oligomer, panel H. | ||
The copper centres of CuNiRs in the resting state
All CuNiRs characterised to date contain an electron-providing T1Cu centre and a catalytic T2Cu centre. At the T1Cu site, the Cu is coordinated by two His, one Cys and an axial Met forming a tetrahedral geometry, Fig. 2.6 The structural changes within T1Cu sites between the Cu(II) and Cu(I) states are small (consistent with an entactic/rack state), and close to the level of precision at the typical resolutions of crystal structures.Atomic resolution structures provide more accuracy for bond length parameters but usually at the expense of higher X-ray dose which can readily reduce the T1Cu to the Cu(I) state. EXAFS studies suggest an increase in the Cu–Cys bond length on reduction of the blue copper protein azurin of some 0.07 Å.11
As the main chromophore of CuNiR, the T1Cu site is responsible for the colour of the protein in the cupric state. In general, T1Cu sites in copper proteins can vary widely in colour, including blue, green, red and purple.12,13 The relationship between T1Cu colour and geometry has been systematically studied. CuNiRs from different species appear blue, green or ‘bluish-green’ despite possessing an identical set of ligand residues. Insights into the relationship between T1Cu structure, colour and redox potential have been obtained by comparison of structures of blue and green CuNiRs and by mutagenesis of first and second coordination shell residues. Blue CuNiRs typically exhibit a long/weak Cu–Met bond ∼2.9 Å while in green CuNiRs this bond is much shorter.
Work from Solomon and co-workers revealed that in green CuNiRs, two spectral forms were present, with the ratio between these being strongly temperature dependent.14 At low temperatures, a green form with a strong Met–Cu interaction is dominant, while at higher temperatures, a blue form becomes more populated, with a weaker Cu–Met interaction. The T1Cu geometry, colour and redox potential are profoundly affected by mutation of ligand residues with consequent effects on enzyme activity.
The catalytic T2Cu is separated from the T1Cu by ∼12.6 Å and the centres are connected by a Cys–His bridge (for rapid electron transfer) comprised of the T1Cu ligand Cys and a T2Cu ligating His.6 The T2Cu has (His)3–H2O ligation in the resting state with a tetrahedral geometry, Fig. 2 and 3A. One of the three His ligands is provided from an adjacent monomer of the CuNiR trimer. Upon reduction (in the absence of substrate), the T2Cu loses the coordinated H2O resulting in Cu–(His)3 ligation.15 The T2Cu centre possesses only a weak UV-visible absorption spectrum that is obscured by the much stronger absorbance of the T1Cu centre.16 The T2Cu ligand binding pocket contains several conserved residues with proposed functional roles related to substrate access, specificity, activity, electron transfer & proton transfer.16 The Cu-coordinated water molecule is H-bonded to an aspartic acid residue (AspCAT) and, via a water bridge, to a conserved His (HisCAT).6 AspCAT has been proposed to play roles in substrate guidance/stabilisation and to act as a proton donor,17 while HisCAT acts as a second proton donor. Crystal structures show that AspCAT can adopt two different conformations, named ‘gatekeeper’ – pointed away from T2Cu and ‘proximal’ – oriented towards T2Cu and H-bonding to the bound water molecule in the resting state.18 Mutation of AspCAT to asparagine (Asn) in AxNiR eliminated catalytic activity providing strong evidence for its role in proton transfer.19 The properties of AspCAT and HisCAT may provide the rationale for the observed pH dependence of CuNiR activity. Solomon and coworkers have reported a pKa of 6.4 for AspCAT (in AxNiR) when NO2− is bound.20 Finally, an isoleucine (IleCAT) residue that lies above the T2Cu binding site has been shown by structural studies21 to profoundly influence substrate orientation and consequently the catalytic activity.
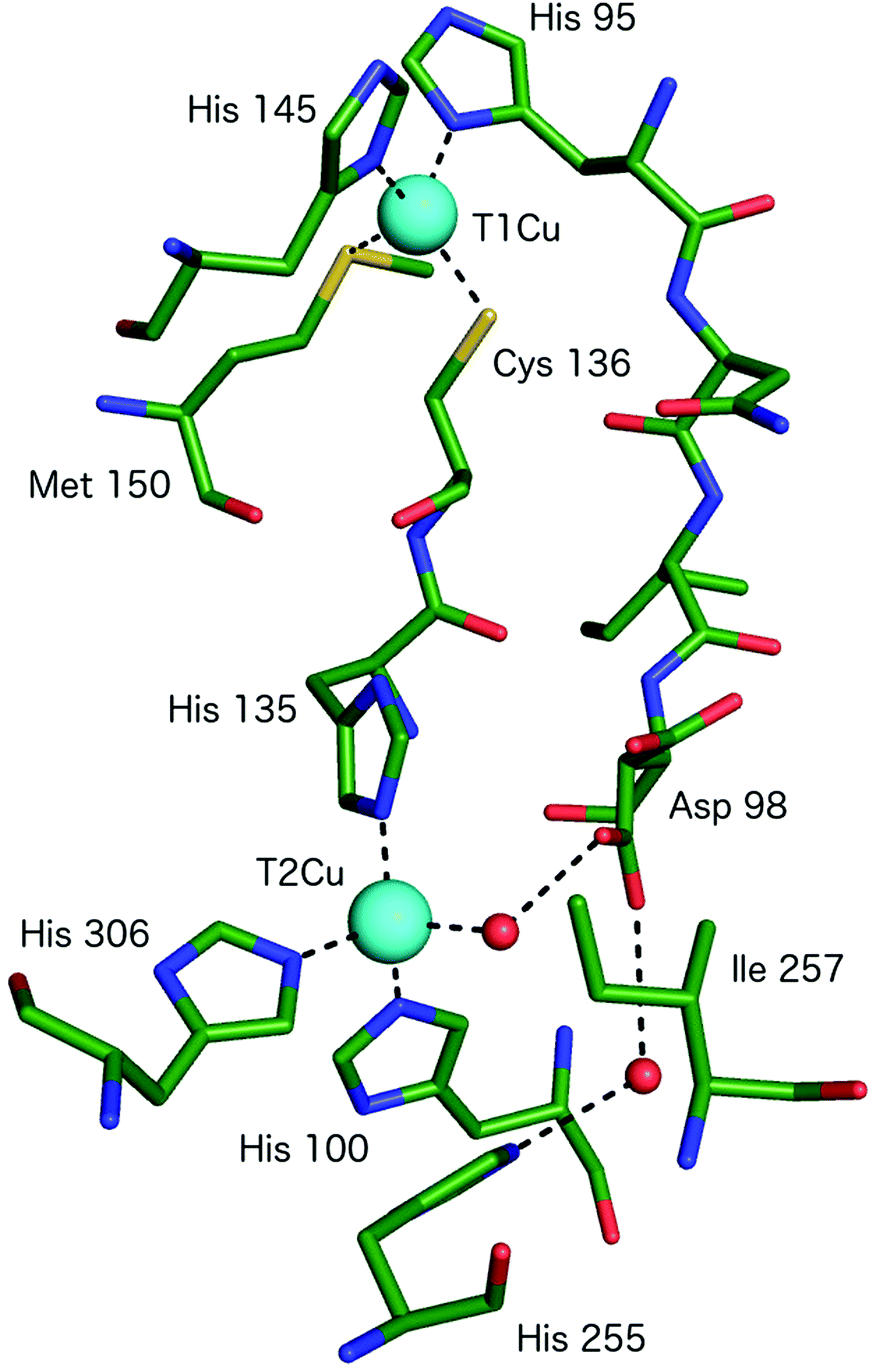 | ||
| Fig. 2 The T1Cu and T2Cu centres of AcNiR and the protein links between them. The T1Cu atom has (His)2CysMet ligation and is linked to the catalytic T2Cu centre by a Cys-His bridge. A second link is made between the T1Cu ligand His95 and T2Cu site residue Asp98, proposed to be a sensor loop, enabling intramolecular T1–T2 electron transfer. The T2Cu coordination is completed by three more His residues, one of which originates from an adjacent monomer of the trimer and a water molecule. Image generated from the structure of resting state AcNiR at 0.9 Å resolution (PDB 2BW4).17 | ||
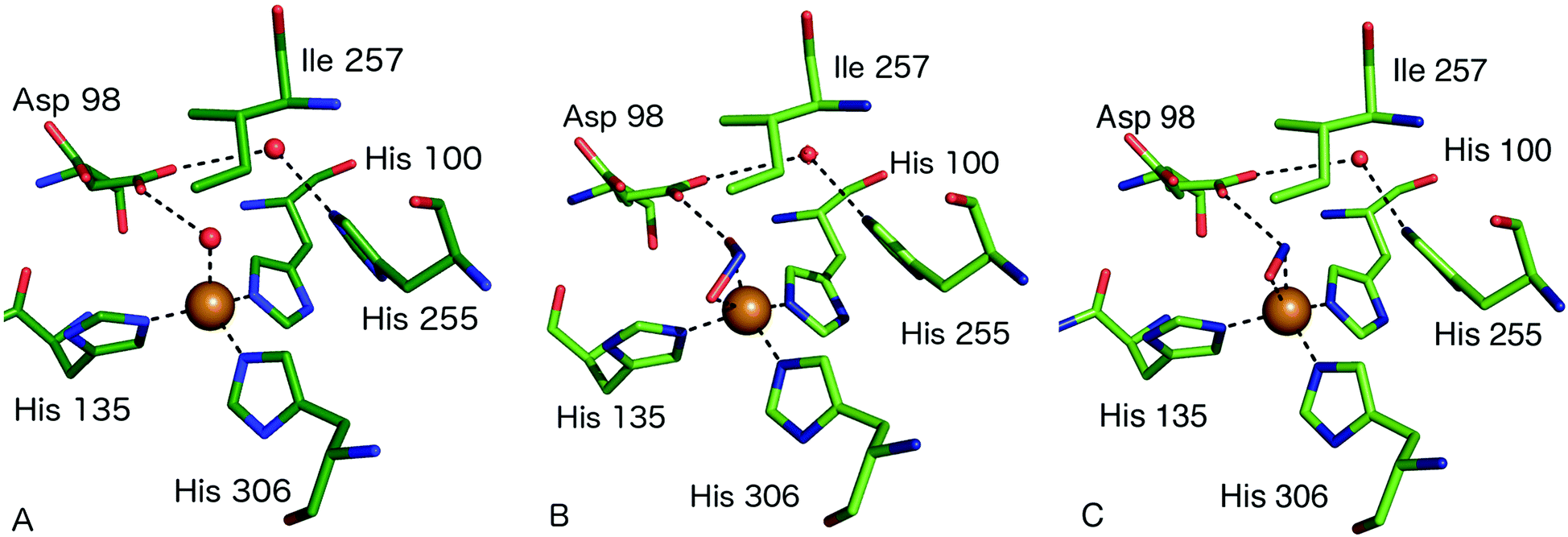 | ||
| Fig. 3 Type 2 Cu site structures (A) resting state AcNiR with a water molecule bound to the T2Cu (PDB 2BW4); (B) nitrite bound to the T2Cu site in a side-on bidentate O manner (PDB 5AKR); (C) the T2Cu site of AcNiR bound to NO at 1.22 Å resolution (PDB 5I6N). The NO ligand is bound in a side-on manner with near-equidistant Cu–N and Cu–O bonds. Residue Asp98 forms a H-bond to the N atom of NO and is oriented toward the proton delivery channel. Panels A and B are from the structures described in ref. 18 while panel C is from ref. 55. | ||
Enzymatic mechanism of nitrite reduction: structural insights
The consensus mechanism for two domain CuNiRs begins with the resting state enzyme, with a water bound to the T2Cu centre, Fig. 3A. On the basis of X-ray crystallography and EXAFS studies on native and site-selected AxNiR mutants, supported by kinetics measurements, a catalytic mechanism was initially proposed in which NO2− coordinates to the T2Cu(II), displacing the water molecule in the native enzyme, Fig. 3B. In the absence of nitrite at the T2Cu there is a limited potential for electron transfer (ET) from the T1Cu, but following nitrite binding ET from T1Cu(I) to the T2Cu site occurs,15,22,23 which reduces the T2Cu. Two protons required for the catalytic reaction are thought to be provided by residues His254 (HisCAT) and Asp97 (AspCAT) and the proton-coupled electron-transfer (PCET) from T1Cu to T2Cu, involving the HisCAT and AspCAT residues, is thought to be the rate-determining step.24,25 In this PCET process, the two protons are provided to the bound NO2− along with an ET from the reduced T2Cu, to yield the NO product, Fig. 3C. This product then dissociates from the T2Cu, allowing water to rebind to reform the oxidised resting T2Cu state, Fig. 3A. Structural and kinetic studies on a number of other CuNiRs gave apparently conflicting evidence on whether the intramolecular ET occurred before or after NO2− binding [e.g.ref. 15, 26 and 27]. These conflicts have been mostly reconciled by a kinetic scheme for catalytic turnover in which two alternative mechanistic pathways are possible, varying in the sequence of ET and binding of nitrite, depending on the pH and nitrite concentration.28 The catalytic mechanism at the T2Cu site of CuNiRs has also been extensively explored using theoretical methods based on the crystal structures, though as yet no clear consensus has been reached on the precise nature and order of the proposed PCET reaction.29–34Key features relevant to this mechanism that are informed by structures include the modes of substrate and product binding, the likely electron and proton transfer pathways and steps and the conserved water structure within the active site pocket.
Substrate binding to the T2Cu centre and specificity
The first structure of a nitrite-bound CuNiR complex adduct was determined in 1991.6 Since then, structures have been determined of NO2− and NO-bound complexes at a variety of resolutions and pH of native and mutant CuNiRs from different organisms.18,19,21–23,27,35–56 Nitrite has typically been observed to bind either in an asymmetric bidentate binding geometry with its O1 and O2 atoms ligating to T2Cu(II) or in a ‘side-on’ geometry with similar distances from T2Cu to the three atoms of nitrite. One of the highest resolution structures of a CuNiR–nitrite complex is that of AcNiR, to 1.10 Å resolution,18 where nitrite was observed with bidentate binding to T2Cu and dual conformations of Asp98CAT were present, with the proposal that the gatekeeper conformation of Asp98 (AspCAT) may be important for guiding NO2− to its binding site at the T2Cu. The proximal conformation, on the contrary, is significant for proton transfer, bond-cleavage and formation and release of the product, NO. The resting state has water bound to oxidised T2Cu and forms a H-bond to the negatively charged AspCAT residue. Nitrite is observed to bind to the T2Cu ion via a O-coordinate bidentate mode, displacing the bound water molecule and forming H-bonds between the Oδ2 atom of AspCAT and the O2 atom of nitrite. Proton transfer occurs from the AspCAT to the nitrite to form the intermediate T2Cu+NOOH on reduction of the T2Cu site. His255 (HisCAT) facilitates bond cleavage of the N–ON bond to form the product, NO, by supplying the second proton in the widely-accepted mechanism. The NO product dissociates and leaves the resting water-bound oxidised T2Cu ion. Structures have been determined of ascorbate-reduced and reduced nitrite-soaked AfNiR at 2.0 [PDB 1AQ8] and 1.85 Å [PDB 1AS8] resolutions, respectively, with nitrite bound to reduced T2Cu, revealing few structural changes relative to the oxidised form.41In the different structures of nitrite complexes, bidentate nitrite has been modelled in both ‘top-hat’ and ‘side-on’ positions. XFEL SFX54 data have revealed that the ‘top-hat’ mode is the binding geometry initially present in the T2Cu(II) form of the enzyme and that the side-on may be the result of T2Cu reduction. This highlights that great care must be taken in crystallographic studies of nitrite-bound CuNiRs due to the propensity for X-ray generated photoelectrons to initiate catalysis within the crystal, leading to the conversion of substrate to NO at the T2Cu site. Structures determined from CuNiR nitrite complexes using high X-ray doses in many cases reveal a significant population of the T2Cu-NO product.27 Most recently this phenomenon has been exploited to drive in crystallo reactions to generate ‘structural movies’ of enzyme catalysis in green AcNiR55,57 and thermostable Geobacillus thermodenitrificans (GtNiR).58
T2Cu site mutations in AfNiR significantly altered the binding mode of nitrite21 and these were related to changes in enzyme activity. Mutation of this IleCAT to Val or Leu led to asymmetric bidentate NO2− binding while mutation to Met, Thr, Gly or Ala resulted in monodentate coordination, allowing a water molecule to also bind to T2Cu in a position close to that usually occupied by the O2 atom of nitrite. Monodentate nitrite binding was associated with greatly reduced catalytic activity, pointing to a key role for IleCAT in promoting a nitrite binding geometry conducive during catalysis. Crystal structures were determined of the oxidised and chemically-reduced AspCAT to Asn mutant in AfNiR.35 In these structures, nitrite was bound in a side-on manner with an increased level of disorder, ascribed to a shift of Asn relative to Asp producing an unfavourable geometry for H-bonding with the bound protonated nitrite. However, this disorder could simply represent a mixture of top-hat and side-on nitrite molecules within the crystal. The HisCAT to Asn mutant of AfNiR showed no displacement of the water molecule and the nitrite is bound to the T2Cu in a monodentate manner via an O atom. Nitrite is oriented away from Ile257 and towards an Ala residue and the pocket H-bonding network is drastically altered in the H255N mutant, indicating an essential role for nitrite binding and function.35
A somewhat different binding mode for nitrite was observed in a structure of the C135A mutant (which eliminates T1Cu binding) of the thermophile GtNiR,52 a close homologue of GkNiR. The 1.90 Å structure showed nitrite bound in a monodentate manner via its O2 atom with this atom being positioned to form a H-bond to AspCAT. HisCAT adopted a rotated conformation likely due to the formation of a H-bond to a Gln residue rather than the Thr which interacts with HisCAT in other CuNiRs. Interestingly, in this enzyme, the gatekeeper conformation of AspCAT and the ‘vertical’ conformer of HisCAT are sterically restricted by the unusual position of a Phe residue near to the catalytic pocket which provides additional rigidity to the active site in this enzyme from a thermophile, Fig. 4A. Intriguingly, when the structure of the nitrite complex of GtNiR was determined at 320 K, the observed substrate binding mode was side-on, similar to that observed in other CuNiRs,53 a result ascribed to increased dynamic flexibility of the pocket at the higher temperature.
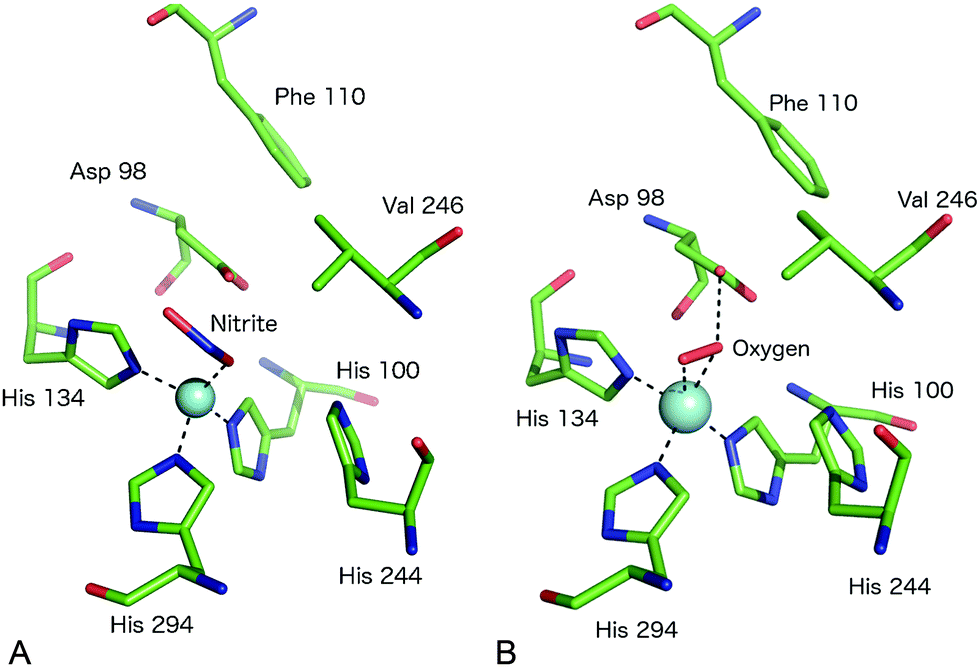 | ||
| Fig. 4 The T2Cu site environment of GtNiR. (A) The nitrite-complex (PDB code 3WKP),52 note the monodentate binding of nitrite via a single O atom, the substitution of IleCAT with a Val residue and the steric restriction of the gatekeeper AspCAT (Asp98) conformation (seen in other CuNiRs) by Phe110, resulting in only the proximal conformation being observed; (B) oxygen binding to GtNiR (PDB code 3WNJ).61 Note that partially occupied water molecules are omitted for clarity. Cu atoms are shown as cyan spheres. | ||
Substrate and copper access to the T2Cu site
The T2Cu site, as mentioned previously, makes up the active substrate-binding site and is coordinated by three His and one H2O/OH− ligand.38 In the resting state nitrite has access to the T2Cu centre via a channel some ∼12 Å deep from the protein surface and displaces the H2O ligand to initiate catalysis. Intriguingly, mutation of the surface-exposed Phe306 to a Cys residue some 12 Å away from T2Cu in AxNiR resulted in a change to the second coordination sphere of the Cu with consequent effects on the rate-limiting step of the enzyme reaction.59 In this F306C mutant, the substrate access channel is more open and contains an additional water bound to the T2Cu-coordinating water molecule. The specific activity was remarkably increased four-fold while the apparent nitrite affinity and the rate of intramolecular ET were lower than in native AxNiR. In this mutant, intramolecular ET became the rate-limiting step.Binding of small molecule ligands to the T2Cu centre
Native CuNiR can bind small molecules including acetate, nitrate, formate, azide and N2O, of which complex structures have all been determined crystallographically. The binding mode of such ligands60 is strongly influenced by the pocket residues IleCAT and AspCAT and these, together with the dimensions of the substrate access channel, act to prevent the binding of larger ligands. Monoatomic species such as chloride have also been shown to bind to T2Cu.42,54A recent study of GtNiR at 1.20 Å resolution61 revealed electron density consistent with the interpretation that an oxygen ligand was bound at the T2Cu site, in the absence of nitrite or NO, Fig. 4B. CuNiRs have the ability to catalyse the two-electron reduction of O2 to hydrogen peroxide (H2O2) but no structural data for this molecular oxygen ligand has previously been observed. In the GtNiR structure, the O2 ligand was modelled in a side-on manner with one short (2.1 Å) and one longer (2.4 Å) Cu–O bond length. Oxygen forms H-bonds with AspCAT and a water molecule and also makes van der Waals contacts with ValCAT. This latter hydrophobic interaction appears to be related to the side on binding mode as previously proposed for NO binding. Oxygen binding has not been observed in other CuNiRs and was proposed to be observable in GtNiR due to the presence of a Val residue in place of IleCAT, which creates an unusually open substrate binding pocket and/or the slower O2-reduction rate in the CuNiR from this species. Interestingly, the structure of a CuNiR from Nitrosomonas europaea50 revealed a much more restricted substrate channel than in other CuNiRs, proposed to be the basis of the unusual O2 tolerance of this enzyme enabling it to function in an aerobic environment. In addition, a second water channel was observed and assigned as a water exit channel in this enzyme.
It is a notable feature of CuNiRs that they possess a significant superoxide dismutase activity,15,62 for example AxNiR at a remarkable ∼56% activity compared to bovine Cu, Zn superoxide dismutase (CuZnSOD), one of the most efficient enzymes known. The structural similarity between the Cu(I) form of the T2Cu site and that of CuZnSOD has been noted from both EXAFS and crystallographic data.15 Interestingly, CuZnSOD does not display NiR activity.
Identification of intramolecular electron and proton transfer pathways
The CuNiR enzyme reaction requires that one electron and two protons are supplied to the catalytic T2Cu centre and it has been demonstrated that this occurs in a coupled (PCET) manner.25 Spectroscopic and mutational studies showed that electrons are transferred from physiological redox partner proteins such as azurin, pseudoazurin or cytochrome c551 to reduce the T1Cu centre. Intramolecular ET occurs between the Cu sites to deliver the electron to the T2Cu centre, in the process reoxidising the T1Cu. The pathway for intramolecular ET was identified as a Cys–His bridge between ligating residues of the two Cu centres, providing a facile through-bond pathway.T1Cu site mutations have been characterised revealing effects upon enzyme colour, redox potential and ET. A M150E mutation in AfNiR resulted in a protein binding Zn to the T1Cu site39 with a 1000-fold loss of activity, while a C130A mutation in AxNiR led to a vacant T1Cu site23 and abolition of activity.
Analysis of high resolution crystal structures led to proposals for proton channels connecting the protein surface (and bulk solvent) with the T2Cu site. A ‘primary’ proton channel in AxNiR linked the T2Cu site residue AspCAT with the surface via a well-ordered network of water molecules.42 A ‘secondary’ channel was later identified in a structure at high pH where residues Asn90, Asn107 and Ala131 together with water molecules also provide a H-bonding network to the surface.43 Supporting evidence was provided from activity and structural work on mutated enzymes. For example, a H254F mutation abolished the ‘primary’ channel yet retained near-native levels of activity. In contrast the N90S variant targeting the secondary channel lowered activity by 70%. These data led to the proposal that this secondary channel was in fact the most important route for protons.22,25 Similarly, the ET pathway has been investigated by structural and related studies on mutants. As well as the C130A AxNiR mutation described above, a H129V mutation in AxNiR resulted in an unprecedented bis-His T2Cu binding site63 while disrupting the Cys–His bridge leading to a complete loss of enzyme activity.
The structure of the T2Cu-NO species
All crystal structures of CuNiR–NO complexes have revealed a side-on binding mode to T2Cu, Fig. 3C.18,45,55 Structures have been determined of NO complexes by several approaches: (i) NO-soaking into AfNiR crystals,45 (ii) endogenously present NO, in crystals grown from protein isolated from Achromobacter cycloclastes cells grown under denitrifying conditions18 and (iii) produced from nitrite in situ by X-ray driven catalysis using ‘multiple-structure one crystal’ (MSOX) serial crystallography.55 The structures determined by each method consistently show side-on binding with near-equidistant bond lengths to Cu for the N and O atoms and reveal that bound NO interacts with active site residues. Several issues regarding the NO complex remain challenging to address from the electron density alone. The electron density for the N and O atoms is very similar and structures have been determined with different assignments of the density peaks modelled.18,45,55 Consideration of bonding further suggests that the NO molecule interacts with AspCAT which adopts its ‘proximal’ position in the product complex. Similarly, crystal structures are not yet able to give a definitive answer to the identification of the side-on NO complex as Cu(I)–NO˙ or Cu(II)–NO−.An outstanding mystery is the suggestion that solution spectroscopic data supports an end-on mode of NO binding [Cu–N–O] rather than side-on binding. In particular, ENDOR data64 of native and the I289V and I257A mutants of RsNiR suggests a Cu–N–O angle of 160° in native RsNiR with magnetic interaction of O with the Cδ1 atom of Ile257. Simulations of the complex show 3–10 kcal mol−1 energetic preference for the end-on form.65 A subsequent EPR study of model compound analogues found a clear dependence of gz upon the Cu–N–O angle.66 Taken together with the previous EPR data of RsNiR,64 this work suggested that a strongly bent (∼136°) Cu–N–O binding geometry is feasible in CuNiRs. One proposal from computational simulation was that Ile257 dictates the side-on binding mode observed in structures with the proposal that a small structural change when the protein is in solution would allow the end-on binding mode to be sterically feasible.67
Surface properties and intermolecular electron transfer
The electrostatic surfaces of CuNiRs are variable, with distinct differences between the blue and green classes of 2 domain enzymes. This, together with comparable analysis of the electrostatic surfaces of the partner proteins68 provides a rationale for the observed specificity of CuNiRs from different species for their cognate electron donors.68 Azurins reacted rapidly with blue CuNiRs but slowly with green CuNiRs. In contrast, pseudoazurin reacted effectively with either colour of CuNiR. Notably AfNiR (green) has a strong surface negative charge complementing the positive charge of the pseudoazurin complementary surface.68 Moreover, a common structural feature of CuNiRs is a hydrophobic patch on the enzyme surface near to the location of the T1Cu centre, which has been proposed to form part of the charged protein–protein interface in the ET complex.69,70Identifying the precise route(s) of intermolecular ET between the redox centre of the electron donor protein (T1Cu or haem Fe) and the CuNiR T1Cu centre is more challenging, in part due to the difficulty of obtaining crystal structures of transient ET complexes. Mutational analysis of putative pathway residues have provided insights. For example, a W138H mutation in AxNiR produced a large drop in activity with the physiological redox partner azurin but retained full activity with the artificial electron donor methyl viologen.36 This supported a role for Trp138 in intermolecular ET, consistent with a docking model of the azurin–AxNiR complex which positioned Tyr197 close to the azurin T1Cu ligand His117. Tyr197 is close to Trp138 which is adjacent in sequence to a T1Cu His of AxNiR.36
Structural characterisation of complexes between CuNiRs and redox partners
Structural characterisation of the ET complexes between CuNiRs and their partner proteins have remained elusive due to the transient nature of the complexes. Computational docking and mutagenesis have attempted to identify interface regions between CuNiRs and their specific azurin, pseudoazurin or c-type cytochrome partners e.g.67 Paramagnetic nuclear magnetic resonance (NMR) data have been used to characterise the interacting region in the complex between AfNiR and its partner pseudoazurin [Paz].71 In this case, distance restraints derived from NMR data of gadolinium added to engineered surface cysteines were used to restrain computational docking simulations. The resulting structural model positions the T1Cu atoms of Paz and AfNiR some 15.5 (±0.5) Å apart.One crystal structure available for such an ET complex is that between AxNiR and a functional partner protein, Cytc551, at 1.7 Å resolution49 (PDB 2ZON). This CuNiR has been shown to accept electrons either from azurin, psuedoazurin or a cytochrome electron donor. In the binary AxNiR–Cytc551 complex, the edge of the haem moiety lies some 10.5 Å from T1Cu in an arrangement likely to promote efficient intermolecular ET, Fig. 5A. The protein–protein interface involves the previously proposed hydrophobic patch surrounded by a semicircle of interface water molecules.49 Analysis of the structure suggested the main ET route to be from the haem CBC methyl carbon through space to Pro88 of the CuNiR. Through-bond ET then proceeds to the adjacent T1Cu ligand His89.
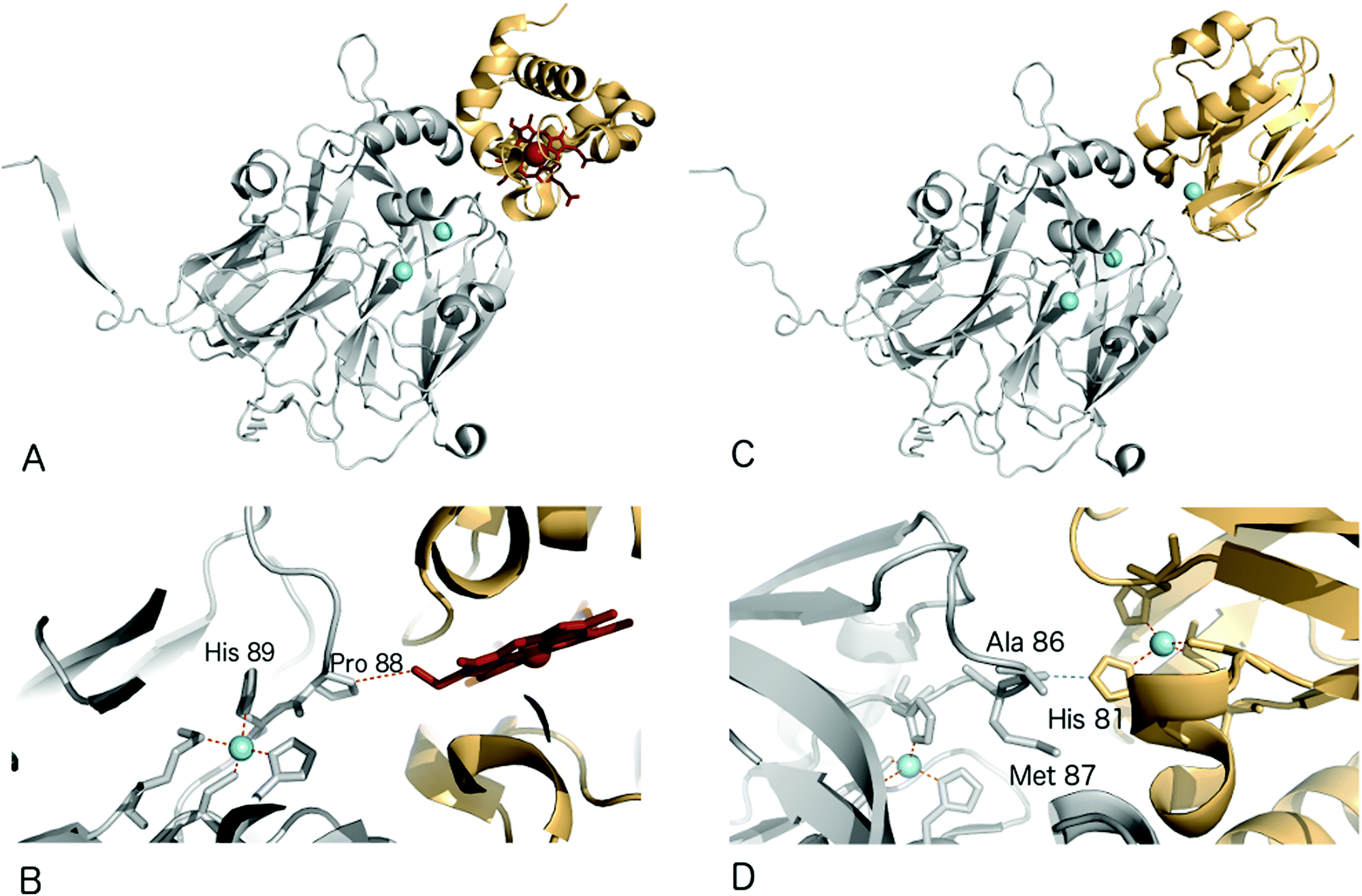 | ||
| Fig. 5 Structures of electron transfer complexes between CuNiRs and electron transfer proteins. Cu atoms are shown as cyan spheres, while the haem group of cyt. c551 is represented using red sticks (A) structure of the proposed electron transfer complex between AxNiR (gray) and cyt. c551 (orange); (B) the interface region of the complex shown in (a), note the close contact between Pro88 of AxNiR and the haem edge of cyt. c551. (C) Structure of the proposed electron transfer complex between AxNiR (gray) and pseudoazurin from H. denitrificans (PAz, orange); (D) interface region of the complex shown in (C) here note the close contact between Ala86 of AxNiR and the T1Cu ligand His81 in Paz. | ||
Recently, a heterogeneous complex between AxNiR and a pseudoazurin from H. denitrificans as the electron donor was solved at 3.0 Å resolution (PDB 5B1J). The intermolecular contact at the hydrophobic patches adjacent to the T1Cu sites positions the Cu centres of the two proteins to within ∼16 Å. The solvent-exposed His81 Cu-ligand of the pseudoazurin lies ∼2.6 Å from the Ala86 oxygen atom of AxNiR, an arrangement that would allow through-space ET between the proteins,72Fig. 5B.
3-Domain fused CuNiRs
A recent development has been the discovery of CuNiRs containing additional domains. The first example was the structure of the hexameric CuNiR from Hyphomicrobium denitrificans (HdNiR),73 determined at 2.2 Å resolution,74Fig. 1. HdNiR has high sequence homology to AniA, while the structure revealed a novel additional N-terminal cupredoxin domain, containing an additional T1Cu(T1CuN) centre. The extended enzyme forms a 340 kDa hexameric prism-shaped oligomer. Interestingly, the additional T1CuN cupredoxin domain has low sequence homology to the other cupredoxin domains of HdNiR but high homology with those of AfNiR. The separation of the T1CuN centres is 14 Å in a head to head arrangement, suggesting that ET proceeds by initial reduction of T1CuN by Cytc550 followed by ET between T1CuN and T1CuC, Fig. 6A. The additional cupredoxin domain in HdNiR may not be involved in the catalytic mechanism74 due to the large distance separating it from the T2Cu centre where catalysis occurs. Subsequent bioinformatics analysis led to the identification of other N- and C-terminal 3-domain CuNiRs.9 The C-terminally-extended enzymes possess an additional haem-cytochrome domain. A crystal structure of a C-terminally extended CuNiR from Pseudoalteromonas haloplanktis (PhNiR) was deposited in the Protein Data Bank in 2008 (PDB 2ZOO) but remains unpublished. The first detailed and published structural analysis of a C-terminally extended CuNiR, came from the atomic resolution (1.01 Å) structure of the 499 residue CuNiR from Ralstonia picketti (RpNiR) by Hasnain and co-workers.75 The 1.01 Å resolution structure of RpNiR75 revealed a domain binding motif that positioned the T1Cu and Fe atoms only 10.6 Å apart, notably closer than the separation of centres in the AxNiR–Cytc551 complex, Fig. 5 and 6. A series of water molecules were present between the cytochrome domain and the surface above the T1Cu site, suggesting a role in electronic coupling across the interface. This hypothesis was tested by structural and functional analysis of site-directed mutants which confirmed the roles of Met92 and Pro93 in interface ET. As well as the differences in domain structure, notable differences are apparent between RpNiR and the 2-domain NiRs in the vicinity of the catalytic T2Cu centre. In contrast to 2-domain NiRs where the T2Cu-bound water is H-bonded to AspCAT and HisCAT, in RpNiR this water instead forms an H-bond to AspCAT and a second water molecule. In PhNiR, the water is only H-bonded to AspCAT.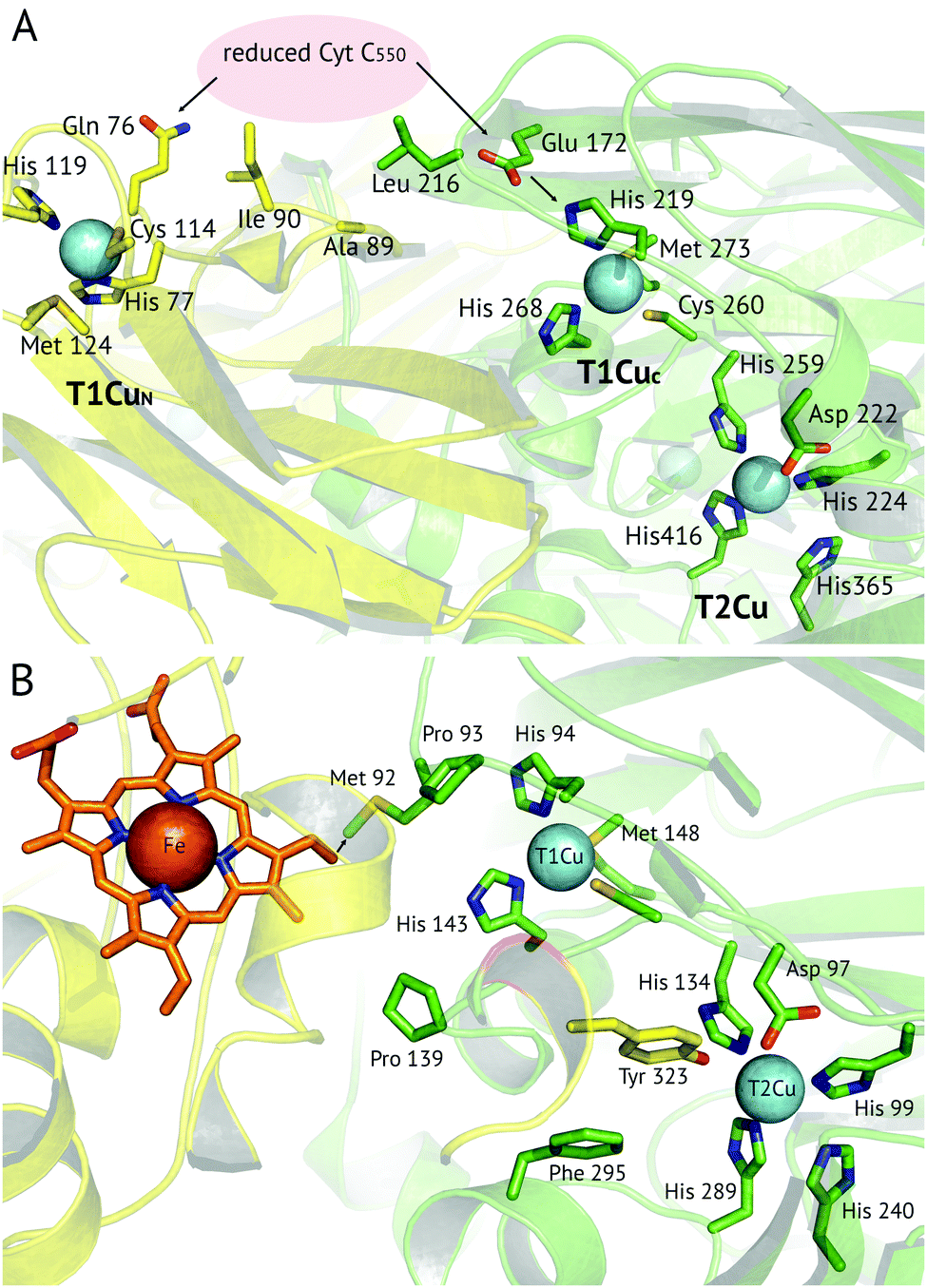 | ||
Fig. 6 Proposed electron transfer routes between the redox centres of 3-domain CuNiRs. In (A) ![[H with combining low line]](https://www.rsc.org/images/entities/i_char_0048_0332.gif) ![[d with combining low line]](https://www.rsc.org/images/entities/i_char_0064_0332.gif) NiR the external electron donor partner protein cyt. c550 is proposed to bind to a hydrophobic patch spanning the N-terminal (near Ala89, Ile90) and cupredoxin domains (near Leu216), where it is positioned for electron transfer to both the T1CuN and T1CuC sites (indicated by arrows).74 In (B) RpNiR, the proposed internal electron transfer route is from the haem (CBC atom) to Met92 and to the T1Cu site.75 Interface and T2Cu water molecules have been omitted from the figures for clarity. NiR the external electron donor partner protein cyt. c550 is proposed to bind to a hydrophobic patch spanning the N-terminal (near Ala89, Ile90) and cupredoxin domains (near Leu216), where it is positioned for electron transfer to both the T1CuN and T1CuC sites (indicated by arrows).74 In (B) RpNiR, the proposed internal electron transfer route is from the haem (CBC atom) to Met92 and to the T1Cu site.75 Interface and T2Cu water molecules have been omitted from the figures for clarity. | ||
Notably, in the substrate binding channel of RpNiR, a Tyr residue acts to ‘plug’ the substrate binding channel, excluding water molecules.75 The atomic resolution structure of RpNiR provided clear evidence for the observed preference for nitrite binding to reduced T2Cu in this enzyme, rather than the preferred oxidised state in 2-domain CuNiRs.
Membrane associated CuNiRs
The crystal structure of the soluble domain of the outer membrane AniA protein from Neisseria gonorrhoeae was determined in 2002.44 The AniA protein has around half the specific activity of AfNiR, is essential for growth in low-oxygen conditions with nitrite present and protects against human-sera killing of the pathogen. The soluble domain has a similar fold to the typical CuNiRs but has two shortened loops, with one loop possibly related to interactions with a membrane anchored azurin electron donor and the other likely to interact with the membrane itself. The Cu centres in AniA are similar to those of 2-domain CuNiRs, but with a visible colour intermediate between that of green and blue sites. While bioinformatics identified related proteins to AniA across a range of species, further structures have not yet been forthcoming.Variations on the classical CuNiR structure and further classification of CuNiRs
In the vicinity of the metal centres, CuNiRs have been divided into classes based on the length of the two loops, the linker loop between the cupredoxin domains forming a monomer and the ‘tower loop’ adjacent to the T1Cu site.44 The majority of structures are of class I enzymes, while class II includes AniA.44 Some recently discovered CuNiRs fall between these two classes with intermediate length loops, e.g. GkNiR. Several CuNiRs from thermophilic bacteria have been structurally characterised. The structure of Geobacillus kaustophilus HTA426 CuNiR (GkNiR)51 revealed a novel 28 residue additional α-helical region close to the N-terminus (residues 41–68). Other notable features of GkNiR include the presence of a Val residue in place of IleCAT and differences in the hydrogen bonding network around the T2Cu centre.Much of the structural data presented above has come from single, static crystal structures. Recent developments in X-ray sources, rapid detectors and data handling have allowed the determination of both ‘damage free’ single structures from large numbers of microcrystals and also multiple (potentially hundreds of) sequential datasets from just one protein crystal, making it possible to follow reactions in crystallo. Both approaches show promise for determining structures of relevant catalytic intermediates.
Insights from multiple-structures one crystal (MSOX) serial crystallography
Multiple Structures from One Crystal (MSOX) is a data collection technique which uses solvated electrons generated during X-ray diffraction experiments to drive reactions requiring electron transfer.76 By taking sequential diffraction datasets over the same volume of one crystal it is possible to observe a reaction, occurring through a series of high resolution crystal structures with progressively increasing X-ray dose, Fig. 7. For NiRs, this approach was used to obtain 3 (low, medium and high-dose) datasets from tobacco assimilatory NiR, a Fe–S cluster and sirohaem containing protein that converts nitrite to ammonium.77 A series of this sort for a CuNiR, comprising 45 consecutive datasets collected in 19 s each for AcNiR, between 1.07 Å and 1.62 Å resolutions, clearly showed conversion of bound NO2−, to side-on NO55 within the crystal. At the end of the 45 structure series, a Cu(II) species with bound water is observed after loss of the NO ligand. Crystal structures are not yet able to give a definitive answer to the identification of the observed side-on NO complex as Cu(I)–NO˙ or Cu(II)–NO˙−.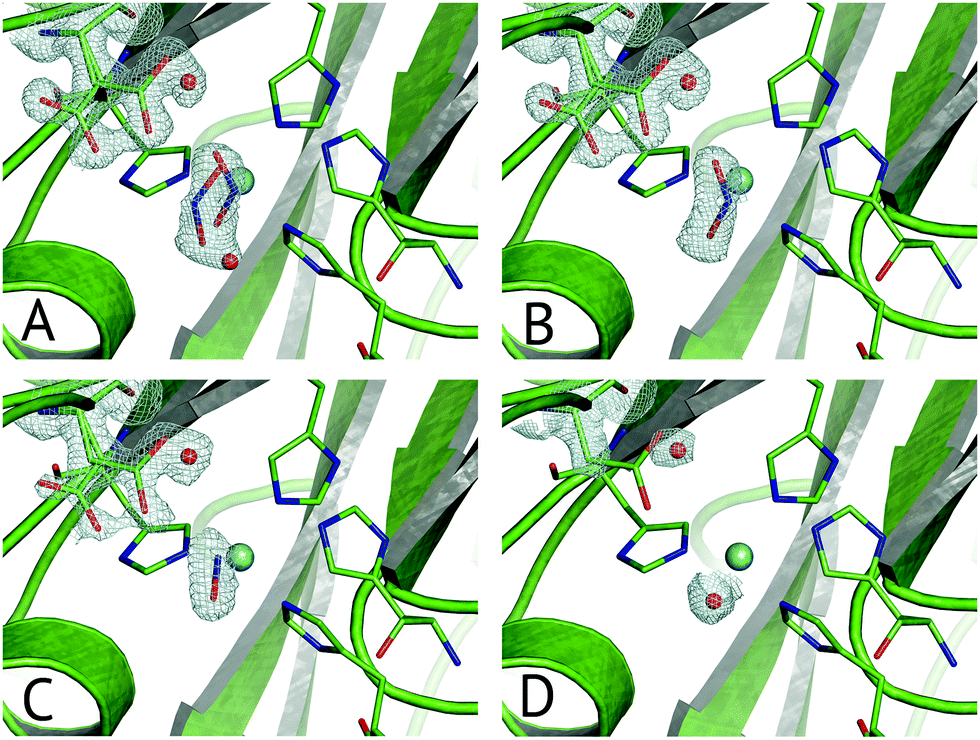 | ||
| Fig. 7 Selected frames from the 45-frame MSOX structural movie of AcNiR catalysis at 100 K.55 (A) The starting nitrite-bound state with partial occupancies in the top-hat and side-on modes (dataset 1, PDB 5i6k); (B) fully side-on nitrite (dataset 4, PDB 5i6l); (C) side-on nitric oxide product complex (dataset 17, PDB 5i6n); (D) water rebinding to the T2Cu following dissociation of NO (dataset 40, PDB 5i6k). Electron density is shown, contoured at 0.53–0.36 e− Å−3, for the ligands and the AspCAT residue, present in its ‘gatekeeper’ and ‘proximal’ orientations. The T2Cu atom is depicted by the cyan sphere and water molecules by red spheres. | ||
Little to no change to the overall fold of the enzyme or in the vicinity of the T1Cu site were seen throughout the MSOX series. The low dose atomic resolution starting structure exhibited a previously unseen dual conformation of NO2−, in “top-hat” and “side-on” bidentate orientations, of which the top-hat conformation matches the vertical substrate coordination from ‘radiation-damage-free’ XFEL structures,54,58 corroborating the idea that this vertical binding mode represents the initial nitrite binding position. Overall, the 45-dataset MSOX series demonstrated several structural changes at the T2Cu site, Fig. 7. Starting with the dual conformation nitrite and both gatekeeper and proximal AspCAT conformations, by dataset four the series showed nitrite adopting a single side-on conformation and increasing proximal AspCAT occupancy, lending credence to the operation of the sensor loop in response to the copper oxidation states during catalysis.23 By dataset eleven a second previously unseen conformation was observed, with either NO2−, NO or a H2O molecule at the T2Cu, indicative of catalytic turnover in the crystal. The changes in gatekeeper and proximal positions of AspCAT provide some structural evidence for the role of AspCAT in substrate delivery and product release.
XFEL structures of intact redox states of CuNiRs by serial femtosecond crystallography
Reduction of metalloproteins by X-ray generated solvated electrons has been well-characterised, presents a major challenge for structural biology of such proteins,78,79 and has been demonstrated to occur in CuNiRs.27 Various online spectroscopic techniques have been developed to monitor the redox state of metals in crystals.80–82 Strategies to mitigate such reduction include merging of composite, low dose partial datasets83 or helical data collection approaches.84 Use of rapid X-ray detectors and cryogenic temperatures – typically 100 K – minimises but does not eliminate the effects of photoreduction or radiation damage to crystals. However, intact fully-oxidised structures may require the use of femtosecond X-ray pulses that can only be provided with sufficient brilliance by an X-ray free electron laser (XFEL).85 The first CuNiR XFEL structures at room temperature were recently obtained using the Spring-8 Compact Free Electron Laser (SACLA).54,58Comparison of the 1.43 Å resolution serial femtosecond crystallography (SFX) structure of resting state GtNiR58 with conventional synchrotron radiation (SRX) structures revealed a 10° rotation of the imidazole ring of HisCAT, in the SRX data in relation to the SFX structure. This led to the suggestion that HisCAT may function as a proton-relay switch, whereby T2Cu reduction causes HisCAT rotation thus altering the H-bonding network of HisCAT with Thr268 and Glu267 to destabilise the positive charge of HisCAT and facilitate proton transfer to the bridging water.
Subsequently, SFX structures were determined of Alcaligenes faecalis NiR (AfNiR) in its resting and nitrite bound (PDB 5D4I) states at 2.03 Å and 1.60 Å resolutions, respectively.54 The SFX structure of intact nitrite bound AfNiR revealed a single, vertical ‘top-hat’ binding mode of NO2− compared to the side on conformation typically observed in SRX structures,18 suggesting the conformational change to side on NO2− results from photoreduction of the metal centres. AfNiR, as in GtNiR, shows a conformational change in HisCAT in photoreduced SRX structures compared to oxidised SFX structures. However, unlike GtNiR where HisCAT rotates 10° pivoting on Nδ1 and Cβ with a 0.3 Å shift in Cβ, in AfNiR, HisCAT rotates 20° about Cγ with no change in Cβ, switching the H-bonding partner of HisCAT Nε2 from the Glu279 carbonyl to the hydroxyl of Thr280 which is both a longer and weaker H-bond, Fig. 8. Based on this conformational change between SFX and SRX structure54 an updated reaction mechanism for CuNiRs was suggested in which nitrite binds to the T2Cu and is protonated by AspCAT. This is followed by intramolecular ET to give side-on nitrite and rotation of HisCAT, where weaker H-bonds facilitate proton transfer from HisCAT to the bridging water by destabilising the positive charge of HisCAT. With this the necessary second proton can be delivered to form NO in either end-on or side-on binding modes. However, the recently published MSOX series55 showing X-ray induced ligand turnover from NO2− to side-on NO to water shows no change in the H-bond distances between HisCAT and Glu279 (2.7 Å) between low and high X-ray doses and a gradual increase in the bond length between HisCAT and Thr280 with increasing dose (2.8 Å at 0.69 MGy and 3.1 Å at 27.60 MGy).
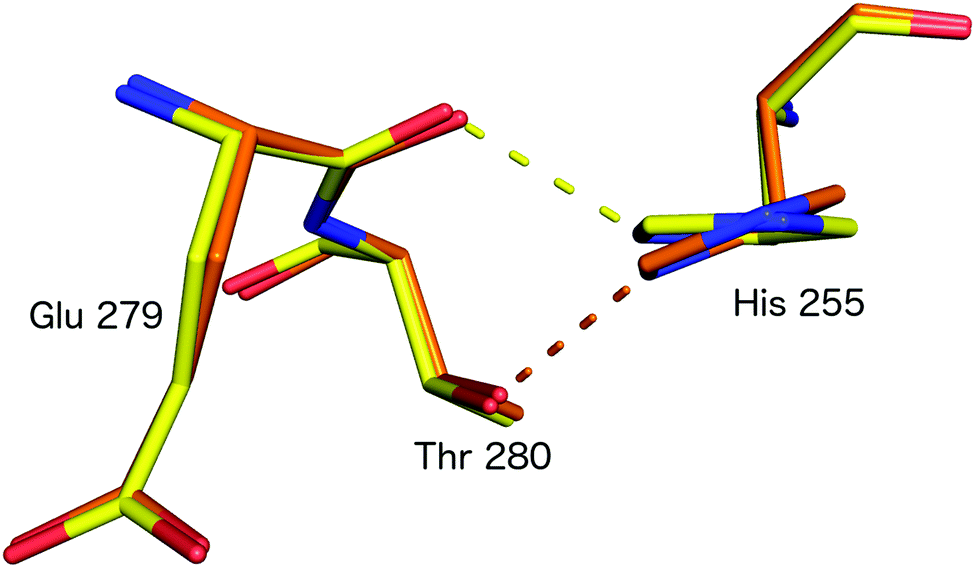 | ||
| Fig. 8 HisCAT rotation in the proposed redox coupled proton transfer mechanism derived from comparison of XFEL (SFX; PDB 4YSC) and synchrotron radiation (SRX; PDB 4YSE) structures of AfNiR.54 Note the H-bond switching of HisCAT (His255) between Glu279 O atom (damage free oxidised XFEL structure, yellow) and Thr280 (reduced SRX structure, orange). | ||
Outlook and remaining questions
Several exciting new discoveries of CuNiR enzymes together with methodological developments have taken place in recent years. The advent of rapid mixing technologies at XFEL sources may allow time-resolved structural analysis of the CuNiR reaction with damage-free structures for reaction intermediates. The ever-increasing number of sequenced genomes promises the discovery of new variations of CuNiRs with differing domain structures. However, several outstanding questions remain that structural analysis will play a key role in addressing. For example, the protonation states of the important ‘catalytic residues’ have yet to be unequivocally determined for all stages of the reaction mechanism. Neutron diffraction – another way to obtain room temperature radiation-damage free structures – may help provide this information in the future and its potential for studying CuNiRs, in combination with high resolution X-ray structures, has already been shown.86 Establishing the precise chemical nature of the side-on NO intermediate also remains challenging as do the routes of substrate access and product escape. While significant insights have been gained from the current structures of protein–protein electron transfer complexes, improved structural resolution is required to clarify details of the emerging picture. The dynamic behaviour of CuNiRs is clearly of vital importance and can be addressed by combining structural data, measured at different temperatures, with molecular dynamics and QM/MM simulations.Acknowledgements
The work was supported by BBSRC grant awards BB/M020924/1 to RWS, BB/M022714/1 to MH and by Leverhulme Trust grant RPG-2014-355 to MH and RWS. We acknowledge Professors S. S. Hasnain and R. R. Eady and Dr S. Antonyuk for fruitful discussions of CuNiRs over many years.References
- R. Knowles, Denitrification, Microbiol. Rev., 1982, 46, 43–70 CAS.
- W. G. Zumft, Cell biology and molecular basis of denitrification, Microbiol. Mol. Biol. Rev., 1997, 61, 533–616 CAS.
- R. R. Eady and S. S. Hasnain, in Comprehensive Coordination Chemistry II: Bio-Coordination Chemistry, ed. L. T. W. Que, Elsevier, Oxford, 2003, vol. 8, pp. 759–786 Search PubMed.
- A. J. Thomson, G. Giannopoulos, J. Pretty, E. M. Baggs and D. J. Richardson, Biological sources and sinks of nitrous oxide and strategies to mitigate emissions, Philos. Trans. R. Soc., B, 2012, 367, 1157–1168 CrossRef CAS PubMed.
- Metalloenzymes in Denitrification: Applications and Environmental Impacts, ed. I. Moura, J. Moura, S. Pauleta and L. Maia, The Royal Society of Chemistry, 2017 Search PubMed.
- J. W. Godden, S. Turley, D. C. Teller, E. T. Adman, M. Y. Liu, W. J. Payne and J. LeGall, The 2.3 Å X-ray structure of nitrite reductase from Achromobacter cycloclastes, Science, 1991, 253, 438–442 CAS.
- E. T. Adman, in Topics in Molecular and Structural Biology: Metalloproteins, Part 1: Metal proteins with redox roles, ed. P. M. Harrison, MacMillan, New York, 1985, vol. 6, pp. 1–42 Search PubMed.
- A. C. Merkle and N. Lehnert, Binding and activation of nitrite and nitric oxide by copper nitrite reductase and corresponding model complexes, Dalton Trans., 2012, 41, 3355–3368 RSC.
- M. J. Ellis, J. G. Grossmann, R. R. Eady and S. S. Hasnain, Genomic analysis reveals widespread occurrence of new classes of copper nitrite reductases, J. Biol. Inorg. Chem., 2007, 12, 1119–1127 CrossRef CAS PubMed.
- R. R. Eady, S. V. Antonyuk and S. S. Hasnain, Fresh insight to functioning of selected enzymes of the nitrogen cycle, Curr. Opin. Chem. Biol., 2016, 31, 103–112 CrossRef CAS PubMed.
- K. C. Cheung, R. W. Strange and S. S. Hasnain, 3D EXAFS refinement of the Cu site of azurin sheds light on the nature of structural change at the metal centre in an oxidation-reduction process: an integrated approach combining EXAFS and crystallography, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2000, 56, 697–704 CrossRef CAS.
- E. I. Solomon, Spectroscopic methods in bioinorganic chemistry: blue to green to red copper sites, Inorg. Chem., 2006, 45, 8012–8025 CrossRef CAS PubMed.
- P. Hosseinzadeh, S. Tian, N. M. Marshall, J. Hemp, T. Mullen, M. J. Nilges, Y. G. Gao, H. Robinson, D. A. Stahl, R. B. Gennis and Y. Lu, A Purple Cupredoxin from Nitrosopumilus maritimus Containing a Mononuclear Type 1 Copper Center with an Open Binding Site, J. Am. Chem. Soc., 2016, 138, 6324–6327 CrossRef CAS PubMed.
- S. Ghosh, X. Xie, A. Dey, Y. Sun, C. P. Scholes and E. I. Solomon, Thermodynamic equilibrium between blue and green copper sites and the role of the protein in controlling function, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 4969–4974 CrossRef CAS PubMed.
- R. W. Strange, L. M. Murphy, F. E. Dodd, Z. H. Abraham, R. R. Eady, B. E. Smith and S. S. Hasnain, Structural and kinetic evidence for an ordered mechanism of copper nitrite reductase, J. Mol. Biol., 1999, 287, 1001–1009 CrossRef CAS PubMed.
- E. Adman and M. E. Murphy, Copper Nitrite Reductase, Handbook of Metalloproteins, 2006, vol. 6 Search PubMed.
- E. T. Adman, J. W. Godden and S. Turley, The structure of copper-nitrite reductase from Achromobacter cycloclastes at five pH values, with NO2− bound and with type II copper depleted, J. Biol. Chem., 1995, 270, 27458–27474 CrossRef CAS PubMed.
- S. V. Antonyuk, R. W. Strange, G. Sawers, R. R. Eady and S. S. Hasnain, Atomic resolution structures of resting-state, substrate- and product-complexed Cu-nitrite reductase provide insight into catalytic mechanism, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 12041–12046 CrossRef CAS PubMed.
- M. J. Ellis, M. Prudencio, F. E. Dodd, R. W. Strange, G. Sawers, R. R. Eady and S. S. Hasnain, Biochemical and crystallographic studies of the Met144Ala, Asp92Asn and His254Phe mutants of the nitrite reductase from Alcaligenes xylosoxidans provide insight into the enzyme mechanism, J. Mol. Biol., 2002, 316, 51–64 CrossRef CAS PubMed.
- E. I. Solomon, D. E. Heppner, E. M. Johnston, J. W. Ginsbach, J. Cirera, M. Qayyum, M. T. Kieber-Emmons, C. H. Kjaergaard, R. G. Hadt and L. Tian, Copper active sites in biology, Chem. Rev., 2014, 114, 3659–3853 CrossRef CAS PubMed.
- M. J. Boulanger and M. E. Murphy, Directing the mode of nitrite binding to a copper-containing nitrite reductase from Alcaligenes faecalis S-6: characterization of an active site isoleucine, Protein Sci., 2003, 12, 248–256 CrossRef CAS PubMed.
- M. A. Hough, R. R. Eady and S. S. Hasnain, Identification of the proton channel to the active site type 2 Cu center of nitrite reductase: structural and enzymatic properties of the His254Phe and Asn90Ser mutants, Biochemistry, 2008, 47, 13547–13553 CrossRef CAS PubMed.
- M. A. Hough, M. J. Ellis, S. Antonyuk, R. W. Strange, G. Sawers, R. R. Eady and S. S. Hasnain, High resolution structural studies of mutants provide insights into catalysis and electron transfer processes in copper nitrite reductase, J. Mol. Biol., 2005, 350, 300–309 CrossRef CAS PubMed.
- N. G. Leferink, R. R. Eady, S. S. Hasnain and N. S. Scrutton, Laser-flash photolysis indicates that internal electron transfer is triggered by proton uptake by Alcaligenes xylosoxidans copper-dependent nitrite reductase, FEBS J., 2012, 279, 2174–2181 CrossRef CAS PubMed.
- N. G. H. Leferink, C. Han, S. V. Antonyuk, D. J. Heyes, S. E. J. Rigby, M. A. Hough, R. R. Eady, N. S. Scrutton and S. S. Hasnain, Proton-Coupled Electron Transfer in the Catalytic Cycle of Alcaligenes xylosoxidans Copper-Dependent Nitrite Reductase, Biochemistry, 2011, 50, 4121–4131 CrossRef CAS PubMed.
- H. J. Wijma, L. J. C. Jeuken, M. P. Verbeet, F. A. Armstrong and G. W. Canters, A random-sequential mechanism for nitrite binding and active site reduction in copper-containing nitrite-reductase, J. Biol. Chem., 2006, 281, 16340–16346 CrossRef CAS PubMed.
- M. A. Hough, S. V. Antonyuk, R. W. Strange, R. R. Eady and S. S. Hasnain, Crystallography with online optical and X-ray absorption spectroscopies demonstrates an ordered mechanism in copper nitrite reductase, J. Mol. Biol., 2008, 378, 353–361 CrossRef CAS PubMed.
- Ł. Krzemiński, L. Ndamba, G. W. Canters, T. J. Aartsma, S. D. Evans and L. J. C. Jeuken, Spectroelectrochemical Investigation of Intramolecular and Interfacial Electron-Transfer Rates Reveals Differences Between Nitrite Reductase at Rest and During Turnover, J. Am. Chem. Soc., 2011, 133, 15085–15093 CrossRef PubMed.
- M. Lintuluoto and J. M. Lintuluoto, DFT Study on Nitrite Reduction Mechanism in Copper-Containing Nitrite Reductase, Biochemistry, 2016, 55, 210–223 CrossRef CAS PubMed.
- M. Lintuluoto and J. M. Lintuluoto, DFT Study on Enzyme Turnover Including Proton and Electron Transfers of Copper-Containing Nitrite Reductase, Biochemistry, 2016, 55, 4697–4707 CrossRef CAS PubMed.
- Y. Li, M. Hodak and J. Bernholc, Enzymatic Mechanism of Copper-Containing Nitrite Reductase, Biochemistry, 2015, 54, 1233–1242 CrossRef CAS PubMed.
- S. Ghosh, A. Dey, Y. Sun, C. P. Scholes and E. I. Solomon, Spectroscopic and computational studies of nitrite reductase: proton induced electron transfer and backbonding contributions to reactivity, J. Am. Chem. Soc., 2009, 131, 277–288 CrossRef CAS PubMed.
- S. A. De Marothy, M. R. Blomberg and P. E. Siegbahn, Elucidating the mechanism for the reduction of nitrite by copper nitrite reductase-A contribution from quantum chemical studies, J. Comput. Chem., 2007, 28, 528–539 CrossRef CAS PubMed.
- R. Silaghi-Dumitrescu, Copper-containing nitrite reductase: A DFT study of nitrite and nitric oxide adducts, J. Inorg. Biochem., 2006, 100, 396–402 CrossRef CAS PubMed.
- M. J. Boulanger and M. E. Murphy, Alternate substrate binding modes to two mutant (D98N and H255N) forms of nitrite reductase from Alcaligenes faecalis S-6: structural model of a transient catalytic intermediate, Biochemistry, 2001, 40, 9132–9141 CrossRef CAS PubMed.
- M. L. Barrett, R. L. Harris, S. Antonyuk, M. A. Hough, M. J. Ellis, G. Sawers, R. R. Eady and S. S. Hasnain, Insights into redox partner interactions and substrate binding in nitrite reductase from Alcaligenes xylosoxidans: crystal structures of the Trp138His and His313Gln mutants, Biochemistry, 2004, 43, 16311–16319 CrossRef CAS PubMed.
- F. Jacobson, A. Pistorius, D. Farkas, W. De Grip, O. Hansson, L. Sjolin and R. Neutze, pH dependence of copper geometry, reduction potential, and nitrite affinity in nitrite reductase, J. Biol. Chem., 2007, 282, 6347–6355 CrossRef CAS PubMed.
- E. T. Adman, J. E. Godden and S. Turley, The Structure of copper nitrite reductase from Achromobactor cycloclastes at 5 pH values, with NO2− bound, and with type-2 Cu(II) depleted, J. Biol. Chem., 1995, 270, 27458–27474 CrossRef CAS PubMed.
- M. E. P. Murphy, S. Turley, M. Kukimoto, M. Nishiyama, S. Horinouchi, H. Sasaki, M. Tanokura and E. T. Adman, Structure of Alcaligenes faecalis Nitrite Reductase and a Copper Site Mutant, M150E, That Contains Zinc, Biochemistry, 1995, 34, 12107–12117 CrossRef CAS PubMed.
- F. E. Dodd, S. S. Hasnain, Z. H. L. Abraham, R. R. Eady and B. E. Smith, Structures of a Blue-Copper Nitrite Reductase and its Substrate-Bound Complex, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1997, D53, 406–418 CrossRef CAS PubMed.
- M. E. P. Murphy, S. Turley and E. T. Adman, Structure of nitrite bound to copper-containing nitrite reductase from Alcaligenes faecalis, J. Biol. Chem., 1997, 272, 28455–28460 CrossRef CAS PubMed.
- F. E. Dodd, J. Van Beeumen, R. R. Eady and S. S. Hasnain, X-ray structure of a blue-copper nitrite reductase in two crystal forms. The nature of the copper sites, mode of substrate binding and recognition by redox partner, J. Mol. Biol., 1998, 282, 369–382 CrossRef CAS PubMed.
- M. J. Ellis, F. E. Dodd, R. W. Strange, M. Prudencio, G. Sawers, R. R. Eady and S. S. Hasnain, X-ray structure of a blue copper nitrite reductase at high pH and in copper-free form at 1.9 A resolution, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2001, 57, 1110–1118 CrossRef CAS.
- M. J. Boulanger and M. E. Murphy, Crystal structure of the soluble domain of the major anaerobically induced outer membrane protein (AniA) from pathogenic Neisseria: a new class of copper-containing nitrite reductases, J. Mol. Biol., 2002, 315, 1111–1127 CrossRef CAS PubMed.
- E. I. Tocheva, F. I. Rosell, A. G. Mauk and M. E. Murphy, Side-on copper-nitrosyl coordination by nitrite reductase, Science, 2004, 304, 867–870 CrossRef CAS PubMed.
- F. Jacobson, H. Guo, K. Olesen, M. Okvist, R. Neutze and L. Sjolin, Structures of the oxidized and reduced forms of nitrite reductase from Rhodobacter sphaeroides 2.4.3 at high pH: changes in the interactions of the type 2 copper, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2005, 61, 1190–1198 Search PubMed.
- K. Paraskevopoulos, M. A. Hough, R. G. Sawers, R. R. Eady and S. S. Hasnain, The structure of the Met144Leu mutant of copper nitrite reductase from Alcaligenes xylosoxidans provides the first glimpse of a protein-protein complex with azurin II, J. Biol. Inorg. Chem., 2007, 12, 789–796 CrossRef CAS PubMed.
- E. I. Tocheva, F. I. Rosell, A. G. Mauk and M. E. Murphy, Stable copper-nitrosyl formation by nitrite reductase in either oxidation state, Biochemistry, 2007, 46, 12366–12374 CrossRef CAS PubMed.
- M. Nojiri, H. Koteishi, T. Nakagami, K. Kobayashi, T. Inoue, K. Yamaguchi and S. Suzuki, Structural basis of inter-protein electron transfer for nitrite reduction in denitrification, Nature, 2009, 462, 117–120 CrossRef CAS PubMed.
- T. J. Lawton, K. E. Bowen, L. A. Sayavedra-Soto, D. J. Arp and A. C. Rosenzweig, Characterization of a nitrite reductase involved in nitrifier denitrification, J. Biol. Chem., 2013, 288, 25575–25583 CrossRef CAS PubMed.
- Y. Fukuda, H. Koteishi, R. Yoneda, T. Tamada, H. Takami, T. Inoue and M. Nojiri, Structural and functional characterization of the Geobacillus copper nitrite reductase: involvement of the unique N-terminal region in the interprotein electron transfer with its redox partner, Biochim. Biophys. Acta, 2014, 1837, 396–405 CrossRef CAS PubMed.
- Y. Fukuda, K. M. Tse, M. Lintuluoto, Y. Fukunishi, E. Mizohata, H. Matsumura, H. Takami, M. Nojiri and T. Inoue, Structural insights into the function of a thermostable copper-containing nitrite reductase, J. Biochem., 2014, 155, 123–135 CrossRef CAS PubMed.
- Y. Fukuda and T. Inoue, High-temperature and high-resolution crystallography of thermostable copper nitrite reductase, Chem. Commun., 2015, 51, 6532–6535 RSC.
- Y. Fukuda, K. M. Tse, T. Nakane, T. Nakatsu, M. Suzuki, M. Sugahara, S. Inoue, T. Masuda, F. Yumoto, N. Matsugaki, E. Nango, K. Tono, Y. Joti, T. Kameshima, C. Song, T. Hatsui, M. Yabashi, O. Nureki, M. E. Murphy, T. Inoue, S. Iwata and E. Mizohata, Redox-coupled proton transfer mechanism in nitrite reductase revealed by femtosecond crystallography, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 2928–2933 CrossRef CAS PubMed.
- S. Horrell, S. V. Antonyuk, R. R. Eady, S. S. Hasnain, M. A. Hough and R. W. Strange, Serial crystallography captures enzyme catalysis in copper nitrite reductase at atomic resolution from one crystal, IUCrJ, 2016, 3, 271–281 CAS.
- B. A. Wallace, Protein characterisation by synchrotron radiation circular dichroism spectroscopy, Q. Rev. Biophys., 2009, 42, 317–370 CrossRef CAS PubMed.
- J. Spence and E. Lattman, Imaging enzyme kinetics at atomic resolution, IUCrJ, 2016, 3, 228–229 CrossRef CAS PubMed.
- Y. Fukuda, K. M. Tse, M. Suzuki, K. Diederichs, K. Hirata, T. Nakane, M. Sugahara, E. Nango, K. Tono, Y. Joti, T. Kameshima, C. Song, T. Hatsui, M. Yabashi, O. Nureki, H. Matsumura, T. Inoue, S. Iwata and E. Mizohata, Redox-coupled structural changes in nitrite reductase revealed by serial femtosecond and microfocus crystallography, J. Biochem., 2016, 159, 527–538 CrossRef CAS PubMed.
- N. G. Leferink, S. V. Antonyuk, J. A. Houwman, N. S. Scrutton, R. R. Eady and S. S. Hasnain, Impact of residues remote from the catalytic centre on enzyme catalysis of copper nitrite reductase, Nat. Commun., 2014, 5, 4395 CAS.
- E. I. Tocheva, L. D. Eltis and M. E. P. Murphy, Conserved active site residues limit inhibition of a copper-containing nitrite reductase by small molecules, Biochemistry, 2008, 47, 4452–4460 CrossRef CAS PubMed.
- Y. Fukuda, K. M. Tse, Y. Kado, E. Mizohata, H. Matsumura and T. Inoue, Insights into unknown foreign ligand in copper nitrite reductase, Biochem. Biophys. Res. Commun., 2015, 464, 622–628 CrossRef CAS PubMed.
- W. P. Michalski and D. J. D. Nicholas, Molecular characterization of a copper-containing nitrite reductase from Rhodopseudomonas sphaeroides from sp. denitrificans, Biochim. Biophys. Acta, 1985, 828, 130–137 CAS.
- M. J. Ellis, S. V. Antonyuk, R. W. Strange, G. Sawers, R. R. Eady and S. S. Hasnain, Observation of an unprecedented Cu Bis-His site: crystal structure of the H129V mutant of nitrite reductase, Inorg. Chem., 2004, 43, 7591–7593 CrossRef CAS PubMed.
- O. M. Usov, Y. Sun, V. M. Grigoryants, J. P. Shapleigh and C. P. Scholes, EPR-ENDOR of the Cu(I)NO complex of nitrite reductase, J. Am. Chem. Soc., 2006, 128, 13102–13111 CrossRef CAS PubMed.
- S. Ghosh, A. Dey, O. M. Usov, Y. Sun, V. M. Grigoryants, C. P. Scholes and E. I. Solomon, Resolution of the spectroscopy versus crystallography issue for NO intermediates of nitrite reductase from Rhodobacter sphaeroides, J. Am. Chem. Soc., 2007, 129, 10310–10311 CrossRef CAS PubMed.
- K. Fujisawa, A. Tateda, Y. Miyashita, K. Okamoto, F. Paulat, V. K. Praneeth, A. Merkle and N. Lehnert, Structural and spectroscopic characterization of mononuclear copper(I) nitrosyl complexes: end-on versus side-on coordination of NO to copper(I), J. Am. Chem. Soc., 2008, 130, 1205–1213 CrossRef CAS PubMed.
- A. C. Merkle and N. Lehnert, The side-on copper(I) nitrosyl geometry in copper nitrite reductase is due to steric interactions with isoleucine-257, Inorg. Chem., 2009, 48, 11504–11506 CrossRef CAS PubMed.
- L. M. Murphy, F. E. Dodd, F. K. Yousafzai, R. R. Eady and S. S. Hasnain, Electron donation between copper containing nitrite reductases and cupredoxins: the nature of protein–protein interaction in complex formation, J. Mol. Biol., 2002, 315, 859–871 CrossRef CAS PubMed.
- M. Kukimoto, M. Nishiyama, M. E. Murphy, S. Turley, E. T. Adman, S. Horinouchi and T. Beppu, X-ray structure and site-directed mutagenesis of a nitrite reductase from Alcaligenes faecalis S-6: roles of two copper atoms in nitrite reduction, Biochemistry, 1994, 33, 5246–5252 CrossRef CAS PubMed.
- M. Kukimoto, M. Nishiyama, T. Ohnuki, S. Turley, E. T. Adman, S. Horinouchi and T. Beppu, Identification of interaction site of pseudoazurin with its redox partner, copper-containing nitrite reductase from Alcaligenes faecalis S-6, Protein Eng., 1995, 8, 153–158 CrossRef CAS PubMed.
- M. D. Vlasie, R. Fernandez-Busnadiego, M. Prudencio and M. Ubbink, Conformation of pseudoazurin in the 152 kDa electron transfer complex with nitrite reductase determined by paramagnetic NMR, J. Mol. Biol., 2008, 375, 1405–1415 CrossRef CAS PubMed.
- M. Nojiri, in Metalloenzymes in Denitrification: Applications and Environmental Impacts, ed. I. Moura, J. Moura, S. Pauleta and L. Maia, The Royal Society of Chemistry, 2017, ch. 5, pp. 91–113 10.1039/9781782623762-00091.
- S. Suzuki, T. Kohzuma, S. Shidara, K. Okhki and T. Aida, Novel spectroscopic aspects of type 1 copper in Hyphomicrobium nitrite reductase, Inorg. Chim. Acta, 1993, 208, 107–109 CrossRef CAS.
- M. Nojiri, Y. Xie, T. Inoue, T. Yamamoto, H. Matsumura, K. Kataoka, W. Deligeer, K. Yamaguchi, Y. Kai and S. Suzuki, Structure and function of a hexameric copper-containing nitrite reductase, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 4315–4320 CrossRef CAS PubMed.
- S. V. Antonyuk, C. Han, R. R. Eady and S. S. Hasnain, Structures of protein-protein complexes involved in electron transfer, Nature, 2013, 496, 123–126 CrossRef CAS PubMed.
- I. Schlichting, J. Berendzen, K. Chu, A. M. Stock, S. A. Maves, D. E. Benson, R. M. Sweet, D. Ringe, G. A. Petsko and S. G. Sligar, The catalytic pathway of cytochrome p450cam at atomic resolution, Science, 2000, 287, 1615–1622 CrossRef CAS PubMed.
- S. Nakano, M. Takahashi, A. Sakamoto, H. Morikawa and K. Katayanagi, The reductive reaction mechanism of tobacco nitrite reductase derived from a combination of crystal structures and ultraviolet–visible microspectroscopy, Proteins, 2012, 80, 2035–2045 CAS.
- T. Beitlich, K. Kuhnel, C. Schulze-Briese, R. L. Shoeman and I. Schlichting, Cryoradiolytic reduction of crystalline heme proteins: analysis by UV-Vis spectroscopy and X-ray crystallography, J. Synchrotron Radiat., 2007, 14, 11–23 CrossRef CAS PubMed.
- E. F. Garman and M. Weik, X-ray radiation damage to biological macromolecules: further insights, J. Synchrotron Radiat., 2017, 24, 1–6 CrossRef CAS PubMed.
- J. McGeehan, R. B. Ravelli, J. W. Murray, R. L. Owen, F. Cipriani, S. McSweeney, M. Weik and E. F. Garman, Colouring cryo-cooled crystals: online microspectrophotometry, J. Synchrotron Radiat., 2009, 16, 163–172 CrossRef CAS PubMed.
- D. von Stetten, T. Giraud, P. Carpentier, F. Sever, M. Terrien, F. Dobias, D. H. Juers, D. Flot, C. Mueller-Dieckmann, G. A. Leonard, D. de Sanctis and A. Royant, In crystallo optical spectroscopy (icOS) as a complementary tool on the macromolecular crystallography beamlines of the ESRF, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2015, 71, 15–26 CrossRef CAS PubMed.
- R. L. Owen, B. A. Yorke, J. A. Gowdy and A. R. Pearson, Revealing low-dose radiation damage using single-crystal spectroscopy, J. Synchrotron Radiat., 2011, 18, 367–373 CrossRef CAS PubMed.
- D. Kekilli, F. S. Dworkowski, G. Pompidor, M. R. Fuchs, C. R. Andrew, S. Antonyuk, R. W. Strange, R. R. Eady, S. S. Hasnain and M. A. Hough, Fingerprinting redox and ligand states in haemprotein crystal structures using resonance Raman spectroscopy, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2014, 70, 1289–1296 CAS.
- I. Polsinelli, M. Savko, C. Rouanet-Mehouas, L. Ciccone, S. Nencetti, E. Orlandini, E. A. Stura and W. Shepard, Comparison of helical scan and standard rotation methods in single-crystal X-ray data collection strategies, J. Synchrotron Radiat., 2017, 24, 42–52 CAS.
- I. Schlichting, Serial femtosecond crystallography: the first five years, IUCrJ, 2015, 2, 246–255 CrossRef CAS PubMed.
- M. P. Blakeley, S. S. Hasnain and S. V. Antonyuk, Sub-atomic resolution X-ray crystallography and neutron crystallography: promise, challenges and potential, IUCrJ, 2015, 2, 464–474 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |