
Targeting βCys93 in hemoglobin S with an antisickling agent possessing dual allosteric and antioxidant effects
Abstract
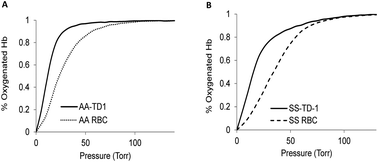
Sickle cell disease (SCD) is an inherited blood disorder caused by a β globin gene mutation of hemoglobin (HbS). The polymerization of deoxyHbS and its subsequent aggregation (into long fibers) is the primary molecular event which leads to red blood cell (RBC) sickling and ultimately hemolytic anemia. We have recently suggested that HbS oxidative toxicity may also contribute to SCD pathophysiology due to its defective pseudoperoxidase activity. As a consequence, a persistently higher oxidized ferryl heme is formed which irreversibly oxidizes “hotspot” residues (particularly βCys93) causing protein unfolding and subsequent heme loss. In this report we confirmed first, the allosteric effect of a newly developed reagent (di(5-(2,3-dihydro-1,4-benzodioxin-2-yl)-4H-1,2,4-triazol-3-yl)disulfide) (TD-1) on oxygen affinity within SS RBCs. There was a considerable left shift in oxygen equilibrium curves (OECs) representing treated SS cells. Under hypoxic conditions, TD-1 treatment of HbS resulted in an approximately 200 s increase in the delay time of HbS polymerization over the untreated HbS control. The effect of TD-1 binding to HbS was also tested on oxidative reactions by incrementally treating HbS with increasing hydrogen peroxide (H2O2) concentrations. Under these experimental conditions, ferryl levels were consistently reduced by approximately 35% in the presence of TD-1. Mass spectrometric analysis confirmed that upon binding to βCys93, TD-1 effectively blocked irreversible oxidation of this residue. In conclusion, TD-1 appears to shield βCys93 (the end point of radical formation in HbS) and when coupled with its allosteric effect on oxygen affinity may provide new therapeutic modalities for the treatment of SCD.
- This article is part of the themed collection: Iron in Biology
 Please wait while we load your content...
Please wait while we load your content...