
Intracellular iron and heme trafficking and metabolism in developing erythroblasts
 *abcd
*abcd
Abstract
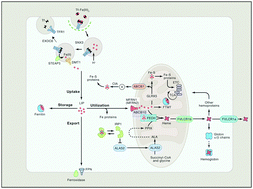
Vertebrate red blood cells (RBCs) arise from erythroblasts in the human bone marrow through a process known as erythropoiesis. Iron uptake is a crucial hallmark, essential for heme biosynthesis in the differentiating erythroblasts, which are dedicated to producing hemoglobin. Erythropoiesis is facilitated by a network of intracellular transport proteins, chaperones, and circulating hormones. Intracellular iron is targeted to the mitochondria for incorporation into a porphyrin ring to form heme and cytosolic iron–sulfur proteins, including Iron Regulatory Protein 1 (IRP1). These processes are tightly regulated to prevent both excess and insufficient levels of iron and heme precursors. Crosstalk between the heme and iron–sulfur synthesizing pathways has been demonstrated to serve as a regulatory feedback mechanism. The activity of δ-aminolevulinic acid synthase (ALAS), the first and rate-limiting enzyme of heme biosynthesis, is a fundamental node of this regulation. Recently, the mitochondrial unfoldase, ClpX, has received attention as a novel key player that modulates this step in heme biogenesis, implicating a role in the pathophysiology of anemic diseases. This chapter reviews the canonical pathways in intracellular iron and heme trafficking and recent findings of iron and heme metabolism in vertebrate red cells. A discussion of the molecular approaches to studying iron and heme transport is provided to highlight opportunities for revealing therapeutic targets.
- This article is part of the themed collections: Recent Review Articles and Iron in Biology
 Please wait while we load your content...
Please wait while we load your content...