
Photocatalytic fixation of nitrogen to ammonia: state-of-the-art advancements and future prospects
Xingzhu
Chen
a,
Neng
Li
*ab,
Zhouzhou
Kong
a,
Wee-Jun
Ong
 *c and
Xiujian
Zhao
a
*c and
Xiujian
Zhao
a
aState Key Laboratory of Silicate Materials for Architectures, Wuhan University of Technology, Hubei, 430070, China. E-mail: lineng@whut.edu.cn
bDepartment of Materials Science & Metallurgy, University of Cambridge, Cambridge, CB3 0FS, UK
cInstitute of Materials Research and Engineering (IMRE), Agency for Science, Technology and Research (A*STAR), 2 Fusionopolis Way, Innovis, Singapore 138634, Singapore. E-mail: ongwj@imre.a-star.edu.sg; ongweejun@gmail.com; Web: https://sites.google.com/site/wjongresearch/
First published on 29th August 2017
Abstract
The burgeoning development of ammonia (NH3) synthesis technology addresses the urgency of food intake required to sustain the population growth of the last 100 years. To date, NH3 has mostly been synthesized by the Haber–Bosch process in industry. Under the ever-increasing pressure of the fossil fuel depletion crisis and anthropogenic global climate change with continuous CO2 emission in the 21st century, research targeting the synthesis of NH3 under mild conditions in a sustainable and environment friendly manner is vigorous and thriving. Therefore, the focus of this review is the state-of-the-art engineering of efficient photocatalysts for dinitrogen (N2) fixation toward NH3 synthesis. Strenuous efforts have been devoted to modifying the intrinsic properties of semiconductors (i.e. poor electron transport, rapid electron–hole recombination and sluggish reaction kinetics), including nanoarchitecture design, crystal facet engineering, doping and heterostructuring. Herein, this review provides insights into the most recent advancements in understanding the charge carrier kinetics of photocatalysts with respect to charge transfer, migration and separation, which are of fundamental significance to photocatalytic N2 fixation. Subsequently, the challenges, outlooks and future prospects at the forefront of this research platform are presented. As such, it is anticipated that this review will shed new light on photocatalytic N2 fixation and NH3 synthesis and will also provide a blueprint for further investigations and momentous breakthroughs in next-generation catalyst design.
 Xingzhu Chen | Xingzhu Chen received her BS in Materials Science and Engineering from Wuhan University of Technology in 2016. She is presently working on her PhD at Wuhan University of Technology. Her research interests include photoelectrochemistry, photocatalysis, and the development of photocatalysts, related materials and devices. |
 Neng Li | Neng Li received his PhD degree in condensed matter physics from Huahzong University of Science and Technology in 2011. He then joined the Shenzhen Institutes of Advanced Technology, Chinese Academy of Sciences as a Research Fellow in 2011. He worked at the Department of Physics and Astronomy, University of Missouri-Kansas City as a postdoctoral researcher from 2012 to 2014. In 2015, he joined Wuhan University of Technology as a Full Professor. Dr Li was a Visiting Research Fellow at the Department of Materials Science & Metallurgy, University of Cambridge, UK from 2016 to 2017. His research interests focus on energy and environmental applications such as photocatalysis, photocatalytic hydrogen production, CO2 reduction, N2 fixation, and the development of hybrid functional materials and related devices. |
 Wee-Jun Ong | Wee-Jun Ong received his BE and PhD degrees in Chemical Engineering from Monash University. In 2015, he was a Visiting Research Fellow at the University of New South Wales and Monash University Clayton Campus, Australia. He is currently a Research Scientist at IMRE, A*STAR in Singapore. At present, he serves as the Associate Editor of Frontiers in Materials, an Editorial Board Member of Scientific Reports, Nanotechnology and Nano Futures, and a Community Board Member of Materials Horizons. He has also been the Lead Guest Editor of several Special Issues. His research interests primarily focus on photocatalytic, photoelectrochemical and electrochemical H2O splitting, CO2 reduction and N2 fixation for energy conversion and storage using two-dimensional (2D)-based nanomaterials, transition metal dichalcogenides/phosphides and carbonaceous-based hybrid nanocomposites. For more details, see http://https://sites.google.com/site/wjongresearch/. |
1. Introduction
It is a well known fact that nitrogen is one of the starting building blocks for the synthesis of amino acids, nucleotides and other major biological compounds in all organisms.1,2 The main source of nitrogen, dinitrogen (N2), is the largest single component of the Earth's atmosphere (around 78% by volume). As an important precursor for nitrogen-containing compounds, ammonia (NH3) could be directly synthesized from dinitrogen by a small group of organisms named diazotrophs in nature.3 However, this could scarcely meet the large needs of the fertilizer industry today.The fixation of N2 to NH3 is thermodynamically accessible: N2(g) + 3H2(g) → 2NH3(g), ΔH298K = −92.2 kJ mol−1. However, this reaction cannot occur spontaneously under ambient conditions. The reduction reactions of N2 to its partial reduzates, such as diazene (N2H2) or hydrazine (N2H4), are also non-spontaneous due to the positive standard enthalpies of formation of +212.9 and +95.35 kJ mol−1 for N2H2 and N2H4, respectively.4 There are many factors hindering the cleavage and hydrogenation of dinitrogen in nature. First, the thermodynamically strong cleavage energy of the first bond in N2 (410 kJ mol−1) manifests the critical challenge in the full dissociation of N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N, which explains the chemical inactivity of N2 compared to other triple-bonded molecules.5 Additionally, from the aspect of kinetics, the large energy gap between the HOMO (the σg2p bonding orbital) and LUMO (the πg*2p anti-bonding orbital) explains the high chemical stability of N2. Other factors, such as low proton affinity, also account for the difficulty of direct protonation of N2. Detailed descriptions can be found in the review article by Jia and Quadrelli.4
N, which explains the chemical inactivity of N2 compared to other triple-bonded molecules.5 Additionally, from the aspect of kinetics, the large energy gap between the HOMO (the σg2p bonding orbital) and LUMO (the πg*2p anti-bonding orbital) explains the high chemical stability of N2. Other factors, such as low proton affinity, also account for the difficulty of direct protonation of N2. Detailed descriptions can be found in the review article by Jia and Quadrelli.4
The Haber–Bosch process, which involves the reaction of N2 and hydrogen (H2) over iron or ruthenium-based catalysts, has been estimated to be one of the most intriguing discoveries of the last century.6,7 After 100 years of development, the temperature is controlled to near 450 °C and the reaction pressure has decreased to 15 to 30 MPa from the original 50 to 100 MPa, while the efficiency is limited to 10% to 15%.8,9 Inevitably, this process accounts for 1.6% of total global CO2 emissions; this is attributed to the presence of hydrocarbons in the raw materials (as the main hydrogen source and energy supplier in this process).10
Parallel to the optimization of the thermocatalytic process,11–14 several other methods have aroused widespread attention in the synthesis of ammonia to date. Before we engage in our topic of photocatalysis for the fixation of nitrogen to ammonia, we would like to briefly mention the progress of the other two traditional approaches. Owing to the variable coordination number of transition metals, organometallic complexes can participate in the activation of N2. The ground-breaking work of the first nitrogen complex [Ru(NH3)5(N2)]+ in 1965 challenged the traditional belief that dinitrogen cannot form complexes.15 Since then, nitrogen complexes have been developed to generate ammonia under particular conditions, in which central elements of various transition metals, such as molybdenum,16–18 iron and titanium,19–22 were tested as the sites of dinitrogen coordination and reduction. Furthermore, the model and action mechanism of iron molybdenum complexes as active sites in nitrogenase enzymes have greatly fascinated many chemists and biologists.23–25 On the other hand, inspired by electron and proton transfer processes, numerous researchers have carried out investigations of electrochemical and electrocatalytic approaches.26 Since a solid electrolyte cell was first used in the electrochemical synthesis of ammonia in 1998,27 various conducting ceramic membranes have been tested to produce NH3 from N2.28–31 Since then, polymer electrolytes such as Nafion and sulfonated polysulfone have been deemed to provide excellent proton conductivity under low temperature.32–34 Later, molten salts such as chlorides,35 oxide-carbonates or hydroxides were employed as electrolytes.36–39 Very recently, Chen et al. reported an electrolyte-less cell with the highest faradaic efficiency of 95.1% and a relatively low rate of 3.60 × 10−12 mol s−1 cm−2 at room temperature, wherein the electrocatalyst was based on Fe2O3 nanoparticles supported on conductive carbon nanotubes (Fe2O3-CNT).40
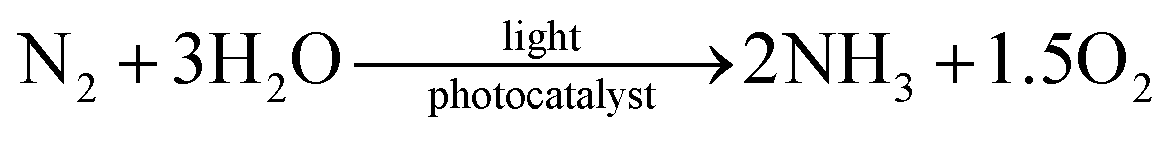
With regard to the inexhaustible and clean solar energy from sunlight,41–47 photocatalysis has been thrust into the limelight in the realms of energy and chemical fuel production; thus, blossoming interest in the design of myriad semiconductors has been witnessed in recent years.48–60 Generally, the photocatalytic process makes use of photons as the driving force to propel the activation of N2. Compared with electrochemical approaches, photon-driven fixation of N2 has reached a stumbling block stemming from the difficulties of N2 chemisorption and activation.61 The magnitudes of production rates for most studied photocatalysts remain below par,62 distinctly hindering the success of the solar fixation of N2. In the initial studies, N2 photoreduction was considered to take place in nature over abundant minerals on the surface of the earth; thus, earlier studies on the photocatalytic fixation of N2 mainly focused on soil minerals and sand in nature.63–65 This was introduced in the previous work of Schrauzer.66 Since the first exploratory study on as-synthesized TiO2-based photocatalysts for N2 fixation under UV light in 1977,67 a flurry of research activities have been devoted to the development of photocatalytic N2 fixation, especially in the 21st century, due to the fact that the driving force (light) and ingredients (water and air) of this process are relatively clean, cheap and accessible, as shown in the following equation.
To the best of our knowledge, there is no literature pertaining to the latest research progress of photocatalytic N2 fixation with respect to the classification of photocatalysts. In this review article, we will present and update the state-of-the-art research advancements in this growing field of N2 fixation, as depicted in Fig. 1. Briefly, the classification of pristine photocatalysts is firstly discussed, followed by the rational development of hybrid heterojunction nanocomposites via various synthesis strategies for application in the fixation of N2 to NH3. Finally, the review is concluded with a summary, invigorating perspectives and future prospects on this research horizon. As such, it is projected that this article will markedly promote the importance of this research platform and help to provide new insights for the exploration of future investigations toward a sustainable future.
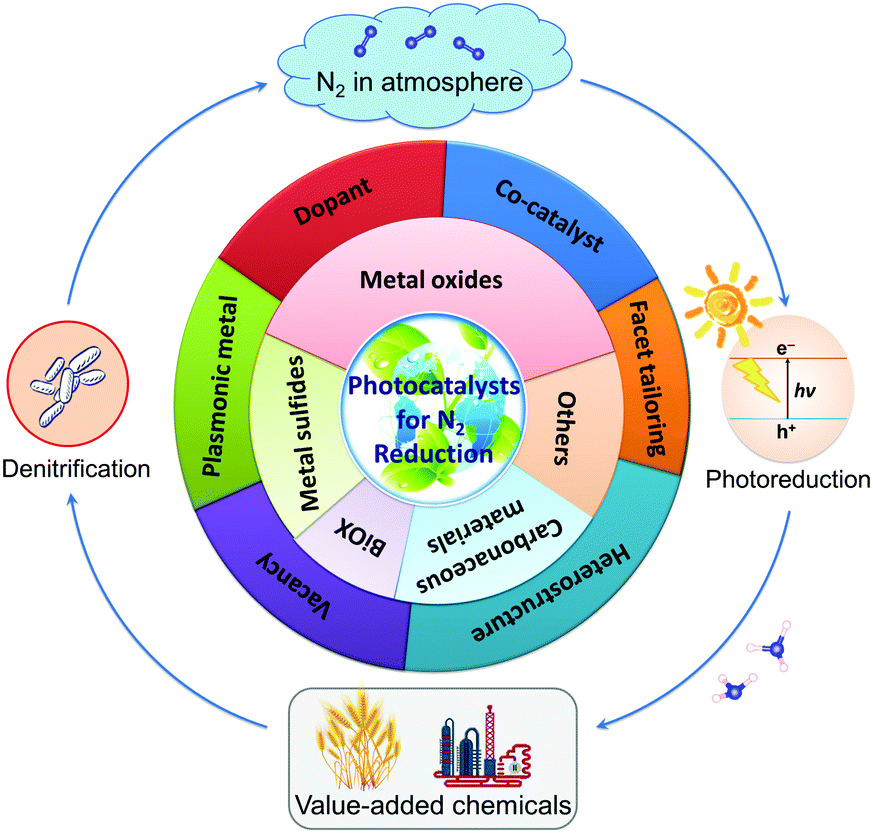 | ||
| Fig. 1 Illustration of the main topics covered in this review article, exemplifying the latest research in the development of photocatalysts for N2 reduction. | ||
2. Fundamental process of N2 photoreduction
2.1. Basic photocatalytic principles over semiconductors
The photocatalytic process of N2 fixation can be divided into several steps. First, the photogenerated electrons are promoted to the conduction band, leaving vacant holes in the valance band. Afterwards, some of the electrons and holes recombine with each other; meanwhile, others migrate to the surface of the catalyst and participate in the redox reaction. Specifically, H2O can be oxidized to O2 by the holes, whereas N2 is reduced to NH3 after a series of multi-step injections of photogenerated electrons and water-derived protons.The hydrogenation reactions related to this process and the corresponding reduction potentials are summarized in Table 1.68–71 As is known to all, whether the photocatalytic redox reaction can occur largely depends on the reduction potential of the adsorbate and the position of the energy band in the semiconductor (Fig. 2).72–85 For example, the position of the semiconductor conduction band should be higher (more negative) than the reduction potential of the N2 hydrogenation, while the valance band should be lower (more positive) than the oxygen evolution potential. The maximum energy transition state is located in the very first electron transfer (−4.16 V vs. NHE) and proton-coupled electron transfer (−3.2 V vs. NHE) processes (Table 1), hampering the overall kinetic reaction.86 On this basis, these are the main two limitations that must be overcome to activate N2 molecules for NH3 formation. It is of utmost importance to maintain a small band gap of the semiconductor, preferably in the visible light region, which can still satisfy the thermodynamic reduction potentials of N2 to NH3. Additionally, it is necessary to suppress the recombination of charge carriers in semiconductor photocatalysts in order to enhance the solar conversion efficiency and apparent quantum yield (AQY) of the reaction.
| Reaction | E 0 (V) |
|---|---|
| a Reduction potential E0vs. NHE at pH 7. b Reduction potential E0vs. RHE. | |
| H2O → ½O2 + 2H+ + 2e− | 0.81a |
| 2H+ + 2e− → H2 | −0.42a |
| N2 + e− → N2− | −4.16 |
| N2 + H+ + e− → N2H | −3.2 |
| N2 + 2H+ + 2e− → N2H2 | −1.10b |
| N2 + 4H+ + 4e− → N2H4 | −0.36 |
| N2 + 5H+ + 4e− → N2H5+ | −0.23 |
| N2 + 6H+ + 6e− → 2NH3 | 0.55 |
| N2 + 8H+ + 8e− → 2NH4+ | 0.27 |
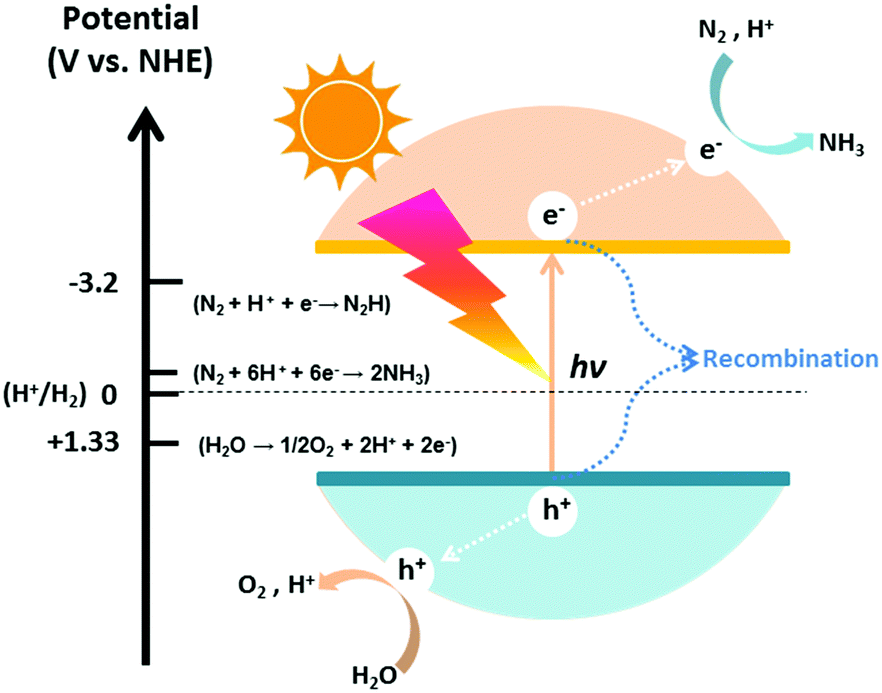 | ||
| Fig. 2 Schematic of semiconductor-based photocatalysts used for the conversion of N2 to NH3. The redox potentials (V vs. NHE at pH = 0) of water splitting and dinitrogen hydrogenation are marked on the left. | ||
2.2. N2 reduction mechanism
Recently, a number of investigations from the viewpoint of experimental and density functional theory (DFT) calculations have been carried out to demystify N2 reduction mechanisms over various catalysts.87–89 As is mentioned above, in the case of photo-driven NH3 synthesis, elementary electrochemical reactions can be employed to help decipher the hydrogenation process in greater detail. For example, at the very first proton-coupled electron transfer, the N2 adsorbed on the catalyst surface (*N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N) gains one proton (H+) from the environment and one photo-generated electron (e−) from the catalyst to generate an adsorbed chemical species (*NNH) as shown: *NN + H+ + e− → *NNH˙
N) gains one proton (H+) from the environment and one photo-generated electron (e−) from the catalyst to generate an adsorbed chemical species (*NNH) as shown: *NN + H+ + e− → *NNH˙
For the following transfer of H+/e− pairs, 5 routes were proposed by Azofra et al. in their DFT study (Fig. 3).87 There are two widely accepted mechanisms for the conversion of N2 to NH3. These are commonly called distal and alternating mechanisms.19 In the distal mechanism, H+/e− pairs are proposed to consecutively attach to one N atom of N2 to form a terminal nitride intermediate, liberating the first NH3 and leaving a single N, which finally converts into another NH3 (Fig. 3, path 1). In contrast, the alternating mechanism postulates that H+/e− pairs occur alternately on the two N atoms of N2 (Fig. 3, path 2).
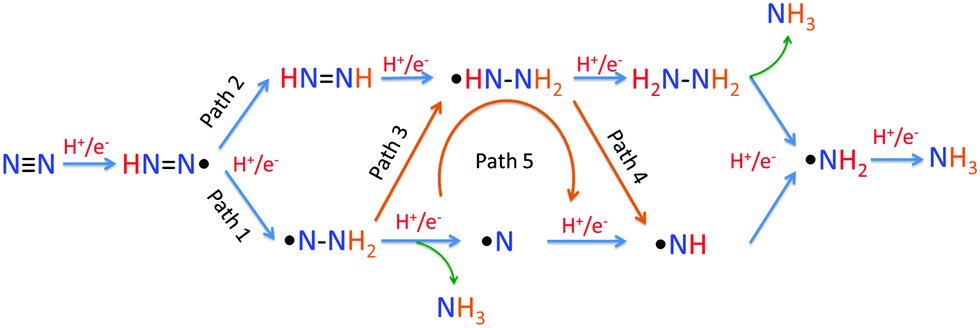 | ||
| Fig. 3 Five proposed routes for the conversion of N2 to NH3. Reproduced with permission.87 Copyright 2016, Royal Society of Chemistry. | ||
In the study of the N2 fixation mechanism, the combination of experimental investigation with computational simulation will help us to further understand the reaction routes: the computational simulation gives a prospective direction for guidance of further experiments, while the experimental research provides feedback and confirmation of the optimization of the theoretical model.
3. Classification of photocatalysts for N2 fixation to NH3
Most traditional unmodified semiconductors cannot meet the energy standards for the reduction potentials of the intermediate reactions. Hitherto, many concerted efforts have been made regarding the choice and modification of semiconductors, such as doping, introducing vacancies, plasmon induction, facet tailoring and heterostructure assembly, to improve the photocatalytic performance. In the subsequent sections, the photocatalysts are classified based on their elemental compositions.3.1. Metal oxide-based materials
N bond. With several transformations of surficial Ti from Ti3+ to Ti4+, the electrons were naturally injected into N2. Meanwhile, Ti3+ could be regenerated under UV irradiation. The groundbreaking exploration on TiO2-based materials dates back to 1977. Schrauzer and Guth synthesized iron-doped TiO2 by heating iron(III) sulfate-impregnated anatase TiO2; they testified to its great ability to reduce N2 under UV irradiation.67 In their work, ammonia and a small quantity of hydrazine were detected, with the iron content in TiO2 varying from 0% to 1%. Interestingly, the NH3 yields reached a maximum value over 0.2% Fe2O3-doped TiO2 under UV. The facile impregnation method described in this research has thus enlightened and inspired research endeavors by multitudinous worldwide researchers.
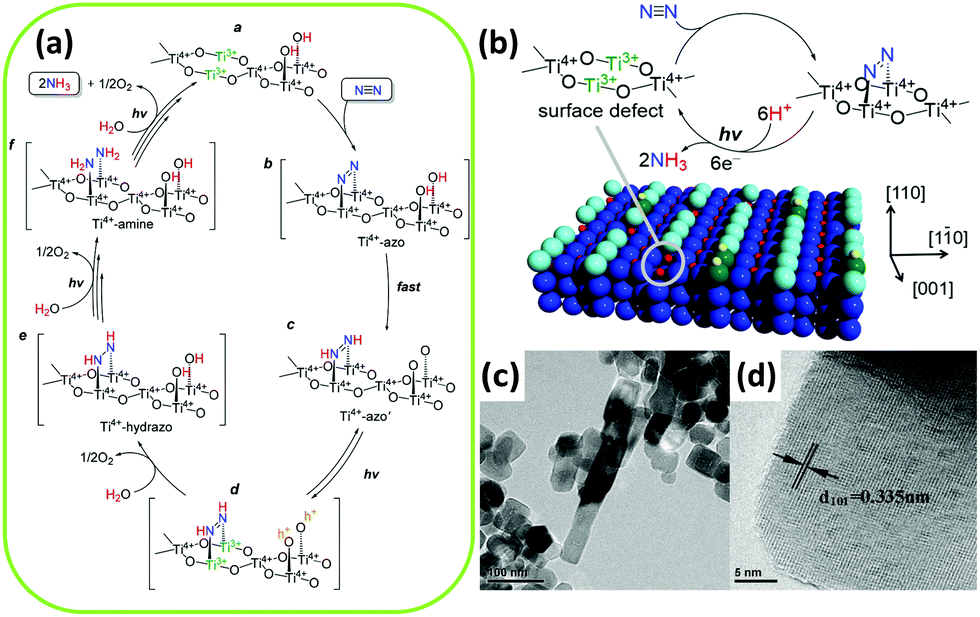 | ||
| Fig. 4 (a) Mechanism for N2 conversion to NH3 over the surface oxygen vacancies of TiO2. (b) A photocatalytic cycle for N2 reduction at the surface of rutile TiO2(110). Reproduced with permission.92 Copyright 2017, American Chemical Society. (c) Transmission electron microscopy (TEM) and (d) high-resolution TEM (HRTEM) images of Fe3+-doped TiO2. Reproduced with permission.99 Copyright 2014, Elsevier. | ||
After that, Augugliaro et al. utilized an identical impregnation technique to prepare Fe2O3-hybridized TiO2 and supported it on Al2O3. An improved NH3 production rate was gained in gas–solid fluidized bed reactors.93 According to literature reports, the effect of iron ions facilitated the transformation of the crystallinity of TiO2, which noticeably affected the photocatalytic efficiency. Notably, the incorporation of Fe accelerated the phase transformation of anatase into rutile and also promoted grain growth during calcination. In this regard, the ratio of anatase to rutile played a prevailing role in the efficiency of the catalysts by maintaining a constant concentration of iron.94,95 After a prolonged calcination process, the increased content and growing grain size of rutile phase as well as the loss of surface Ti–OH groups led to a decrease of the active surface area, resulting in inferior photocatalytic performance.67
Several authors have employed diversified methods in the preparation of iron-doped TiO2. In Radford and Francis's research, both the anatase and rutile phase of TiO2 were sampled for doping with iron in metal vapor without high-heat treatment.96 Aqueous suspensions of samples were irradiated under UV light. Undoped samples demonstrated minimal or no activity under light irradiation. It should be noted that the activity of doped anatase was higher than that of doped rutile, which can be attributed to the more negative flat band potential of anatase.97 The author investigated whether iron was the sole active species involved in the photoactivity because the yield of ammonia over iron-doped anatase was far beyond their anticipation. However, Shrauzer indicated later that the NH3 may have formed on a photo-assisted organoiron compound.66 As for the role of Fe3+, Soria et al. demonstrated that iron ions could temporarily trap photogenerated electrons and promote the separation of charge carriers.98 They coprecipitated an aqueous solution of TiCl3, which contained Fe3+ ions, in an ammonia solution; they then heated the resulting solid for a day. The residual chloride ions were noted to exhibit poor activity of the coprecipitation specimens. Recently, Fe-doped TiO2 with highly exposed (101) facets obtained via a hydrothermal method by adsorbing Fe3+ on a precursor of hydrogen titanate nanotubes was reported by Zhao et al. (Fig. 4c and d).99 The N2 photoreduction performance was enhanced by exposing the (101) facets of Fe-doped TiO2 with ethanol as the scavenger. A low doping concentration of Fe3+ on the TiO2 surface enhanced the trapping of electrons and holes by forming Fe2+ and Fe4+ to inhibit charge recombination. The unstable Fe2+ and Fe4+ transferred electrons and holes to Ti4+ and OH−, generating Ti3+ (as mentioned in the preceding section, Ti3+ plays an essential role in the photocatalytic activation)92 and ˙OH, respectively. Similar to the findings by Soria et al.,98 the authors mentioned that no nitrite was formed in the presence of ethanol solution because ethanol functioned as a scavenger of ˙OH to prevent the oxidation of ammonia.
Apart from iron-doped TiO2, other transition metals have been used as dopants in TiO2.100–103 The reports of these investigations, while significant, were popular in the last century; however, they are receiving little attention at present because there have been few breakthroughs in their activity in N2 photoreduction.100,101 In the original study, Schrauzer and Guth tested eleven metal dopants in addition to iron, including Co, Mo, Ni, Pd, V, Cr, Cu and the noble metals Pt, Ag, Au, and Pb.67 Compared to the control group, the Co, Mo and Ni dopants contributed to enhancement of the NH3 yield of TiO2. However, eight other metals presented inhibitive effects on NH3 formation. This phenomenon was attributed to the different influences of the metal dopants on the phase transformation of TiO2. Apparently, the samples containing Co, Mo and Ni accelerated the anatase to rutile transformation during heat treatment; meanwhile, no acceleration of phase transformation was readily observed on doping with eight other metals. In contrast, a different phenomenon was manifested by Palmisano and his co-workers, where chromium-ion-doped TiO2 was found to be effective in N2 photoreduction.100 They clarified that compared with iron-ion-doped TiO2, the chromium-ion-doped TiO2 demonstrated shorter diffusion lengths of minority carriers, accounting for its higher rate of hole–electron recombination. Another study suggested that chromium ions were mainly dispersed on the surface in the formation of the solid solution. The increase of Cr ions reduced the OH groups on the surface, which explains its lower activity at higher contents.101 Dopants such as Mg, Ce and V have also been investigated in TiO2-based photocatalysts by Ileperuma et al.102,103 In their work, catalysts were suspended in double-distilled water under irradiation. The results implied that the ammonia yield increased with increasing pH value. At the same time, higher nitrate content was detected in production.102 Similar studies involved doping TiO2 with a relatively high concentration of 10 wt% Ce or V. The V-doped TiO2 catalysts were shown to possess n-type semiconductor behavior at pH = 3, while the Ce-doped TiO2 catalysts displayed p-type behaviour at pH = 12.5.103 However, there was no substantial difference in the NH3 yields of both systems.
In addition to the modification of metal doping, transition metals can also be loaded on the TiO2 surface as a co-catalyst. For example, Miyami et al. have reported the enhancement of N2 photoreduction using Ru-loaded TiO2.104 Khan et al. used a Ru(III) complex as a photosensitizer in a Pt–TiO2 semiconductor.105 Ru(III) complex could be excited and subsequently inject electrons into the conduction band of TiO2 to form a Ru(IV) complex. A systematic study on noble metal-loaded TiO2 was investigated by Ranjit et al.106 By comparing four noble metal catalysts as co-catalysts of TiO2, they found the order of photoactivity was Ru > Rh > Pd > Pt, which is closely associated with the strength of the M–H bond (M = noble metal). In other words, the incorporation of noble metals, which exhibit a high barrier for H2 evolution, resulted in high NH3 yields. In 2008, Linnik and Kisch designed a Ru-modified TiO2 film with conducting glass as the substrate and humic acid as the sacrificial agent.107 However, the activity of the catalysts was not greatly improved compared to other doping systems.
In 2000, Hoshino et al. designed a TiO2/conducting polymer system with an organic/inorganic heterojunction.108 The photogenerated carriers at the organic/inorganic interface led to the synthesis of ammonia. In their work, a composite of poly(3-methylthiophene) (P3MeT) and TiO2 was irradiated under a fluorescent lamp, and NH4+ClO4− needle crystals were obtained. A rigorous comparison with Schrauzer's method was performed in the following year, and a comparable ammonia production rate was obtained.109 It was deduced that the separation of photogenerated charge carriers at the interface and the presence of P3MeT inhibited the accumulation of NH3 in TiOx. A further study was conducted by substituting TiO2 crystal with its amorphous phase in 2007.110 These investigations undoubtedly provide a new concept and future direction of catalyst modification.
Most earlier studies suggested that bare Fe2O3 has no photocatalytic activity for N2 cleavage.67,98,117 It is worth noting that iron is useful in the Haber–Bosch process and also functions similarly to a nitrogen enzyme because of its good interaction with dinitrogen.6,19,112 As expected, later studies have proven that ferric oxide is capable of photofixating nitrogen to ammonia.111,113 However, in a pure form, neither Fe2O3 nor its reduced product Fe3O4 had N2 photocatalytic activity. To overcome this bottleneck, Khader et al. successfully designed α-Fe2O3, which was effective in photo-activating nitrogen by partially reducing α-Fe2O3 to Fe3O4.111 Fascinatingly, in the presence of 3 to 5 at% iron in the form of Fe(II) in the partially reduced Fe2O3, NH3 was detected in an aqueous slurry of the catalyst under UV irradiation. Most recently, another comparison of Fe2O3 and TiO2 was performed by Lashgari and Zeinalkhani.113 The 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio nanocomposites of Fe2O3–TiO2 and Pd-loaded Fe2O3–TiO2 were taken into consideration. It is noteworthy that the rate of NH3 production was in the order of TiO2 < Fe2O3–TiO2 < Pd/Fe2O3–TiO2. Therefore, with the incorporation of Fe2O3 into TiO2, the coupled heterojunction nanocomposites played a profound role in the N2 photoreduction process owing to the extended photoabsorption of visible light in Fe2O3.
:1 ratio nanocomposites of Fe2O3–TiO2 and Pd-loaded Fe2O3–TiO2 were taken into consideration. It is noteworthy that the rate of NH3 production was in the order of TiO2 < Fe2O3–TiO2 < Pd/Fe2O3–TiO2. Therefore, with the incorporation of Fe2O3 into TiO2, the coupled heterojunction nanocomposites played a profound role in the N2 photoreduction process owing to the extended photoabsorption of visible light in Fe2O3.
Furthermore, tungsten oxide with oxygen vacancies (WO3−x) was reported to have poor activity in the gas phase.114 ZnO was reported to have photocatalytic activity for nitrogen fixation.118 In the case of commercial ZnO, the ammonia yield of unmodified ZnO was greater than that of Pt-loaded ZnO, which is consistent with a previous report by Miyama in 1980.104 However, for ZnO prepared by means of wet etching or precipitation methods (Fig. 5a and b), the situation was reversed. For a wide band gap semiconductor, mesoporous β-Ga2O3 nanorods were employed in N2 photoreduction by Zhao et al. in 2015.115 The photoactivity of β-Ga2O3 was ameliorated in the presence of different alcohols, including methanol, ethanol, and tert-butanol (TBA), which formed ˙CO2−in situ. The reducing ability of ˙CO2− dramatically enhanced the reduction of N2 to NH3. Moreover, the presence of O2 in the N2 facilitated the formation of ˙CO2−, which enhanced the reducing power (Fig. 5c). Very recently, a low-valent bismuth monoxide (BiO) without additional reducing agents was employed for N2 photoreduction because the low-valent bismuth with empty 6d orbitals provided excellent N2 chemisorption and activation centres (the synthesis procedure is shown in Fig. 5d). The N2 was activated by three arranged Bi atoms by donating electrons to the 6d orbitals of Bi and accepting lone pairs of electrons from the three Bi atoms to its anti-bonding orbitals (σ*2px, π*2py and π*2pz) (Fig. 3e).116
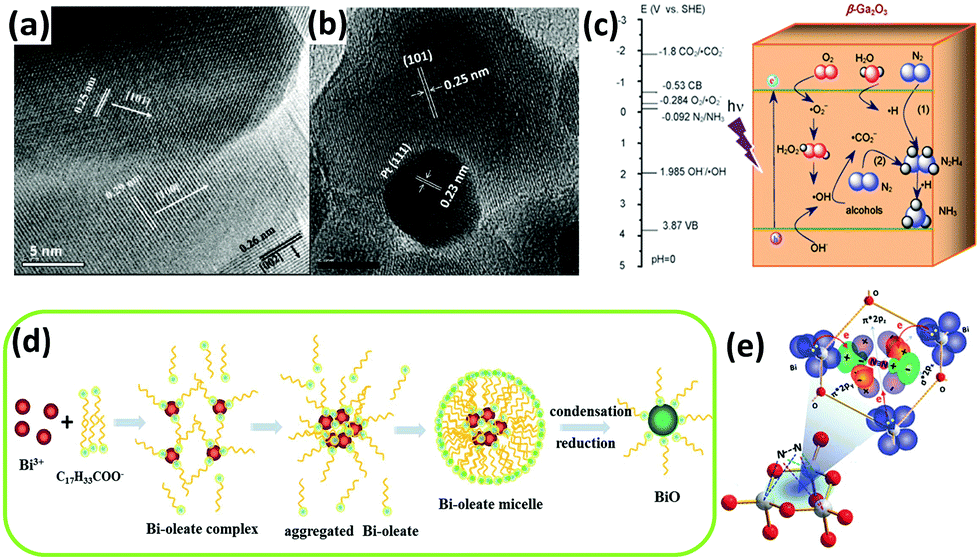 | ||
| Fig. 5 HRTEM images of (a) the fusion of three prominent lattice planes along the wet etching ZnO grain boundaries and (b) a Pt cluster at the grain boundary. Reproduced with permission.118 Copyright 2010, American Chemical Society. (c) Possible electron transfer pathways for the conversion of N2 to NH3 over β-Ga2O3. Reproduced with permission.115 Copyright 2015, Royal Society of Chemistry. (d) Schematic of the synthesis process for BiO quantum dots. (e) A 1N2–3Bi(II) side-on bond structure formed by electron sharing. Reproduced with permission.116 Copyright 2017, Royal Society of Chemistry. | ||
In addition to the above-discussed TiO2 photocatalyst, as reported in Section 3.1.1., ferric oxide has been shown to be a compelling and effective photocatalyst for N2 photoreduction. Importantly, the narrow band gap of Fe2O3 guarantees its photocatalytic activity even in the visible light region.119 Although semiconductors with wide band gaps are favorable in the activation of N2 as a result of their high redox abilities, they remain limited in their UV response. It is worth mentioning that bismuth-containing systems may be useful in N2 fixation; this will be further corroborated in Section 3.3.
However, there are some controversial opinions on the possible use of iron titanate in N2 fixation, as reported in 2001.121 The dominant hypothesis for the conflicting results in Fe systems is mainly accredited to the different approaches to catalyst preparation.122 Using the sol–gel method, Rusina and his coworkers prepared iron titanate films with relatively high Fe contents by reacting iron chloride and tetraisopropyl titanate, leading to the successful development of a new Fe2Ti2O7 phase.121 After 90 min of light irradiation (λ ≥ 320 nm), a maximum ammonia yield of 17 μM was obtained at 75% alcohol content when Fe/Ti was fixed at 1:1. The produced ammonia was subsequently oxidized to nitrite in the presence of ethanol. Meanwhile, in another report, Zhao et al. revealed that ethanol could prevent the oxidation of ammonia.99 In a continuation of their work, further investigations on iron titanate films were conducted by Kisch and Linnik.123 The electron transfer mechanism of N2 fixation on the Fe2Ti2O7 thin films involved a series of photochemical processes of nitrogen → diazene → hydrazine → ammonia → nitrate. In addition, the essential role of chloride ions was discussed. Although Schrauzer and Soria have noted that the generation of TiO2 using chloride ions is unfavorable,98,124 both the inhibiting and accelerating effects of iron chloride as the precursor were corroborated.
In addition to iron titanate systems, titanate-like SrTiO3 and BaTiO3 have also been investigated under UV irradiation for N2 photoreduction. Judging from the results of the experiments, undoped SrTiO3 and BaTiO3 produced minimal NH3 yields (0.82 and 1.74 μmol gcat−1 after 2 h of reaction, respectively). Their yields of NH3 reached 5 to 5.2 μmol gcat−1 in 2 h when doped with RuO2 and NiO.125 In another paper, plasmonic-metal/semiconductor photocatalysts elicited great attention for their high light-harvesting properties.126 Using gold nanoparticles (Au-NPs) as the plasmonic nanostructure, Oshikiri and his co-workers designed Au-NPs/Nb-SrTiO3/Ru (Fig. 6a) and Au-NPs/Nb-SrTiO3/Zr/ZrOx (Fig. 6b–d) semiconductor photoelectrodes in 2014 and 2016, respectively.127,128 Au-NPs were loaded on a 0.05 wt% niobium-doped strontium titanate (Nb-SrTiO3) single crystalline substrate, and Ru or Zr film as a co-catalyst was deposited on the opposite side of Nb-SrTiO3 (Fig. 6c and d). Zr was oxidized to ZrOx. Ethanol was added as a sacrificial electron donor in the anodic chamber. In later reports, the production rates of Zr/ZrOx and Ru systems were compared to evaluate the production selectivity. Interestingly, H2 was the main product in the case of Ru systems, indicating that proton reduction is the primary reaction on the Ru co-catalyst. In contrast, in the case of Au-NPs/Nb-SrTiO3/Zr/ZrOx, the production rate of NH3 was conspicuously higher because Zr and ZrOx bind N more favorably than H.129 However, its NH3 production rate was still obviously lower than that of the electrosynthesis of ammonia.130 Recently, sparked by the unique features of Mo element in the Mo-cofactor of nitrogenase enzymes, Hao et al. fabricated hydrogenated bismuth molybdate (H-Bi2MoO6) which was capable of reducing N2 under sunlight.131 In this context, the hydrogenation reaction induced oxygen vacancies on Bi2MoO6 (Fig. 6e–h), endowing H-Bi2MoO6 with activity under visible light for N2 fixation. Using air instead of pure nitrogen, the NH3 production rate under sunlight was as high as 1.3 mmol g−1 h−1. This high nitrogen activation could not be attained if molybdenum was replaced with tungsten as a counterpart or if the hydrogenation process was omitted.
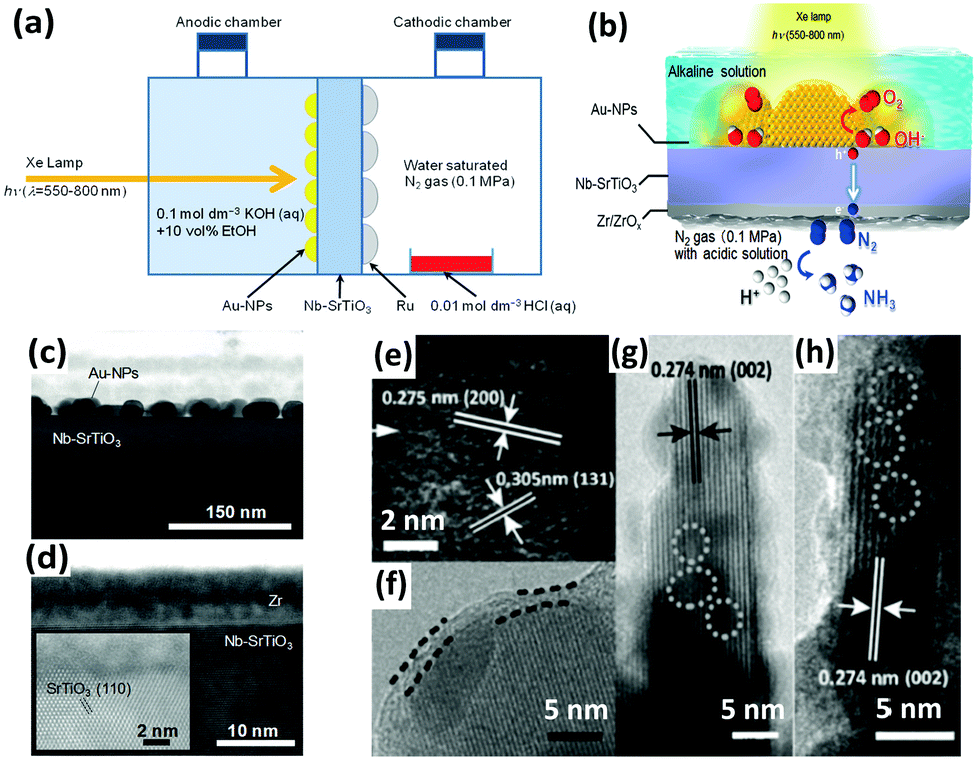 | ||
| Fig. 6 (a) Schematic of the NH3 photosynthesis device using an Au-NPs/SrTiO3 photoelectrode. Reproduced with permission.127 Copyright 2014, Wiley-VCH. (b) Diagram of NH3 synthesis on the Au-NPs/Nb-SrTiO3/Zr/ZrOx photoelectrode. Cross-sectional view of the bright field scanning TEM (BF-STEM) images of (c) the Au-NPs/Nb-SrTiO3 interface and (d) the Zr film deposited onto Nb-SrTiO3. Reproduced with permission.128 Copyright 2016, Wiley-VCH. (e–h) HRTEM images of H-Bi2MoO6 (the superficial and lattice disorders induced by defects are marked by parallel dashed lines and circles, respectively). Reproduced with permission.131 Copyright 2016, Wiley-VCH. | ||
Judging from the existing literature, we can conclude that most of the investigations of ternary metal oxides are focused on titanate systems.121–123,125,127,128 Iron titanate exhibits relatively high photoactivity in N2 fixation under UV irradiation, in a pure nitrogen flow, and in the presence of organic scavengers. On the other hand, bismuth molybdate has been reported to enable impressive NH3 production rates after performing the hydrogenation process under mild conditions. However, for N2 activation, strontium titanate does not reach the general level of other catalysts even after modification by doping or plasmon induction.
In the initial study by Tennakone et al., amorphous Fe2O3 (H2O)n was prepared by gradually adding KOH solution to FeCl3 solution. After saturation with N2, the solution was irradiated under a 100 W tungsten lamp (IR and UV filtered off). A maximum NH3 yield of ca. 4 μM was detected after 40 min.132 In 1991, ultrafine particles of Fe(O)OH were prepared from photohydrolysis of iron(II) bicarbonate by the same author. Nitrate was also detected in the solution after prolonged irradiation. The initial quantum yield of NH3 was about 10−2, which is higher than that of hydrous ferric oxide (∼10−3).133 Ileperuma et al. claimed that a hydrous ferric oxide-loaded bentonite system produced more NH3 and less nitrate than the parent hydrous ferric oxide under UV irradiation.137 In contrast with pure hydrous ferric oxide, a system of coprecipitating Fe(III) and Ti(IV) hydrous oxides (Ti to Fe ratio = ∼8%) was found to be more effective in the generation of NH3, while the rate of nitrate production using this composite was comparatively low.138 Hydrous ferric oxide was believed to play roles in nitrogen chemisorption and reduction, while TiO2 provided hole accumulation centres. Although the authors revealed the predominant effect of the heterojunction interface in the mechanism of electron–hole separation, they still questioned the meaning of these concepts in a colloidal system. In 1992, the yield of vanadium-substituted hydrous ferric oxides was ameliorated under UV irradiation. V3+ sites demonstrated distinctive attributes in capturing photogenerated holes in the mixed hydroxide.139 With the incorporation of V3+, a maximum NH3 yield of about 200 μM was reached in about 24 h when the vanadium-to-iron ratio was at unity. The subsequent decrease of NH3 concentration was ascribed to the inactivation of catalyst followed by the photocatalytic decomposition of NH3. This reason for the decrease of ammonia was also suggested in 1987 because of the continuous generation of H2 and the unchanged content of O2.132 Conversely, Ileperuma et al. pointed out that NH3 was oxidized to nitrate because NO2− and NO3− were detected in the final products along with a decrease in NH3 yield.137
Additionally, hydrous cuprous oxide was able to photoreduce N2 to NH3 after impregnation with cuprous chloride, which underwent sacrificial oxidation to form cupric chloride. The high ammonia yield presumably resulted from the separation of oxidation and reduction sites as well as the chemisorption of N2 on Cu2O·xH2O, which served as reduction sites.134 Pursuing the exciting idea in a previous study of coprecipitated hydrous oxides of Fe and Ti,138 a coprecipitate of samarium(III) and vanadium(III) was subsequently explored.135 The maximum NH3 yield was obtained within 1 h when the V-to-Sm ratio was ∼1. When employing this composite, the ammonia production rate was strikingly higher than that of previously reported vanadium-substituted hydrous ferric oxide.139 Very recently, highly efficient N2 photoreduction was achieved over carbon-WO3·H2O (HWO-C) in water.136 WO3·H2O was selected as the photocatalyst due to its extraordinary electron and proton conductivity. As a result, the rapid transfer of photogenerated electrons and water-derived protons to N2 was enhanced. In this circumstance, carbon modification prominently enhanced the NH3 production rate of HWO by increasing its surface activation and simultaneously promoting the separation and transport of charge carriers to retard the electron–hole recombination rate. A similar phenomenon in the use of other carbon-modified photocatalysts for potential energy applications has also been reported by our research group.140 Therefore, it is evident that the modification of carbon plays a decisive role in suppressing the recombination of electron–hole pairs by prolonging the lifetime of charge carriers for enhanced photoactivity.
3.2. Metal sulfide-based materials
In addition to the large family of metal oxide-based photocatalysts, the exploration of metal sulfides has also experienced a tremendous upsurge in the scientific community, especially in the field of photocatalysis.141–143 The narrow band gap of metal sulfides is conducive to strong absorption of visible light, which results in highly efficient solar utilization efficiency.In 1980, Miyama et al. employed CdS semiconductors for the fixation of nitrogen under UV irradiation; the recorded yield of NH3 in 5 h was 10.67 μmol gcat−1.104 They constructed CdS/Pt binary wafered catalysts whose NH3 yield was drastically higher than that of pristine CdS. The remarkable performance of the loading of Pt co-catalysts has heightened research interest in other potential modifications of CdS photocatalysts. In 1988, Khan et al. reported the fixation of N2 with a CdS/Pt/RuO2 semiconductor particulate system under the illumination of visible light (λ = 505 nm).144 Dinitrogen was activated to react with [Ru(Hedta)H2O]− to form the Ru(II) dinitrogen complex. The holes in the valence band trapped the electrons released from RuO2. Two years later, a similar research team conducted the stepwise reduction of dinitrogen to ammonia on visible-light-responsive Pt/CdS·Ag2S/RuO2 photocatalysts.145 The addition of silver facilitated the migration of electrons from the conduction band to minimize the photocorrosion of CdS. The production rate of NH3 was twice that in the absence of silver. In view of the obtained results, it is clear that noble metals such as Pt, Ag and Ru work well with CdS. The decrease in the yield of NH3 over time may originate from the photocorrosion of CdS to S and Cd2+.144 To retain the high photocatalytic activity and stability of CdS for the reduction of N2 to NH3, in 2017, Ye et al. demonstrated the fixation of N2 using Cd0.5Zn0.5S solid solution for the first time.146 To enhance the photocatalytic performance, a transition metal phosphide (Ni2P) was employed as a co-catalyst to rapidly transfer the photoinduced electrons to Ni2P via well-contacted heterointerfaces to diminish the charge recombination.
On the other hand, owing to the recent interdisciplinary interest in material science and biology, organic-sulfide catalysts have also been designed for enhanced N2 fixation activity. In 2016, Brown et al. reported nitrogen reduction by the MoFe protein, which is the active site of nitrogenase, adsorbed onto CdS nanorods to form biohybrid complexes.147 The NH3 production rate reached 315 nmol per mg MoFe protein per min under visible light. In this regard, CdS nanocrystals were used to photosensitize the MoFe protein so that the photon energy was derived from ATP (Fig. 7a). Furthermore, the rates were comparable to that of physiological NH3 production by nitrogenase with energy provided by ATP. Because the active sites of nitrogenase contain the elements Fe, Mo and S, a synthetic complex of Fe, Mo and S could be taken into account. By exploiting the advantage of the FeMo cofactor in nitrogenases, Banergee et al. inferred that biomimetic chalcogels comprising FeMoS inorganic clusters could reduce N2 to NH3 in aqueous media under light.148 The Fe2Mo6S8 chalcogel was an analog to the MoFe active site in the enzyme and was linked by Sn2S6 ligands to form an amorphous network (Fig. 7b). This study proved that structural analogues of nitrogenase can be functional and can even confer better properties than nitrogenase. In 2016, to make a better mimic of nitrogenases, not only MoFe protein but also Fe protein was considered by Liu et al.149 They employed a chalcogel system consisting of Fe2Mo6S8(SPh)3 and Fe3S4 biomimetic clusters linked by Sn2S6. Iron was believed to be more vital than molybdenum for the light-driven reduction of nitrogen due to the fact that a weak bonding orbital between nitrogen and iron was manifested through localized orbital analysis. More importantly, their conclusion that iron is a better active site for N2 binding than Mo has also been revealed by recent biochemical and spectroscopic data.19,150
 | ||
| Fig. 7 (a) Reaction scheme for N2 reduction to NH3 by CdS:MoFe protein biohybrids. Reproduced with permission.147 Copyright 2016, American Association for the Advancement of Science. (b) Schematic of the composition of Mo2Fe6S8–Sn2S6 biomimetic chalcogel. Reproduced with permission.148 Copyright 2015, American Chemical Society. (c) HRTEM image of g-C3N4/ZnMoCdS. (d) Schematic of the charge transfer process at the heterojunction interface of g-C3N4/ZnMoCdS. Reproduced with permission.155 Copyright 2016, Royal Society of Chemistry. (e) Schematic of the trion-induced multi-electron N2 reduction process. Reproduced with permission.71 Copyright 2017, Elsevier. | ||
Motivated by the successful fixation of N2 under visible light with oxygen vacancies on the surface of BiOBr,151 a number of researchers wondered if sulfur vacancies could eventually promote photoactivity. Inspired by this idea, Hu et al. obtained Zn0.1Sn0.1Cd0.8S with sulfur vacancies formed by multi-component metal sulfides as catalysts for reducing N2 under visible light.152 Surprisingly, the sulfur vacancies introduced chemical adsorption sites on the surface, which aided the activation of N2 molecules by extending the bond distance between the nitrogen atoms in N2. It was also implied that the generation rate of NH3 was linearly related to the concentration of sulfur vacancies. Additionally, the sulfur vacancies also trapped electrons, thus promoting the separation of photoinduced electrons and holes. In the same year, a similar quaternary system of Mo0.1Ni0.1Cd0.8S, which contributed to the reduction of N2 under visible light, was published by Cao et al.153 At the same time, tri-component metal sulfides were studied by this group. Two similar heterostructures of g-C3N4/ZnSnCdS and g-C3N4/ZnMoCdS were employed for N2 photoreduction.154,155 In the hybrid of g-C3N4/ZnMoCdS (Fig. 7c), a tight heterojunction coupling between g-C3N4 and ZnMoCdS was pivotal for efficient charge transfer. The photogenerated electrons were transferred from g-C3N4 to the metal sulfide, while the holes were transported in the opposite direction (Fig. 7d).155 This clearly accounted for the improved visible light absorption after hybridization. Furthermore, the redistribution of electrons and holes on each side of the heterojunction established an internal electric field to impede the recombination of charge carriers.
As a transition metal dichalcogenide (TMD), it is well-known that MoS2 possesses excellent electrical,156 optical,157 and optoelectronic properties.158 In 2017, Sun et al. reported photocatalytic N2 reduction to NH3 with ultrathin MoS2.159 In this paper, the photocatalytic performances of MoS2 samples under different preparation conditions were studied; it was found that the sonicated ultrathin MoS2 sample generated a large amount of NH3 with favorably high stability. In ultrathin TMDs, the tightly bound excitons could facilely capture additional electrons to form charged excitons with more than two electrons in one bound state.160 Thus, it is believed that these charged excitons functioned as electron-rich species to facilitate the multi-electron reduction process of molecular N2 (Fig. 7e). Particularly, a six-electron transfer profess was responsible for the acceleration of photocatalytic N2 reduction to NH3.
3.3. Bismuth oxyhalides
Bismuth oxyhalides, BiOX (X = Cl, Br, and I), have recently become well known for their superior optical properties; they are also propitious for industrial applications, namely the photodecomposition of organic pollutants.161 The layered structure of BiOX provides adequate space for the polarization of atoms, and the as-formed internal electric field will contribute to the efficient separation and transfer of charge carriers.162,163In recent years, BiOX has also been reported for applications in solar nitrogen fixation.61,86,88,151,164 In 2015, Li et al. employed {001}-faceted BiOBr nanosheets with oxygen vacancies (OVs) to reduce N2 under visible light.151 In this scenario, the N2 fixation rate was estimated to be 104.2 μmol h−1 (per gram of BOB-001-OV). Remarkably, it was a quantum leap for N2 fixation under visible light in the absence of organic scavengers. Theoretical simulations revealed that the OVs of BiOBr could elongate the NN triple bond of adsorbed N2 from 1.078 to 1.133 Å (Fig. 8a), which subsequently promoted the activation of N2 molecules. Moreover, the large number of OVs on the surface of BiOBr formed a defect state lying near the bottom of the conduction band of the photocatalyst, hampering the recombination of electron–hole pairs. However, the surface OVs of BiOBr are naturally oxidized, leading to reduced activity. Li's group also examined the photocatalytic activity of BiOCl with OVs on different facets.88 They claimed that the kinetics and mechanism of N2 fixation varied due to the dissimilar facets of BiOCl nanosheets. They highlighted that nitrogen fixation with OVs on the {001} facets followed a distal pathway (N2 → N-NH3 → N + NH3 → 2NH3), whereas the reaction on the {010} facets followed an alternative pathway (N2 → N2H3 → N2H4) (Fig. 8b). The quantum yields were 1.8% h−1 and 4.3% h−1 for OVs on the {001} and {010} facets of BiOCl, respectively, under UV light (λ = 254 nm). In the most recent work by Ye's group, ultrafine light-switchable OVs of Bi5O7Br nanotubes with a diameter of ca. 5 nm and large exposed surface sites were synthesized via a water-assisted low-temperature wet chemical approach (Fig. 8c–e).164 The NH3 generation rate of nanotubes (1.38 mmol h−1 g−1) was 2.5 times higher than that of nanosheets, with an apparent quantum efficiency (AQE) as high as 2.3% at 420 nm.
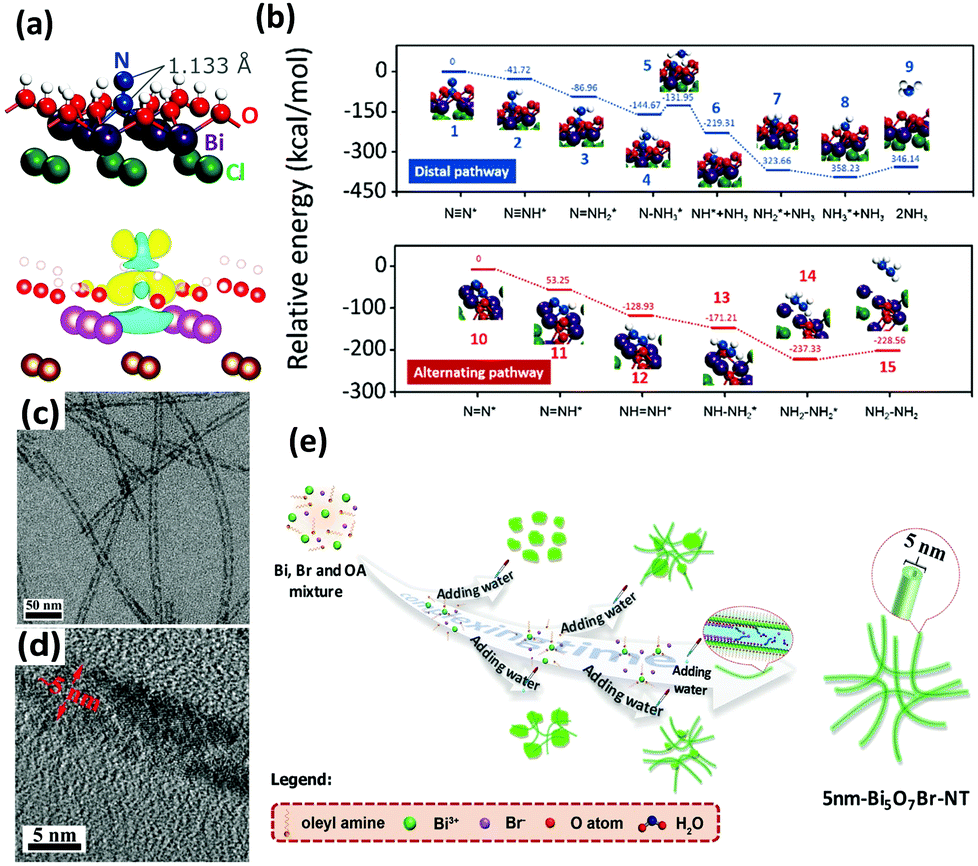 | ||
| Fig. 8 (a) The adsorption geometry and the charge density difference of N2 on the OVs of the BiOBr (001) surface (the yellow and blue isosurfaces represent charge accumulation and depletion, respectively). Reproduced with permission.151 Copyright 2015, American Chemical Society. (b) Free energy profiles of OV-mediated N2 fixation on the (001) and (010) surfaces of BiOCl. Reproduced with permission.88 Copyright 2016, Royal Society of Chemistry. (c and d) TEM images of Bi5O7Br nanotubes at different magnifications. (e) Schematic of the synthesis procedure of ultrafine Bi5O7Br nanotubes. Reproduced with permission.164 Copyright 2017, Wiley-VCH. | ||
As a continuation of the previous research by Zhang's and Ye's groups,88,164 Bai et al. utilized bismuth-rich bismuth oxyhalide (Bi5O7I) nanosheets with different dominant {001} and {101} facets.89 The N2 fixation rates of Bi5O7I-001 and Bi5O7I-100 were 111.5 μmol L−1 h−1 and 47.6 μmol L−1 h−1, respectively. After 5 cycles, the photocatalytic properties and structures of Bi5O7I-001 and Bi5O7I-100 elucidated high stabilities for the N2 fixation process. The more negative conduction band potential of Bi5O7I-001 (−1.45 V) compared to that of Bi5O7I-100 (−0.85 V), explains the higher NH3 production rate of Bi5O7I-001. In short, although most literature studies place significant emphasis on the exposure of different facets, it is conceivable that the combined effects of OVs and facet-dependent studies will open a new outlook and provide inspiration for the development of advanced photocatalysts for N2 photoreduction. Additionally, the combination of experimental results and theoretical simulations is highly necessary to fully elucidate the mechanism of NN triple bond activation and the NH3 formation pathway. Thus, more emphasis on facet-controlled and vacancy-mediated bismuth oxyhalides should be devoted in the future to accentuate the scientific aspects and unravel the exact reaction steps for N2 photofixation.
3.4. Carbonaceous materials
Metal-free semiconductor photocatalysts have triggered a renaissance of great interest since the advent of H-terminated boron-doped (B-doped) diamond as a solid-state source of electrons in water for photo-driven N2 fixation in 2013.165 It was demonstrated that the UV excitation of B-doped diamond induced electron emissions and their ejection into liquids to produce solvated electrons which reacted quickly with protons to form neutral atomic hydrogen, H˙, and finally facilitated the subsequent reaction with N2 to yield NH3. In this context, the photocatalytic activity decreased with time because the oxidation of the H-terminated surface to O-terminated resulted in a loss of electron affinity. The authors also revealed that photocatalytic behaviour could be observed using the natural diamond dispersed in water. Further study of the mechanism of action of aqueous solvated electrons was methodically reported in the following year.166 Three possible reaction steps, including (1) electron transfer (N2 + e− → N2−), (2) proton-coupled electron transfer (N2 + e− + H2O → N2H + OH−) and (3) hydrogen atom addition (N2 + H˙ → N2H), were proposed with the help of kinetic modelling. It was concluded that the reduction of N2 to NH3 involved hydrogen atom addition at the initial steps and protonation/electron transfer by solvated electrons at the later steps. In 2016, metal-diamond heterostructures (i.e. diamond thin films grown on Mo, Ni and Ti metal substrates) were reported. It was noted that some electrons excited from the metal substrates would be injected into the conduction band of the diamond film, followed by emission into water.167In the present literature, most research has focused a spotlight on carbonaceous nanomaterials. Benefiting from the rational significance of the oxygen vacancies in bismuth oxyhalides, as described in Section 3.3.,151 graphitic carbon nitride (g-C3N4) with nitrogen vacancies (NV-g-C3N4) has become a focal area in materials science since 2015.73 Because they have the same shape and size as the nitrogen atoms in dinitrogen, nitrogen vacancies (NVs) are beneficial in the selective chemisorption and activation of N2. This explains why the nitrogen fixation rate remained unchanged when N2 was replaced by air as the N2 source. In addition, NVs prominently improved the separation of charge carriers by trapping photogenerated electrons and promoting electron transfer to the adsorbed N2.62 Following this astounding discovery, NV-g-C3N4 was extensively investigated based on manifold aspects, namely the modification of preparation routes for large-scale production and the enlargement of the specific surface area.168–171 Furthermore, modifications such as iron doping and ruthenium loading have been exhaustively explored in recent years.172,173 Similar to diamond, the H-termination of Ru-loaded g-C3N4 played an indispensable role in the activation of N2.170
Recently, g-C3N4 has been widely employed to construct heterostructure junctions with another component, including the hybridization of g-C3N4 with metal sulfides,154,155 metal oxides,174–176 and reduced graphene oxide (rGO).177 As a proof of concept, multi-metal oxides with tunable band structures match favorably with other semiconductors when engineering intact heterojunction interfaces.178 After assembly, the g-C3N4/MgAlFeO nanorod heterostructure was effective in N2 photoreduction. In particular, a Z-scheme heterojunction was established, as depicted in Fig. 9a. In this instant, the photoexcited electrons in the conduction band of MgAlFeO were transferred to the valance band of g-C3N4. Due to this phenomenon, the electrons in the conduction band of g-C3N4 did not recombine with the holes, instead participating in the N2 reduction.174 A similar Z-scheme heterojunction mechanism for N2 fixation was reported in other studies: (1) W18O49/g-C3N4 catalyst (Fig. 9b and c) was active under near-infrared (NIR) irradiation175 because of the coherent oscillations of the surface free electrons induced by the oxygen vacancies on W18O49. (2) 3,4-Dihydroxybenzaldehyde-functionalized Ga2O3/graphitic carbon nitride (Ga2O3-DBD/g-C3N4), synthesized by incorporating Ga2O3-DBD NPs into g-C3N4 networks (Fig. 9d),176 was reported to be capable of converting N2 to NH3 with the aid of the strong reducing power of ˙CO2−, which was formed by the oxidation of methanol by active oxygen species (Fig. 9e). In essence, this is similar to the N2 reduction mechanism of β-Ga2O3, as discussed earlier in Fig. 5c.
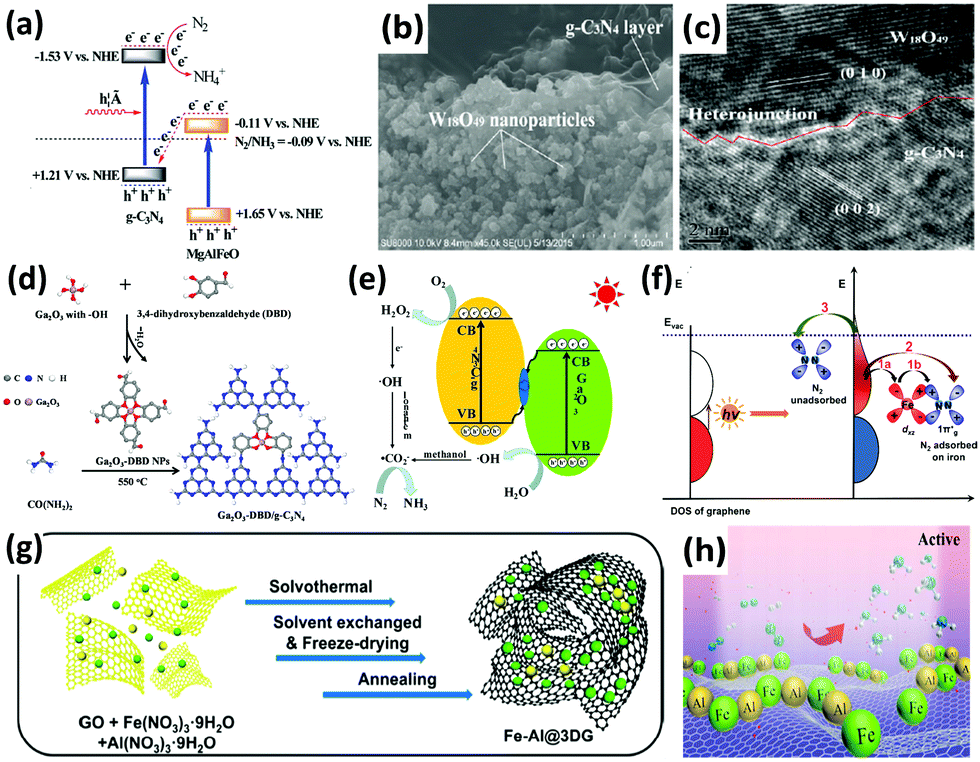 | ||
| Fig. 9 (a) Tentative Z-scheme heterojunction interface of g-C3N4/MgAlFeO. Reproduced with permission.174 Copyright 2017, Royal Society of Chemistry. (b) SEM and (c) HRTEM images of WO18O49/g-C3N4. Reproduced with permission.175 Copyright 2017, Royal Society of Chemistry. (d) The formation process and (e) photocatalytic N2 reduction mechanism of Ga2O3-DBD/g-C3N4. Reproduced with permission.176 Copyright 2017, Elsevier. (f) Possible paths of photo-generated electrons from graphene for the activation of N2 molecules. Reproduced with permission.183 Copyright 2016, American Chemical Society. (g) Schematic of the synthesis procedure of the Fe-Al@3DG and (h) NH3 synthesis over Fe-Al@3DG. Reproduced with permission.184 Copyright 2017, Elsevier. | ||
As for two-dimensional (2D) graphene, a metal-free 2D/2D hybrid heterostructure of g-C3N4/rGO was designed by coupling rGO and protonated g-C3N4, in which rGO served as a conducting substrate.177 Protonated g-C3N4 exhibited 5 times the NH3 production rate of pristine g-C3N4, and the activity was further ameliorated after assembly with rGO. It was deduced that the face-to-face contact area on the g-C3N4/rGO heterointerface for enhanced charge transfer substantially decreased the electron–hole recombination rate of g-C3N4.179 In essence, this finding is analogous to our previous studies on metal-free 2D/2D graphene/g-C3N4 for CO2 photoreduction, in which graphene acts as an excellent electron reservoir.180–182 There is another report on the loading of iron oxide on bulk three-dimensional (3D) cross-linked graphene (Fe@3DG) for the reduction of N2 to NH3 under light irradiation.183 Graphene played the role of generating highly energetic electrons. A significant enhancement of photocatalytic activity was observed by increasing the electron density of Fe due to its electron donation from the Fe d orbital to the anti-bonding orbital of N2 (Fig. 9f).
Because the photoactivity of the Fe@3DG nanocomposite significantly decreased after 5 hours of reaction, in 2017, the same research group modified the existing Fe@3DG nanocomposite by incorporating Al2O3 to form Fe-Al@3DG photocatalysts (Fig. 9g).184 Al2O3 functioned as an excellent dispersing agent and barrier to prevent the aggregation of Fe2O3 nanoparticles on the 3D graphene (Fig. 9h). Fascinatingly, the NH3 production yield of 20Fe-2Al@3DG (20 wt% Fe and 2 wt% Al) remained steady even after 60 hours of light-driven reaction, which was more than two times that of the parent 20Fe@3DG (20 wt% Fe), demonstrating the salient role of Al2O3 as a structural promoter. In general, the employment of 2D/2D heterojunction interfaces and 3D nanoarchitecture design for photocatalysis based on the motivation of effective charge carrier transfer and separation is still in the infant stage. Furthermore, most existing literature studies focus on 0D/2D hybrid nanostructures. Therefore, extensive studies in this related field are imperative to achieve excellent prospects in addressing the limitations of this process for scientific merit and to enhance fundamental research knowledge toward realizing industrialization in the future.
3.5 Other potential materials
Gallium phosphide is a typical semiconductor that has been researched in the photocatalytic field for a long time.185,186 In 1978, Dickson et al. employed a photoelectrochemical cell to perform N2 fixation.187 The system consisted of a p-GaP cathode and an aluminum metal anode immersed in a non-aqueous electrolyte of titanium tetraisopropoxide and AlCl3 dissolved in glyme. In this system, the p-GaP electrode absorbed light and provided the activation energy for the photoenhanced reduction of N2. In 1980, Miyama et al. reported the performance of GaP and GaP/Pt for the reduction of N2. In fact, a few researchers have already implemented the reduction of N2 on GaP nanoparticles without light.104Zeolites have also been applied in the photochemical synthesis of NH3 from N2. Khan et al. reported the production of ammonia with titanium-exchanged zeolites under visible light.188 Two years later, they tested the exchange of different types of zeolites with titanium ions. It is noteworthy that the Ti3+ ions contributed to the reduction of N2 for the oxidation of Ti3+ to Ti4+.189 Furthermore, the NH3 production was reported to increase with increasing re-exchange time of the zeolites by Ti3+.
As mentioned above,127,128 localized surface plasmon resonance (LSPR) of metal particles has also been utilized to drive the photoabsorption of other catalysts. In 2014, Zeng et al. applied catalytically active metal (Os) directly to LSPR Au particles (Os–Au nanocomposites) for the NH3 synthesis reaction.190 It was found that the Au nanoparticles with superior LSPR absorbance and photoabsorption did not show any catalytic activity, while Os nanoparticles with high catalytic activity could not absorb photoenergy. By allying the concept of LSPR Au particles with the active metal (Os), the Os–Au nanohybrids outperformed the individual components, thus improving the solar light utilization for the chemical synthesis of NH3. In 2016, a solar-driven photoelectrochemical cell based on plasmon-enhanced black silicon (bSi) was documented by MacFarlane's group.191 The plasmon-enhanced bSi was decorated with Au nanoparticles (GNPs) and a layer of Cr (Fig. 10a–c). In the GNP/bSi/Cr photoelectrochemical cell, bSi, GNPs and Cr performed as photoabsorber, reduction catalysis sites and hole-sink layer, respectively. The bSi ameliorated the scattering and absorption of light and provided a sufficient surface area for the decoration of a large amount of GNPs. From the charge transfer mechanism, the GNPs accumulated photogenerated electrons, whereas Cr acted as a sacrificial anode to gather holes. This research resulted in hundreds of times improvement in the NH3 production rate compared with plasmon-induced photoelectrodes (Au NPs/Nb-SrTiO3/Ru and Au NPs/Nb-SrTiO3/Zr/ZrOx), as mentioned in Section 3.1.3.127,128
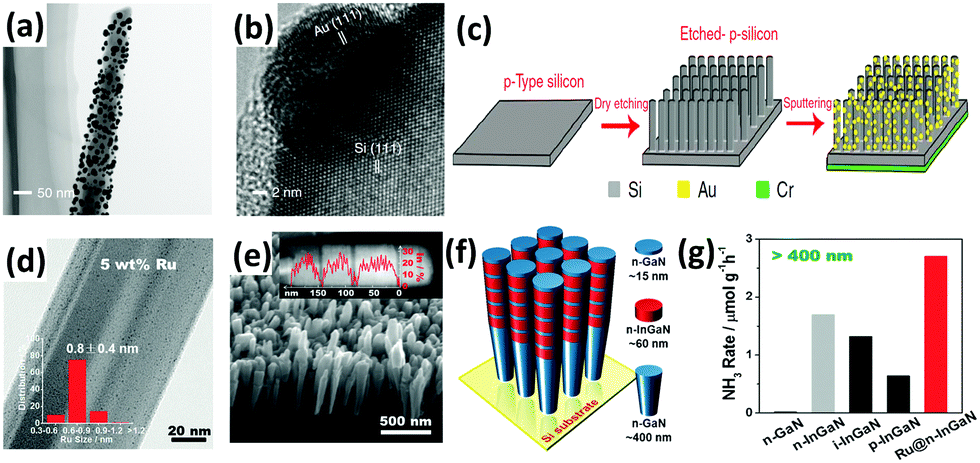 | ||
| Fig. 10 (a) TEM and (b) HRTEM images of a GNPs-coated p-type silicon nanowire. (c) Schematic of the fabrication of the GNP/bSi/Cr photoelectric cell. Reproduced with permission.191 Copyright 2016, Nature Publishing Group. (d) TEM image of the 5 wt% Ru-modified GaN NWs (inset plot shows the diameter distributions of the Ru sub-nanoclusters). (e) SEM image of the developed InGaN/GaN nanowires on the Si (111) substrate. (f) Schematic of the InGaN/GaN nanowire structure. (g) Rate of NH3 generation over various III-nitride semiconductors under visible light irradiation (λ > 400 nm). Reproduced with permission.192 Copyright 2017, Wiley-VCH. | ||
A similar one-dimensional (1D) structure of gallium nitride nanowires (GaN NWs) grown on a silicon (Si) substrate was very recently designed by Li and co-workers.192 The Ge-doped n-type GaN NWs exhibited improved N2 photofixation activity under UV irradiation with the aid of ultrasmall Ru clusters of ca. 0.8 nm (Fig. 10d). After the incorporation of Ru, the photoexcited electrons were facilely transferred from the III-nitride semiconductors to Ru as a result of an effective interfacial metal/semiconductor Schottky junction with a barrier height of 0.94 eV, thus demonstrating the role of Ru as an electron reservoir to facilitate the cleavage of NN triple bonds. Furthermore, the band gap was shifted to the visible light region (2.34 eV) by introducing In (ca. 25% composition) into GaN NWs, forming 5 segments of n-type InGaN NWs on the GaN NW template (Fig. 10e and f). Assisted by Ru sub-nanoclusters, the photoactivity of n-type InGaN/n-GaN toward NH3 synthesis was the highest compared to other reference samples under visible light irradiation (λ > 400 nm) (Fig. 10g). For ease of comparison of the selected literature studies, Tables 2 and 3 summarize most of the photocatalysts, including metal oxide-based, metal sulfide-based, bismuth oxyhalides (BiOX), and metal-free carbonaceous nanomaterials.
| Year | Catalyst | T (K) | Light source | Nitrogen source | Organic scavenger | NH3 average rNH3 | Author | Ref. | |
|---|---|---|---|---|---|---|---|---|---|
| a The unit mM h−1 cm−2 is not standardized because the volume of the solution was not given in the literature. b The catalyst cannot be weighed because it denatures upon drying. c rt denotes room temperature. d ϕ = apparent quantum yield. e 2-PrOH denotes 2-propanol. f EDTA denotes ethylenediaminetetraacetic acid. g TBA denotes tert-butanol. | |||||||||
| TiO2-Based Materials | 1997 | 0.2 wt% Fe-doped TiO2 | 313 | 390–420 nm | N2 | None | 10 μmol g−1 h−1 | Schrauzer | 67 |
| 1988 | TiO2 | 500 | UV | N2 | None | 0.83 μmol g−1 h−1 | Bourgeois | 91 | |
| 1991 | 0.5 wt% Fe-doped TiO2 | 353 | UV | N2 | None | 6 μmol g−1 h−1 | Soria | 98 | |
| 2014 |
Fe-doped TiO2
(Fe/Ti = 10−4) |
298 | λ = 254 nm | Air | Ethanol | 1 gcat L−1; 400 μM h−1 | Zhao | 99 | |
| 2017 | JRC-TIO-6 (rutile) | 313 | λ > 254 nm | N2 | 2-PrOHe | 1 gcat L−1; 2.5 μM h−1 | Hirakawa | 92 | |
| 1977 | 0.4 wt% Co-doped TiO2 | 313 | UV | N2 | None | 6.3 μmol g−1 h−1 | Schrauzer | 67 | |
| 1977 | 0.4 wt% Mo-doped TiO2 | 313 | UV | N2 | None | 6.7 μmol g−1 h−1 | Schrauzer | 67 | |
| 1977 | 0.4 wt% Cr-doped TiO2 | 313 | UV | N2 | None | 0.37 μmol g−1 h−1 | Schrauzer | 67 | |
| 1988 | 0.5 wt% Cr-doped TiO2 | 353 | UV | N2 | None | 2.6 μmol g−1 h−1 | Palmisano | 100 | |
| 1990 | 2 wt% Mg-doped TiO2 | — | UV | N2 | None | 0.67 gcat L−1; 6.9 μM h−1 | Ileperuma | 102 | |
| 1993 | 10 wt% V-doped TiO2 | — | UV | N2 | None | 0.8 gcat L−1; 4.9 μM h−1 | Ileperuma | 103 | |
| 1993 | 10 wt% Ce-doped TiO2 | — | UV | N2 | None | 0.8 gcat L−1; 3.4 μM h−1 | Ileperuma | 103 | |
| 1996 | 0.24 wt% Ru-loaded TiO2 | — | UV-vis | N2 | Ethanol | 22.7 μmol g−1 h−1 | Ranjit | 106 | |
| 2001 | TiO2/P3MeT | 293 | UV | N2 | None | — | Hoshino | 109 | |
| 2008 | 5% RuCl3-modified TiO2 | rtc | UV-vis | N2 | Humic acid | 4 μM h−1 cm−2a |
Linnik | 109 | |
| Other binary oxides | 1987 | Partially reduced Fe2O3 | 303 | UV-vis | N2 | None | 10 μmol g−1 h−1 | Khader | 111 |
| 2010 | Pt-loaded ZnO | — | UV | N2 | Na-EDTAf | 860 μmol g−1 h−1 | Janet | 118 | |
| 2015 | Mesoporous β-Ga2O3 nanorods | 298 | λ = 254 nm | N2/O2 | TBAg |
ϕ = 36.1%d |
Zhao | 115 | |
| 2017 | Fe2O3 | 298 | UV-vis | N2 | Ethanol | 0.5 gcat L−1; 1362.5 μM h−1 | Lashgari | 113 | |
| 2017 | BiO quantum dots | 298 | UV-vis | N2 | None | 0.25 gcat L−1; 50.5 μM h−1 | Sun | 116 | |
| Ternary metal oxides | 1983 | BaTiO3 | 313 | UV | N2 | None | 0.87 μmol g−1 h−1 | Li | 125 |
| 1983 | RuO2–NiO–BaTiO3 | 313 | UV | N2 | None | 2.6 μmol g−1 h−1 | Li | 125 | |
| 2001 | Iron titanate films (Fe/Ti = 1:1) |
— | λ ≥ 320 nm | N2 | Ethanol | 0.57 μM h−1 cm−2a |
Rusina | 121 | |
| 2014 | Au NPs/Nb-SrTiO3/Ru | rtc | 550–800 nm | N2 | Ethanol | 1.1 nmol h−1 cm−2 | Oshikiri | 127 | |
| 2016 | Au NPs/Nb-SrTiO3/Zr/ZrOx | rtc | 550–800 nm | N2 | Ethanol | 6.5 nmol h−1 cm−2 | Oshikiri | 128 | |
| 2016 | H-Bi2MoO6 | rtc | Sunlight | Air | None | 1300 μmol g−1 h−1 | Hao | 131 | |
| Hydrous oxides | 1987 | Fe2O3(H2O)n | — | Vis | N2 | None | 6 μM h−1b |
Tennakone | 132 |
| 1988 | Hydrous oxides of Fe(III) and Ti(IV) (Ti:Fe = 8%) |
— | Vis | N2 | None | 22 μM h−1b |
Tennakone | 138 | |
| 1989 | Cu2O·xH2O·CuCl | — | UV | N2 | None | 70 μM h−1b |
Tennakone | 134 | |
| 1991 | Fe(O)OH | — | UV | N2 | None | 9.25 μM h−1b |
Tennakone | 133 | |
| 1991 | Bentonite-hydrous ferric oxide | — | UV | N2 | None | 2 gcat L−1; 1.33 μM h−1 | Ilepruma | 137 | |
| 1992 | V(III)-substituted hydrous ferric oxide | 299 | UV | N2 | None | 8.33 μM h−1b |
Tennakone | 139 | |
| 1993 | Sm2O3·xH2O/V2O3·xH2O | 299 | UV | N2 | None | 100 μM h−1b |
Tennakone | 135 | |
| 2016 | Carbon-tungstic acid hybrids | — | UV-vis | N2 | None | 205 μmol g−1 h−1b |
Li | 136 | |
| Year | Catalyst | T (K) | Light source | Nitrogen source | Organic scavenger | NH3 average rNH3 | Author | Ref. | |
|---|---|---|---|---|---|---|---|---|---|
| a The catalyst cannot be weighed because it denatures upon drying. b rt denotes room temperature. c EDTA denotes ethylenediaminetetraacetic acid. d Using optical filters, light of λ > 800 nm and λ < 800 nm was removed for the 300 W Xenon lamp and 200 W infrared light source, respectively, to simulate full-spectrum light. | |||||||||
| BiOX | 2015 | BiOCl | 298 | UV-vis | N2 | Methanol | 0.67 gcat L−1; 46.2 μM h−1 | Li | 88 |
| 2015 | BiOBr | 298 | Vis | N2 | None | 0.5 gcat L−1; 1042 μM h−1 | Li | 151 | |
| 2016 | Bi5O7I | 293 | UV-vis | N2 | Methanol | 0.5 gcat L−1; 111.5 μM h−1 | Bai | 89 | |
| 2017 | Bi5O7Br nanotubes | rtb | λ > 400 nm | N2 | None | 1380 μmol g−1 h−1 | Wang | 164 | |
| Metal sulfides | 1980 | CdS/Pt | 311 | UV | N2 | None | 3.26 μmol g−1 h−1 | Miyama | 104 |
| 1988 | CdS/Pt/RuO2 | 303 | λ ≥ 505 nm | N2 | None | 4 gcat L−1; 620 μm h−1 | Khan | 144 | |
| 1990 | Pt/CdS·Ag2S/RuO2 | 303 | UV-vis | N2 | None | 2 gcat L−1; 1260 μM h−1 | Khan | 145 | |
| 2015 | [Mo2Fe6S8(SPh)3]3+-[Sn2S6]4− | rtb | UV-vis | N2 | — | 10.1 μM h−1a |
Banerjee | 148 | |
| 2016 | Mo2Fe6S8(SPh)3-Fe4S4-[Sn2S6]4− | rtb | UV-vis | N2 | — | 18.82 μM h−1a |
Liu | 149 | |
| 2016 | CdS/MoFe protein | — | λ = 405 nm | — | None | 315 μmol gprotein−1 min−1 | Brown | 147 | |
| 2016 | Zn0.1Sn0.1Cd0.8S | 303 | 400–800 nm | N2 | Ethanol | 0.4 gcat L−1; 105.2 μM h−1 | Hu | 152 | |
| 2016 | Mo0.1Ni0.1Cd0.8S | 303 | 400–800 nm | N2 | Ethanol | 0.4 gcat L−1; 71.2 μM h−1 | Cao | 153 | |
| 2016 | g-C3N4/ZnSnCdS | 303 | 400–800 nm | N2 | Ethanol | 0.4 gcat L−1; 167.6 μM h−1 | Hu | 154 | |
| 2016 | g-C3N4/ZnMoCdS | 298 | 400–800 nm | Air | Ethanol | 0.4 gcat L−1; 77.6 μM h−1 | Zhang | 155 | |
| 2016 | Ni2P/Cd0.5Zn0.5S | 293 | λ ≥ 400 nm | N2 | None | 0.4 gcat L−1; 101.5 μM h−1 | Ye | 146 | |
| 2016 | MoS2 | 298 | λ ≥ 420 nm | N2 | None | 325 μmol g−1 h−1 | Sun | 159 | |
| Carbonaceous materials | 2013 | Boron-doped diamond | — | UV | N2 | None | — | Zhu | 165 |
| 2015 | g-C3N4 | — | λ ≥ 420 nm | Air | Methanol | 1 gcat L−1; 160 μM h−1 | Dong | 62 | |
| 2016 | g-C3N4/rGO | 303 | 400–800 nm | Air | Na-EDTAc | 0.4 gcat L−1; 206 μM h−1 | Hu | 177 | |
| 2016 | Fe-loaded 3D graphene | 473 | UV | N2/H2 | None | 24 μmol g−1 h−1 | Lu | 183 | |
| 2017 | Fe-Al-loaded 3D graphene | 473 | UV | N2/H2 | None | 25.3 μmol g−1 h−1 | Yang | 184 | |
| 2017 | Fe-doped g-C3N4 | 303 | 400–800 nm | N2 | Ethanol | 0.4 gcat L−1; 120 μM h−1 | Hu | 172 | |
| 2017 | g-C3N4/MgAlFeO | 303 | 400–800 nm | N2 | Ethanol | 0.4 gcat L−1; 166.8 μM h−1 | Wang | 174 | |
| 2017 | W18O49/g-C3N4 | — | UV-vis-NIRd | N2 | Ethanol | 0.4 gcat L−1; 57.8 μM h−1 | Liang | 175 | |
| 2017 | Ga2O3-DBD/g-C3N4 | — | UV-vis | N2 | Ethanol | 0.4 gcat L−1; 112.5 μM h−1 | Cao | 176 | |
| Other system | 1980 | GaP/Pt | 311 | UV | N2 | None | 5 μmol g−1 h−1 | Miyama | 104 |
| 1981 | Ti3+-exchanged zeolites | — | Vis | N2 | None | 20.6 μmol g−1 h−1 | Khan | 188 | |
| 1983 | Ti3+-exchanged zeolites | ∼305 | Vis | N2 | None | 32.9 μmol g−1 h−1 | Khan | 189 | |
| 2015 | Cs2O-promoted Os–Au | 333 | λ ≥ 450 nm | N2/H2 | None | 2685 μmol g−1 h−1 | Zeng | 190 | |
| 2016 | GNP/bSi/Cr | — | UV-vis | N2 | None | 0.78 μmol h−1 cm−2 | Ali | 191 | |
| 2017 | 5% Ru@n-GaN NWs | 283 | 290–380 nm | N2 | None | 120 μmol g−1 h−1 | Li | 192 | |
4. Concluding remarks
The photoreduction of N2 to NH3 is regarded as a scientifically challenging yet environmentally friendly technology for the sustainable growth of the human population. NH3 has elicited much research fascination and broad interdisciplinary attention as a hydrogen carrier in addition to its wide use as an industrial raw material and fertilizer.193,194 In this review article, we have thoroughly presented and classified a diverse selection of photocatalysts for N2 fixation. The selected literature reports are summarized in Tables 2 and 3. Overall, we genuinely envision that this review will establish benchmarks and mitigate the present obstacles in N2 photoreduction as well as broaden new inroads at the forefront of this research hotspot. In order to optimize the practical schemes, several concluding remarks, prospects and suggestions are elucidated.(I) Pre-treatment
In the study of iron-doped TiO2, it is apparent that the N2 reduction photocatalytic activity is highly dependent on the pre-treatment method of the as-developed catalysts. The different preparation techniques are presented in Table 4. On the one hand, the synthetic routes markedly influence the photocatalytic activity based on a plethora of aspects, including chemical compounds, defects, crystal morphologies and particle sizes. Additionally, heat treatment plays a central role in the physicochemical properties of the nanomaterials. Notably, annealing at a high temperature introduces defect states, which enhance the photoreduction ability.91 In another aspect, prolonged heat treatment adversely affects the phase transformation and the amount of surface ˙OH groups, which is detrimental to the activity of photocatalysts.67 As described earlier, the treatment of catalysts in H2 atmosphere will contribute to an increase of Hads on the surface or even form an H-terminated surface, thus facilitating the hydrogenation of nitrogen. However, the amount of surficial H will decrease with time, leading to suppressed photoactivity.| Year | Author | Preparation procedures |
|---|---|---|
| 1977 | Schrauzer | Calcine iron sulfate impregnated with anatase TiO2 at 1273 K for 1 h in air |
| 1991 | Soria | Coprecipitate solution of TiCl3 and iron ions in an NH3 solution and anneal the resulting solid at 823 K for 24 h |
| 2001 | Rusina | React iron chloride with tetraisopropyl titanate in a sol–gel method and calcine the resulting solid at 923 K for 20 min |
| 2014 | Zhao | Incorporate Fe3+ into hydrogen titanate phase nanotubes via a hydrothermal method followed by performing calcination at 773 K for 3 h |
(II) Reaction mixture
In addition to the gas-phase reaction, aqueous-phase photoreaction has also been widely employed via the suspension of photocatalysts in liquid. An aqueous slurry with an appropriate concentration of photocatalyst enhances the photoreaction by accelerating the dispersion of the photocatalyst for improved mass transfer. However, high concentrations lead to poor penetration of photons.96 It has also been reported that in the presence of photocatalysts, the ˙OH radicals in water oxidize ammonia to nitrate.125 As such, ammonia and nitrate are detected in the final products of photoreaction.133The presence of organic scavengers such as ethanol and methanol can obviously increase the NH3 yield by providing sacrificial electron donors to the holes in the semiconductor.123 As for the side reaction of the sacrificial agent, Rusina et al. inferred that ethanol facilitates the formation of nitrite and nitrate,121 while Zhao and Lashgari claimed that ethanol serves as a superior ˙OH scavenger which protects the NH3 yield from further oxidation to its higher oxidation states of nitrite or nitrate (Fig. 11).99,113 Usually, holes react with hydroxide ions to form hydroxyl radicals (OH− + hVB+ → ˙OH), which can further oxidize NH3. In this regard, when ethanol is present, it can be oxidized to CH3CHO and C2H5OC2H5.99 Along with the consumption of photo-generated holes in an oxidation process, the N2 reduction rates can be markedly increased due to the availability of abundant photo-generated electrons for the reduction reactions. Although ethanol is generally utilized as a typical organic scavenger, it is crucial to comprehensively study different alcohols in order to determine suitable sacrificial agents for specific light-driven catalytic systems.
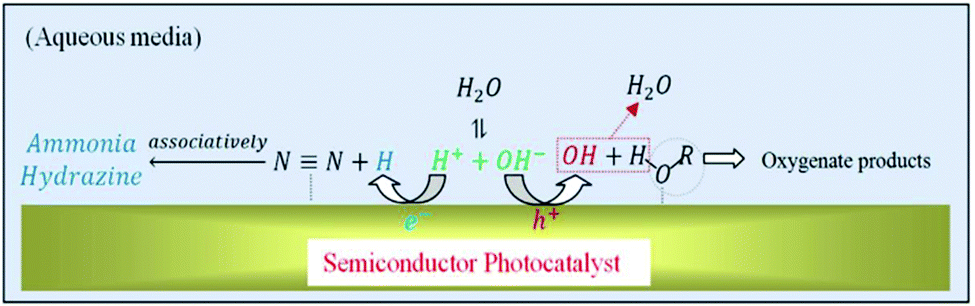 | ||
| Fig. 11 Schematic of the reaction mechanism of N2 fixation at the semiconductor/solution interface in the presence of alcohol as a sacrificial agent. Reproduced with permission.113 Copyright 2017, Elsevier. | ||
(III) Modification of semiconductors
Transition metals are widely used as dopants, co-catalysts and plasmonic nanostructures with the aim of enhancing photocatalytic efficiency. Among these, earth-abundant iron is the most dominant as an efficient metal dopant. In contrast, it has been reported that noble metals as dopants in TiO2 demonstrate inhibiting effects for N2 photoreduction.67 Moreover, this inhibiting effect has been observed on Pt-loaded ZnO compared with pure ZnO.104 However, the incorporation of Ru as a co-catalyst on semiconductors has exhibited an evident positive influence on N2 fixation activity.106 Recently, plasmonic metals were reported to enhance photoabsorption via LSPR. To date, Au has been selected to couple with myriad catalysts, such as SrTiO3,127,128 Os and black silicon,190,191 because its light-harvesting effects can efficiently utilize the visible light region of the solar spectrum.Additionally, the flourishing investigation of oxygen vacancies induced in BiOX has inspired many researchers due to their unique characteristics for the efficient separation of charge carriers. As illustrated in Fig. 12,151 OVs suppress the recombination of electrons and holes by directly trapping electrons from the conduction band and then transferring them to the anti-bonding orbitals of adsorbed N2. Since the first seminal report on oxygen vacancies in BiOBr for N2 fixation in 2015, a number of studies on nitrogen, sulfur and oxygen vacancies have been reported to date.62,152,154 As a proof of concept, a hybrid structure is always desirable for modulating band structures for ameliorated solar energy conversion.195–203 Importantly, the heterostructure will spatially accumulate the separation and transfer of photogenerated charge carriers upon their interactions at the contacted interface.
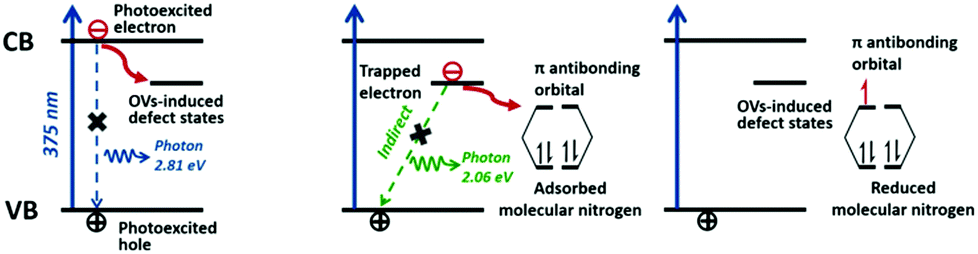 | ||
| Fig. 12 Schematic of the enhancement of OVs-induced photocatalysts for interfacial electron transfer processes. Reproduced with permission.151 Copyright 2015, American Chemical Society. | ||
(IV) Suggestions, outlook and future prospects
As shown in Table 2, the assessments of photocatalytic activities for N2 reduction are not unified. The standard NH3 production rate can be expressed in units of μmol h−1 g−1 for the gas-phase reaction, μM h−1 for the reaction in solution and μmol h−1 cm−2 for photoelectrochemical cells. Therefore, apparent quantum yield (AQY) should be generalized as a scientific tool to account for dissimilar experimental conditions, such as light source, catalyst loading, reaction duration and illumination area.Most previous studies have focused on metal oxide-based nanomaterials, especially in the case of TiO2. However, TiO2 is greatly restricted by its UV excitation, which constitutes less than 5% of the solar light spectrum. Additionally, Cd-containing dichalcogenides are mostly in the form of sulfides, while the stability of CdS urgently requires improvement by forming multi-metal systems. Furthermore, air and visible light are gradually being adopted as nitrogen and light sources instead of pure nitrogen and UV irradiation. In order to use air as the N2 source, the selectivity of the desired products should be taken into consideration to avoid the side product of H2 evolution and the decomposition of NH3. In contrast, for the aqueous-phase reaction, more attention should be paid to the nitrite and nitrate contents in the products. In addition, research on metal-free systems has been primarily dominated by g-C3N4 to date. Although in-depth studies on band structures and charge carrier dynamics are necessary, the exploration of new potential photocatalysts is gaining in momentum to surmount the bottlenecks of the fundamental research and applications of N2 fixation. It is our aim to extend the utilization and optical absorption of solar light up to the NIR in addition to UV and visible light to cater to the entire spectrum of solar light.
In other aspect, a robust device for assembling different components is overwhelmingly necessary to develop a multifunctional platform for artificial photosynthesis – N2 fixation. The engineering of an intact “machine” should comprise a N2 reduction catalyst, an oxidation catalyst, a light-harvesting absorber and an electron-transporting bridge to attain high photocatalytic efficiency of N2 fixation. Therefore, the synergetic interactions of various moieties within an individual matrix must be taken into consideration. Good examples of such versatile “machines” include porous metal–organic frameworks (MOFs) and covalent organic frameworks (COFs), to which further exploration should be devoted in the future. Apart from photon-driven catalysis, with the aid of external bias, a photoelectrocatalytic system is another smart avenue to separate the redox reactions (i.e. N2 reduction and H2O oxidation) at the photocathode and photoanode, respectively. Unequivocally, there is an extreme paucity of literature reports on photoelectrochemical N2 fixation at the moment. Thus, a combined light-driven and electro-driven reaction would undeniably greatly facilitate N2 fixation activity. To this end, dissimilar photocatalysts at the cathode and anode sides could be designed. Essentially, with the present knowledge on the electrochemical reduction of N2 based on a wealth of research activities to date, this provides a scientific guiding star for photoelectrochemical N2 reduction systems.
For heterogeneous photocatalytic systems, insights into their reaction pathways are essential, despite the complexity of the N2 fixation reaction. It is prominent that tuning the size of photocatalysts can remarkably alter the ratio of exposed atoms and active sites on the corners, edges and surfaces with different coordination numbers. As a result, this influences the adsorption ability of the reactant N2 molecules as well as the intermediates during the photoreduction process, giving rise to dissimilar performance and activity of the shape- and size-controlled photocatalysts. To comprehend the atomic insights in reactivity, advanced in situ or operando characterization techniques are indispensable. For example, in situ FTIR analysis is beneficial to examine the adsorbed nitrogen species on the surface of photocatalysts. Meanwhile, environmental/in situ TEM and X-ray absorption spectroscopy can provide information on the structure evolution and the modification of the surface coordination number during the photocatalytic reaction process. Equally importantly, time-resolved spectroscopy, femtosecond transient absorption spectroscopy and terahertz time-domain spectroscopy would be helpful to study the excited-state dynamics in a typical photoinduced electron-transfer pathway. Additionally, computer-aided catalyst design via DFT calculations for N2 fixation would be a robust tool for simulating band structures and reaction mechanisms to achieve excellent sunlight absorption and unprecedented catalysis performance. This could in turn provide auspicious references for the rational experimental design of photocatalysts. Therefore, the innovative engineering of photocatalysts from experimental and theoretical aspects is crucial to systematically tune the band gap for effective absorption of solar photon flux as well as high photoactivity for solar energy conversion.
In summary, it is evident that the product yield of N2 photoreduction has gradually increased from a magnitude of μmol g−1 to mmol g−1. Without a doubt, the goal of achieving a magnitude of mol g−1 for the product yield will no longer be a dream; instead, this dream will be transformed into reality and practicality. Judging from the amount of existing literature on this research platform at this juncture, N2 photoreduction has ignited interest and attention in the research community in recent years. However, it should be emphasized that the quantity of samples obtained from each batch is still at the laboratory scale. To divert from the laboratory-scale level to industrial applications, amplifying the yield of catalysts while retaining their intrinsic original structures is a global challenge. More thought should be given to translating these studies from academic research to practicality. Despite the aforementioned impediments to the photoactivation of N2, the future direction of this area is indisputably promising and prospective with the synergetic cooperation of international researchers from diversified disciplines, such as computational scientists, chemists, materials scientists and physicists. Overall, we strongly believe that solar energy conversion for N2 fixation will be enriched to begin a revolution of renewable energy for practical benefits and future commercialization.
Regarding the long-term prospects, there is an infinite scope of daunting challenges and opportunities for global researchers to expand the fundamental science at the forefront of this research hotspot. We are certain that this review article will provide an excellent basis for the next research era not only in photocatalytic N2 fixation specifically, but also in the interdisciplinary fields of chemistry, materials science, energy conversion and energy storage. On the whole, with ceaseless determination and efforts from a diverse range of scientific communities and accomplishments in years to come, the research that has been comprehensively discussed in this review article will unquestionably be surpassed.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
N. Li is grateful for financial support from China Scholarship Council (CSC) with No. 201606955033, National Natural Science Foundation of China (NSFC) with No. 51461135004, Natural Science Foundation of Hubei Province with No. 2015CFB227, Fundamental Research Funds for the Central Universities, and the research board of the State Key Laboratory of Silicate Materials for Architectures. W.-J. Ong is also thankful for support from the Institute of Materials Research and Engineering (IMRE), Agency for Science, Technology and Research (A*STAR) in Singapore.References
- V. Rosca, M. Duca, M. T. de Groot and M. T. M. Koper, Chem. Rev., 2009, 109, 2209–2244 CrossRef CAS PubMed.
- D. E. Canfield, A. N. Glazer and P. G. Falkowski, Science, 2010, 330, 192 CrossRef CAS PubMed.
- B. K. Burgess and D. J. Lowe, Chem. Rev., 1996, 96, 2983–3012 CrossRef CAS PubMed.
- H.-P. Jia and E. A. Quadrelli, Chem. Soc. Rev., 2014, 43, 547–564 RSC.
- A. Shilov, Russ. Chem. Bull., 2003, 52, 2555–2562 CrossRef CAS.
- V. Smil, Nature, 1999, 400, 415 CrossRef CAS.
- B. M. Hoffman, D. Lukoyanov, Z.-Y. Yang, D. R. Dean and L. C. Seefeldt, Chem. Rev., 2014, 114, 4041–4062 CrossRef CAS PubMed.
- C. N. Satterfield, Heterogeneous Catalysis in Practice, McGraw-Hill Companies, 1980 Search PubMed.
- R. Schlögl, Angew. Chem., Int. Ed., 2003, 42, 2004–2008 CrossRef PubMed.
- H. Liu, Chin. J. Catal., 2014, 35, 1619–1640 CrossRef CAS.
- M. Temkin and V. Pyzhev, Acta Physiochim. U. R. S. S., 1940, 12, 217–222 Search PubMed.
- C. Liang, Z. Wei, Q. Xin and C. Li, Appl. Catal., A, 2001, 208, 193–201 CrossRef CAS.
- A. Vojvodic, A. J. Medford, F. Studt, F. Abild-Pedersen, T. S. Khan, T. Bligaard and J. K. Nørskov, Chem. Phys. Lett., 2014, 598, 108–112 CrossRef CAS.
- Y. Inoue, M. Kitano, K. Kishida, H. Abe, Y. Niwa, M. Sasase, Y. Fujita, H. Ishikawa, T. Yokoyama and M. Hara, ACS Catal., 2016, 6, 7577–7584 CrossRef CAS.
- A. D. Allen and C. V. Senoff, Chem. Commun., 1965, 621–622 RSC.
- D. V. Yandulov and R. R. Schrock, Science, 2003, 301, 76–78 CrossRef CAS PubMed.
- G. Schwarz, R. R. Mendel and M. W. Ribbe, Nature, 2009, 460, 839–847 CrossRef CAS PubMed.
- K. Arashiba, Y. Miyake and Y. Nishibayashi, Nat. Chem., 2011, 3, 120–125 CrossRef CAS PubMed.
- J. S. Anderson, J. Rittle and J. C. Peters, Nature, 2013, 501, 84–87 CrossRef CAS PubMed.
- S. E. Creutz and J. C. Peters, J. Am. Chem. Soc., 2014, 136, 1105–1115 CrossRef CAS PubMed.
- J. Rittle and J. C. Peters, J. Am. Chem. Soc., 2016, 138, 4243–4248 CrossRef CAS PubMed.
- T. Shima, S. Hu, G. Luo, X. Kang, Y. Luo and Z. Hou, Science, 2013, 340, 1549–1552 CrossRef CAS PubMed.
- B. M. Hoffman, D. R. Dean and L. C. Seefeldt, Acc. Chem. Res., 2009, 42, 609–619 CrossRef CAS PubMed.
- R. Bjornsson, F. Neese, R. R. Schrock, O. Einsle and S. DeBeer, J. Biol. Inorg. Chem., 2015, 20, 447–460 CrossRef CAS PubMed.
- K. C. MacLeod and P. L. Holland, Nat. Chem., 2013, 5, 559–565 CrossRef CAS PubMed.
- V. Kyriakou, I. Garagounis, E. Vasileiou, A. Vourros and M. Stoukides, Catal. Today, 2017, 286, 2–13 CrossRef CAS.
- G. Marnellos and M. Stoukides, Science, 1998, 282, 98–100 CrossRef CAS PubMed.
- Z. Li, R. Liu, Y. Xie, S. Feng and J. Wang, Solid State Ionics, 2005, 176, 1063–1066 CrossRef CAS.
- E. Vasileiou, V. Kyriakou, I. Garagounis, A. Vourros, A. Manerbino, W. G. Coors and M. Stoukides, Top. Catal., 2015, 58, 1193–1201 CrossRef CAS.
- E. Vasileiou, V. Kyriakou, I. Garagounis, A. Vourros and M. Stoukides, Solid State Ionics, 2015, 275, 110–116 CrossRef CAS.
- Y. Kobayashi, N. Shimoda, Y. Kimura and S. Satokawa, ECS Trans., 2017, 75, 43–52 CrossRef.
- V. Kordali, G. Kyriacou and C. Lambrou, Chem. Commun., 2000, 1673–1674 RSC.
- G. Xu, R. Liu and J. Wang, Sci. China, Ser. B: Chem., 2009, 52, 1171–1175 CrossRef CAS.
- R. Lan and S. Tao, RSC Adv., 2013, 3, 18016–18021 RSC.
- T. Murakami, T. Nishikiori, T. Nohira and Y. Ito, J. Am. Chem. Soc., 2003, 125, 334–335 CrossRef CAS PubMed.
- T. Murakami, T. Nohira, Y. Araki, T. Goto, R. Hagiwara and Y. H. Ogata, Electrochem. Solid-State Lett., 2007, 10, E4–E6 CrossRef CAS.
- I. A. Amar, R. Lan and S. Tao, RSC Adv., 2015, 5, 38977–38983 RSC.
- S. Licht, B. Cui, B. Wang, F.-F. Li, J. Lau and S. Liu, Science, 2014, 345, 637–640 CrossRef CAS PubMed.
- F.-F. Li and S. Licht, Inorg. Chem., 2014, 53, 10042–10044 CrossRef CAS PubMed.
- S. Chen, S. Perathoner, C. Ampelli, C. Mebrahtu, D. Su and G. Centi, Angew. Chem., Int. Ed., 2017, 56, 2699–2703 CrossRef CAS PubMed.
- A. B. Djurisic, Y. H. Leung and A. M. Ching Ng, Mater. Horiz., 2014, 1, 400–410 RSC.
- M. Marszewski, S. Cao, J. Yu and M. Jaroniec, Mater. Horiz., 2015, 2, 261–278 RSC.
- Y. Ma, X. Wang, Y. Jia, X. Chen, H. Han and C. Li, Chem. Rev., 2014, 114, 9987–10043 CrossRef CAS PubMed.
- J. L. White, M. F. Baruch, J. E. Pander Iii, Y. Hu, I. C. Fortmeyer, J. E. Park, T. Zhang, K. Liao, J. Gu, Y. Yan, T. W. Shaw, E. Abelev and A. B. Bocarsly, Chem. Rev., 2015, 115, 12888–12935 Search PubMed.
- K. Wenderich and G. Mul, Chem. Rev., 2016, 116, 14587–14619 CrossRef CAS PubMed.
- Y. Zhao, X. Jia, G. I. N. Waterhouse, L.-Z. Wu, C.-H. Tung, D. O'Hare and T. Zhang, Adv. Energy Mater., 2016, 6, 1501974 CrossRef.
- X. Liu, J. Iocozzia, Y. Wang, X. Cui, Y. Chen, S. Zhao, Z. Li and Z. Lin, Energy Environ. Sci., 2017, 10, 402–434 CAS.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- L. K. Putri, B.-J. Ng, W.-J. Ong, H. W. Lee, W. S. Chang and S.-P. Chai, ACS Appl. Mater. Interfaces, 2017, 9, 4558–4569 CAS.
- W.-J. Ong, L.-L. Tan, S.-P. Chai, S.-T. Yong and A. R. Mohamed, Nanoscale, 2014, 6, 1946–2008 RSC.
- J. Di, J. Xia, H. Li and Z. Liu, Nano Energy, 2017, 35, 79–91 CrossRef CAS.
- J. Yang, D. Wang, H. Han and C. Li, Acc. Chem. Res., 2013, 46, 1900–1909 CrossRef CAS PubMed.
- X. Chen, S. Shen, L. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503–6570 CrossRef CAS PubMed.
- S. Chen, T. Takata and K. Domen, Nat. Rev. Mater., 2017, 2, 17050 CrossRef CAS.
- Q. Lu, Y. Yu, Q. Ma, B. Chen and H. Zhang, Adv. Mater., 2016, 28, 1917–1933 CrossRef CAS PubMed.
- G. A. M. Hutton, B. C. M. Martindale and E. Reisner, Chem. Soc. Rev., 2017 10.1039/c7cs00235a.
- G. Zhang, Z.-A. Lan and X. Wang, Chem. Sci., 2017, 8, 5261–5274 RSC.
- X. Chang, T. Wang and J. Gong, Energy Environ. Sci., 2016, 9, 2177–2196 CAS.
- S. J. A. Moniz, S. A. Shevlin, D. J. Martin, Z.-X. Guo and J. Tang, Energy Environ. Sci., 2015, 8, 731–759 CAS.
- X. Meng, L. Liu, S. Ouyang, H. Xu, D. Wang, N. Zhao and J. Ye, Adv. Mater., 2016, 28, 6781–6803 CrossRef CAS PubMed.
- J. Li, H. Li, G. Zhan and L. Zhang, Acc. Chem. Res., 2017, 50, 112–121 CrossRef CAS PubMed.
- G. Dong, W. Ho and C. Wang, J. Mater. Chem. A, 2015, 3, 23435–23441 CAS.
- N. Dhar and S. Chowdhry, Proc. Natl. Acad. Sci., India, 1968, 38, 485–490 Search PubMed.
- G. N. Schrauzer, N. Strampach, L. N. Hui, M. R. Palmer and J. Salehi, Proc. Natl. Acad. Sci. U. S. A., 1983, 80, 3873–3876 CrossRef CAS.
- A. J. Medford and M. C. Hatzell, ACS Catal., 2017, 7, 2624–2643 CrossRef CAS.
- G. N. Schrauzer, Energy Efficiency and Renewable Energy Through Nanotechnology, Springer, 2011, pp. 601–623 Search PubMed.
- G. N. Schrauzer and T. D. Guth, J. Am. Chem. Soc., 1977, 99, 7189–7193 CrossRef CAS.
- T. Bazhenova and A. Shilov, Coord. Chem. Rev., 1995, 144, 69–145 CrossRef CAS.
- C. J. van der Ham, M. T. Koper and D. G. Hetterscheid, Chem. Soc. Rev., 2014, 43, 5183–5191 RSC.
- L.-j. Guo, Y.-j. Wang and T. He, Chem. Rec., 2016, 16, 1918–1933 CrossRef CAS PubMed.
- S. Sun, X. Li, W. Wang, L. Zhang and X. Sun, Appl. Catal., B, 2017, 200, 323–329 CrossRef CAS.
- W. J. Ong, L. L. Tan, S. P. Chai, S. T. Yong and A. R. Mohamed, ChemSusChem, 2014, 7, 690–719 CrossRef CAS PubMed.
- W.-J. Ong, L.-L. Tan, Y. H. Ng, S.-T. Yong and S.-P. Chai, Chem. Rev., 2016, 116, 7159–7329 CrossRef CAS PubMed.
- L. K. Putri, L.-L. Tan, W.-J. Ong, W. S. Chang and S.-P. Chai, Appl. Mater. Today, 2016, 4, 9–16 CrossRef.
- A. Fuertes, Mater. Horiz., 2015, 2, 453–461 RSC.
- F. K. Kessler, Y. Zheng, D. Schwarz, C. Merschjann, W. Schnick, X. Wang and M. J. Bojdys, Nat. Rev. Mater., 2017, 2, 17030 CrossRef CAS.
- K. Sivula and R. van de Krol, Nat. Rev. Mater., 2016, 1, 16010 CrossRef.
- I. Roger, M. A. Shipman and M. D. Symes, Nat. Rev. Chem., 2017, 1, 0003 CrossRef.
- Y. Tachibana, L. Vayssieres and J. R. Durrant, Nat. Photonics, 2012, 6, 511–518 CrossRef CAS.
- J. Low, J. Yu, M. Jaroniec, S. Wageh and A. A. Al-Ghamdi, Adv. Mater., 2017, 29, 1601694 CrossRef PubMed.
- S. Cao, J. Low, J. Yu and M. Jaroniec, Adv. Mater., 2015, 27, 2150–2176 CrossRef CAS PubMed.
- H. Li, Y. Zhou, W. Tu, J. Ye and Z. Zou, Adv. Funct. Mater., 2015, 25, 998–1013 CrossRef CAS.
- L. K. Putri, W.-J. Ong, W. S. Chang and S.-P. Chai, Appl. Surf. Sci., 2015, 358(part A), 2–14 CrossRef CAS.
- S. Hoang and P.-X. Gao, Adv. Energy Mater., 2016, 6, 1600683 CrossRef.
- X. Li, J. Yu and M. Jaroniec, Chem. Soc. Rev., 2016, 45, 2603–2636 RSC.
- H. Li, J. Li, Z. Ai, F. Jia and L. Zhang, Angew. Chem., 2017 DOI:10.1002/ange.201705628.
- L. M. Azofra, N. Li, D. R. MacFarlane and C. Sun, Energy Environ. Sci., 2016, 9, 2545–2549 CAS.
- H. Li, J. Shang, J. Shi, K. Zhao and L. Zhang, Nanoscale, 2016, 8, 1986–1993 RSC.
- Y. Bai, L. Ye, T. Chen, L. Wang, X. Shi, X. Zhang and D. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 27661–27668 CAS.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- S. Bourgeois, D. Diakite and M. Perdereau, React. Solids, 1988, 6, 95–104 CrossRef CAS.
- H. Hirakawa, M. Hashimoto, Y. Shiraishi and T. Hirai, J. Am. Chem. Soc., 2017, 139, 10929–10936 CrossRef CAS PubMed.
- V. Augugliaro, A. Lauricella, L. Rizzuti, M. Schiavello and A. Sclafani, Int. J. Hydrogen Energy, 1982, 7, 845–849 CrossRef CAS.
- Y. Iida and S. Ozaki, J. Am. Ceram. Soc., 1961, 44, 120–127 CrossRef CAS.
- J. Navio, M. Macias, M. Gonzalez-Catalan and A. Justo, J. Mater. Sci., 1992, 27, 3036–3042 CrossRef CAS.
- P. P. Radford and C. G. Francis, J. Chem. Soc., Chem. Commun., 1983, 1520–1521 RSC.
- M. Rao, K. Rajeshwar, V. P. Verneker and J. DuBow, J. Phys. Chem., 1980, 84, 1987–1991 CrossRef CAS.
- J. Soria, J. Conesa, V. Augugliaro, L. Palmisano, M. Schiavello and A. Sclafani, J. Phys. Chem., 1991, 95, 274–282 CrossRef CAS.
- W. Zhao, J. Zhang, X. Zhu, M. Zhang, J. Tang, M. Tan and Y. Wang, Appl. Catal., B, 2014, 144, 468–477 CrossRef CAS.
- L. Palmisano, V. Augugliaro, A. Sclafani and M. Schiavello, J. Phys. Chem., 1988, 92, 6710–6713 CrossRef CAS.
- C. Martin, I. Martin, V. Rives, L. Palmisano and M. Schiavello, J. Catal., 1992, 134, 434–444 CrossRef CAS.
- O. Ileperuma, K. Tennakone and W. Dissanayake, Appl. Catal., 1990, 62, L1–L5 CrossRef CAS.
- O. Ileperuma, C. Thaminimulla and W. Kiridena, Sol. Energy Mater. Sol. Cells, 1993, 28, 335–343 CrossRef CAS.
- H. Miyama, N. Fujii and Y. Nagae, Chem. Phys. Lett., 1980, 74, 523–524 CrossRef CAS.
- M. M. Taqui Khan, D. Chatterjee and M. Bala, J. Photochem. Photobiol., A, 1992, 67, 349–352 CrossRef.
- K. T. Ranjit, T. K. Varadarajan and B. Viswanathan, J. Photochem. Photobiol., A, 1996, 96, 181–185 CrossRef CAS.
- O. P. Linnik and H. Kisch, Mendeleev Commun., 2008, 18, 10–11 CrossRef CAS.
- K. Hoshino, M. Inui, T. Kitamura and H. Kokado, Angew. Chem., Int. Ed., 2000, 39, 2509–2512 CrossRef CAS PubMed.
- K. Hoshino, Chem. – Eur. J., 2001, 7, 2727–2731 CrossRef CAS.
- K. Hoshino, R. Kuchii and T. Ogawa, Appl. Catal., B, 2008, 79, 81–88 CrossRef CAS.
- M. M. Khader, N. N. Lichtin, G. H. Vurens, M. Salmeron and G. A. Somorjai, Langmuir, 1987, 3, 303–304 CrossRef CAS.
- C. J. Jacobsen, S. Dahl, B. S. Clausen, S. Bahn, A. Logadottir and J. K. Nørskov, J. Am. Chem. Soc., 2001, 123, 8404–8405 CrossRef CAS PubMed.
- M. Lashgari and P. Zeinalkhani, Appl. Catal., A, 2017, 529, 91–97 CrossRef CAS.
- E. Endoh, J. K. Leland and A. J. Bard, J. Phys. Chem., 1986, 90, 6223–6226 CrossRef CAS.
- W. Zhao, H. Xi, M. Zhang, Y. Li, J. Chen, J. Zhang and X. Zhu, Chem. Commun., 2015, 51, 4785–4788 RSC.
- S. Sun, Q. An, W. Wang, L. Zhang, J. Liu and W. A. Goddard Iii, J. Mater. Chem. A, 2017, 5, 201–209 CAS.
- V. Augugliaro, F. D'alba, L. Rizzuti, M. Schiavello and A. Sclafani, Int. J. Hydrogen Energy, 1982, 7, 851–855 CrossRef CAS.
- C. M. Janet, S. Navaladian, B. Viswanathan, T. K. Varadarajan and R. P. Viswanath, J. Phys. Chem. C, 2010, 114, 2622–2632 CAS.
- H. Pan, X. Meng and G. Qin, Phys. Chem. Chem. Phys., 2014, 16, 25442–25448 RSC.
- D. Cordischi, N. Burriesci, F. D'Alba, M. Petrera, G. Polizzotti and M. Schiavello, J. Solid State Chem., 1985, 56, 182–190 CrossRef CAS.
- O. Rusina, A. Eremenko, G. Frank, H. P. Strunk and H. Kisch, Angew. Chem., Int. Ed., 2001, 40, 3993–3995 CrossRef CAS PubMed.
- O. Rusina, O. Linnik, A. Eremenko and H. Kisch, Chem. – Eur. J., 2003, 9, 561–565 CrossRef CAS PubMed.
- O. Linnik and H. Kisch, Photochem. Photobiol. Sci., 2006, 5, 938–942 CAS.
- G. N. Schrauzer, T. D. Guth, J. Salehi, N. Strampach, N. H. Liu and M. R. Palmer, in Homogeneous and Heterogeneous Photocatalysis, ed. E. Pelizzetti and N. Serpone, Reidel, Hingham, MA, 1986, vol. 174, pp. 509–518 Search PubMed.
- Q.-s. Li, K. Domen, S. Naito, T. Onishi and K. Tamaru, Chem. Lett., 1983, 321–324 CrossRef CAS.
- S. Linic, P. Christopher and D. B. Ingram, Nat. Mater., 2011, 10, 911–921 CrossRef CAS PubMed.
- T. Oshikiri, K. Ueno and H. Misawa, Angew. Chem., Int. Ed., 2014, 53, 9802–9805 CrossRef CAS PubMed.
- T. Oshikiri, K. Ueno and H. Misawa, Angew. Chem., 2016, 128, 4010–4014 CrossRef.
- E. Skulason, T. Bligaard, S. Gudmundsdóttir, F. Studt, J. Rossmeisl, F. Abild-Pedersen, T. Vegge, H. Jónsson and J. K. Nørskov, Phys. Chem. Chem. Phys., 2012, 14, 1235–1245 RSC.
- M. A. Shipman and M. D. Symes, Catal. Today, 2017, 286, 57–68 CrossRef CAS.
- Y. Hao, X. Dong, S. Zhai, H. Ma, X. Wang and X. Zhang, Chem. – Eur. J., 2016, 22, 18722–18728 CrossRef CAS PubMed.
- K. Tennakone, S. Wickramanayake, C. Fernando, O. Ileperuma and S. Punchihewa, J. Chem. Soc., Chem. Commun., 1987, 1078–1080 RSC.
- K. Tennakone, J. Bandara, C. Thaminimulla, W. Jayatilake, U. Ketipearachchi, O. Ileperuma and M. Priyadarshana, Langmuir, 1991, 7, 2166–2168 CrossRef CAS.
- K. Tennakone, S. Punchihewa and R. Tantrigoda, Sol. Energy Mater., 1989, 18, 217–221 CrossRef CAS.
- K. Tennakone, C. Thaminimulla and W. Kiridena, Langmuir, 1993, 9, 723–726 CrossRef CAS.
- X. Li, W. Wang, D. Jiang, S. Sun, L. Zhang and X. Sun, Chem. – Eur. J., 2016, 22, 13819–13822 CrossRef CAS PubMed.
- O. Ileperuma, W. Kiridena and W. Dissanayake, J. Photochem. Photobiol., A, 1991, 59, 191–197 CrossRef CAS.
- K. Tennakone, C. Fernando, S. Wickramanayake, M. Damayanthi, L. Silva, W. Wijeratne, O. Illeperuma and S. Punchihewa, Sol. Energy Mater., 1988, 17, 47–53 CrossRef CAS.
- K. Tennakone, C. Thaminimulla and J. Bandara, J. Photochem. Photobiol., A, 1992, 68, 131–135 CrossRef CAS.
- W.-J. Ong, L. K. Putri, Y.-C. Tan, L.-L. Tan, N. Li, Y. H. Ng, X. Wen and S.-P. Chai, Nano Res., 2017, 10, 1673–1696 CrossRef CAS.
- X. Zong, H. Yan, G. Wu, G. Ma, F. Wen, L. Wang and C. Li, J. Am. Chem. Soc., 2008, 130, 7176–7177 CrossRef CAS PubMed.
- H. Yan, J. Yang, G. Ma, G. Wu, X. Zong, Z. Lei, J. Shi and C. Li, J. Catal., 2009, 266, 165–168 CrossRef CAS.
- S. Navalón, A. Dhakshinamoorthy, M. Álvaro and H. Garcia, ChemSusChem, 2013, 6, 562–577 CrossRef PubMed.
- M. M. T. Khan, R. C. Bhardwaj and C. Bhardwaj, Angew. Chem., Int. Ed., 1988, 27, 923–925 CrossRef.
- M. T. Khan and N. N. Rao, J. Mol. Catal. A: Chem., 1990, 58, 323–329 CrossRef CAS.
- L. Ye, C. Han, Z. Ma, Y. Leng, J. Li, X. Ji, D. Bi, H. Xie and Z. Huang, Chem. Eng. J., 2017, 307, 311–318 CrossRef CAS.
- K. A. Brown, D. F. Harris, M. B. Wilker, A. Rasmussen, N. Khadka, H. Hamby, S. Keable, G. Dukovic, J. W. Peters and L. C. Seefeldt, Science, 2016, 352, 448–450 CrossRef CAS PubMed.
- A. Banerjee, B. D. Yuhas, E. A. Margulies, Y. Zhang, Y. Shim, M. R. Wasielewski and M. G. Kanatzidis, J. Am. Chem. Soc., 2015, 137, 2030–2034 CrossRef CAS PubMed.
- J. Liu, M. S. Kelley, W. Wu, A. Banerjee, A. P. Douvalis, J. Wu, Y. Zhang, G. C. Schatz and M. G. Kanatzidis, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 5530–5535 CrossRef CAS PubMed.
- B. M. Hoffman, D. R. Dean and L. C. Seefeldt, Acc. Chem. Res., 2009, 42, 609–619 CrossRef CAS PubMed.
- H. Li, J. Shang, Z. Ai and L. Zhang, J. Am. Chem. Soc., 2015, 137, 6393–6399 CrossRef CAS PubMed.
- S. Hu, X. Chen, Q. Li, Y. Zhao and W. Mao, Catal. Sci. Technol., 2016, 6, 5884–5890 CAS.
- Y. Cao, S. Hu, F. Li, Z. Fan, J. Bai, G. Lu and Q. Wang, RSC Adv., 2016, 6, 49862–49867 RSC.
- S. Hu, Y. Li, F. Li, Z. Fan, H. Ma, W. Li and X. Kang, ACS Sustainable Chem. Eng., 2016, 4, 2269–2278 CrossRef CAS.
- Q. Zhang, S. Hu, Z. Fan, D. Liu, Y. Zhao, H. Ma and F. Li, Dalton Trans., 2016, 45, 3497–3505 RSC.
- S. Wu, J. S. Ross, G.-B. Liu, G. Aivazian, A. Jones, Z. Fei, W. Zhu, D. Xiao, W. Yao, D. Cobden and X. Xu, Nat. Phys., 2013, 9, 149–153 CrossRef CAS.
- K. F. Mak, K. He, J. Shan and T. F. Heinz, Nat. Nanotechnol., 2012, 7, 494–498 CrossRef CAS PubMed.
- E. A. A. Pogna, M. Marsili, D. De Fazio, S. Dal Conte, C. Manzoni, D. Sangalli, D. Yoon, A. Lombardo, A. C. Ferrari, A. Marini, G. Cerullo and D. Prezzi, ACS Nano, 2016, 10, 1182–1188 CrossRef CAS PubMed.
- S. Sun, X. Li, W. Wang, L. Zhang and X. Sun, Appl. Catal., B, 2017, 200, 323–329 CrossRef CAS.
- C. H. Lui, A. J. Frenzel, D. V. Pilon, Y. H. Lee, X. Ling, G. M. Akselrod, J. Kong and N. Gedik, Phys. Rev. Lett., 2014, 113, 166801 CrossRef CAS PubMed.
- H. Cheng, B. Huang and Y. Dai, Nanoscale, 2014, 6, 2009–2026 RSC.
- W. L. Huang and Q. Zhu, J. Comput. Chem., 2009, 30, 183–190 CrossRef CAS PubMed.
- X. Y. Kong, W. Lee, W. J. Ong, S. P. Chai and A. R. Mohamed, ChemCatChem, 2016, 8, 3074–3081 CrossRef CAS.
- S. Wang, X. Hai, X. Ding, K. Chang, Y. Xiang, X. Meng, Z. Yang, H. Chen and J. Ye, Adv. Mater., 2017, 29, 1701774 CrossRef PubMed.
- D. Zhu, L. Zhang, R. E. Ruther and R. J. Hamers, Nat. Mater., 2013, 12, 836–841 CrossRef CAS PubMed.
- J. R. Christianson, D. Zhu, R. J. Hamers and J. R. Schmidt, J. Phys. Chem. B, 2014, 118, 195–203 CrossRef CAS PubMed.
- J. A. Bandy, D. Zhu and R. J. Hamers, Diamond Relat. Mater., 2016, 64, 34–41 CrossRef CAS.
- H. Ma, Z. Shi, S. Li and N. Liu, Appl. Surf. Sci., 2016, 379, 309–315 CrossRef CAS.
- S. Li, X. Chen, S. Hu, Q. Li, J. Bai and F. Wang, RSC Adv., 2016, 6, 45931–45937 RSC.
- H. Ma, Z. Shi, Q. Li and S. Li, J. Phys. Chem. Solids, 2016, 99, 51–58 CrossRef CAS.
- G. Wu, Y. Gao and B. Zheng, Ceram. Int., 2016, 42, 6985–6992 CrossRef CAS.
- S. Hu, X. Chen, Q. Li, F. Li, Z. Fan, H. Wang, Y. Wang, B. Zheng and G. Wu, Appl. Catal., B, 2017, 201, 58–69 CrossRef CAS.
- Z. Ma, S. Zhao, X. Xiong, B. Hu and C. Song, Catal. Lett., 2016, 146, 2324–2329 CrossRef CAS.
- Y. Wang, W. Wei, M. Li, S. Hu, J. Zhang and R. Feng, RSC Adv., 2017, 7, 18099–18107 RSC.
- H. Liang, H. Zou and S. Hu, New J. Chem., 2017, 41, 8920–8926 RSC.
- S. Cao, N. Zhou, F. Gao, H. Chen and F. Jiang, Appl. Catal., B, 2017, 218, 600–610 CrossRef CAS.
- S. Hu, W. Zhang, J. Bai, G. Lu, L. Zhang and G. Wu, RSC Adv., 2016, 6, 25695–25702 RSC.
- S. Hu, F. Li, Z. Fan, F. Wang, Y. Zhao and Z. Lv, Dalton Trans., 2015, 44, 1084–1092 RSC.
- Q. Han, N. Chen, J. Zhang and L. Qu, Mater. Horiz., 2017, 4, 832–850 RSC.
- W.-J. Ong, L.-L. Tan, S.-P. Chai and S.-T. Yong, Chem. Commun., 2015, 51, 858–861 RSC.
- W.-J. Ong, L.-L. Tan, S.-P. Chai, S.-T. Yong and A. R. Mohamed, Nano Energy, 2015, 13, 757–770 CrossRef CAS.
- W.-J. Ong, Front. Mater., 2017, 4, 11, DOI:10.3389/fmats.2017.00011.
- Y. Lu, Y. Yang, T. Zhang, Z. Ge, H. Chang, P. Xiao, Y. Xie, L. Hua, Q. Li and H. Li, ACS Nano, 2016, 10, 10507–10515 CrossRef CAS PubMed.
- Y. Yang, T. Zhang, Z. Ge, Y. Lu, H. Chang, P. Xiao, R. Zhao, Y. Ma and Y. Chen, Carbon, 2017, 124, 72–78 CrossRef CAS.
- P. Dean, Phys. Rev., 1967, 157, 655 CrossRef CAS.
- B. Aurian-Blajeni, M. Halmann and J. Manassen, Sol. Energy Mater., 1983, 8, 425–440 CrossRef CAS.
- C. R. Dickson and A. J. Nozik, J. Am. Chem. Soc., 1978, 100, 8007–8009 CrossRef CAS.
- F. Khan, P.-L. Yue, L. Rizzuti, V. Augugliaro and M. Schiavello, J. Chem. Soc., Chem. Commun., 1981, 20, 1049–1050 RSC.
- F. Khan, P. Yue, L. Rizzuti, V. Augugliaro and A. Brucato, Ind. Eng. Chem. Res., 1983, 22, 238–241 CrossRef CAS.
- H. Zeng, S. Terazono and T. Tanuma, Catal. Commun., 2015, 59, 40–44 CrossRef CAS.
- M. Ali, F. Zhou, K. Chen, C. Kotzur, C. Xiao, L. Bourgeois, X. Zhang and D. R. MacFarlane, Nat. Commun., 2016, 7, 11335 CrossRef CAS PubMed.
- L. Li, Y. Wang, S. Vanka, X. Mu, Z. Mi and C.-J. Li, Angew. Chem., 2017, 129, 8827–8831 CrossRef.
- A. Klerke, C. H. Christensen, J. K. Norskov and T. Vegge, J. Mater. Chem., 2008, 18, 2304–2310 RSC.
- R. Lan, J. T. S. Irvine and S. Tao, Int. J. Hydrogen Energy, 2012, 37, 1482–1494 CrossRef CAS.
- W.-J. Ong, L. K. Putri, L.-L. Tan, S.-P. Chai and S.-T. Yong, Appl. Catal., B, 2016, 180, 530–543 CrossRef CAS.
- D. Zeng, W.-J. Ong, H. Zheng, M. Wu, Y. Chen, D.-L. Peng and M. Han, J. Mater. Chem. A, 2017, 5, 16171–16178 CAS.
- L. K. Putri, W.-J. Ong, W. S. Chang and S.-P. Chai, Catal. Sci. Technol., 2016, 6, 744–754 CAS.
- D. Zeng, L. Xiao, W.-J. Ong, P. Wu, H. Zheng, Y. Chen and D.-L. Peng, ChemSusChem, 2017 DOI:10.1002/cssc.201701345.
- W.-J. Ong, L.-L. Tan, S.-P. Chai and S.-T. Yong, Dalton Trans., 2015, 44, 1249–1257 RSC.
- D. Zeng, W. Xu, W.-J. Ong, J. Xu, H. Ren, Y. Chen, H. Zheng and D.-L. Peng, Appl. Catal., B, 2017 DOI:10.1016/j.apcatb.2017.08.041.
- W.-J. Ong, L.-L. Tan, S.-P. Chai, S.-T. Yong and A. R. Mohamed, Nano Res., 2014, 7, 1528–1547 CrossRef CAS.
- L. j. Guo, Y. j. Wang and T. He, Chem. Rec., 2016, 16, 1918–1933 CrossRef CAS PubMed.
- P. Qiu, C. Xu, N. Zhou, H. Chen and F. Jiang, Appl. Catal., B, 2017 DOI:10.1016/j.apcatb.2017.09.010.
| This journal is © The Royal Society of Chemistry 2018 |