
Reversible switching of solid-state luminescence by heat-induced interconversion of molecular packing†
Karattu Chali
Naeem
ab and
Vijayakumar C.
Nair
 *ab
*ab
aPhotosciences and Photonics Section, CSIR-National Institute for Interdisciplinary Science and Technology (NIIST), Trivandrum 695 019, India. E-mail: cvijayakumar@niist.res.in
bAcademy of Scientific and Innovative Research (AcSIR), CSIR-NIIST Campus, Thiruvananthapuram 695019, Kerala, India
First published on 23rd October 2017
Abstract
Dynamic switching of solid-sate luminescence with high contrast and reproducibility is a challenging task but important for several optoelectronic applications. One promising approach towards this end is to control the mode of solid-state packing of luminescent organic chromophores with an external stimulus. Herein, we present the heat-induced interconversion of molecular packing in acceptor–donor–acceptor type divinylbenzene derivatives. This interconversion is associated with the switching of solid-state luminescence from red to greenish-yellow (Δλ = 95 nm). A detailed investigation of the photophysical properties provided molecular and supramolecular level comprehension of the factors guiding the luminescence switching. The transition between different packing modes is associated with differential excited state coupling and excitation energy migration efficiencies due to the variance in the chromophore organization. Viewed more broadly, our findings illustrate that subtle control over energy migration processes in molecular assemblies by heating may result in functional organic materials with switchable luminescence.
Design, System, ApplicationThe present paper describes the design of acceptor–donor–acceptor type linear pi-conjugated systems with switchable luminescence properties. The self-assembly assisted molecular packing undergoes interconversion on application of heat and mechanical stimuli. Tailor-made donor–acceptor interaction is the key to the design strategy, which is not easy to achieve in chromophore systems but judicious selection of supramolecular tools could help achieve the purpose. It is also necessary that the materials have at least two stable crystalline emissive states which are interconvertible with temperature stimuli. These types of materials may have immediate applications in the area of optical recording media for high density information storage. They may have potential to replace traditional rewritable optical media based on the change in reflectivity associated with heat-induced phase-change technology. |
Introduction
Switching of the solid-state luminescence of organic functional chromophores is a fundamentally important topic of research due to its practical applications such as in optical recording and information storage.1 Development of organic compounds having at least two different molecular packing states, which are luminescent, stable and reversible with an external stimulus, is a successful approach in this direction.2 A variety of solid-state luminescent materials, whose emission properties change in response to external stimuli such as chemical, electrical, light, heat, pH and mechanical force, have been reported by materials scientists.3 The luminescence characteristics of these molecules in the solid state are strongly dependent on their self-assembly properties and supramolecular packing. Intrigued by the possibility of tuning luminescence over a wide range by merely changing the temperature, in the present work, we explored the possibility of controlling the crystalline solid-state packing of organic chromophore assemblies through heat-mode interconversions.In 2005, Araki et al. successfully demonstrated the switching of the solid-state luminescence of a terpyridine derivative by heat-induced interconversion from needle to plate crystals.4 Later, Park and co-workers reported cyanostilbene-based materials exhibiting two-color fluorescence switching in response to pressure, temperature, and solvent vapor.5 The origin of the switching is the two-dimensional sliding capability of molecular layers, which are formed through intermolecular C–H⋯N and C–H⋯O hydrogen bonds. Recently, Weder et al. reported another cyanostilbene derivative that forms five different solid-state molecular packings with distinct luminescence properties induced by thermal and mechanical stimulation.6 Very recently, the same group has extended this strategy to supramolecular polymers.7 Though all these eminent works illustrated the dynamic switching of solid-state luminescence in chromophore assemblies, the design of novel molecular systems with switchable luminescence and an in-depth understanding of the underlying photophysical processes associated with the switching are necessary for further advancements in sensor and data storage devices.
In the present work, we studied the luminescence switching of two acceptor–donor–acceptor type linear π-conjugated oligomers induced by heat stimuli. The molecules consist of 1,4-bis(dodecyloxy)-2,5-divinylbenzene as a donor and benzoxazole or benzothiazole (DBBO and DBBT, respectively) as acceptors connected through vinyl linkage (Scheme 1). Both compounds exhibited transition between two polymorphic states by applying heat with stable and visually distinguishable fluorescence for each state. The luminescence of the polymorphic form obtained as a result of heating can be further tuned by mechanical stimuli. Interestingly, phase changes caused by the external stimuli were also associated with changes in the excitation energy migration properties of the assemblies. Our findings clearly establish the link between molecular packing and fundamental photophysical properties associated with solid-state luminescence switching. These findings may help the development of novel organic solid-state luminescent materials that can be switched by guiding the molecular packing through stimuli-mediated interconversions.
 | ||
| Scheme 1 Chemical structures of DBBO and DBBT derivatives under study. | ||
Experimental
Materials
Reagents and solvents for the synthesis were purchased either from local suppliers or from Sigma-Aldrich, Alfa Aesar or TCI and used as received. Air and water sensitive synthetic procedures were conducted in an inert atmosphere using standard Schlenk techniques.Measurements
Melting points were measured using a Mel-Temp-II melting point apparatus and uncorrected. FT-IR spectra were recorded on a Shimadzu IRPrestige-21 Fourier transform infrared spectrophotometer. 1H and 13C-NMR spectra of the compounds were recorded on a 500 MHz Bruker Avance II spectrometer. All the chemical shifts were referenced to (CH3)4Si (TMS; δ = 0 ppm) for 1H or CDCl3 (δ = 77 ppm) for 13C. High resolution mass spectral (HRMS) analysis was carried out using a JEOL JM AX 505 HA instrument.Differential scanning calorimetry
Differential scanning calorimetry was conducted on a Perkin-Elmer Pyris 6 DSC instrument using sealed aluminium pans. The samples were heated from room temperature to 130 °C under a nitrogen flow, at a heating/cooling rate of 5 °C min−1.Absorption and fluorescence spectroscopy
Absorption spectra were measured using a Shimadzu UV-visible-3101 PC NIR scanning spectrophotometer using a quartz cell with a 1 cm path length. Fluorescence spectra were recorded using a SPEX-Fluorolog F112X spectrofluorometer equipped with a 450 W xenon arc lamp. The spectra were corrected using the program installed by the manufacturer.Fluorescence quantum yield and lifetime
The fluorescence quantum yields in the solution state were determined relative to a standard compound, quinine sulphate (0.1 M H2SO4; ϕF = 0.546), using optically matching solutions. The fluorescence quantum yield of the powder samples was calculated using a calibrated integrating sphere in the SPEX Fluorolog spectrofluorometer. The samples were excited at 390 nm using the Xe-arc lamp as the source of excitation. The absolute quantum yield was determined on the basis of the de Mello method.8 Fluorescence lifetimes were determined using an IBH (FluoroCube) time-correlated picosecond single photon counting (TCSPC) system. The samples were excited at a wavelength of 410 nm (NanoLED-11) with a repetition rate of 1 MHz using a pulsed diode laser (<100 ps pulse duration).Field-emission scanning electron microscopy (FE-SEM)
For the FE-SEM measurements, the samples were drop cast and air-dried on a flat surface of a SEM brass grid and subjected to thin gold coating using a JEOL JFC-1200 fine coater. The samples were analysed with a JEOL JSM-5600 LV scanning electron microscope.X-ray diffraction measurements
Wide angle X-ray diffraction patterns of the samples were recorded using an XEUSS WAXS system by Xenocs, operated at 50 kV and 0.60 mA. The XEUSS WAXS system is equipped with a Linkam THMS 600 hot stage and a Linkam TMS 93 programmable temperature controller in order to perform the temperature-dependent XRD measurements.Fluorescence microscopy
Fluorescence microscopy images were taken by using a Leica DM 2500P microscope with a UV excitation source.Synthesis and characterization
The synthesis and characterization of 2,5-bis(dodecyloxy)terephthalaldehyde have been reported earlier.9 Synthesis of the target compounds DBBO and DBBT was performed as follows.Results and discussion
Synthesis and characterization
DBBO and DBBT were synthesised by the Knoevenagel condensation reaction between 2,5-didodecylterephthalaldehyde and 2-methylbenzoxazole or 2-methylbenzothiazole, respectively. Both compounds were characterized by analytical techniques such as 1H NMR, 13C NMR, FT-IR and high-resolution mass spectrometry. They were soluble in common organic solvents like hexane, toluene, tetrahydrofuran (THF), chloroform and dimethylformamide (DMF).Photophysical properties in solution
The absorption and emission properties of DBBO and DBBT in hexane, toluene, THF and DMF are summarized in Table S1† (conc. = 10−5 M; l = 10 mm; λex = 390 nm for fluorescence studies; λex = 410 nm for lifetime studies). The absorption spectra of both molecules red-shifted only marginally with increasing solvent polarity (Fig. S1a and c†), indicating that the dipole moments of the molecules in the ground state are fairly insensitive to solvent polarity. Nevertheless, about 10–13 nm blue-shift was observed in hexane compared to other solvents, which was attributed to aggregation. The emission spectra were more sensitive to solvent polarity and showed a bathochromic shift of ∼40 nm when changing from hexane to DMF (Fig. S1b and d†). This observation suggests that the dipole moments of the excited states are slightly higher compared to those of the ground states. The linking of donor and acceptor moieties to form a conjugated backbone results in the localization of π-electrons towards acceptors in more polar solvents leading to an internal charge transfer (ICT) state. Solvent relaxation processes in more polar solvents perturb the vibrational levels of the molecules resulting in broadening of the emission spectra. In other words, the direct excitation of these molecules leads to a locally excited state (LE) having nearly the same dipole moment as the ground state, which subsequently transforms into a more polar emissive state via an intramolecular charge transfer (ICT) process. This was accompanied by a slight increase of lifetimes (Fig. S1e and f†), but fluorescence quantum yields remained the same (Table S1†).Thermoresponsive luminescence behavior
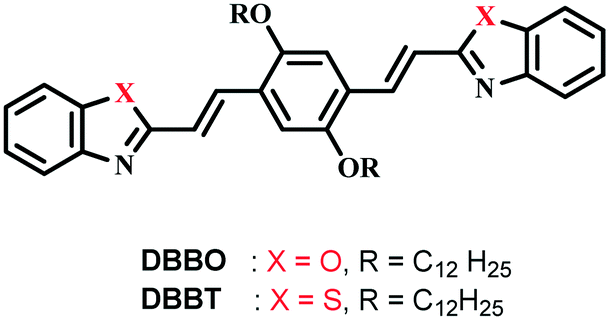
DBBO was observed to exist in two polymorphic forms with significantly different fluorescence emission colors. The pristine form (P-form, pure sample precipitated from CH2Cl2 on addition of cold hexane) was stable and can be converted to another stable form on heating at ∼80 °C followed by cooling to room temperature under ambient conditions (H-form). The switching was associated with a blue-shift of the emission from red to greenish-yellow. The emission maximum of the P-form was at 636 nm, whereas that of the H-form was at 541 nm. The corresponding emission spectra are shown in Fig. 1a. The absolute quantum yield of the P-form was found to be 16%, which was nearly doubled to 30% on switching to the H-form. Though the H-form was very stable under ambient conditions, it can be reverted back to the P-form upon solvent fuming (dichloromethane; HF-form), restoring the original red emission. For this purpose, the H-form of the compound was exposed to the vapors of dichloromethane for about 2 minutes by keeping it in a closed Petri dish containing saturated vapors of the solvent. It could be assumed that the solvent vapors act as a medium for rearranging the H-form to the P-form. However, dissolution of the sample was not observed during the fuming process. Similar changes were observed when exposed to vapors of chloroform, toluene and tetrahydrofuran. However, slightly more time (about 3 minutes) was required for the complete conversion of the sample, probably due to the low volatility of these solvents when compared to that of dichloromethane. It was also noted that the well-powdered sample has a relatively faster recovery rate than the less powdered one. This indicates that the speed of recovery depends on the rate of diffusion of solvent vapors through the sample. In order to study the role of oxygen and moisture in luminescence switching, we have carried out the heating of the P-forms of both compounds in a glovebox under an inert atmosphere and the emission properties were monitored. The emission changes were exactly similar to those under the ambient conditions. This observation proved that ambient oxygen and moisture did not have any effect on the luminescence switching phenomenon.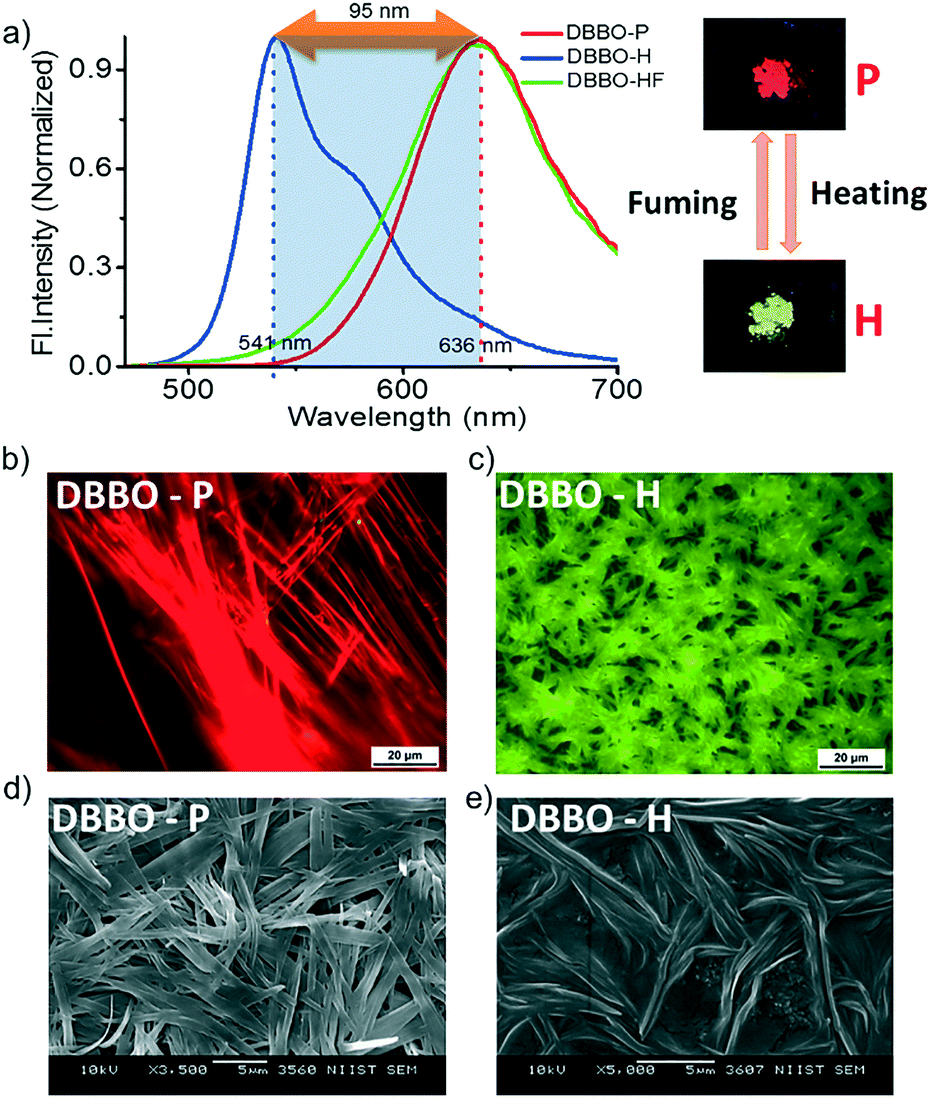 | ||
| Fig. 1 (a) Photoluminescence spectra of DBBO under different conditions; P – pristine powder sample, H – sample heated at 80 °C followed by air cooling to room temperature, and HF – sample H on fuming with dichloromethane. Photographs of the pristine and heated samples under 365 nm UV light are shown at the right side. Fluorescence microscopy images of the (b) pristine and (c) heated samples of DBBO under excitation by a UV source. FE-SEM images of the self-assembled structures of (d) pristine and (e) heated samples of DBBO. | ||
The toluene solution of DBBO drop cast on a microscope cover glass showed rigid, long, needle-like microstructures with brilliant red emission when viewed through a fluorescence microscope under excitation with UV light. (Fig. 1b). On heating the sample at ∼80 °C, the texture was changed to a short fibrous-like morphology with excellent greenish-yellow emission (Fig. 1c). The aggregate morphology was also studied by FE-SEM; the P-form was found to have a rigid, tape-like morphology (Fig. 1d), which got converted to a flexible one in the H-form (Fig. 1e). Both fluorescence microscopy and SEM analysis revealed that the transition from the P-form to the H-form was associated not only with luminescence changes but also with a visible change at the macroscopic level. In order to prove that there is no chemical change occurring in the molecules during heat-induced transformation, both the P-form and H-form were dissolved in THF and fluorescence analysis was conducted (Fig. S3†). Observation of the same spectra reveals that the basic chemical formula of the compounds remains the same after heating. No chemical decomposition was observed on several cycles of luminescence switching. The large difference in the fluorescence of both states makes the material useful for the preparation of a fast response optical storage medium (Fig. S4†). In the case of DBBT, comparable changes were obtained, which are summarized in the ESI.†
Phase transition behavior
The phase transition associated with luminescence switching was studied by differential scanning calorimetry (DSC). The first heating cycle of DBBO showed a broad endothermic peak at around 79 °C, which corresponds to the phase transition from the P-form to the H-form. However, such a transition was absent in the second cycle, which implies the irreversibility of the H-form to P-form transition upon cooling (Fig. 2a). The endothermic transition at 116 °C corresponds to the melting temperature, whereas, the exothermic peak at 105 °C could be attributed to the cold crystallization (regeneration of the H-form) of the molecules from their melt state. The heat-induced crystalline phase transition of the material was further proved by temperature-dependent XRD analysis (Fig. 2b). DBBO showed sharp and intense peaks in the P-form, which started transforming to the H-form above 60 °C and completed by 80 °C. The peaks around 2θ = 10° disappeared, while other peaks either disappeared or shifted significantly. Above 116 °C, all peaks disappeared due to the melting of the material.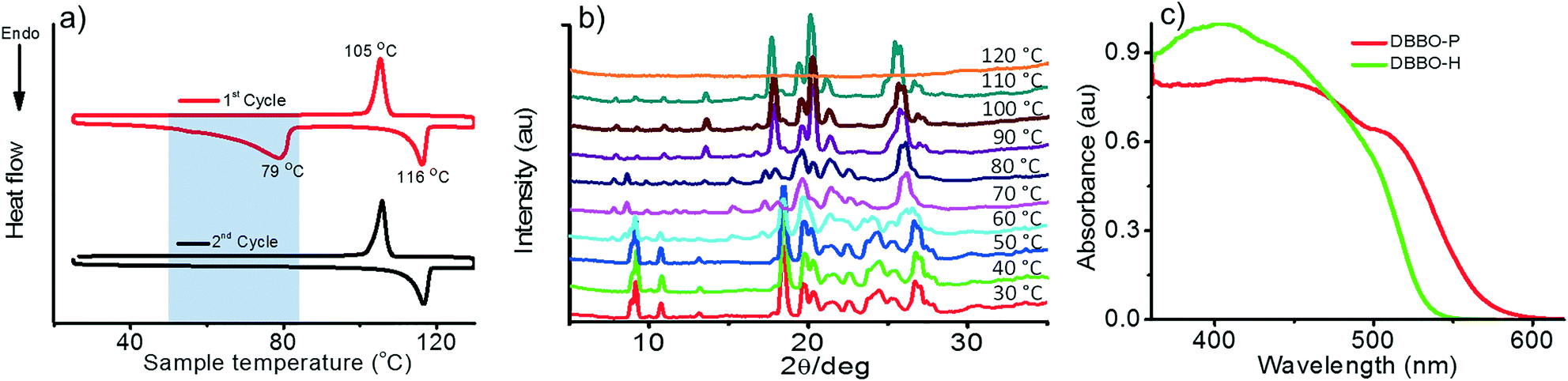 | ||
| Fig. 2 a) DSC thermogram showing the first and second heating cycles, b) temperature-dependent XRD patterns and c) solid-state absorption spectra of the P-form and H-form of DBBO. | ||
It must be noted that the emission spectrum of the P-form (Fig. 1a) does not have any vibrational features, whereas, the H-form has a vibrational shoulder at around 575 nm. The absence of vibrational features and the red-shifted emission maximum imply that the P-form has a stronger donor–acceptor interaction resulting in ICT behavior. On the other hand, emission from the H-form is more of an LE in nature. The solid-state UV-vis absorption spectrum of the P-form and H-form was measured (Fig. 2c) to investigate this aspect further. The former has a clear shoulder corresponding to the ICT state at 525 nm, whereas, it disappeared in the H-form indicating the absence of the ICT phenomenon.
Excitation energy migration
Time-resolved fluorescence lifetime measurement was carried out to obtain a better understanding about the underlying photophysical properties at the supramolecular level associated with the luminescence switching. The fluorescence lifetime was measured for the P-form, H-form, and HF-form and compared with that of the solution state (S-form; Fig. 3). The average fluorescence lifetime of DBBO in the S-state (in toluene) was found to be 1.55 ns, while it substantially increased to 10.77 ns in the P-form. However, the lifetime decreased to 6.03 ns in the H-form and reverted back to >10 ns on fuming with the solvent (HF-form), which is in accordance with the fluorescence studies. The longer lifetime of the P-form could be attributed to efficient energy migration due to the effective orbital overlap and electronic communication between the neighboring molecules.10 As a result, the excitons move through a large number of chromophores before recombination. During this movement, they lose some amount of energy resulting in red-shifted emission and lower fluorescence quantum yields. In the H-form, the orbital overlap between donor and acceptor moieties of the neighboring molecules is weak. Hence, the excitons move through less chromophores. The reduction in energy migration efficiency is reflected in the blue-shift of the emission maximum and increases the quantum yield. The molecularly dissolved state has a very short lifetime and the highly blue-shifted emission confirms the absence of energy migration between the chromophores in solution. These studies imply that heat-induced molecular packing transition basically modifies the energy migration efficiencies in the molecular assemblies, which in turn control the color and intensity of the luminescence.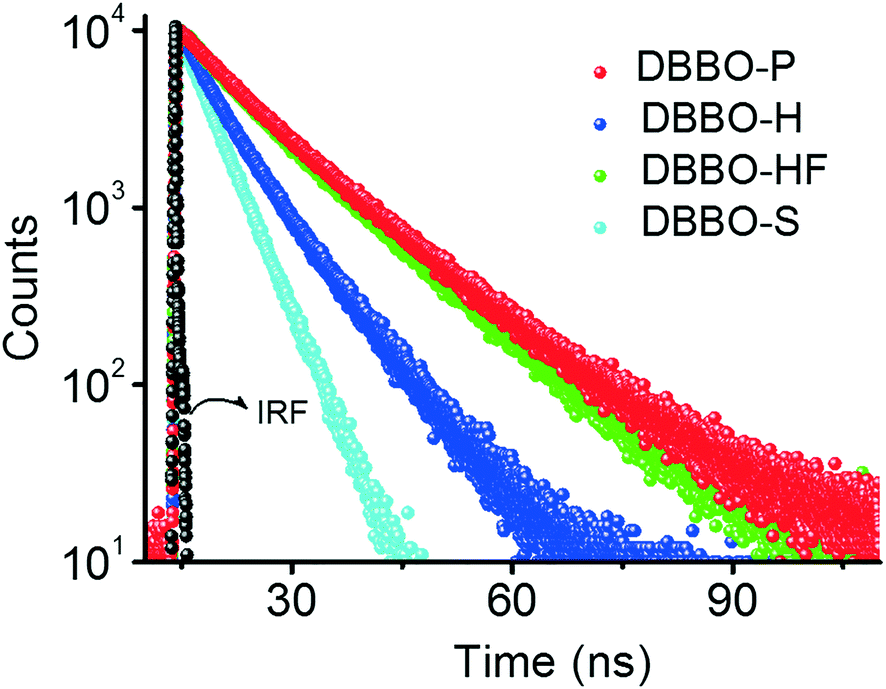 | ||
| Fig. 3 Time-resolved fluorescence decay profile of DBBO at different states; P – pristine powder sample; H – heated sample; HF – H powder on fuming with dichloromethane; S – molecularly dissolved state (IRF – instrument response factor). | ||
Mechanoresponsive luminescence behaviour
We hypothesized that forcing DBBO and DBBT molecules to come closer by applying mechanical force may improve the intermolecular donor–acceptor interactions and the regeneration of the ICT state. In other words, regeneration of P-form emission from the H-form would be possible by applying mechanical stimuli. However, this process is completely different from the interconversion achieved by solvent fuming, which is a spontaneous reorganization of the molecules in a solvent medium. As anticipated, the H-form exhibits mechanoresponsive luminescence switching; upon grinding, the greenish-yellow emission (λ = 541 nm) changes to broad red emission (λ = 613 nm; Fig. 4a). The resulting emission was comparable with that of the P-state, albeit not exactly similar. The spectrum was broader with a remarkable tailing to the higher energy side and the fluorescence quantum yield was lower (6%). The tailing implies that the conversion is not complete and a significant amount of the material remains in the H-state. The lower quantum yield indicates the increase in the non-radiative recombination of excitons in the sample. Nevertheless, the overall energy migration efficiency improved, as is evident from the increase of the lifetime from 6.03 ns to 7.76 ns (Fig. 4b). Powder X-ray diffraction analysis proved that the diffraction peaks observed in the H-form disappear upon grinding (Fig. S7†). This implies that grinding causes the disruption of the long-range order in the molecular assembly. However, the enhancement of donor–acceptor interactions at the molecular level was enough to obtain the red-shifted emission similar to that of the P-state. It is also worth noting that the mechano-stimuli do not have any effect on the P-form.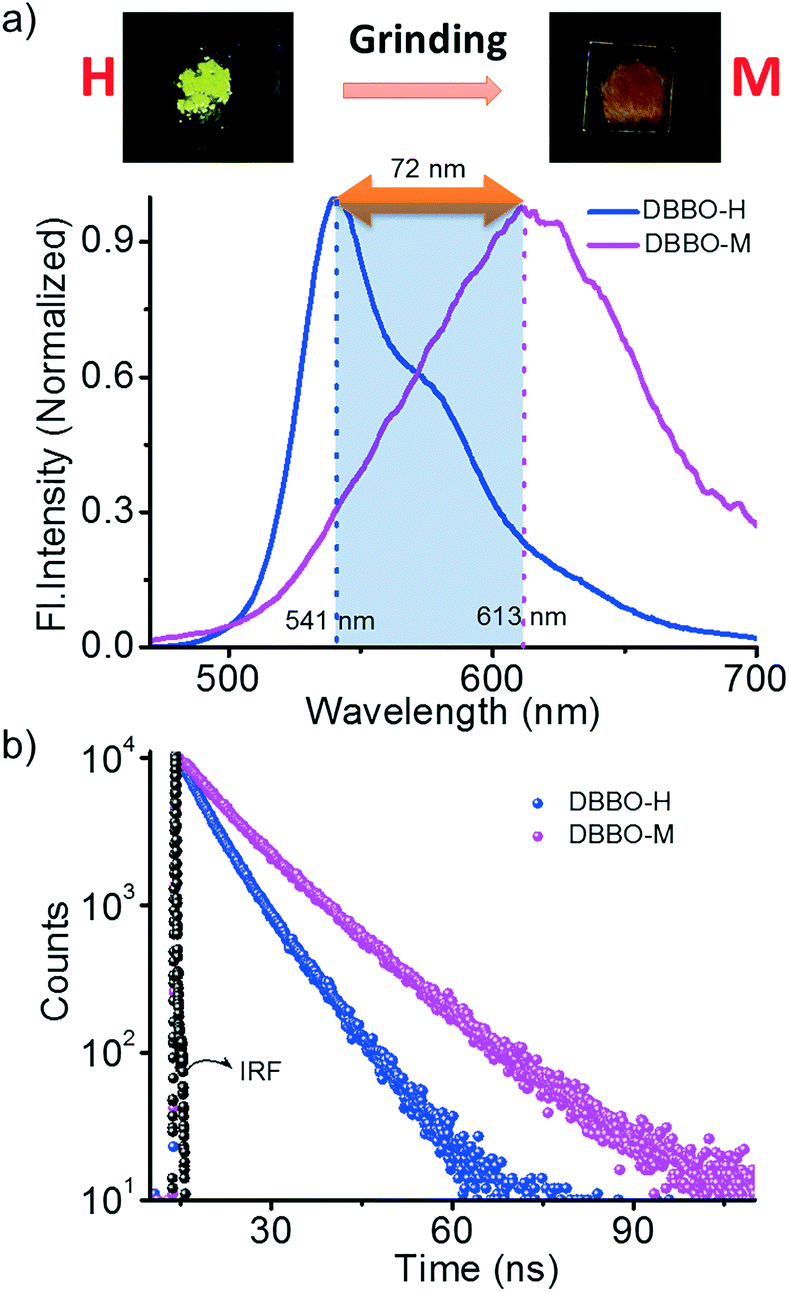 | ||
| Fig. 4 a) Photoluminescence spectra and b) time-resolved fluorescence decay profile of DBBO under different conditions; H – heated sample and M – H sample after applying mechanical force; (IRF – instrument response factor). | ||
Though DBBT exhibited comparable thermoresponsive and energy migration properties to those of DBBO, the mechanoresponsive nature was different. Compared to the 72 nm red-shift exhibited by DBBO, only 37 nm shift was observed for DBBT. Moreover, the intensity of the emission spectrum in the blue region is substantially higher. This observation is in accordance with our previous work on anthracene appended benzoxazole and benzothiazole derivatives.11 Due to the stronger π-stacking interaction in the latter, it showed only very weak mechanochromic luminescence response. The luminescence spectral changes and corresponding photographs of the H-form and M-form samples are shown in Fig. 5a. In accordance with the fluorescence spectral changes, the improvement in the lifetime was also minimal (2.22 ns to 3.18 ns) as evident from Fig. 5b.
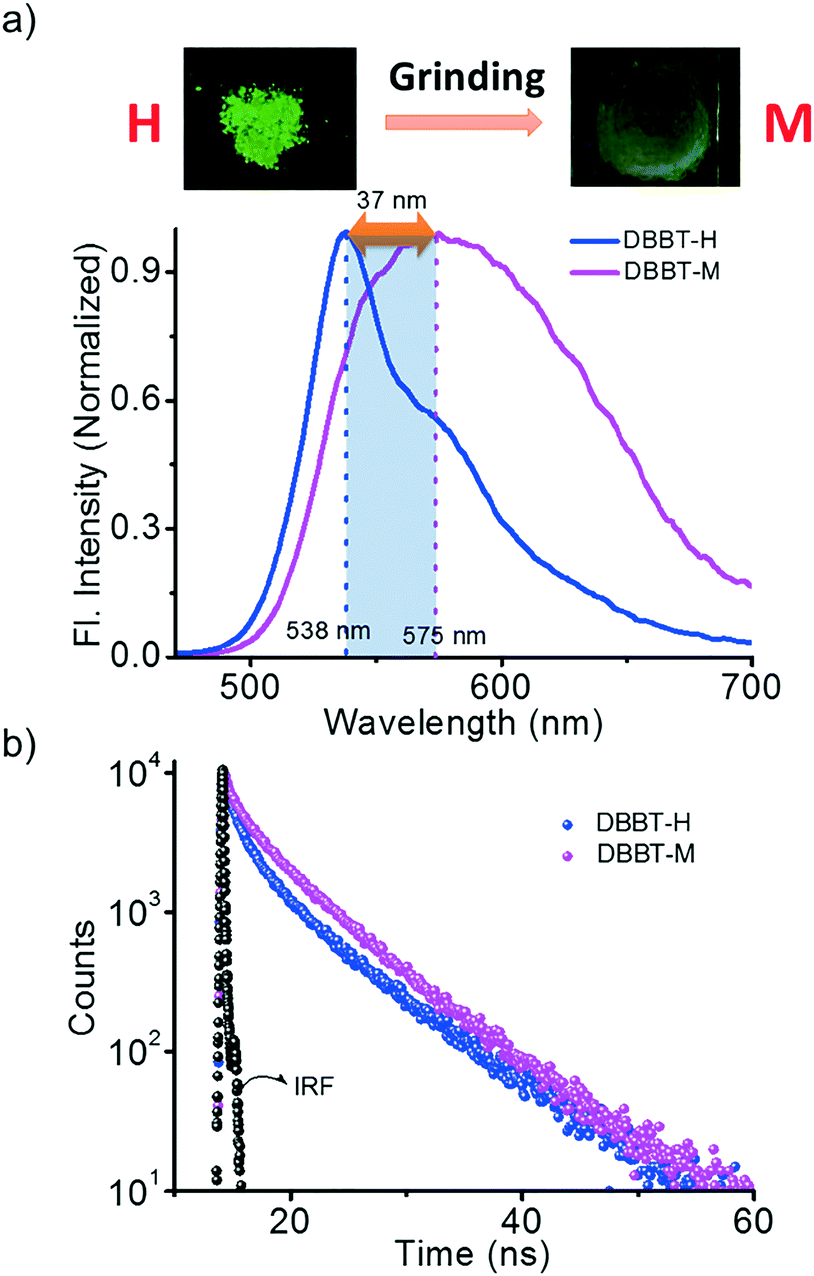 | ||
| Fig. 5 a) Photoluminescence spectra and b) time-resolved fluorescence decay profile of DBBT under different conditions; H – heated powder sample and M – H sample after grinding; (IRF – instrument response factor). | ||
Heat-induced phase-change technology has become one of the major methods for developing rewritable optical media.12 Traditionally, the change in reflectivity associated with the phase change is used as the output. Though the use of the change in luminescence has many advantages such as high signal-to-noise ratios, excellent sensitivity and easy operation, the availability of a very limited number of potential materials is a bottleneck. Our results prove that benzoxazole or benzothiazole containing divinylbenzene derivatives are excellent materials for rewritable optical media and information storage with high contrast luminescence states. We illustrated that the variation in fundamental photophysical properties such as excitation energy migration associated with different molecular packings is responsible for the luminescence color and intensity switching. Consequently, switching to the initial emission characteristics was achieved not only by restoring the molecular packing through solvent fuming but also by forcing the molecules to interact closely by applying mechanical stimuli. Hence, our findings pave the way for developing novel organic materials with switchable luminescence through multiple stimuli.
Conclusions
We have synthesized and characterized two novel benzoxazole or benzothiazole substituted 1,4-bis(dodecyloxy)-2,5-divinylbenzene derivatives showing excellent solid-state emission and thermochromic luminescence response properties. DSC and XRD analyses confirmed that heating of these materials results in a phase transformation between two crystalline states having different molecular packing modes. These two polymorphic states of each derivative possess reversible and high contrast visually distinguishable fluorescence (Δλ > 90 nm). Pristine samples with red-shifted emission and longer lifetimes indicate highly excited state coupling between the adjacent molecules due to close interaction leading to efficient energy migration. Heating causes molecular rearrangement resulting in another crystalline state with greenish-yellow emission and a short lifetime, which signifies the weak excited state molecular coupling and reduced energy migration. This large difference in fluorescence color and intensity could be utilized for developing optical rewritable media.Conflicts of interest
There are no conflicts to declare.Acknowledgements
V. C. N thanks DST for the Ramanujan Fellowship (SR/S2/RJN-133/2012). K. C. N. is grateful to UGC for the Senior Research Fellowship. We thank Dr. E. Bhoje Gowd for XRD measurements.Notes and references
- (a) X. Hou, C. Ke, C. J. Bruns, P. R. McGonigal, R. B. Pettman and J. F. Stoddart, Nat. Commun., 2015, 6, 6884–6893 CrossRef PubMed; (b) S. Yagai, S. Okamura, Y. Nakano, M. Yamauchi, K. Kishikawa, T. Karatsu, A. Kitamura, A. Ueno, D. Kuzuhara, H. Yamada, T. Seki and H. Ito, Nat. Commun., 2014, 5, 4013–4023 CrossRef CAS PubMed; (c) S. Hirata and T. Watanabe, Adv. Mater., 2006, 18, 2725–2729 CrossRef CAS; (d) A. Kishimura, T. Yamashita, K. Yamaguchi and T. Aida, Nat. Mater., 2005, 4, 546–549 CrossRef CAS PubMed; (e) X. Wang, O. S. Wolfbeis and R. J. Meier, Chem. Soc. Rev., 2013, 42, 7834–7869 RSC; (f) F. Ciardelli, G. Ruggeri and A. Pucci, Chem. Soc. Rev., 2013, 42, 857–870 Search PubMed; (g) Y. Sagara, T. Mutai, I. Yoshikawa and K. Araki, J. Am. Chem. Soc., 2007, 129, 1520–1521 CrossRef CAS PubMed.
- (a) Y. Sagara and T. Kato, Nat. Chem., 2009, 1, 605–610 CrossRef CAS PubMed; (b) Y. Sagara, S. Yamane, M. Mitani, C. Weder and T. Kato, Adv. Mater., 2016, 28, 1073–1095 CrossRef CAS PubMed; (c) Z. Chi, X. Zhang, B. Xu, X. Zhou, C. Ma, Y. Zhang, S. Liu and J. Xu, Chem. Soc. Rev., 2012, 41, 3878–3896 RSC; (d) J. N. Zhang, H. Kang, N. Li, S. M. Zhou, H. M. Sun, S. W. Yin, N. Zhao and B. Z. Tang, Chem. Sci., 2017, 8, 577–582 RSC; (e) S. K. Park, I. Cho, J. Gierschner, J. H. Kim, J. H. Kim, J. E. Kwon, O. K. Kwon, D. R. Whang, J. H. Park, B. K. An and S. Y. Park, Angew. Chem., Int. Ed., 2016, 55, 203–207 CrossRef CAS PubMed; (f) S.-J. Yoon and S. Park, J. Mater. Chem., 2011, 21, 8338–8346 RSC; (g) S. Xue, X. Qiu, Q. Sun and W. J. Yang, J. Mater. Chem. C, 2016, 4, 1568–1578 RSC; (h) H. Wu, C. Hang, X. Li, L. Yin, M. Zhu, J. Zhang, Y. Zhou, H. Ågren, Q. Zhang and L. Zhu, Chem. Commun., 2016, 53, 2661–2664 RSC; (i) X. Du, F. Xu, M.-S. Yuan, P. Xue, L. Zhao, D.-E. Wang, W. Wang, Q. Tu, S.-W. Chen and J. Wang, J. Mater. Chem. C, 2016, 4, 8724–8730 RSC; (j) M. Okazaki, Y. Takeda, P. Data, P. Pander, H. Higginbotham, A. P. Monkman and S. Minakata, Chem. Sci., 2017, 8, 2677–2686 RSC; (k) X. Luo, W. Zhao, J. Shi, C. Li, Z. Liu, Z. Bo, Y. Q. Dong and B. Z. Tang, J. Phys. Chem. C, 2012, 21967–21972 CrossRef CAS; (l) Y. Sagara, Y. C. Simon, N. Tamaoki and C. Weder, Chem. Commun., 2016, 52, 5694–5697 RSC; (m) M. Sen Yuan, D. E. Wang, P. Xue, W. Wang, J. C. Wang, Q. Tu, Z. Liu, Y. Liu, Y. Zhang and J. Wang, Chem. Mater., 2014, 26, 2467–2477 CrossRef; (n) Y. Xie, Y. Ge, Q. Peng, C. Li, Q. Li and Z. Li, Adv. Mater., 2017, 29 DOI:10.1002/adma.201606829; (o) J.-X. Qiu, Y.-X. Li, X.-F. Yang, Y. Nie, Z.-W. Zhang, Z.-H. Chena and G.-X. Sun, J. Mater. Chem. C, 2014, 2, 5954–5962 RSC.
- (a) H.-J. Yen and G.-S. Liou, Chem. Commun., 2013, 49, 9797–9799 RSC; (b) A. Beneduci, S. Cospito, M. La Deda, L. Veltri and G. Chidichimo, Nat. Commun., 2014, 5, 3105 Search PubMed; (c) W. R. Browne, M. M. Pollard, B. De Lange, A. Meetsma and B. L. Feringa, J. Am. Chem. Soc., 2006, 128, 12412–12413 CrossRef CAS PubMed; (d) E. Takahashi, H. Takaya and T. Naota, Chem. – Eur. J., 2010, 16, 4793–4802 CrossRef CAS PubMed; (e) Y. Gong, Y. Tan, J. Liu, P. Lu, C. Feng, W. Z. Yuan, Y. Lu, J. Z. Sun, G. He and Y. Zhang, Chem. Commun., 2013, 49, 4009–4011 RSC; (f) Q. Zhu, W. Yang, S. Zheng, H. H. Y. Sung, I. D. Williams, S. Liu and B. Z. Tang, J. Mater. Chem. C, 2016, 4, 7383–7386 RSC; (g) J. Chen, S. Ma, J. Zhang, L. Wang, L. Ye, B. Li, B. Xu and W. Tian, J. Phys. Chem. Lett., 2014, 5, 2781–2784 CrossRef CAS PubMed; (h) J. Zhang, J. Chen, B. Xu, L. Wang, S. Ma, Y. Dong, B. Li, L. Ye and W. Tian, Chem. Commun., 2013, 49, 3878–3880 RSC; (i) K. Wang, S. Huang, Y. Zhang, S. Zhao, H. Zhang and Y. Wang, Chem. Sci., 2013, 4, 3288–3293 RSC; (j) Y.-Q. Li, T. Huang, B.-Q. Jin, Y. Shi, Z.-T. Liu and X.-G. Jin, Acta Polym. Sin., 2016, 6, 750–759 Search PubMed.
- T. Mutai, H. Satou and K. Araki, Nat. Mater., 2005, 4, 685–687 CrossRef CAS PubMed.
- S. J. Yoon, J. W. Chung, J. Gierschner, K. S. Kim, M. G. Choi, D. Kim and S. Y. Park, J. Am. Chem. Soc., 2010, 132, 13675–13683 CrossRef CAS PubMed.
- Y. Sagara, A. Lavrenova, A. Crochet, Y. C. Simon, K. M. Fromm and C. Weder, Chem. – Eur. J., 2016, 22, 4374–4378 CrossRef CAS PubMed.
- A. Lavrenova, D. W. R. Balkenende, Y. Sagara, S. Schrettl, Y. C. Simon and C. Weder, J. Am. Chem. Soc., 2017, 139, 4302–4305 CrossRef CAS PubMed.
- J. C. de Mello, H. F. Wittmannn and R. H. Friend, Adv. Mater., 1997, 9, 230–232 CrossRef CAS.
- (a) S. J. George and A. Ajayaghosh, Chem. – Eur. J., 2005, 11, 3217–3227 CrossRef CAS PubMed; (b) V. K. Praveen, S. J. George, R. Varghese, C. Vijayakumar and A. Ajayaghosh, J. Am. Chem. Soc., 2006, 128, 7542–7550 CrossRef CAS PubMed; (c) B. Wang and M. R. Wasielewski, J. Am. Chem. Soc., 1997, 119, 12–21 CrossRef CAS.
- (a) C. Vijayakumar, V. K. Praveen, K. K. Kartha and A. Ajayaghosh, Phys. Chem. Chem. Phys., 2011, 13, 4942–4949 RSC; (b) C. Vijayakumar, V. K. Praveen and A. Ajayaghosh, Adv. Mater., 2009, 21, 2059–2063 CrossRef CAS; (c) A. Ajayaghosh, V. K. Praveen and C. Vijayakumar, Chem. Soc. Rev., 2008, 37, 109–122 RSC.
- K. C. Naeem, A. Subhakumari, S. Varughese and V. C. Nair, J. Mater. Chem. C, 2015, 3, 10225–10231 RSC.
- (a) A. Seeboth, D. Lötzsch, R. Ruhmann and O. Muehling, Chem. Rev., 2014, 114, 3037–3068 CrossRef CAS PubMed; (b) S. Abraham, V. A. Mallia, K. V. Ratheesh, N. Tamaoki and S. Das, J. Am. Chem. Soc., 2006, 128, 7692–7698 CrossRef CAS PubMed; (c) M. Louis, A. Brosseau, R. Guillot, F. Ito, C. Allain and R. Métivier, J. Phys. Chem. C, 2017, 121, 15897–15907 CrossRef CAS; (d) S. J. Yoon, J. H. Kim, K. S. Kim, J. W. Chung, B. Heinrich, F. Mathevet, P. Kim, B. Donnio, A. J. Attias, D. Kim and S. Y. Park, Adv. Funct. Mater., 2012, 22, 61–69 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis scheme, absorption spectra, emission spectra, lifetime decay profile and photophysical parameters of DBBO and DBBT in the solution state, solid state emission spectra, SEM images, fluorescence microscopy images, DSC analysis, temperature dependent XRD of DBBT, PXRD patterns, solid state absorption spectra and fluorescence lifetime of DBBO and DBBT in the solid state. See DOI: 10.1039/c7me00089h |
| This journal is © The Royal Society of Chemistry 2018 |