
Beyond native block copolymer morphologies
Gregory S.
Doerk
 * and
Kevin G.
Yager
*
* and
Kevin G.
Yager
*
Center for Functional Nanomaterials, Brookhaven National Laboratory, Upton, NY, USA. E-mail: gdoerk@bnl.gov; kyager@bnl.gov
First published on 20th September 2017
Abstract
Block copolymers are a well-studied and canonical self-assembly paradigm. Despite active research, new methods are continually emerging that allow the formation of yet more diverse and complex morphologies. In this review, we analyze and comment on the state-of-the-art with respect to exploiting block copolymers in order to form ‘non-native’ morphologies; that is, structures that deviate from the bulk equilibrium morphologies observed in the simplest block copolymer systems. The range of reviewed strategies can be organized with respect to where the final structural information is encoded. Self-assembly involves intrinsic encoding within the molecules themselves, while external fields or templates can be used to externally encode a desired structure. Hybrid strategies, which exploit the responsiveness of self-assembly but control it through external forces or process history, are particularly promising.
Design, System, ApplicationBlock copolymers are self-assembling materials that spontaneously phase separate to generate a variety of simple shapes and patterns, such as hexagonally-packed cylinders. This article reviews methods for exploiting block copolymer materials to generate more complex nanostructures. In particular, the intrinsic morphologies of copolymers can be directed using external templates or processing history, allowing a variety of ‘non-native’ morphologies to be created. By broadening the range of patterns that self-assembly can form, a broader range of applications become viable. |
1. Introduction
Self-assembly is an elegant and powerful paradigm for generating nanostructures. The desired final structure is encoded within the architecture of the molecular building blocks, with the actual fabrication step occurring spontaneously through inter-molecular and intra-molecular interactions and stochastic, hierarchical assembly of the components.1 The underlying fundamental principle is energy-minimization, wherein the constituent species explore their local state-space, evolving towards equilibrium. Because the assembly is spontaneous and simultaneous throughout the material, self-assembly holds the promise of rapid, inexpensive, and scalable fabrication of precise nano-architectures. Block copolymers—long-chain molecules whose chemical identity varies in a prescribed manner through the macromolecular architecture—are a versatile and well-studied class of self-assembling materials.2–6 The chemically distinct segments along the polymer chains experience a driving force towards phase separation. The covalent links between the chemically incompatible blocks frustrate this tendency; the compromise is local demixing of chain segments—“microphase separation”—which thus generates a nanostructure whose size and shape directly reflects the chain architecture.2 Thus, a host of different nanoscale morphologies can be easily formed by synthesizing the appropriate block copolymer (BCP) macromolecules. While the local ordering motif is defined by the material's intrinsic packing, the long-range order is strongly influenced by coarsening kinetics; experimentally one typically observes an isotropic distribution of grains within which the intrinsic morphology is well-defined, or long-range undulations/distortions of the local motif (e.g. ‘fingerprint’ patterns).The grand promise of self-assembly is the rapid formation of arbitrary, complex three-dimensional objects with structural precision from the molecular to the macroscale, simply by mixing together appropriate building blocks. In this sense, the range of structures readily accessible via self-assembly may seem disappointingly limited.7–9 In the canonical case of linear diblock copolymers, the easily obtained morphologies are those that minimize interfacial area between the blocks and curvature of interfaces: thus one can only form alternating lamellae (equal volume fraction of the two blocks), bicontinuous gyroid phases (slight asymmetry in volume fractions), hexagonally-packed cylinders (moderate asymmetry), or spheres arranged in a body-centered cubic lattice (greater asymmetry).10–12 Similar morphologies are frequently observed in the self-assembly of molecular (e.g. liquid crystalline) materials. In other words, self-assembly generically creates simple structures that minimize surface area and maximize symmetry. Great effort has gone into increasing the diversity and complexity of self-assembled structures. Supramolecular building blocks can be designed to encode non-trivial shapes,13,14 nanoparticles can be induced into assembling into lattices,15–18 and DNA can be used to form nano-shapes19–21 or as a programmable linker to control the organization of nanoparticles22,23 into complex superlattices24–29 and finite-sized meso-objects.30–32 The increasing sophistication of self-assembled structures is impressive; yet more elaborate assemblies require correspondingly more elaborate building blocks, in order to encode the requisite organizational information.27,33–35 This seemingly inescapable correlation between component complexity and assembly complexity severely limits the utility of self-assembly methods. It is in this context that a variety of alternative strategies to control structure formation have been presented. In particular, some aspect of the required target structure can be enforced externally, through applied fields or environmental conditions. In this area, block copolymer researchers have led the way, demonstrating the power of external influences—from the finesse of processing history to the strict guidance of templates—in controlling the morphology, orientation, and degree-of-order of self-assembling materials.36,37
Here, we discuss existing and emerging strategies for controlling structure formation in block copolymer thin films; in particular focusing on methods for generating “non-native” morphologies that go beyond the conventional structures observed in the bulk equilibrium diblock copolymer phase diagram (lamellae, gyroid, hexagonally-packed cylinders, spheres on a cubic lattice). We focus on methods to control the local ordering motif (i.e. the minimal unit cell in the idealized extended lattice), while noting that extensive research has also gone into the extremely important issues of improving long-range order36,38,39 and accelerating assembly kinetics.37 The enormous variety of methods can be loosely organized along an “information encoding” axis (Fig. 1), where conventional self-assembly is one extreme, with all requisite information encoded in the macromolecular building blocks themselves (intrinsic encoding), and top–down lithographic patterning represents the opposite extreme, with structural information entirely enforced in a top–down manner (external enforcement). Between these extremes, a variety of hybrid strategies have emerged, which direct the intrinsic ordering of the BCP material, and thereby combine the advantages of self-assembly (rapid spontaneous pattern formation) with the advantages of external control (precise and tunable selection of ordering). These hybrid methods hold enormous promise. By exploiting aspects of processing history to control structure formation, the same BCP materials can be used to generate a variety of different final morphologies. This thus expands the utility and impact of any particular synthesized copolymer chain architecture. On the other hand, these hybrid methods necessarily exploit history-dependent phenomena, and thus non-equilibrium aspects of ordering. This invites increased experimental complexity, owing to the vast state-spaces associated with processing histories, and the challenges of handling kinetic effects, such as trapping in metastable states, degenerate morphologies, and pathway-dependent assembly.37,40–43 Despite these challenges, the promise of improved methods is clear: by better understanding fundamental aspects of self-assembly, ever more sophisticated nanostructures can be fabricated using relatively simple and well-understood macromolecular building blocks.
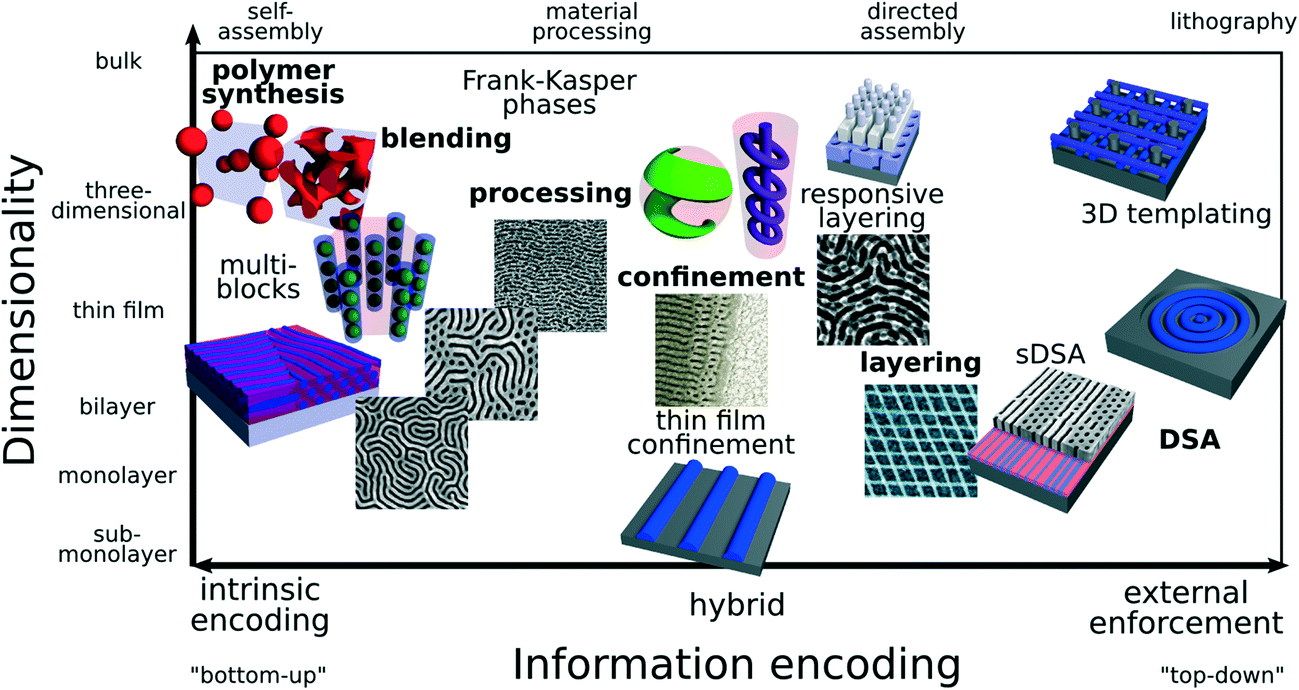 | ||
| Fig. 1 Methods for generating block copolymer (BCP) patterns beyond conventional equilibrium morphologies. Methods can be loosely organized according to how structural information is encoded. Traditional self-assembly involves synthesizing materials with a desired equilibrium morphology; i.e. the desired structure is encoded intrinsically into the molecules (bottom-up method). On the opposite extreme, lithography involves prescribing a nano-pattern externally, through an impinging light-field or electron-beam raster (top–down method). A host of methods combine the intrinsic ordering of self-assembly with some external control. Material processing history can be used to select non-equilibrium structures. Confinement perturbs natural BCP ordering, giving rise to unconventional frustrated phases. Layering of BCP phases can be used to generate new lattice types. This can be done in an enforced manner, or in a responsive manner where the natural ordering of the BCP is leveraged. Lithographic templates can be used for directed self-assembly (DSA), where the defined topographic or chemical features strongly control the ordering of a BCP phase. As the strength of the external enforcement increases, the ability to finely select a particular ordering motif increases. However, there is a tradeoff since the directing method becomes more complex and costly; the spontaneous ordering of the BCP is in some sense being wasted. Thus, hybrid methods, which direct ordering but exploit the responsiveness of self-assembly, hold great promise for the facile fabrication of nanostructures. | ||
2. Synthesis
The most fundamental means of expanding the diversity of self-assembled morphologies is to design and synthesize new molecules that assemble into new structures. Linear diblock copolymers are the canonical materials in the BCP community. The main morphologies observed in this phase diagram are alternating lamellae (when the block volume fractions are roughly equal), hexagonally-packed cylinders (asymmetric volume fractions), and spheres on a cubic lattice (greater asymmetry).10–12 Thus, the morphology can be controlled simply by the polymer chain architecture. The theoretical diblock-copolymer phase diagram also includes regions with more exotic morphologies, such as gyroid or perforated lamellae;2,10,44 experimentally such morphologies exist over a narrow range and can be difficult to access, owing to effects such as fluctuations, material polydispersity, and kinetic trapping.44–47 However, appropriate BCP compounds can stabilize particular motifs. For instance, including charged blocks can broaden the HPL and gyroid regions,48 or form percolated phases.49 Charges change the effective segregation strength (shifting the overall phase diagram) and can also introduce “electrostatic cohesion” within one of the blocks, which distorts the phase diagram asymmetrically, making one set of phases stable over a broader composition range (thus more easily accessible).The range of possible morphologies can be extended by considering chain architectures beyond the linear coil–coil diblocks. The most natural extension is to consider multi-block copolymers, allowing three or more materials to be combined and nano-structured.3 The formation of morphologies follows the same rules as for linear diblocks (material segregation and minimization of interfacial area), with the multiplicity of materials expanding the range of possible structures. Triblock copolymers can stabilize some of the exotic diblock phases (gyroid, perforated lamellae),50 and can form a wide variety of morphologies beyond the diblock phase diagram, including cylinder of alternating types, bi- and tri-continuous phases, coaxial cylinders or core–shell spheres, spheres organized along lamellae or inside cylinders, cylinders ribbed with toruses, helices, and many others (Fig. 2).12,51–55 Triblock copolymers can be used to form square packing motifs,56 or tetragonally-packed “cylinders” with rectangular cross-sections.57 Clearly, multi-block architectures can broaden the range of accessible morphologies. The space of possibilities could be further expanded by more investigations of non-linear copolymer architectures. For instance, star triblock copolymers are predicted to form a range of non-trivial Archimedean tilings.58
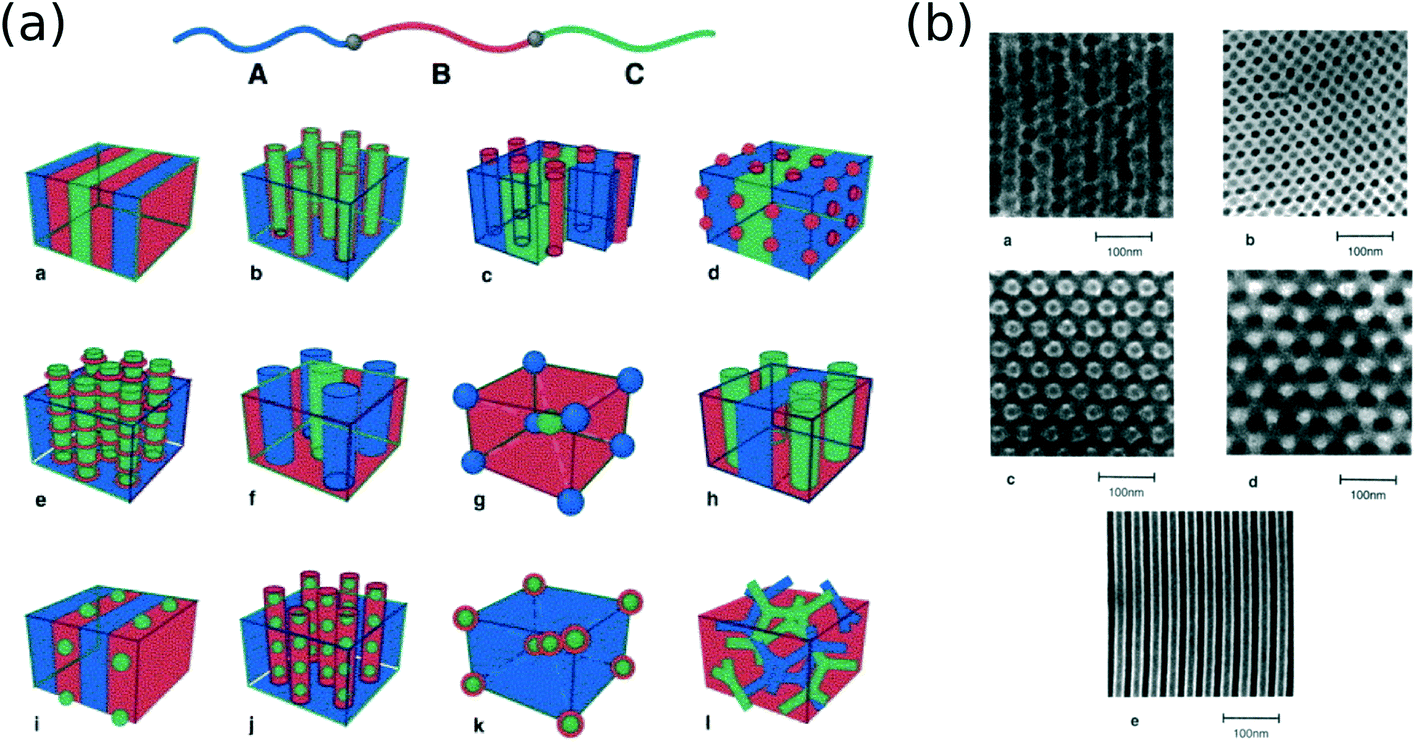 | ||
| Fig. 2 (a) Example morphologies formed by linear triblock copolymers formed from three different materials.12 Reproduced with permission from Elsevier 2004. The macromolecular architecture dictates the equilibrium morphology; thus improved synthetic methods can broaden the range of possible BCP morphologies. (b) Electron microscopy images of select triblock copolymer morphologies.52 Reproduced with permission from the American Chemical Society 1994. Micrograph (b)a shows a multi-sphere morphology (corresponding to schematic (a)g), with a body-centered cubic (BCC) arrangement of spheres of alternating chemical identify. (b)b shows a multi-cylinder morphology (schematic (a)f), with two cylinder types packed in a tetragonal lattice. (b)c and (b)d shows an ordered tricontinuous double-diamond (OTDD) phase (schematic (a)l), which is the three-component analog of the traditional gyroid phase. (b)e shows a multi-lamellar phase (schematic (a)a), where the three material components are stacked. | ||
A key parameter in polymer synthesis is the distribution of chain lengths; i.e. the polydispersity index (PDI). Monodisperse materials (PDI = 1.0) are conceptually simplest, yet real materials nearly always have some distribution in chain lengths. Higher PDI typically pushes the order–disorder transition temperature higher, owing to the presence of a small number of much larger chains throughout;59 while also broadening interblock interfaces, owing to localization of shorter chains close to this interface, which reduces surface tension.60 At sufficient PDI, coexisting phases and macrophase separation may occur.59 Polydispersity normally occurs as a continuous unimodal distribution of chain lengths. However, more complex chain length distributions can be engineered by blending of components (either during synthesis or assembly), which can be exploited to control assembly behavior, as discussed in the next section.
Bottlebrush copolymers61 exhibit several beneficial phenomena—such as reduced chain entanglements—making them interesting candidates for generating variants of phases observed in the diblock or triblock phase diagram; including conventional phases that deviate from the usual volume fractions (e.g. alternating lamellae where the volume fraction of the two phases is quite different), and known morphologies assembling at much larger length-scales. Rod–coil and rod–rod BCPs62–67 can form conventional BCP and liquid crystal phases, as well as unconventional phases (double-hexagonal, zigzag lamellae, helical ribbons, branched cylinders, etc.).
There are a variety of self-assembling materials that are closely-related to BCPs in the sense that the material architecture is similar, and the phase diagram is analogous, even if the materials themselves are not strictly block copolymers. For instance, dendrons and dendrimers,68–72 giant surfactants,73,74 and sugar–polyolefin conjugates75,76 exhibit ordering highly analogous to BCP materials. There is a tremendous untapped opportunity in applying the wealth of methods that have been optimized for controlling BCP ordering (field or shear alignment, zone annealing, directed/templated assembly, etc.) to these novel materials.
There is obviously enormous potential for the creation of new, non-trivial morphologies by designing the correct macromolecular chain architecture. Appropriate combination of material blocks and chain branching can theoretically yield extremely complex periodic patterns. On the other hand, increasingly branched architectures may frustrate ordering. Hutchings et al. developed synthesis methods to generate hyperbranched structures from macromonomers (HyperMacs)77 and corresponding hyperbranched copolymers (HyperBlocks).78 These architectures strongly influence mechanical properties, and exhibit local phase separation, yet the chain architecture evidently frustrates long-range ordering.78–80 Thus, architectural branching must be carefully designed in order to enforce a tailored well-ordered structure.
The obvious challenge in creating more complex nanoscale architectures via synthesis is the labor involved in synthesizing new compounds, where reaction sequences must be tested, synthesis conditions optimized, products carefully purified, and appropriate casting/processing conditions identified. Synthesis schemes that emphasize easy variation of architecture by combining building blocks can aid in exploring the vast combinatorial spaces more quickly. There is also an opportunity for theory and simulation methods to guide future synthesis efforts. A detailed survey of possible morphologies that can be obtained through control of block copolymer architecture would be instrumental in selecting future synthesis targets that give rise to new morphologies. Even more powerful would be a series of theory-informed design rules, such that for a target nano-structure, the (simplest) required copolymer chain architecture can be predicted. Given the labor involved in polymer synthesis, it is crucial to have detailed guidance so that synthesis of only the highest-priority architectures is attempted. The recent development of inverse design of morphologies is a promising step in this direction.81,82
3. Processing
Inducing the formation of ordered domains in BCPs requires that the polymer chains have sufficient mobility to diffuse and rearrange, a requirement typically met through thermal annealing or by plasticization (e.g. swelling in solvent vapors). At equilibrium, the minimum energy state for BCP thin films is a single defect-free grain of well-ordered, uniform domains oriented as dictated by global boundary conditions.83,84 However, a polygrain structure is observed experimentally unless special measures taken to promote long-range order. The energetic degeneracy of grain orientations, nucleation and propagation of domain order from sample boundaries,85,86 and the statistical distribution of grain sizes and shapes43,87–90 reflect the significant importance of initial ordering pathways. Moreover, once microphase separation has taken place, both domain and grain boundaries themselves create large energy barriers to further chain rearrangement,90,91 inhibiting defect annihilation and leading to kinetically trapped states.These aspects highlight a general pathway-dependence to ordering in BCPs that renders defectivity, domain orientation, and even morphology sensitive to the details of the process method used to promote ordering, and the processing history. For instance, thermally-reversible sphere-like surface structures may form in registry with underlying parallel cylinder morphologies;92i.e. a particular structural motif appears within a narrow process window. Domain packing and morphological orientation can change over time,43 permitting selection of a particular morphology orientation by truncating the annealing at a specific interval, though this depends on the film thickness and interfacial conditions.85 Moreover, short but ultra-rapid photothermal annealing has been shown to promote domain ordering and alignment to a degree that is unattainable at lower annealing temperatures in practical time scales.37,42,93–101 The plethora of results demonstrating pathway-dependent ordering highlights both a challenge and opportunity. Kinetic aspects of assembly are often a nuisance, limiting one's ability to achieve good order. On the other hand, this dependence on processing can be exploited to access otherwise inaccessible morphologies.
Bates et al. in particular have demonstrated that new spherical BCP phases can emerge based on careful control over the thermal annealing process. Upon cooling from a disordered state or from a temperature near the order–disorder transition (ODT) where the well-known body-centered cubic (BCC) lattice is the equilibrium bulk phase, compositionally asymmetric diblock and some tetrablock copolymers can nucleate and grow a Frank–Kasper σ phase (Fig. 3a and b), which can be considered to be a crystal approximant to dodecagonal quasicrystals.102 Formation of this phase is mediated by packing frustration for spherical macromolecular particles within a uniform space-filling lattice and is indeed more stable than the BCC phase away from the order disorder transition.103 Strikingly, polyisoprene-block-polylactide diblock copolymers supercooled from the disordered state and then heated slowly can form a variety of nearly-degenerate, long-lived, low-symmetry metastable Frank Kasper phases akin to ones observed in metal alloys,104 and even dodecagonal quasicrystals that nucleate from spherical domain clusters with local tetrahedral or icosahedral symmetry.105
 | ||
| Fig. 3 (a) Transmission electron microscopy (TEM) image of a tetrablock copolymer morphology and (b) schematic of a Frank–Kasper σ phase consistent with the TEM image.102 Reproduced with permission from American Association for the Advancement of Science 2010. (c) Scanning electron microscopy (SEM) images of morphologies produced from a PS-b-PDMS diblock copolymer that forms an equilibrium bulk cylindrical phase through solvent vapor annealing with varying ratios of toluene in toluene/heptane solvent mixtures.113 Reproduced with permission from the American Chemical Society 2012. (d) TEM images of morphologies (top) with overlaid tiling patterns (bottom) produced by binary blends of PI-b-PS-b-P2VP triblock copolymers with varying lengths of end blocks. Scale bars represent 100 nm.128 Reproduced with permission from the American Chemical Society 2015. (e) TEM image of Au nanoparticles filling chiral pores derived from an achiral poly(1,4-butadiene)-b-poly(ethylene oxide) diblock copolymer prepared using D-tartaric acid.135 Reproduced with permission from the American Chemical Society 2017. | ||
Swelling BCPs with solvent vapors offers an alternative to purely thermal annealing that is especially valuable for BCPs with high segregation strengths (χ) due to their inherently slow ordering kinetics.106,107 The solvent molecules screen polymer interactions within swollen BCPs, thereby reducing χ and the concomitant energy barriers to polymer mobility and morphology reorganization.108,109 In the case of a solvent that swells both blocks equally, the equilibrium morphology is preserved, and surface energy differences between the two blocks that prevent perpendicular domain orientation can be mitigated.109,110 Rapid solvent removal predominantly traps the BCP in the morphology of the swollen state. Solvent vapor annealing (SVA) with a solvent that selectively swells one block will increase that block's effective volume fraction, enabling in some cases a shift from the bulk equilibrium morphology in the swollen state. For example, poly(α-methylstyrene)-block-poly(hydroxystyrene) (PαMS-b-PHOST) can form ordered cylinder domains by SVA using tetrahydrofuran (THF), a non-selective solvent, or spheres when using acetone, a selective solvent for PHOST.111 Similarly, SVA using mixtures of solvents with opposing selectivities, such as toluene and heptane for the case of PS-b-PDMS, allows access to a variety of self-assembled morphologies including spheres, cylinders (in-plane and perpendicular), lamellae, and perforated lamellae, some of which are not formed in the bulk (Fig. 3c).112–114 Domain spacing and relative width are also tunable through the degree of swelling, and the selectivity of the solvent used, respectively.108
Blending BCPs with homopolymers, small molecules, nanomaterials, and other BCPs provides a way to adjust many characteristics of a BCP solution or melt while avoiding the time, complexity, and cost associated with polymer synthesis.115–118 Polymer blending has been studied extensively, outlining various ways in which the properties of a BCP can be tuned. Blending diblock copolymers of the same type with similar volume fractions but different molecular weight enables monotonic tuning of the domain width and spacing within the intermediate range of the two BCPs, while preserving the morphology.119–124 Substantial disparity in the polymer chain lengths of mixed BCPs can result in macrophase separation into large and small domain phases. Binary blends of diblock copolymers with different compositions may form single phases exhibiting a conventional morphology (lamellar, cylinders, spheres, etc.) or two coexisting phases with uniform domain spacing.125–127 Since all polymer chains in blends of copolymers having blocks with the same chemical identity will share common domain interfaces, the morphologies are sensitive to the distribution of chain lengths, which highlights by analogy the impact of polydispersity on the morphological domain structure and stability of BCPs.
Recent work by Matsushita et al. presents a more complicated and interesting situation for binary blends of polyisoprene-block-polystyrene-block-poly(2-vinyl pyridine) (PI-b-PS-b-P2VP) ABC terpolymers with constant molecular weight but varying lengths of the end blocks. In the bulk, these blends have been shown to self-assemble into a variety of morphologies inaccessible to neat triblocks, including tetragonally packed rectangular cylinders,57 cylindrical lattices with large unit cells and triangular/pentagonal tiling128 (Fig. 3d), asymmetric double tetragonal domains,129 and tricontinuous double diamond networks.130 The short and long chains in the end blocks tend to localize the copolymer junction points within the morphology in order to maximize conformational entropy of the end block chains. The localization of junction points in turn relieves packing frustration and forms domain interfaces with non-constant mean curvatures that are required to stabilize these exotic morphologies,130 showcasing how engineered chain polydispersity can dramatically expand the range of stable morphologies in BCP self-assembly.
Further intriguing possibilities are opened up by including hydrogen bonding polymers or small molecules. In blends of BCPs with one block from each BCP that can interact through hydrogen bonding with a single block from the other BCP, the highly favorable hydrogen bonding interactions can overcome typical configurational entropy penalties (chain stretching/compression), permitting self-assembly into novel morphologies like thin film square arrays,131 asymmetric lamellae,132 or “kaleidoscopic” tiling pattern.133 Alternatively, small molecules can be selectively loaded into a domain in which they hydrogen bond with the constituent polymer block. For example, 3-pentadecylphenol (PDP) can be selectively loaded into poly(4-vinyl pyridine) (P4VP) domains of PS-b-P4VP via hydrogen bonding, where the packing of the PDP molecules generates a smaller-scale secondary lamellae morphology within the swollen P4VP domains.134 In another recent example, blends of a poly(1,4-butadiene)-block-poly(ethylene oxide) (PB-b-PEO) BCP with enantiomers of tartaric acid self-assembled to form chiral helical cylinders (Fig. 3e), where the chirality of the tartaric acid induced conformational chirality in the blend through hydrogen bonding with the PEO.135,136
Homopolymers of the constituent blocks blended with BCPs selectively swell and redistribute within domains and can therefore also be used to adjust domain spacing,137,138 make the BCP responsive to templates,139,140 or change the effective volume fraction of either block and hence shift the position of the blend in the morphological phase diagram.141–144 Blending BCPs with ceramic precursors or inorganic nanoparticles that selectively associate with one block enables the formation of technologically-relevant nanocomposites, whose phase behavior can be predicted in analogy to conventional BCP-homopolymer binary blends.145,146 Addition of the inorganic component allows a traversal through the morphological space of the parent BCP, and in some cases gives access to complex geometries. Early work suggested that this includes a “plumber's nightmare morphology,” though later re-evaluation indicated that this morphology is consistent with a distorted double gyroid.147 Merging the tunability afforded by block-selective additives like small molecules or ligand-coated nanoparticles with the thermodynamic constraints imposed by BCP chain topologies can also give rise to unconventional morphologies. These include asymmetric lamellae in a brush block copolymer blended with Au nanoparticles whose ligands hydrogen bond with one block,148 and alternating gyroid morphologies with structural chirality in co-assembled nanoparticle-triblock copolymer blends, whose self-assembly is dominated by enthalpic interactions.149,150
It is clear that control of processing conditions is crucial in order to obtain a desired structure from a BCP material. Moreover, judicious selection of material and process history can be used to form a particular non-native morphology. Control of structure formation through processing is attractive since it does not require any new materials synthesis or complex fabrication. Processing parameters can be easily and rapidly varied (including in realtime in response to structural measurements), allowing the vast combinatorial spaces of assembly to be explored. On the other hand, there is a limit to how far one can push a self-assembling material away from its intrinsic ordering using processing conditions alone. Thus, as discussed in subsequent sections, there is considerable value in investigating extreme directing methods, which strongly enforce order using boundary conditions or iterative assembly.
4. Confinement
The assembly of block copolymers can be strongly influenced by local confinement, due to both chemical affinity (e.g. one block preferentially wetting an interface) and commensurability (e.g. selecting a morphology or orientation that fills a given nano-volume without defects).58,151–160 The simplest form of confinement is the thin film thickness dimension; even here, a great amount of morphological control can be exercised.4,5,161–164 BCPs can form a variety of thin film morphologies beyond those observed in the bulk, including half-lamellae, hybrid orientations (i.e. morphologies with competing parallel and perpendicular orientations of morphology), hexagonally perforated lamellae, and hemispheres.4,11,161,162,164–169 The phase diagram for ordering within a thin film is remarkably rich, since conventional morphologies must frequently distort in order to accommodate the thickness dimension (Fig. 4a).170 Thus, one observes elongated spheres, squashed cylinders, hemi-cylinders, necked cylinders, crossed cylinders,171etc.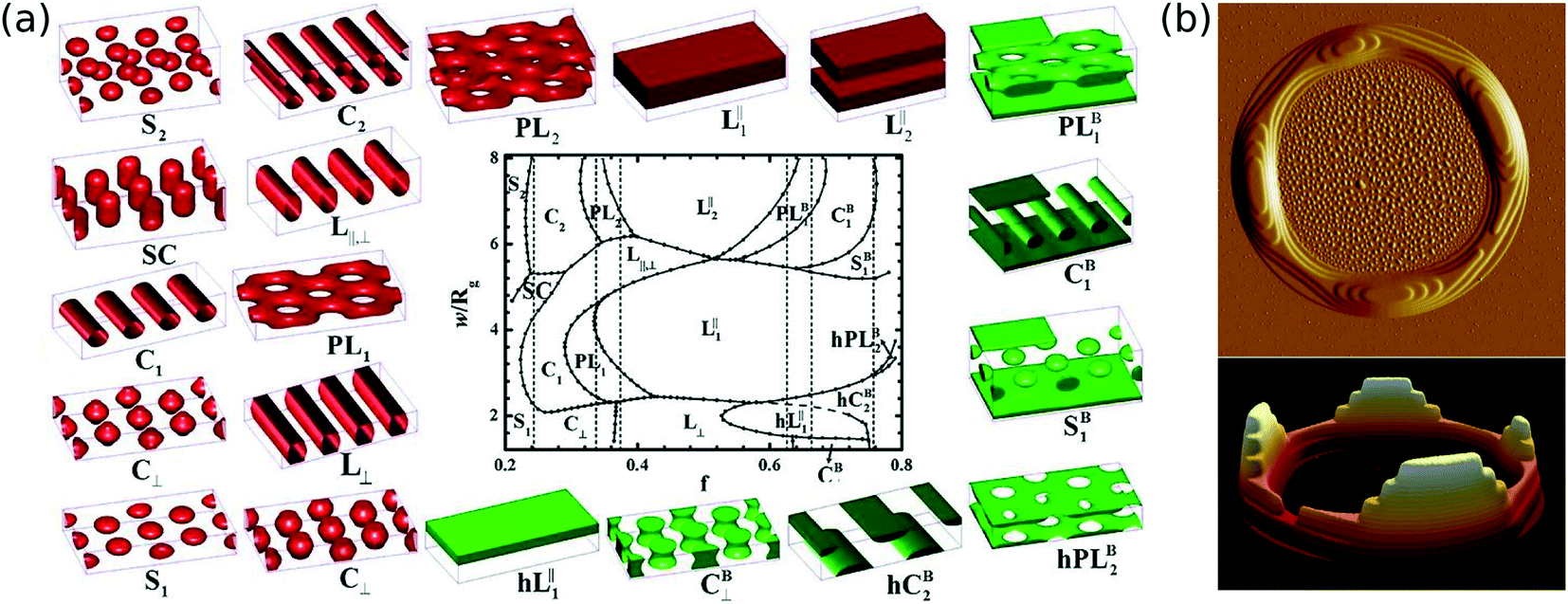 | ||
| Fig. 4 Selected BCP morphologies that arise due to thin film confinement. (a) A host of morphologies not observed in the bulk arise in thin films, owing to confinement, commensurability, and preferential wetting of interfaces (substrate and free surface).170 Reproduced with permission from the American Chemical Society 2013. (b) Non-trivial structures can arise as the BCP morphology attempt to order within a droplet. In this case, the ordering of a lamellar morphology couples to a Plateau–Rayleigh-like instability, leading to a terraced ring.177 Reproduced with permission from Springer 2011. | ||
For certain orientations of morphologies (lamellae sheets or cylinder long-axes parallel to substrate), commensurability induces the formation of islands and/or holes on the film surface.172–174 That is, for thicknesses that do not allow for an integer number of morphological layers, the free surface will spontaneously partition laterally into domains of commensurate thickness, giving rise to surface features with relatively sharp perimeters. These islands/holes (combined with their internally-registered BCP morphology) can be thought of as new, designed nanostructures. The height and internal organization of such structures can be cleverly controlled by selecting top and bottom interfacial energies.175 Although seemingly simple, this terracing effect can be combined with finite-size effects (droplets of BCP material) to yield non-trivial three-dimensional hierarchical structures (Fig. 4b).176,177 One can also exploit BCP assembly within a solution-bound droplet to control both the BCP ordering and the droplet shape.178–184 There is considerable untapped potential for the creation of hierarchical nano-structures by coupling the ordering of a BCP morphology with the deformable shape of the enclosing film or droplet surface. That is, by carefully controlling the structure of the polymer material prior to ordering, self-assembly will develop a morphology, and restructure the polymer droplet in a concerted way. If design rules can be developed, such a method could allow the creation of a wide variety of novel structures.
When BCPs are confined within rigid nano-volumes, the morphology must adapt so as to follow the shape, curvature, and size of the confining volume. Owing to the strong driving force for a BCP to retain its equilibrium repeat-spacing and local ordering motif, this usually results in a distorted version of the BCP morphology that curves and rearranges so as to fit an integer number of repeats within the volume. In two-dimensional (2D) confining wells, the morphology typically aligns with the sidewalls.155 That is, aligning with parallel trenches, forming ring motifs in circular wells, and sphere or cylinder phases packing based on the symmetry of confinement.154,157,185–188 Three-dimensional (3D) nano-confinement has been studied extensively using theory and simulation methods.158,159 Confinement of lamellar morphologies within cylindrical volumes is predicted to form concentric/coaxial layering.151 Ordering of lamellae within spherical cavities induces a high degree of frustration. For relatively large confining volumes, the lamellae layering is maintained, forming a striped spherical nano-object. For more extreme confinement, unexpected phases are predicted, including Janus, onion layering, wheel-like, mushroom-like, tennis-ball ordering, and screw-like or helical phases (Fig. 5a).189 Cylinder morphologies confined to spherical or ellipsoidal volumes generate a host of exotic structures, including stacked toruses, single and double helices, and cross-stitched motifs (Fig. 5b and c).156,171,190 Cylinder phases ordering within nano-pores are predicted to generate stacked toruses, rings around a central cylinder, or helical/multi-helical patterns whose pitch depends on the confinement size.153
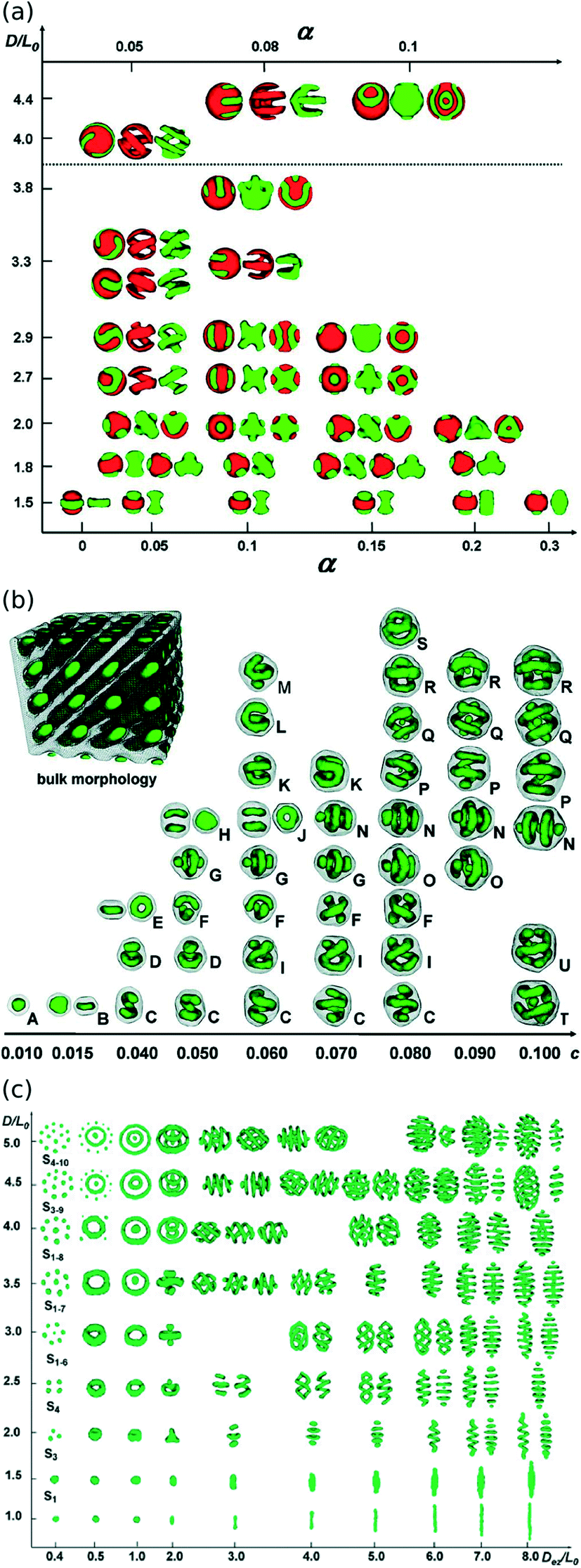 | ||
| Fig. 5 Complex morphologies form as BCP phases order within a confining volume. (a) A selection of morphologies predicted to arise when a lamellar BCP orders within a spherical volume.189 Reproduced with permission from the American Chemical Society 2007. (b) Select morphologies for a cylinder-forming BCP ordering within a spherical volume.156 Reproduced with permission from the American Chemical Society 2011. (c) Select morphologies for a cylinder-forming BCP ordering within ellipsoidal volumes.171 Reproduced with permission from the Royal Society of Chemistry 2011. | ||
A subset of these predicted phases have been confirmed experimentally.159,178,180,184,191–194 Wu et al. confined a copolymer system that forms a hexagonally-packed cylinder phase in the bulk.152 When confined within cylindrical pores (in an alumina membrane) of diameter ∼4× the BCP repeat-spacing, they observed stacked torus phases, as well as the formation of chiral helical structures, depending on confinement size-scale. There is obviously considerable potential for exploiting yet more complex 3D confinement volumes. Rider et al. exploited the free volume of colloidal crystals to control the ordering of BCPs.195 Lamellar phases within the spherical volumes of an inverse colloidal crystal generated the expected onion-like motifs, while cylinder phases formed complex wrapped perforated motifs. Lamellae ordering within the continuous free volume of a colloidal crystal generated three-armed junctions. In all these cases, the morphology results from the competing requirements to maintain material connectivity, minimize interfacial area, and accommodate the surrounding confinement. Studies such as these point towards the enormous untapped potential of frustrating BCP assembly using unconventional confinement volumes. With respect to confinement, theory and simulation are clearly leading the way, having predicted rich phase diagrams that as of yet have not been adequately explored experimentally. Thus, a crucial missing component to unlocking the potential of confinement to control BCP morphologies is a simple method to generate 3D confinement volumes, and fill these with BCP materials.
5. Layering
A conceptually simple but potentially powerful method to generate new nanostructures is to layer conventional self-assembled phases. The combined 3D structure of such multi-layers can exhibit symmetry and structural motifs entirely absent in any of the constituent layers. This thus naturally leverages BCP self-assembly, while providing enormous combinatorial control over the final structure, by simply selecting the sequence of materials. Osuji et al. have demonstrated how electrospray deposition can be used to continuously grow wide-area BCP films.196–199 A unique capability of this film formation method is that the composition of the material can be varied along the film normal direction, including layering different BCP materials or growing a film with a continuous composition gradient (Fig. 6a).200 Such films are intrinsically non-equilibrium and can exhibit highly non-native ordering; e.g. different repeat-periods in the top vs. bottom of the film. This technique holds great promise for both forming non-trivial morphologies during deposition (with appropriate choice of substrate temperature, ordering can co-occur with deposition), or forming tailored non-equilibrium layerings that are used as starting points for annealing and controlled reordering. | ||
| Fig. 6 Examples of novel structures that can be formed by layering BCP materials. (a) Electrospray deposition can be used to grow layers of polymer material, where the makeup of the film can be varied continuously or discontinuously during deposition.200 This allows different BCP materials to be stacked (electron micrographs shown in lower-left). Reproduced with permission from Wiley 2016. (b) Different nanoscale meshes can be fabricated by shear-aligning successive BCP layers, and converting them to inorganic replicas.8 Examples from all possible two-dimensional Bravais symmetries are shown. Adapted with permission from Nature Publishing Group 2015. (c) New morphologies can arise when BCP bilayers are ordered in a responsive manner.9 The second layer aligns, registers, and even distorts in order to accommodate the subtle surface topography of the first. The combined unit cell exhibits motifs not found in either constituent. Reproduced with permission from Nature Publishing Group 2016. (d) Responsive layering can be used to iteratively construct more complex three-dimensional structures. An example of trilayer ordering is shown.9 Adapted with permission from Nature Publishing Group 2016. (e) Responsive assembly of incommensurate layers can give rise to new unit cells (Moiré superstructures).221 Reproduced with permission from the American Chemical Society 2017. | ||
Another strategy for creating new structures is to layer independently-ordered BCP films. Film transfer methods have been used to stack nanostructured layers. For instance, BCP films were ordered and aligned on separate substrates, and then transferred using soft elastic (polydimethylsiloxane, PDMS) molds.201–204 By controlling the crossing angle during the transfer process, the shape of the new combined lattice can be selected. Multi-layered structures can be fabricated iteratively on a single substrate by fixing layers using crosslinking or material conversion. Woo et al. used crosslinkable materials to allow a second BCP layer to be cast and ordered on top of a post-anneal (ordered) BCP phase; for instance demonstrating a combined “vertical lamellae over vertical cylinders” structure.205 Kim et al. used shear to align BCP phases, and UV crosslinking to immobilize them, allowing square and rhombic arrays to be formed by layering. Majewski et al. demonstrated how photothermal annealing95 with “soft shear”97,206,207 effects can be used to align BCP phases.42,95,97 Combined with conversion of the BCP morphology to an inorganic replica,185,208 customized lattices can be rapidly fabricated over wide areas.8 For instance, a monolayer of BCP cylinders can be ordered and aligned via photothermal shearing, and converted into a metallic nanowire array. A second BCP material is cast atop, and similarly aligned and converted. The entire processing occurs on a single substrate, and exploits rapid and efficient wide-area ordering. By selecting the BCP materials in each layer, the size of the final unit cell can be controlled, while the angle between the two shear-alignment steps controls the shape of the final unit cell (Fig. 6b). In this way, all possible 2D Bravais lattice symmetries were realized: square (p4mm crystallographic group), rhombic (c2mm), rectangular (p2mm), oblique (p2), and hexagonal (p6mm). These examples demonstrate the power of layering BCP morphologies, wherein lattice symmetries that the constituent BCPs do not natively form can be constructed. Even relatively simple two-layer nanostructures have a range of potential applications; inorganic grids may be useful for nano-filtration, tuning optical properties,209,210 sensing,211 or as transparent electrodes.212,213
While layering of independently-ordered morphologies is clearly useful, such strategies do not take full advantage of the self-assembling nature of BCP materials. An extremely attractive alternative is to exploit the responsive nature of self-assembly, such that subsequent layers spontaneous order—in response to underlying layers—to generate a desired combined 3D structure. A simple example can be seen in the natural propensity for BCP line phases (horizontal cylinders or vertical lamellae) to orient orthogonal to underlying topographic undulations.169,214–216 Thus, a BCP ordering on top of an underlying BCP line pattern will generate local square or rectangular lattice symmetries, because of the natural tendency for the second layer to align orthogonally.217 It is worth emphasizing that such symmetries are not natively seen in the diblock copolymer phase diagram. Rahman et al. demonstrated how a startling range of new ordering motifs can be accessed by exploiting responsive layering.9 A first layer of BCP material was cast and ordered using conventional thermal annealing. This layer was infiltrated with an organometallic species from the vapor phase,218,219 which selectively loads only one of the BCP blocks. This selective infiltration serves multiple roles: it immobilizes the first layer, allowing additional layers to be cast atop; it slightly swells the infiltrated block, generating a subtle surface topography that replicates the BCP morphology; and it allows the overall final structure (if desired) to be converted into an inorganic replica. A second BCP layer cast atop the immobilized first layer was found to order in a templated manner, with the second-layer morphology orienting and registering with respect to the first. This templated ordering occurs because the morphology must accommodate the undulations of the underlying layer; in particular aligning the interblock interface atop height variations, since this minimizes chain stretching penalties. Because of this responsive assembly of the second layer, a host of new morphologies—with respect to the combined bilayer structure—were discovered (Fig. 6c), including square and rectangular packing, dots-on-lines, dots zigzagging across lines, dots bridging lines, undulating lines connecting hexagonally-arranged dots, lines layered between nanopores, and cylinders localized between nanopores. This responsive layering can be iterated, generating complex, bespoke three-dimensional nanostructures by selecting the sequence of BCP films (Fig. 6d). Buriak et al. demonstrated how layering can be used to generate high-density morphologies, or to create Moiré superstructures (Fig. 6e).220,221 Here again, the critical point is that the intrinsic responsiveness of the BCP is exploited, with the second layer spontaneously aligning and registering with respect to the first. By correctly the selecting the morphology and size of the layered materials, particular morphologies can be accessed. Responsive layering holds enormous promise for the fabrication of desired nanostructures, in particular because it allows one to rapidly explore combinatorial spaces without new materials synthesis. Thus, relatively simple BCP materials can be used to fabricate rather exotic lattices. On the other hand, it is clear that these methods would benefit strongly by including more sophisticated BCP materials in their libraries. Thus, a compelling avenue for future study is to investigate multi-layered structures that include more complex self-assembling materials, such as rod–coil copolymers, triblocks, or dendrimers.
6. Directed self-assembly templates
Lithographic templates present opportunities to rationally dictate boundary conditions of chemical affinity and spatial confinement to direct BCP self-assembly. Templates for BCP directed self-assembly (DSA) come in two distinct flavors, using either topographical templates that penetrate or confine the BCP thin film (graphoepitaxy)222–224 or templates with patterned differences in chemical affinity that underlie the BCP (chemoepitaxy).225–227 The ordering of the BCP is thereby directed by a propensity to minimize both morphological distortion and chemical mismatch between the template and the BCP. Considerable effort has been aimed at leveraging the periodicity and uniformity of BCP patterns as a resolution enhancement to existing lithography processes by subdividing and rectifying imperfections in lithographic patterns.226,227 For these applications, the BCP is primarily intended to produce an at-scale or downscaled replica of the lithographic templates; in turn, the templates are designed to match the natural symmetry of the native BCP morphology, with feature sizes that satisfy commensurability constraints with respect to the BCP repeat-spacing.228,229Nevertheless, the rectilinear layouts of many device designs have motivated research into extending DSA to produce patterns with square or rectangular symmetries. One straightforward approach is to use established DSA methods to align thin film BCP patterns on two separate substrates, and then to combine them, as for example by double nanoimprint, where the second pattern is rotated a predetermined angle (e.g. 90°) with respect to the first pattern.230 Other DSA approaches use square or rectangular symmetry chemical patterns or topographical templates. Commensurability between the template lattice and the native BCP dimensions (width and period) plays a critical role in determining the observed morphologies; adjusting template dimensions can result in competition between template–domain interactions due to confinement and the intrinsic drive for the BCP to minimize block interfacial area and curvature. This frustration of the normal domain ordering can give rise to patterns and morphologies not observed in the bulk BCP, such as loops or semi-cylinders231 as well as square symmetry spheres or perforated lamellae, bimodal perforated lamellae, and periodic superstructures232,233 (Fig. 7a). Both of these examples use a BCP cylinder phase, which traditionally packs into a hexagonal lattice. As an alternative, Ji et al. showed that PS-b-PMMA spheres, which adopt a body-centered cubic (BCC) lattice in the bulk but a hexagonal lattice in thin films, can be directed into a square or rectangular packings in thin films using commensurate chemical patterns. Through molecular transfer printing, the surface projection of this sphere lattice can, furthermore, be used to template the self-assembly of rectangular arrays of perpendicularly oriented PMMA cylinders in a PS-b-PMMA thin film.234
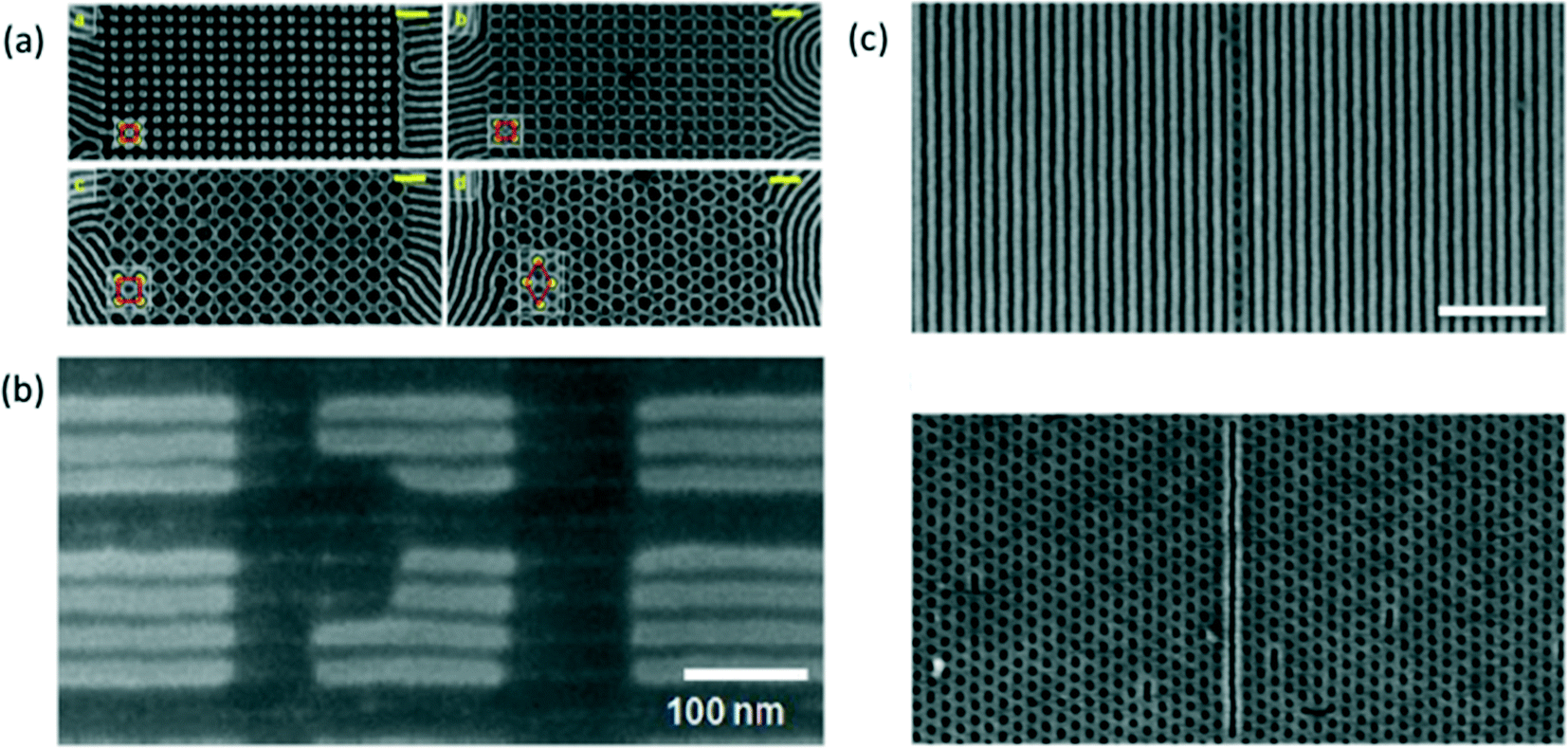 | ||
Fig. 7 (a) Scanning electron microscopy (SEM) images of monolayer nanostructures fabricated through DSA of a PS-b-PDMS diblock copolymer that forms an equilibrium bulk cylindrical phase using square symmetry sacrificial post templates.233 Scale bars represent 100 nm. Reproduced with permission from Wiley-VCH 2013. (b) Customized line-space pattern fabricated through DSA of a lamellae-forming PS-b-PMMA BCP using a hybrid organic/inorganic prepattern.244 Reproduced with permission from SPIE 2015. (c) A single line of cylindrical holes amid a regular lamellar grating pattern (top) and a single line amid a regular hexagonal array of holes (bottom) produced through sDSA of a 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 blend of cylinder and lamellae forming PS-b-PMMA diblock copolymers.247 The scale bars represents 400 nm, and applies to both images. Reproduced with permission from Nature Publishing Group 2016. :50 blend of cylinder and lamellae forming PS-b-PMMA diblock copolymers.247 The scale bars represents 400 nm, and applies to both images. Reproduced with permission from Nature Publishing Group 2016. | ||
Lateral confinement has also been investigated experimentally235 and in simulations236 as a means to generate BCP patterns with non-native symmetries. The propagation of order from confinement pattern edges strongly determines domain placement, pattern symmetry, and ordering quality.237 Using square confinement has been successful in producing groups of cylinders with square rather than hexagonal symmetry. In the limit of small topographically confinement, the position of single BCP domains can be controlled, with domain densities determined by the template rather than the BCP. However, for high density merged confinement features, the energy balance must account for both the confinement geometry and the natural BCP periodicity in determining template design rules that minimize defectivity.238
A grand challenge for templated DSA is to create complex and customizable nanopatterns while retaining the periodicity and uniformity inherent to BCP self-assembly. Significant control can be imposed through a high degree of external encoding by using templates with features congruent with the BCP dimensions, such as 1:1 chemical patterns.225 Even with precise guide features, pattern formation can be frustrated, as the natural tendency for the BCP to minimize domain curvature and interfacial area can lead to defects at features such as bends. Addressing this problem, Stoykovich et al. have shown that in PS-b-PMMA/PS/PMMA ternary blends, the homopolymer can redistribute to alleviate local energy penalties, enabling directed assembly without defects onto 1:1 chemical patterns incorporating device-relevant features like isolated lines, bends, T-junctions and jogs.139,140 Subsequent work showed that this approach is extendable to chemical patterns at twice the BCP period.239
A growing body of work has since elaborated other ways to achieve complex, customizable patterns through BCP DSA. One prominent strategy developed by Ross et al. is to locally adjust the conditions for optimal commensurability between a BCP and the template, for example by using sparsely spaced HSQ posts to direct a cylinder-forming PS-b-PDMS BCP. By controlling the shape or arrangement of posts, the cylinders long-axis alignment direction can be controlled, owing to required commensurability match between the BCP and the template. Combining experimental results with field theoretic simulations, a set of design rules were developed.240,241 Other customization strategies have focused on templates that produce unidirectional BCP line patterns accompanied by predetermined “line cuts” where individual lines are removed or terminated, so as to more closely resemble state-of-the-art unidirectional circuit layouts. Tsai et al., used graphoepitaxy including “2D geometry” to truncate individual domains along the template wall, while allowing adjacent domains to continue unperturbed.242 Another strategy introduced by IBM researchers uses underlying hybrid organic/inorganic prepatterns for BCP DSA. The prepatterns include both guiding features (e.g. lines) and non-guiding elements (e.g. rectangles), where global free energy minimization allows the BCP to filter out the effect of non-guiding information in the prepattern. The final pattern after etch transfer is the union or the intersection of the prepattern and the self-assembled pattern (depending on the type of hybrid prepattern), where the inclusion of guiding and non-guiding features in the prepattern permits a wealth of possible customizations (Fig. 7b).243,244
Many more complex patterns could be created if different self-assembled morphologies could be combined during DSA. One way to achieve this leverages the fact that SVA can induce different morphologies in the same BCP based on differential swelling of the polymer blocks in the chosen solvent. Self-assembled domain morphologies formed through SVA may be “frozen” via selective-area cross-linking induced by patternwise ultraviolet or electron beam irradiation. Subsequent SVA using a solvent with different block selectivity can induce the formation of a separate morphology in the unexposed areas.245,246 While these approaches are compatible with DSA, the domain morphology is encoded through processing and not through the template itself. More recently, our group has demonstrated how DSA can be combined with multiple morphologies by exploiting blends of BCPs. Blends of cylinder- and lamellae-forming PS-b-PMMA diblock copolymers were found to form coexisting phases in thin films; that is, some parts of the film exhibit lamellar line patterns, while other areas exhibit hexagonal cylinder patterns.127 Strikingly, the morphology that forms can be locally selected by choosing an appropriate underlying chemical pattern. Using purely chemical line-patterns, the BCP can be induced into forming a lamellar or hexagonal pattern, based on the width and/or spacing of the lines, which in turn enforce competing chain distortion and interfacial energy constraints. This selective form of DSA (sDSA) enables prescriptive positional control over both the self-assembled morphology, and the precise alignment/registry of this morphology, as for example in the creation of a single line of cylindrical holes amid a lamellar grating pattern, or its opposite (Fig. 7c).247
Templated DSA of complex, customizable structures and morphologies has focused mainly on fabricating two-dimensional patterns. Yet templates produced using traditional two-dimensional lithography can still direct BCP self-assembly toward a wide array of three-dimensional structures. Commensurability between the template and BCP is again critical. For example, lamellae assembled on incommensurate chemical patterns can assemble into non-bulk three dimensional structures based on the size of the template features and their chemical affinities for either block.248 The sparse post strategy established by the Ross group can also be applied in this context, where energetic competition between the favored bilayer stacking arrangement in a cylinder-forming PS-b-PDMS thin films and the local commensurability between the template post period and the BCP equilibrium period produce a large array of crossed cylinder motifs.249
7. Conclusion
Block copolymers are a canonical example of a self-assembling material. In some sense, BCPs represent a relatively simple example of self-assembly; e.g. lacking the detailed programmability of DNA-based self-assembly methods. While this obviously limits the complexity of structures BCPs can intrinsically form, the simplicity of BCP assembly is in part what makes these materials excellent model systems for exploring fundamental aspects of self-assembly, especially the increasingly important role of external influences in controlling structure formation. The combined use of intrinsic ordering—defined via synthesis—and imposed constraints—defined via processing and material environment—allows a wide variety of nanoscale motifs to be accessed. Despite being well-studied, block copolymers continue to hold enormous untapped potential for generating customized nanostructures.Many recent advances that expand the landscape of self-assembled nanoscale structures, patterns and morphologies use more than one of the strategies described above, thus combining different modes of morphological differentiation. For instance, researchers have combined the complex morphologies of triblocks with structural control via confinement;58,160 others have combined substrate patterning with tuning of film thickness to control morphology.169 Further exploration of multi-modal strategies promises the discovery of many new self-assembled structural motifs. For instance, while triblock copolymers already broaden the bulk equilibrium morphological set substantially, additional structural motifs with a high degree of regularity may become possible as we deepen our understanding of fundamental aspects, such as polydispersity and hydrogen bonding in triblock blends. Blending can be used to engineer the chain length distribution, giving rise to unique morphologies and coexisting phases not possible for monodisperse single-component systems.127 Using DSA as one mode in a multi-modal strategy is particularly enticing as it offers the highest degree of external encoding for the self-assembled structure; with thermal annealing of diblock copolymers, however, DSA is typically used to align the equilibrium, bulk morphology. Selective DSA is one multi-modal example where morphology can be chosen in a BCP blend by external encoding using chemical epitaxy,247 but a graphoepitaxial version of sDSA could engender many exciting new possibilities through the combination of templating, confinement, and chain heterogeneity. Other new avenues for expanding the morphology space using DSA involve a shift from two-dimensional to three-dimensional control. Though still two-dimensional patterns, post arrays are an example that begins to penetrate this space by enforcing different commensurability constraints for monolayer and bilayer BCP films.249 Creating true three-dimensional templates (3D periodic lattices) for DSA would impose full spatial confinement constraints, simultaneously controlling both domain structure and positional registration. The formidable challenge of template fabrication should be acknowledged as it is difficult to lithographically create well-defined, three-dimensional structures that strongly confine BCPs at a scale in which they are not kinetically trapped. Nevertheless, commercial two-photon lithography tools, and hierarchical self-assembly250 present possible paths toward three-dimensional DSA. It is likely that yet other multi-modal assembly strategies—which have not yet been envisioned—will bring us closer to the goal of sophisticated nanostructural synthesis using simple polymeric building blocks.
A key theme in the BCP community is the exploitation of the inherent responsiveness in self-assembly. This fundamental advantage of self-assembling materials holds continued promise for controlling structural order. For instance, while layering of pre-ordered self-assembled structures is a powerful way to create designed structures, responsive layering9 leverages the intrinsic structure formation of the combined materials, allowing new—and often surprising—morphologies to emerge. Future research should ideally exploit this responsiveness even more deeply, fully exploring the “hybrid” regime (c.f. Fig. 1) where intrinsic and extrinsic forces are combined. For instance, responsive self-assembling layers can be ordered on top of designed (via DSA) first-layer pre-patterns.233,249 This combination of templating and responsive assembly could be used to propagate a selected substrate pattern into higher multi-layered structures, to organize known BCP shapes into non-native lattices, or to coax BCPs to form entirely new three-dimensional morphologies. Furthermore, responsive self-assembly methods, such as selective DSA and responsive layering, should be investigated in a broader range of materials, including rod–coil diblocks, triblock copolymers, and bottlebrush materials. In addition to broadening the landscape of morphologies, such studies could shed light on fundamental aspects of assembly by comparing the responsiveness of different macromolecular architectures. Studying the responsive layering assembly on blended materials would similarly uncover important motifs in assembly. On the one hand, blended materials can be predicted to respond more strongly to environmental forces, since they can tolerate greater morphological distortion (through rearrangement and partition of the different chain types), and thereby generate morphologies that are very different from the unperturbed morphologies. On the other hand, the ability of blended materials to rearrange and tolerate defects could be argued to make them less responsive, in the sense that they can rearrange in order to relax towards a morphology that minimizes surface area and maximizes symmetry. The exact material response will of course depend on the balance of energy between intrinsic relaxation and external drivers.
The block copolymer community has demonstrated a startling diversity of structures by using elegant macromolecular building blocks. Despite this extensive research, there remain many opportunities to create more elaborate nanostructures. More elaborate BCP materials, obtained through creative synthesis, continues to broaden the range of possible starting morphologies. Growing insight into the responsiveness of BCPs has enabled many new structures to be created by cleverly combining intrinsic information in encoding with environmental constraints. Finally, continued effort via theory and simulations is a powerful way to rapidly explore the enormous combinatorial space of possibilities, and is crucial in order to develop concrete design rules for the formation of a target structure.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work took place at the Center for Functional Nanomaterials, which is a U.S. DOE Office of Science Facility, at Brookhaven National Laboratory, funded under Contract No. DE-SC0012704.References
- D. Philp and J. F. Stoddart, Self-Assembly in Natural and Unnatural Systems, Angew. Chem., Int. Ed. Engl., 1996, 35, 1154–1196 CrossRef.
- F. S. Bates and G. H. Fredrickson, Block Copolymer Thermodynamics: Theory and Experiment, Annu. Rev. Phys. Chem., 1990, 41, 525–557 CrossRef CAS PubMed.
- F. S. Bates and G. H. Fredrickson, Block Copolymers—Designer Soft Materials, Physics Today, 1999, 32 CrossRef CAS.
- M. J. Fasolka and A. M. Mayes, Block Copolymer Thin Films: Physics and Applications, Annu. Rev. Mater. Res., 2001, 31, 323–355 CrossRef CAS.
- J. N. L. Albert and T. H. Epps, Self-assembly of block copolymer thin films, Mater. Today, 2010, 13, 24–33 CrossRef CAS.
- Y. Mai and A. Eisenberg, Self-assembly of block copolymers, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC.
- S. Y. Kim, A. Nunns, J. Gwyther, R. L. Davis, I. Manners, P. M. Chaikin and R. A. Register, Large-Area Nanosquare Arrays from Shear-Aligned Block Copolymer Thin Films, Nano Lett., 2014, 14, 5698–5705 CrossRef CAS PubMed.
- P. W. Majewski, A. Rahman, C. T. Black and K. G. Yager, Arbitrary lattice symmetries via block copolymer nanomeshes, Nat. Commun., 2015, 6, 7448 CrossRef PubMed.
- A. Rahman, P. W. Majewski, G. Doerk, C. T. Black and K. G. Yager, Non-native three-dimensional block copolymer morphologies, Nat. Commun., 2016, 7, 13988 CrossRef PubMed.
- L. Leibler, Theory of Microphase Separation in Block Copolymers, Macromolecules, 1980, 13, 1602–1617 CrossRef CAS.
- M. W. Matsen and F. S. Bates, Unifying Weak- and Strong-Segregation Block Copolymer Theories, Macromolecules, 1996, 29, 1091–1098 CrossRef CAS.
- V. Castelletto and I. W. Hamley, Morphologies of block copolymer melts, Curr. Opin. Solid State Mater. Sci., 2004, 8, 426–438 CrossRef CAS.
- J. V. Barth, G. Costantini and K. Kern, Engineering atomic and molecular nanostructures at surfaces, Nature, 2005, 437, 671–679 CrossRef CAS PubMed.
- R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Supramolecular Coordination: Self-Assembly of Finite Two- and Three-Dimensional Ensembles, Chem. Rev., 2011, 111, 6810–6918 CrossRef CAS PubMed.
- E. V. Shevchenko, D. V. Talapin, N. A. Kotov, S. O'Brien and C. B. Murray, Structural diversity in binary nanoparticle superlattices, Nature, 2006, 439, 55–59 CrossRef CAS PubMed.
- M. Grzelczak, J. Vermant, E. M. Furst and L. M. Liz-Marzán, Directed Self-Assembly of Nanoparticles, ACS Nano, 2010, 4, 3591–3605 CrossRef CAS PubMed.
- M. H. Lash, M. V. Fedorchak, J. J. McCarthy and S. R. Little, Scaling up self-assembly: bottom-up approaches to macroscopic particle organization, Soft Matter, 2015, 11, 5597–5609 RSC.
- M. A. Boles, M. Engel and D. V. Talapin, Self-Assembly of Colloidal Nanocrystals: From Intricate Structures to Functional Materials, Chem. Rev., 2016, 116, 11220–11289 CrossRef CAS PubMed.
- P. W. K. Rothemund, Folding DNA to create nanoscale shapes and patterns, Nature, 2006, 440, 297–302 CrossRef CAS PubMed.
- A. V. Pinheiro, D. Han, W. M. Shih and H. Yan, Challenges and opportunities for structural DNA nanotechnology, Nat. Nanotechnol., 2011, 6, 763–772 CrossRef CAS PubMed.
- I. Saaem and T. H. LaBean, Overview of DNA origami for molecular self-assembly, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2013, 5, 150–162 CrossRef CAS PubMed.
- L. H. Tan, H. Xing and Y. Lu, DNA as a Powerful Tool for Morphology Control, Spatial Positioning, and Dynamic Assembly of Nanoparticles, Acc. Chem. Res., 2014, 47, 1881–1890 CrossRef CAS PubMed.
- X. Zhang, R. Wang and G. Xue, Programming macro-materials from DNA-directed self-assembly, Soft Matter, 2015, 11, 1862–1870 RSC.
- D. Nykypanchuk, M. M. Maye, D. van der Lelie and O. Gang, DNA-guided crystallization of colloidal nanoparticles, Nature, 2008, 451, 549–552 CrossRef CAS PubMed.
- S. Y. Park, A. K. R. Lytton-Jean, B. Lee, S. Weigand, G. C. Schatz and C. A. Mirkin, DNA-programmable nanoparticle crystallization, Nature, 2008, 451, 553–556 CrossRef CAS PubMed.
- R. J. Macfarlane, B. Lee, M. R. Jones, N. Harris, G. C. Schatz and C. A. Mirkin, Nanoparticle Superlattice Engineering with DNA, Science, 2011, 334, 204–208 CrossRef CAS PubMed.
- O. Gang and A. V. Tkachenko, DNA-programmable particle superlattices: Assembly, phases, and dynamic control, MRS Bull., 2016, 41, 381–387 CrossRef CAS.
- W. Liu, M. Tagawa, H. L. Xin, T. Wang, H. Emamy, H. Li, K. G. Yager, F. W. Starr, A. V. Tkachenko and O. Gang, Diamond family of nanoparticle superlattices, Science, 2016, 351, 582–586 CrossRef CAS PubMed.
- Y. Tian, Y. Zhang, T. Wang, H. L. Xin, H. Li and O. Gang, Lattice engineering through nanoparticle-DNA frameworks, Nat. Mater., 2016, 15, 654–661 CrossRef CAS PubMed.
- S. Vial, D. Nykypanchuk, K. G. Yager, A. V. Tkachenko and O. Gang, Linear Mesostructures in DNA–Nanorod Self-Assembly, ACS Nano, 2013, 7, 5437–5445 CrossRef CAS PubMed.
- Y. Tian, T. Wang, W. Liu, H. L. Xin, H. Li, Y. Ke, W. M. Shih and O. Gang, Prescribed nanoparticle cluster architectures and low-dimensional arrays built using octahedral DNA origami frames, Nat. Nanotechnol., 2015, 10, 637–644 CrossRef CAS PubMed.
- W. Liu, J. Halverson, Y. Tian, A. V. Tkachenko and O. Gang, Self-organized architectures from assorted DNA-framed nanoparticles, Nat. Chem., 2016, 8, 867–873 CrossRef CAS PubMed.
- A. V. Tkachenko, Theory of Programmable Hierarchic Self-Assembly, Phys. Rev. Lett., 2011, 106, 255501 CrossRef PubMed.
- C. Knorowski and A. Travesset, Materials design by DNA programmed self-assembly, Curr. Opin. Solid State Mater. Sci., 2011, 15, 262–270 CrossRef CAS.
- J. D. Halverson and A. V. Tkachenko, DNA-programmed mesoscopic architecture, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2013, 87, 062310 CrossRef PubMed.
- H. Hu, M. Gopinadhan and C. O. Osuji, Directed self-assembly of block copolymers: a tutorial review of strategies for enabling nanotechnology with soft matter, Soft Matter, 2014, 10, 3867–3889 RSC.
- P. W. Majewski and K. G. Yager, Rapid ordering of block copolymer thin films, J. Phys.: Condens. Matter, 2016, 28, 403002 CrossRef PubMed.
- M. Luo and T. H. Epps, Directed Block Copolymer Thin Film Self-Assembly: Emerging Trends in Nanopattern Fabrication, Macromolecules, 2013, 46, 7567–7579 CrossRef CAS.
- S.-J. Jeong, J. Y. Kim, B. H. Kim, H.-S. Moon and S. O. Kim, Directed self-assembly of block copolymers for next generation nanolithography, Mater. Today, 2013, 16, 468–476 CrossRef CAS.
- W. van Zoelen, T. Asumaa, J. Ruokolainen, O. Ikkala and G. ten Brinke, Phase Behavior of Solvent Vapor Annealed Thin Films of PS-b-P4VP(PDP) Supramolecules, Macromolecules, 2008, 41, 3199–3208 CrossRef CAS.
- E. Mattia and S. Otto, Supramolecular systems chemistry, Nat. Nanotechnol., 2015, 10, 111–119 CrossRef CAS PubMed.
- P. W. Majewski and K. G. Yager, Latent Alignment in Pathway-Dependent Ordering of Block Copolymer Thin Films, Nano Lett., 2015, 15, 5221–5228 CrossRef CAS PubMed.
- P. W. Majewski and K. G. Yager, Reordering transitions during annealing of block copolymer cylinder phases, Soft Matter, 2016, 12, 281–294 RSC.
- F. S. Bates, M. F. Schulz, A. K. Khandpur, S. Forster, J. H. Rosedale, K. Almdal and K. Mortensen, Fluctuations, conformational asymmetry and block copolymer phase behaviour, Faraday Discuss., 1994, 98, 7–18 RSC.
- D. A. Hajduk, H. Takenouchi, M. A. Hillmyer, F. S. Bates, M. E. Vigild and K. Almdal, Stability of the Perforated Layer (PL) Phase in Diblock Copolymer Melts, Macromolecules, 1997, 30, 3788–3795 CrossRef CAS.
- T. M. Beardsley and M. W. Matsen, Monte Carlo phase diagram for diblock copolymer melts, Eur. Phys. J. E: Soft Matter Biol. Phys., 2010, 32, 255–264 CrossRef CAS PubMed.
- T. M. Beardsley and M. W. Matsen, Monte Carlo Phase Diagram for a Polydisperse Diblock Copolymer Melt, Macromolecules, 2011, 44, 6209–6219 CrossRef CAS.
- X. Wang, M. Goswami and R. Kumar, G. Sumpter, B. and Mays, J. Morphologies of block copolymers composed of charged and neutral blocks, Soft Matter, 2012, 8, 3036–3052 RSC.
- C. E. Sing, J. W. Zwanikken and M. Olvera de la Cruz, Electrostatic control of block copolymer morphology, Nat. Mater., 2014, 13, 694–698 CrossRef CAS PubMed.
- M. W. Matsen and R. B. Thompson, Equilibrium behavior of symmetric ABA triblock copolymer melts, J. Chem. Phys., 1999, 111, 7139–7146 CrossRef CAS.
- Y. Mogi, H. Kotsuji, Y. Kaneko, K. Mori, Y. Matsushita and I. Noda, Preparation and morphology of triblock copolymers of the ABC type, Macromolecules, 1992, 25, 5408–5411 CrossRef CAS.
- Y. Mogi, M. Nomura, H. Kotsuji, K. Ohnishi, Y. Matsushita and I. Noda, Superlattice Structures in Morphologies of the ABC Triblock Copolymers, Macromolecules, 1994, 27, 6755–6760 CrossRef CAS.
- Y. Mogi, K. Mori, Y. Matsushita and I. Noda, Tricontinuous morphology of triblock copolymers of the ABC type, Macromolecules, 1992, 25, 5412–5415 CrossRef CAS.
- R. Kumar, S. W. Sides, M. Goswami, B. G. Sumpter, K. Hong, X. Wu, T. P. Russell, S. P. Gido, K. Misichronis, S. Rangou, A. Avgeropoulos, T. Tsoukatos, N. Hadjichristidis, F. L. Beyer and J. W. Mays, Morphologies of ABC Triblock Terpolymer Melts Containing Poly(Cyclohexadiene): Effects of Conformational Asymmetry, Langmuir, 2013, 29, 1995–2006 CrossRef CAS PubMed.
- T. Higuchi, H. Sugimori, X. Jiang, S. Hong, K. Matsunaga, T. Kaneko, V. Abetz, A. Takahara and H. Jinnai, Morphological Control of Helical Structures of an ABC-Type Triblock Terpolymer by Distribution Control of a Blending Homopolymer in a Block Copolymer Microdomain, Macromolecules, 2013, 46, 6991–6997 CrossRef CAS.
- C. Tang, J. Bang, G. E. Stein, G. H. Fredrickson, C. J. Hawker, E. J. Kramer, M. Sprung and J. Wang, Square Packing and Structural Arrangement of ABC Triblock Copolymer Spheres in Thin Films, Macromolecules, 2008, 41, 4328–4339 CrossRef CAS.
- Y. Asai, K. Yamada, M. Yamada, A. Takano and Y. Matsushita, Formation of Tetragonally-Packed Rectangular Cylinders from ABC Block Terpolymer Blends, ACS Macro Lett., 2014, 3, 166–169 CrossRef CAS.
- S. Li, Y. Jiang and J. Z. Y. Chen, Morphologies and phase diagrams of ABC star triblock copolymers confined in a spherical cavity, Soft Matter, 2013, 9, 4843–4854 RSC.
- N. A. Lynd, A. J. Meuler and M. A. Hillmyer, Polydispersity and block copolymer self-assembly, Prog. Polym. Sci., 2008, 33, 875–893 CrossRef CAS.
- A. Noro, M. Okuda, F. Odamaki, D. Kawaguchi, N. Torikai, A. Takano and Y. Matsushita, Chain Localization and Interfacial Thickness in Microphase-Separated Structures of Block Copolymers with Variable Composition Distributions, Macromolecules, 2006, 39, 7654–7661 CrossRef CAS.
- R. Verduzco, X. Li, S. L. Pesek and G. E. Stein, Structure, function, self-assembly, and applications of bottlebrush copolymers, Chem. Soc. Rev., 2015, 44, 2405–2420 RSC.
- S. A. Jenekhe and X. L. Chen, Self-Assembly of Ordered Microporous Materials from Rod-Coil Block Copolymers, Science, 1999, 283, 372–375 CrossRef CAS PubMed.
- M. Lee, B.-K. Cho and W.-C. Zin, Supramolecular Structures from Rod–Coil Block Copolymers, Chem. Rev., 2001, 101, 3869–3892 CrossRef CAS PubMed.
- B. D. Olsen and R. A. Segalman, Self-assembly of rod–coil block copolymers, Mater. Sci. Eng., R, 2008, 62, 37–66 CrossRef.
- C.-L. Liu, C.-H. Lin, C.-C. Kuo, S.-T. Lin and W.-C. Chen, Conjugated rod–coil block copolymers: Synthesis, morphology, photophysical properties, and stimuli-responsive applications, Prog. Polym. Sci., 2011, 36, 603–637 CrossRef CAS.
- R. Verduzco, I. Botiz, D. L. Pickel, S. M. Kilbey, K. Hong, E. Dimasi and S. B. Darling, Polythiophene-block-polyfluorene and Polythiophene-block-poly(fluorene-co-benzothiadiazole): Insights into the Self-Assembly of All-Conjugated Block Copolymers, Macromolecules, 2011, 44, 530–539 CrossRef CAS.
- M. J. Robb, S.-Y. Ku and C. J. Hawker, 25th Anniversary Article: No Assembly Required: Recent Advances in Fully Conjugated Block Copolymers, Adv. Mater., 2013, 25, 5686–5700 CrossRef CAS PubMed.
- V. Percec, J. G. Rudick, M. Peterca, M. E. Yurchenko, J. Smidrkal and P. A. Heiney, Supramolecular Structural Diversity among First-Generation Hybrid Dendrimers and Twin Dendrons, Chem. – Eur. J., 2008, 14, 3355–3362 CrossRef CAS PubMed.
- J. G. Rudick, in Hierarchical Macromolecular Structures: 60 Years after the Staudinger Nobel Prize II, ed. V. Percec, Springer International Publishing, Cham, 2013, pp. 345–362 Search PubMed.
- J. G. Rudick and V. Percec, Induced Helical Backbone Conformations of Self-Organizable Dendronized Polymers, Acc. Chem. Res., 2008, 41, 1641–1652 CrossRef CAS PubMed.
- B. M. Rosen, C. J. Wilson, D. A. Wilson, M. Peterca, M. R. Imam and V. Percec, Dendron-Mediated Self-Assembly, Disassembly, and Self-Organization of Complex Systems, Chem. Rev., 2009, 109, 6275–6540 CrossRef CAS PubMed.
- K. Kwon, J. M. Ok, Y. H. Kim, J.-S. Kim, W.-B. Jung, S.-Y. Cho and H.-T. Jung, Direct Observation of Highly Ordered Dendrimer Soft Building Blocks over a Large Area, Nano Lett., 2015, 15, 7552–7557 CrossRef CAS PubMed.
- X. Yu, K. Yue, I.-F. Hsieh, Y. Li, X.-H. Dong, C. Liu, Y. Xin, H.-F. Wang, A.-C. Shi, G. R. Newkome, R.-M. Ho, E.-Q. Chen, W.-B. Zhang and S. Z. D. Cheng, Giant surfactants provide a versatile platform for sub-10-nm nanostructure engineering, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 10078–10083 CrossRef CAS PubMed.
- T. R. Wilks, J. Bath, J. W. de Vries, J. E. Raymond, A. Herrmann, A. J. Turberfield and R. K. O'Reilly, “Giant Surfactants” Created by the Fast and Efficient Functionalization of a DNA Tetrahedron with a Temperature-Responsive Polymer, ACS Nano, 2013, 7, 8561–8572 CrossRef CAS PubMed.
- T. S. Thomas, W. Hwang and L. R. Sita, End-Group-Functionalized Poly(α-olefinates) as Non-Polar Building Blocks: Self-Assembly of Sugar–Polyolefin Hybrid Conjugates, Angew. Chem., Int. Ed., 2016, 55, 4683–4687 CrossRef CAS PubMed.
- S. R. Nowak, W. Hwang and L. R. Sita, Dynamic Sub-10-nm Nanostructured Ultrathin Films of Sugar–Polyolefin Conjugates Thermoresponsive at Physiological Temperatures, J. Am. Chem. Soc., 2017, 139, 5281–5284 CrossRef CAS PubMed.
- L. R. Hutchings, J. M. Dodds and S. J. Roberts-Bleming, HyperMacs: Highly Branched Polymers Prepared by the Polycondensation of AB2 Macromonomers, Synthesis and Characterization, Macromolecules, 2005, 38, 5970–5980 CrossRef CAS.
- L. R. Hutchings, J. M. Dodds, D. Rees, S. M. Kimani, J. J. Wu and E. Smith, HyperMacs to HyperBlocks: A Novel Class of Branched Thermoplastic Elastomer, Macromolecules, 2009, 42, 8675–8687 CrossRef CAS.
- F. L. Beyer, S. P. Gido, C. Büschl, H. Iatrou, D. Uhrig, J. W. Mays, M. Y. Chang, B. A. Garetz, N. P. Balsara, N. B. Tan and N. Hadjichristidis, Graft Copolymers with Regularly Spaced, Tetrafunctional Branch Points: Morphology and Grain Structure, Macromolecules, 2000, 33, 2039–2048 CrossRef CAS.
- Y. Zhu, E. Burgaz, S. P. Gido, U. Staudinger, R. Weidisch, D. Uhrig and J. W. Mays, Morphology and Tensile Properties of Multigraft Copolymers with Regularly Spaced Tri-, Tetra-, and Hexafunctional Junction Points, Macromolecules, 2006, 39, 4428–4436 CrossRef CAS.
- S. P. Paradiso, K. T. Delaney and G. H. Fredrickson, Swarm Intelligence Platform for Multiblock Polymer Inverse Formulation Design, ACS Macro Lett., 2016, 5, 972–976 CrossRef CAS.
- M. R. Khadilkar, S. Paradiso, K. T. Delaney and G. H. Fredrickson, Inverse Design of Bulk Morphologies in Multiblock Polymers Using Particle Swarm Optimization, Macromolecules, 2017, 50, 6702–6709 CrossRef CAS.
- H. Takahashi, N. Laachi, K. T. Delaney, S.-M. Hur, C. J. Weinheimer, D. Shykind and G. H. Fredrickson, Defectivity in Laterally Confined Lamella-Forming Diblock Copolymers: Thermodynamic and Kinetic Aspects, Macromolecules, 2012, 45, 6253–6265 CrossRef CAS.
- B. Kim, N. Laachi, K. T. Delaney, M. Carilli, E. J. Kramer and G. H. Fredrickson, Thermodynamic and kinetic aspects of defectivity in directed self-assembly of cylinder-forming diblock copolymers in laterally confining thin channels, J. Appl. Polym. Sci., 2014, 131, 40790 Search PubMed.
- S. Ji, C.-C. Liu, W. Liao, A. L. Fenske, G. S. W. Craig and P. F. Nealey, Domain Orientation and Grain Coarsening in Cylinder-Forming Poly(styrene-b-methyl methacrylate) Films, Macromolecules, 2011, 44, 4291–4300 CrossRef CAS.
- A. M. Welander, G. S. W. Craig, Y. Tada, H. Yoshida and P. F. Nealey, Directed Assembly of Block Copolymers in Thin to Thick Films, Macromolecules, 2013, 46, 3915–3921 CrossRef CAS.
- H. J. Dai, N. P. Balsara, B. A. Garetz and M. C. Newstein, Grain Growth and Defect Annihilation in Block Copolymers, Phys. Rev. Lett., 1996, 77, 3677–3680 CrossRef CAS PubMed.
- T. Q. Chastek and T. P. Lodge, Grain Shapes and Growth Kinetics of the Cylinder Phase in a Block Copolymer Solution, Macromolecules, 2004, 37, 4891–4899 CrossRef CAS.
- T. Q. Chastek and T. P. Lodge, Grain shapes and growth kinetics during self-assembly of block copolymers, J. Polym. Sci., Part B: Polym. Phys., 2006, 44, 481–491 CrossRef CAS.
- H. J. Ryu, D. B. Fortner, S. Lee, R. Ferebee, M. De Graef, K. Misichronis, A. Avgeropoulos and M. R. Bockstaller, Role of Grain Boundary Defects During Grain Coarsening of Lamellar Block Copolymers, Macromolecules, 2013, 46, 204–215 CrossRef CAS.
- R. Ruiz, J. K. Bosworth and C. T. Black, Effect of structural anisotropy on the coarsening kinetics of diblock copolymer striped patterns, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 054204–054205 CrossRef.
- X. Zhang, K. G. Yager, N. J. Fredin, H. W. Ro, R. L. Jones, A. Karim and J. F. Douglas, Thermally Reversible Surface Morphology Transition in Thin Diblock Copolymer Films, ACS Nano, 2010, 4, 3653–3660 CrossRef CAS PubMed.
- J. P. Singer, K. W. Gotrik, J.-H. Lee, S. E. Kooi, C. A. Ross and E. L. Thomas, Alignment and reordering of a block copolymer by solvent-enhanced thermal laser direct write, Polymer, 2014, 55, 1875–1882 CrossRef CAS.
- A. G. Jacobs, B. Jung, C. K. Ober and M. O. Thompson, Control of PS-b-PMMA directed self-assembly registration by laser induced millisecond thermal annealing, Proc. SPIE, 2017, 9049, 90492B Search PubMed.
- P. W. Majewski and K. G. Yager, Millisecond Ordering of Block Copolymer Films via Photothermal Gradients, ACS Nano, 2015, 9, 3896–3906 CrossRef CAS PubMed.
- A. G. Jacobs, B. Jung, J. Jiang, C. K. Ober and M. O. Thompson, Control of polystyrene-block-poly(methyl methacrylate) directed self-assembly by laser-induced millisecond thermal annealing, J. Micro/Nanolithogr., MEMS, MOEMS, 2015, 14, 031205–031205 CrossRef.
- P. W. Majewski and K. G. Yager, Block Copolymer Response to Photothermal Stress Fields, Macromolecules, 2015, 48, 4591–4598 CrossRef CAS.
- A. G. Jacobs, C. Liedel, H. Peng, L. Wang, D.-M. Smilgies, C. K. Ober and M. O. Thompson, Kinetics of Block Copolymer Phase Segregation during Sub-millisecond Transient Thermal Annealing, Macromolecules, 2016, 49, 6462–6470 CrossRef CAS.
- H. M. Jin, S. H. Lee, J. Y. Kim, S.-W. Son, B. H. Kim, H. K. Lee, J. H. Mun, S. K. Cha, J. S. Kim, P. F. Nealey, K. J. Lee and S. O. Kim, Laser Writing Block Copolymer Self-Assembly on Graphene Light-Absorbing Layer, ACS Nano, 2016, 10, 3435–3442 CrossRef CAS PubMed.
- J. Jiang, A. G. Jacobs, B. Wenning, C. Liedel, M. O. Thompson and C. K. Ober, Ultrafast Self-Assembly of Sub-10 nm Block Copolymer Nanostructures by Solvent-Free High-Temperature Laser Annealing, ACS Appl. Mater. Interfaces, 2017, 9, 31317–31324 CAS.
- H. M. Jin, D. Y. Park, S.-J. Jeong, G. Y. Lee, J. Y. Kim, J. H. Mun, S. K. Cha, J. Lim, J. S. Kim, K. H. Kim, K. J. Lee and S. O. Kim, Flash Light Millisecond Self-Assembly of High χ Block Copolymers for Wafer-Scale Sub-10 nm Nanopatterning, Adv. Mater., 2017, 1700595 CrossRef PubMed.
- S. Lee, M. J. Bluemle and F. S. Bates, Discovery of a Frank-Kasper σ Phase in Sphere-Forming Block Copolymer Melts, Science, 2010, 330, 349–353 CrossRef CAS PubMed.
- S. Lee, C. Leighton and F. S. Bates, Sphericity and symmetry breaking in the formation of Frank–Kasper phases from one component materials, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 17723–17731 CrossRef CAS PubMed.
- K. Kim, M. W. Schulze, A. Arora, R. M. Lewis, M. A. Hillmyer, K. D. Dorfman and F. S. Bates, Thermal processing of diblock copolymer melts mimics metallurgy, Science, 2017, 356, 520–523 CrossRef CAS PubMed.
- T. M. Gillard, S. Lee and F. S. Bates, Dodecagonal quasicrystalline order in a diblock copolymer melt, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 5167–5172 CrossRef CAS PubMed.
- Y. S. Jung and C. A. Ross, Orientation-Controlled Self-Assembled Nanolithography Using a Polystyrene–Polydimethylsiloxane Block Copolymer, Nano Lett., 2007, 7, 2046–2050 CrossRef CAS PubMed.
- E. Kim, H. Ahn, S. Park, H. Lee, M. Lee, S. Lee, T. Kim, E.-A. Kwak, J. H. Lee, X. Lei, J. Huh, J. Bang, B. Lee and D. Y. Ryu, Directed Assembly of High Molecular Weight Block Copolymers: Highly Ordered Line Patterns of Perpendicularly Oriented Lamellae with Large Periods, ACS Nano, 2013, 7, 1952–1960 CrossRef CAS PubMed.
- J. W. Jeong, W. I. Park, M.-J. Kim, C. A. Ross and Y. S. Jung, Highly Tunable Self-Assembled Nanostructures from a Poly(2-vinylpyridine-b-dimethylsiloxane) Block Copolymer, Nano Lett., 2011, 11, 4095–4101 CrossRef CAS PubMed.
- S. Xiong, L. Wan, Y. Ishida, Y.-A. Chapuis, G. S. W. Craig, R. Ruiz and P. F. Nealey, Directed Self-Assembly of Triblock Copolymer on Chemical Patterns for Sub-10-nm Nanofabrication via Solvent Annealing, ACS Nano, 2016, 10, 7855–7865 CrossRef CAS PubMed.
- T. Ghoshal, A. Chaudhari, C. Cummins, M. T. Shaw, J. D. Holmes and M. A. Morris, Morphological evolution of lamellar forming polystyrene-block-poly(4-vinylpyridine) copolymers under solvent annealing, Soft Matter, 2016, 12, 5429–5437 RSC.
- J. K. Bosworth, M. Y. Paik, R. Ruiz, E. L. Schwartz, J. Q. Huang, A. W. Ko, D.-M. Smilgies, C. T. Black and C. K. Ober, Control of Self-Assembly of Lithographically Patternable Block Copolymer Films, ACS Nano, 2008, 2, 1396–1402 CrossRef CAS PubMed.
- Y. S. Jung and C. A. Ross, Solvent-Vapor-Induced Tunability of Self-Assembled Block Copolymer Patterns, Adv. Mater., 2009, 21, 2540–2545 CrossRef CAS.
- K. W. Gotrik, A. F. Hannon, J. G. Son, B. Keller, A. Alexander-Katz and C. A. Ross, Morphology Control in Block Copolymer Films Using Mixed Solvent Vapors, ACS Nano, 2012, 6, 8052–8059 CrossRef CAS PubMed.
- W. Bai, A. F. Hannon, K. W. Gotrik, H. K. Choi, K. Aissou, G. Liontos, K. Ntetsikas, A. Alexander-Katz, A. Avgeropoulos and C. A. Ross, Thin Film Morphologies of Bulk-Gyroid Polystyrene-block-polydimethylsiloxane under Solvent Vapor Annealing, Macromolecules, 2014, 47, 6000–6008 CrossRef CAS.
- B. Löwenhaupt and G. P. Hellmann, Microphase and macrophase separation in blends with a two-block copolymer, Polymer, 1991, 32, 1065–1076 CrossRef.
- V. Abetz and T. Goldacker, Formation of superlattices via blending of block copolymers, Macromol. Rapid Commun., 2000, 21, 16–34 CrossRef CAS.
- J. Peng, X. Gao, Y. Wei, H. Wang, B. Li and Y. Han, Controlling the size of nanostructures in thin films via blending of block copolymers and homopolymers, J. Chem. Phys., 2005, 122, 114706 CrossRef PubMed.
- A. Haryono and W. H. Binder, Controlled Arrangement of Nanoparticle Arrays in Block-Copolymer Domains, Small, 2006, 2, 600–611 CrossRef CAS PubMed.
- M. W. Matsen, Immiscibility of large and small symmetric diblock copolymers, J. Chem. Phys., 1995, 103, 3268–3271 CrossRef CAS.
- L. Kane, M. M. Satkowski, S. D. Smith and R. J. Spontak, Phase Behavior and Morphological Characteristics of Compositionally Symmetric Diblock Copolymer Blends, Macromolecules, 1996, 29, 8862–8870 CrossRef CAS.
- D. Yamaguchi and T. Hashimoto, A Phase Diagram for the Binary Blends of Nearly Symmetric Diblock Copolymers. 1. Parameter Space of Molecular Weight Ratio and Blend Composition, Macromolecules, 2001, 34, 6495–6505 CrossRef CAS.
- E. Sivaniah, S. Matsubara, Y. Zhao, T. Hashimoto, K. Fukunaga, E. J. Kramer and T. E. Mates, Symmetric Diblock Copolymer Thin Films on Rough Substrates: Microdomain Periodicity in Pure and Blended Films, Macromolecules, 2008, 41, 2584–2592 CrossRef CAS.
- X. Zhang, J. N. Murphy, N. L. Y. Wu, K. D. Harris and J. M. Buriak, Rapid Assembly of Nanolines with Precisely Controlled Spacing from Binary Blends of Block Copolymers, Macromolecules, 2011, 44, 9752–9757 CrossRef CAS.
- L. D. Williamson and P. F. Nealey, Macrophase Separation of Blends of Diblock Copolymers in Thin Films, Macromolecules, 2015, 48, 3997–4003 CrossRef CAS.
- T. Hashimoto, S. Koizumi and H. Hasegawa, Ordered Structure in Blends of Block Copolymers.2.Self-Assembly for Immiscible Lamella-Forming Copolymers, Macromolecules, 1994, 27, 1562–1570 CrossRef CAS.
- R. J. Spontak, J. C. Fung, M. B. Braunfeld, J. W. Sedat, D. A. Agard, L. Kane, S. D. Smith, M. M. Satkowski, A. Ashraf, D. A. Hajduk and S. M. Gruner, Phase Behavior of Ordered Diblock Copolymer Blends: Effect of Compositional Heterogeneity, Macromolecules, 1996, 29, 4494–4507 CrossRef CAS.
- K. G. Yager, E. Lai and C. T. Black, Self-Assembled Phases of Block Copolymer Blend Thin Films, ACS Nano, 2014, 8, 10582–10588 CrossRef CAS PubMed.
- Y. Asai, A. Takano and Y. Matsushita, Creation of Cylindrical Morphologies with Extremely Large Oblong Unit Lattices from ABC Block Terpolymer Blends, Macromolecules, 2015, 48, 1538–1542 CrossRef CAS.
- Y. Asai, A. Takano and Y. Matsushita, Asymmetric Double Tetragonal Domain Packing from ABC Triblock Terpolymer Blends with Chain Length Difference, Macromolecules, 2016, 49, 6940–6946 CrossRef CAS.
- Y. Asai, J. Suzuki, Y. Aoyama, H. Nishioka, A. Takano and Y. Matsushita, Tricontinuous Double Diamond Network Structure from Binary Blends of ABC Triblock Terpolymers, Macromolecules, 2017, 50, 5402–5411 CrossRef CAS.
- C. Tang, E. M. Lennon, G. H. Fredrickson, E. J. Kramer and C. J. Hawker, Evolution of Block Copolymer Lithography to Highly Ordered Square Arrays, Science, 2008, 322, 429–432 CrossRef CAS PubMed.
- S. H. Han, V. Pryamitsyn, D. Bae, J. Kwak, V. Ganesan and J. K. Kim, Highly Asymmetric Lamellar Nanopatterns via Block Copolymer Blends Capable of Hydrogen Bonding, ACS Nano, 2012, 6, 7966–7972 CrossRef CAS PubMed.
- H. Miyase, Y. Asai, A. Takano and Y. Matsushita, Kaleidoscopic Tiling Patterns with Large Unit Cells from ABC Star-Shaped Terpolymer/Diblock Copolymer Blends with Hydrogen Bonding Interaction, Macromolecules, 2017, 50, 979–986 CrossRef CAS.
- S. Valkama, T. Ruotsalainen, A. Nykänen, A. Laiho, H. Kosonen, G. ten Brinke, O. Ikkala and J. Ruokolainen, Self-Assembled Structures in Diblock Copolymers with Hydrogen-Bonded Amphiphilic Plasticizing Compounds, Macromolecules, 2006, 39, 9327–9336 CrossRef CAS.
- X. Lu, D.-P. Song, A. Ribbe and J. J. Watkins, Chiral Arrangements of Au Nanoparticles with Prescribed Handedness Templated by Helical Pores in Block Copolymer Films, Macromolecules, 2017, 50, 5293–5300 CrossRef CAS.
- L. Yao, X. Lu, S. Chen and J. J. Watkins, Formation of Helical Phases in Achiral Block Copolymers by Simple Addition of Small Chiral Additives, Macromolecules, 2014, 47, 6547–6553 CrossRef CAS.
- G. Liu, M. P. Stoykovich, S. Ji, K. O. Stuen, G. S. W. Craig and P. F. Nealey, Phase Behavior and Dimensional Scaling of Symmetric Block Copolymer–Homopolymer Ternary Blends in Thin Films, Macromolecules, 2009, 42, 3063–3072 CrossRef CAS.
- K. O. Stuen, C. S. Thomas, G. Liu, N. Ferrier and P. F. Nealey, Dimensional Scaling of Cylinders in Thin Films of Block Copolymer–Homopolymer Ternary Blends, Macromolecules, 2009, 42, 5139–5145 CrossRef CAS.
- M. P. Stoykovich, M. Muller, S. O. Kim, H. H. Solak, E. W. Edwards, J. J. de Pablo and P. F. Nealey, Directed Assembly of Block Copolymer Blends into Nonregular Device-Oriented Structures, Science, 2005, 308, 1442 CrossRef CAS PubMed.
- M. P. Stoykovich, H. Kang, K. C. Daoulas, G. Liu, C.-C. Liu, J. J. de Pablo, M. Müller and P. F. Nealey, Directed Self-Assembly of Block Copolymers for Nanolithography: Fabrication of Isolated Features and Essential Integrated Circuit Geometries, ACS Nano, 2007, 1, 168–175 CrossRef CAS PubMed.
- D. Broseta and G. H. Fredrickson, Phase equilibria in copolymer/homopolymer ternary blends: Molecular weight effects, J. Chem. Phys., 1990, 93, 2927–2938 CrossRef CAS.
- M. W. Matsen, Phase Behavior of Block Copolymer/Homopolymer Blends, Macromolecules, 1995, 28, 5765–5773 CrossRef CAS.
- P. K. Janert and M. Schick, Phase Behavior of Ternary Homopolymer/Diblock Blends: Influence of Relative Chain Lengths, Macromolecules, 1997, 30, 137–144 CrossRef CAS.
- N. R. Washburn, T. P. Lodge and F. S. Bates, Ternary Polymer Blends as Model Surfactant Systems, J. Phys. Chem. B, 2000, 104, 6987–6997 CrossRef CAS.
- P. F. W. Simon, R. Ulrich, H. W. Spiess and U. Wiesner, Block Copolymer–Ceramic Hybrid Materials from Organically Modified Ceramic Precursors, Chem. Mater., 2001, 13, 3464–3486 CrossRef CAS.
- B. C. Garcia, M. Kamperman, R. Ulrich, A. Jain, S. M. Gruner and U. Wiesner, Morphology Diagram of a Diblock Copolymer–Aluminosilicate Nanoparticle System, Chem. Mater., 2009, 21, 5397–5405 CrossRef CAS.
- G. E. S. Toombes, A. C. Finnefrock, M. W. Tate, R. Ulrich, U. Wiesner and S. M. Gruner, A Re-Evaluation of the Morphology of a Bicontinuous Block Copolymer–Ceramic Material, Macromolecules, 2007, 40, 8974–8982 CrossRef CAS.
- D.-P. Song, Y. Gai, B. M. Yavitt, A. Ribbe, S. Gido and J. J. Watkins, Structural Diversity and Phase Behavior of Brush Block Copolymer Nanocomposites, Macromolecules, 2016, 49, 6480–6488 CrossRef CAS.
- M. Stefik, S. Wang, R. Hovden, H. Sai, M. W. Tate, D. A. Muller, U. Steiner, S. M. Gruner and U. Wiesner, Networked and chiral nanocomposites from ABC triblock terpolymer coassembly with transition metal oxide nanoparticles, J. Mater. Chem., 2012, 22, 1078–1087 RSC.
- Z. Li, K. Hur, H. Sai, T. Higuchi, A. Takahara, H. Jinnai, S. M. Gruner and U. Wiesner, Linking experiment and theory for three-dimensional networked binary metal nanoparticle–triblock terpolymer superstructures, Nat. Commun., 2014, 5, 3247 Search PubMed.
- X. He, M. Song, H. Liang and C. Pan, Self-assembly of the symmetric diblock copolymer in a confined state: Monte Carlo simulation, J. Chem. Phys., 2001, 114, 10510–10513 CrossRef CAS.
- Y. Wu, G. Cheng, K. Katsov, S. W. Sides, J. Wang, J. Tang, G. H. Fredrickson, M. Moskovits and G. D. Stucky, Composite mesostructures by nano-confinement, Nat. Mater., 2004, 3, 816–822 CrossRef CAS PubMed.
- B. Yu, P. Sun, T. Chen, Q. Jin, D. Ding, B. Li and A.-C. Shi, Confinement-Induced Novel Morphologies of Block Copolymers, Phys. Rev. Lett., 2006, 96, 138306 CrossRef PubMed.
- Y. S. Jung, W. Jung and C. A. Ross, Nanofabricated Concentric Ring Structures by Templated Self-Assembly of a Diblock Copolymer, Nano Lett., 2008, 8, 2975–2981 CrossRef CAS PubMed.
- J. Bang, U. Jeong, D. Y. Ryu, T. P. Russell and C. J. Hawker, Block Copolymer Nanolithography: Translation of Molecular Level Control to Nanoscale Patterns, Adv. Mater., 2009, 21, 4769–4792 CrossRef CAS PubMed.
- P. Chi, Z. Wang, B. Li and A.-C. Shi, Soft Confinement-Induced Morphologies of Diblock Copolymers, Langmuir, 2011, 27, 11683–11689 CrossRef CAS PubMed.
- C. Simao, A. Francone, D. Borah, O. Lorret, M. Salaun, B. Kosmala, M. T. Shaw, B. Dittert, N. Kehagias, M. Zelsmann, M. A. Morris and C. M. S. Torres, Soft Graphoepitaxy of Hexagonal PS-b-PDMS on Nanopatterned POSS Surfaces fabricated by Nanoimprint Lithography, J. Photopolym. Sci. Technol., 2012, 25, 239–244 CrossRef CAS.
- Z. Jin and H. Fan, Self-assembly of nanostructured block copolymer nanoparticles, Soft Matter, 2014, 10, 9212–9219 RSC.
- H. Yabu, T. Higuchi and H. Jinnai, Frustrated phases: polymeric self-assemblies in a 3D confinement, Soft Matter, 2014, 10, 2919–2931 RSC.
- B. Yu, J. Deng, B. Li and A.-C. Shi, Patchy nanoparticles self-assembled from linear triblock copolymers under spherical confinement: a simulated annealing study, Soft Matter, 2014, 10, 6831–6843 RSC.
- M. J. Fasolka, P. Banerjee, A. M. Mayes, G. Pickett and A. C. Balazs, Morphology of Ultrathin Supported Diblock Copolymer Films: Theory and Experiment, Macromolecules, 2000, 33, 5702–5712 CrossRef CAS.
- A. Knoll, A. Horvat, K. S. Lyakhova, G. Krausch, G. J. A. Sevink, A. V. Zvelindovsky and R. Magerle, Phase Behavior in Thin Films of Cylinder-Forming Block Copolymers, Phys. Rev. Lett., 2002, 89, 035501 CrossRef CAS PubMed.
- R. A. Segalman, Patterning with block copolymer thin films, Mater. Sci. Eng., R, 2005, 48, 191–226 CrossRef.
- G. E. Stein, E. J. Kramer, X. Li and J. Wang, Layering Transitions in Thin Films of Spherical-Domain Block Copolymers, Macromolecules, 2007, 40, 2453–2460 CrossRef CAS.
- T. L. Morkved and H. M. Jaeger, Thickness-induced morphology changes in lamellar diblock copolymer ultrathin films, Europhys. Lett., 1997, 40, 643–648 CrossRef CAS.
- I. Park, S. Park, H.-W. Park, T. Chang, H. Yang and C. Y. Ryu, Unexpected Hexagonally Perforated Layer Morphology of PS-b-PMMA Block Copolymer in Supported Thin Film, Macromolecules, 2006, 39, 315–318 CrossRef CAS.
- J. L. Carvalho, M. V. Massa, S. L. Cormier, M. W. Matsen and K. Dalnoki-Veress, Reversible sphere-to-lamellar wetting transition at the interface of a diblock copolymer system, Eur. Phys. J. E: Soft Matter Biol. Phys., 2011, 34, 51 CrossRef CAS PubMed.
- R. D. Peters, P. Stasiak, M. W. Matsen and K. Dalnoki-Veress, Morphology Induced Spinodal Decomposition at the Surface of Symmetric Diblock Copolymer Films, ACS Macro Lett., 2013, 2, 441–445 CrossRef CAS.
- M. Peng, S. Ma, J. Hu and R. Wang, Hierarchical nanostructures of diblock copolymer thin films directed by a saw-toothed substrate, Soft Matter, 2015, 11, 6642–6651 RSC.
- W. Li, M. Liu, F. Qiu and A.-C. Shi, Phase Diagram of Diblock Copolymers Confined in Thin Films, J. Phys. Chem. B, 2013, 117, 5280–5288 CrossRef CAS PubMed.
- B. Yu, B. Li, Q. Jin, D. Ding and A.-C. Shi, Confined self-assembly of cylinder-forming diblock copolymers: effects of confining geometries, Soft Matter, 2011, 7, 10227–10240 RSC.
- T. P. Russell, A. Menelle, S. H. Anastasiadis, S. K. Satija and C. F. Majkrzak, Unconventional morphologies of symmetric, diblock copolymers due to film thickness constraints, Macromolecules, 1991, 24, 6263–6269 CrossRef CAS.
- M. Maaloum, D. Ausserre, D. Chatenay and Y. Gallot, Spinodal decomposition-like patterns via metastable state relaxation, Phys. Rev. Lett., 1993, 70, 2577–2580 CrossRef CAS PubMed.
- S. Joly, D. Ausserré, G. Brotons and Y. Gallot, Hole nucleation in thin diblock copolymer films, Eur. Phys. J. E: Soft Matter Biol. Phys., 2002, 8, 355–363 CrossRef CAS PubMed.
- S. Kim, C. M. Bates, A. Thio, J. D. Cushen, C. J. Ellison, C. G. Willson and F. S. Bates, Consequences of Surface Neutralization in Diblock Copolymer Thin Films, ACS Nano, 2013, 7, 9905–9919 CrossRef CAS PubMed.
- A. B. Croll, M. V. Massa, M. W. Matsen and K. Dalnoki-Veress, Droplet Shape of an Anisotropic Liquid, Phys. Rev. Lett., 2006, 97, 204502 CrossRef PubMed.
- J. D. McGraw, I. D. W. Rowe, M. W. Matsen and K. Dalnoki-Veress, Dynamics of interacting edge defects in copolymer lamellae, Eur. Phys. J. E: Soft Matter Biol. Phys., 2011, 34, 1–7 CrossRef CAS PubMed.
- S.-J. Jeon, G.-R. Yi, C. M. Koo and S.-M. Yang, Nanostructures Inside Colloidal Particles of Block Copolymer/Homopolymer Blends, Macromolecules, 2007, 40, 8430–8439 CrossRef CAS.
- S.-J. Jeon, G.-R. Yi and S.-M. Yang, Cooperative Assembly of Block Copolymers with Deformable Interfaces: Toward Nanostructured Particles, Adv. Mater., 2008, 20, 4103–4108 CrossRef CAS.
- S. G. Jang, D. J. Audus, D. Klinger, D. V. Krogstad, B. J. Kim, A. Cameron, S.-W. Kim, K. T. Delaney, S.-M. Hur, K. L. Killops, G. H. Fredrickson, E. J. Kramer and C. J. Hawker, Striped, Ellipsoidal Particles by Controlled Assembly of Diblock Copolymers, J. Am. Chem. Soc., 2013, 135, 6649–6657 CrossRef CAS PubMed.
- D. Klinger, C. X. Wang, L. A. Connal, D. J. Audus, S. G. Jang, S. Kraemer, K. L. Killops, G. H. Fredrickson, E. J. Kramer and C. J. Hawker, A Facile Synthesis of Dynamic, Shape-Changing Polymer Particles, Angew. Chem., Int. Ed., 2014, 53, 7018–7022 CrossRef CAS PubMed.
- K. H. Ku, J. M. Shin, M. P. Kim, C.-H. Lee, M.-K. Seo, G.-R. Yi, S. G. Jang and B. J. Kim, Size-Controlled Nanoparticle-Guided Assembly of Block Copolymers for Convex Lens-Shaped Particles, J. Am. Chem. Soc., 2014, 136, 9982–9989 CrossRef CAS PubMed.
- B. V. K. J. Schmidt, J. Elbert, D. Scheid, C. J. Hawker, D. Klinger and M. Gallei, Metallopolymer-Based Shape Anisotropic Nanoparticles, ACS Macro Lett., 2015, 4, 731–735 CrossRef CAS.
- H. Yang, K. H. Ku, J. M. Shin, J. Lee, C. H. Park, H.-H. Cho, S. G. Jang and B. J. Kim, Engineering the Shape of Block Copolymer Particles by Surface-Modulated Graphene Quantum Dots, Chem. Mater., 2016, 28, 830–837 CrossRef CAS.
- J. Chai and J. M. Buriak, Using Cylindrical Domains of Block Copolymers To Self-Assemble and Align Metallic Nanowires, ACS Nano, 2008, 2, 489–501 CrossRef CAS PubMed.
- X. Y. Bao, Y. He, C. Bencher, L. W. Chang, H. Dai, Y. Chen, P. T. J. Chen and H. S. P. Wong in 2011 International Electron Devices Meeting, 2011, pp. 7.7.1–7.7.4 Search PubMed.
- C. G. Hardy and C. Tang, Advances in square arrays through self-assembly and directed self-assembly of block copolymers, J. Polym. Sci., Part B: Polym. Phys., 2013, 51, 2–15 CrossRef CAS.
- C. Hong Kyoon, C. Jae-Byum, F. H. Adam, K. W. Y. Joel, K. B. Karl, A.-K. Alfredo and A. R. Caroline, Nanoscale spirals by directed self-assembly, Nano Futures, 2017, 1, 015001 CrossRef.
- B. Yu, B. Li, Q. Jin, D. Ding and A.-C. Shi, Self-Assembly of Symmetric Diblock Copolymers Confined in Spherical Nanopores, Macromolecules, 2007, 40, 9133–9142 CrossRef CAS.
- J. Zhang, W. Kong and H. Duan, Soft Confinement-Induced Morphologies of the Blends of AB Diblock Copolymers and C Homopolymers, Langmuir, 2017, 33, 3123–3133 CrossRef CAS PubMed.
- T. Higuchi, A. Tajima, K. Motoyoshi, H. Yabu and M. Shimomura, Frustrated Phases of Block Copolymers in Nanoparticles, Angew. Chem., 2008, 120, 8164–8166 CrossRef.
- L. Li, K. Matsunaga, J. Zhu, T. Higuchi, H. Yabu, M. Shimomura, H. Jinnai, R. C. Hayward and T. P. Russell, Solvent-Driven Evolution of Block Copolymer Morphology under 3D Confinement, Macromolecules, 2010, 43, 7807–7812 CrossRef CAS.
- R. Deng, S. Liu, F. Liang, K. Wang, J. Zhu and Z. Yang, Polymeric Janus Particles with Hierarchical Structures, Macromolecules, 2014, 47, 3701–3707 CrossRef CAS.
- L. M. Pitet, E. Alexander-Moonen, E. Peeters, T. S. Druzhinina, S. F. Wuister, N. A. Lynd and E. W. Meijer, Probing the Effect of Molecular Nonuniformity in Directed Self-Assembly of Diblock Copolymers in Nanoconfined Space, ACS Nano, 2015, 9, 9594–9602 CrossRef CAS PubMed.
- D. A. Rider, J. I. L. Chen, J.-C. Eloi, A. C. Arsenault, T. P. Russell, G. A. Ozin and I. Manners, Controlling the Morphologies of Organometallic Block Copolymers in the 3-Dimensional Spatial Confinement of Colloidal and Inverse Colloidal Crystals, Macromolecules, 2008, 41, 2250–2259 CrossRef CAS.
- H. Hu, S. Rangou, M. Kim, P. Gopalan, V. Filiz, A. Avgeropoulos and C. O. Osuji, Continuous Equilibrated Growth of Ordered Block Copolymer Thin Films by Electrospray Deposition, ACS Nano, 2013, 7, 2960–2970 CrossRef CAS PubMed.
- H. Hu, J. P. Singer and C. O. Osuji, Morphology Development in Thin Films of a Lamellar Block Copolymer Deposited by Electrospray, Macromolecules, 2014, 47, 5703–5710 CrossRef CAS.
- H. Hu, K. Toth, M. Kim, P. Gopalan and C. O. Osuji, Continuous and patterned deposition of functional block copolymer thin films using electrospray, MRS Commun., 2015, 5, 235–242 CrossRef CAS.
- H. Hu, Y. Choo, X. Feng and C. O. Osuji, Physical Continuity and Vertical Alignment of Block Copolymer Domains by Kinetically Controlled Electrospray Deposition, Macromol. Rapid Commun., 2015, 36, 1290–1296 CrossRef CAS PubMed.
- Y. Choo, H. Hu, K. Toth and C. O. Osuji, Sequential deposition of block copolymer thin films and formation of lamellar heterolattices by electrospray deposition, J. Polym. Sci., Part B: Polym. Phys., 2016, 54, 247–253 CrossRef CAS.
- J. W. Jeong, W. I. Park, L.-M. Do, J.-H. Park, T.-H. Kim, G. Chae and Y. S. Jung, Nanotransfer Printing with sub-10 nm Resolution Realized using Directed Self-Assembly, Adv. Mater., 2012, 24, 3526–3531 CrossRef CAS PubMed.
- S.-J. Jeong, H.-S. Moon, J. Shin, B. H. Kim, D. O. Shin, J. Y. Kim, Y.-H. Lee, J. U. Kim and S. O. Kim, One-Dimensional Metal Nanowire Assembly via Block Copolymer Soft Graphoepitaxy, Nano Lett., 2010, 10, 3500–3505 CrossRef CAS PubMed.
- J. Y. Kim, B. H. Kim, J. O. Hwang, S.-J. Jeong, D. O. Shin, J. H. Mun, Y. J. Choi, H. M. Jin and S. O. Kim, Flexible and Transferrable Self-Assembled Nanopatterning on Chemically Modified Graphene, Adv. Mater., 2013, 25, 1331–1335 CrossRef CAS PubMed.
- J. W. Jeong, S. R. Yang, Y. H. Hur, S. W. Kim, K. M. Baek, S. Yim, H.-I. Jang, J. H. Park, S. Y. Lee, C.-O. Park and Y. S. Jung, High-resolution nanotransfer printing applicable to diverse surfaces via interface-targeted adhesion switching, Nat. Commun., 2014, 5, 5387 CrossRef CAS PubMed.
- S. Woo, H. S. Wang, Y. Choe, J. Huh and J. Bang, Three-Dimensional Multilayered Nanostructures from Crosslinkable Block Copolymers, ACS Macro Lett., 2016, 5, 287–291 CrossRef CAS.
- G. Singh, K. G. Yager, B. Berry, H.-C. Kim and A. Karim, Dynamic Thermal Field-Induced Gradient Soft-Shear for Highly Oriented Block Copolymer Thin Films, ACS Nano, 2012, 6, 10335–10342 CrossRef CAS PubMed.
- Z. Qiang, Y. Zhang, J. A. Groff, K. A. Cavicchi and B. D. Vogt, A generalized method for alignment of block copolymer films: solvent vapor annealing with soft shear, Soft Matter, 2014, 10, 6068–6076 RSC.
- J. Chai, D. Wang, X. Fan and J. M. Buriak, Assembly of aligned linear metallic patterns on silicon, Nat. Nanotechnol., 2007, 2, 500–506 CrossRef PubMed.
- Y. Fang, Y. Jiao, K. Xiong, R. Ogier, Z.-J. Yang, S. Gao, A. B. Dahlin and M. Käll, Plasmon Enhanced Internal Photoemission in Antenna-Spacer-Mirror Based Au/TiO2 Nanostructures, Nano Lett., 2015, 15, 4059–4065 CrossRef CAS PubMed.
- N. Bonod, Silicon photonics: Large-scale dielectric metasurfaces, Nat. Mater., 2015, 14, 664–665 CrossRef CAS PubMed.
- Y. S. Jung, W. Jung, H. L. Tuller and C. A. Ross, Nanowire Conductive Polymer Gas Sensor Patterned Using Self-Assembled Block Copolymer Lithography, Nano Lett., 2008, 8, 3776–3780 CrossRef CAS PubMed.
- H. W. Liangbing Hu, Yi Cui. Metal nanogrids, nanowires, and nanofibers for transparent electrodes, MRS Bull., 2011, 36, 760–765 CrossRef.
- S. Ye, A. R. Rathmell, Z. Chen, I. E. Stewart and B. J. Wiley, Metal Nanowire Networks: The Next Generation of Transparent Conductors, Adv. Mater., 2014, 26, 6670–6687 CrossRef CAS PubMed.
- S. W. Hong, J. Huh, X. Gu, D. H. Lee, W. H. Jo, S. Park, T. Xu and T. P. Russell, Unidirectionally aligned line patterns driven by entropic effects on faceted surfaces, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 1402–1406 CrossRef CAS PubMed.
- H.-C. Kim, C. T. Rettner and L. Sundstr, Fabrication of 20nm half-pitch gratings by corrugation-directed self-assembly, Nanotechnology, 2008, 19, 235301 CrossRef PubMed.
- S. M. Park, B. C. Berry, E. Dobisz and H. C. Kim, Observation of surface corrugation-induced alignment of lamellar microdomains in PS-b-PMMA thin films, Soft Matter, 2009, 5, 957–961 RSC.
- K. G. A. Tavakkoli, S. M. Nicaise, K. R. Gadelrab, A. Alexander-Katz, C. A. Ross and K. K. Berggren, Multilayer block copolymer meshes by orthogonal self-assembly, Nat. Commun., 2016, 7, 10518 CrossRef PubMed.
- Q. Peng, Y.-C. Tseng, S. B. Darling and J. W. Elam, Nanoscopic Patterned Materials with Tunable Dimensions via Atomic Layer Deposition on Block Copolymers, Adv. Mater., 2010, 22, 5129–5133 CrossRef CAS PubMed.
- M. Ramanathan, Y.-C. Tseng, K. Ariga and S. B. Darling, Emerging trends in metal-containing block copolymers: synthesis, self-assembly, and nanomanufacturing applications, J. Mater. Chem. C, 2013, 1, 2080–2091 RSC.
- C. Jin, B. C. Olsen, N. L. Y. Wu, E. J. Luber and J. M. Buriak, Sequential Nanopatterned Block Copolymer Self-Assembly on Surfaces, Langmuir, 2016, 32, 5890–5898 CrossRef CAS PubMed.
- C. Jin, B. C. Olsen, E. J. Luber and J. M. Buriak, Preferential Alignment of Incommensurate Block Copolymer Dot Arrays Forming Moiré Superstructures, ACS Nano, 2017, 11, 3237–3246 CrossRef CAS PubMed.
- R. A. Segalman, H. Yokoyama and E. J. Kramer, Graphoepitaxy of spherical domain block copolymer films, Adv. Mater., 2001, 13, 1152–1155 CrossRef CAS.
- J. Y. Cheng, A. M. Mayes and C. A. Ross, Nanostructure engineering by templated self-assembly of block copolymers, Nat. Mater., 2004, 3, 823–828 CrossRef CAS PubMed.
- I. Bita, J. K. W. Yang, Y. S. Jung, C. A. Ross, E. L. Thomas and K. K. Berggren, Graphoepitaxy of Self-Assembled Block Copolymers on Two-Dimensional Periodic Patterned Templates, Science, 2008, 321, 939–943 CrossRef CAS PubMed.
- S. Ouk Kim, H. H. Solak, M. P. Stoykovich, N. J. Ferrier, J. J. de Pablo and P. F. Nealey, Epitaxial self-assembly of block copolymers on lithographically defined nanopatterned substrates, Nature, 2003, 424, 411 CrossRef PubMed.
- R. Ruiz, H. Kang, F. A. Detcheverry, E. Dobisz, D. S. Kercher, T. R. Albrecht, J. J. de Pablo and P. F. Nealey, Density Multiplication and Improved Lithography by Directed Block Copolymer Assembly, Science, 2008, 321, 936–939 CrossRef CAS PubMed.
- J. Y. Cheng, C. T. Rettner, D. P. Sanders, H.-C. Kim and W. D. Hinsberg, Dense Self-Assembly on Sparse Chemical Patterns: Rectifying and Multiplying Lithographic Patterns Using Block Copolymers, Adv. Mater., 2008, 20, 3155–3158 CrossRef CAS.
- C.-C. Liu, A. Ramírez-Hernández, E. Han, G. S. W. Craig, Y. Tada, H. Yoshida, H. Kang, S. Ji, P. Gopalan, J. J. de Pablo and P. F. Nealey, Chemical Patterns for Directed Self-Assembly of Lamellae-Forming Block Copolymers with Density Multiplication of Features, Macromolecules, 2013, 46, 1415–1424 CrossRef CAS.
- G. S. Doerk, C.-C. Liu, J. Y. Cheng, C. T. Rettner, J. W. Pitera, L. E. Krupp, T. Topuria, N. Arellano and D. P. Sanders, Pattern Placement Accuracy in Block Copolymer Directed Self-Assembly Based on Chemical Epitaxy, ACS Nano, 2013, 7, 276–285 CrossRef CAS PubMed.
- L. Wan, R. Ruiz, H. Gao, K. C. Patel, J. Lille, G. Zeltzer, E. A. Dobisz, A. Bogdanov, P. F. Nealey and T. R. Albrecht, Fabrication of templates with rectangular bits on circular tracks by combining block copolymer directed self-assembly and nanoimprint lithography, J. Micro/Nanolithogr., MEMS, MOEMS, 2012, 11, 03145 Search PubMed.
- S.-M. Park, G. S. W. Craig, Y.-H. La, H. H. Solak and P. F. Nealey, Square Arrays of Vertical Cylinders of PS-b-PMMA on Chemically Nanopatterned Surfaces, Macromolecules, 2007, 40, 5084–5094 CrossRef CAS.
- K. G. A. Tavakkoli, A. F. Hannon, K. W. Gotrik, A. Alexander-Katz, C. A. Ross and K. K. Berggren, Rectangular Symmetry Morphologies in a Topographically Templated Block Copolymer, Adv. Mater., 2012, 24, 4249–4254 CrossRef PubMed.
- K. G. A. Tavakkoli, S. M. Nicaise, A. F. Hannon, K. W. Gotrik, A. Alexander-Katz, C. A. Ross and K. K. Berggren, Sacrificial-Post Templating Method for Block Copolymer Self-Assembly, Small, 2014, 10, 493–499 CrossRef PubMed.
- S. Ji, U. Nagpal, W. Liao, C.-C. Liu, J. J. de Pablo and P. F. Nealey, Three-dimensional Directed Assembly of Block Copolymers together with Two-dimensional Square and Rectangular Nanolithography, Adv. Mater., 2011, 23, 3692–3697 CrossRef CAS PubMed.
- J. Xu, T. P. Russell, B. M. Ocko and A. Checco, Block copolymer self-assembly in chemically patterned squares, Soft Matter, 2011, 7, 3915–3919 RSC.
- S.-M. Hur, C. J. García-Cervera, E. J. Kramer and G. H. Fredrickson, SCFT Simulations of Thin Film Blends of Block Copolymer and Homopolymer Laterally Confined in a Square Well, Macromolecules, 2009, 42, 5861–5872 CrossRef CAS.
- H. Deng, N. Xie, W. Li, F. Qiu and A.-C. Shi, Perfectly Ordered Patterns via Corner-Induced Heterogeneous Nucleation of Self-Assembling Block Copolymers Confined in Hexagonal Potential Wells, Macromolecules, 2015, 48, 4174–4182 CrossRef CAS.
- H. Yi, X.-Y. Bao, J. Zhang, C. Bencher, L.-W. Chang, X. Chen, R. Tiberio, J. Conway, H. Dai, Y. Chen, S. Mitra and H. S. P. Wong, Flexible Control of Block Copolymer Directed Self-Assembly using Small, Topographical Templates: Potential Lithography Solution for Integrated Circuit Contact Hole Patterning, Adv. Mater., 2012, 24, 3107–3114 CrossRef CAS PubMed.
- G. Liu, C. S. Thomas, G. S. W. Craig and P. F. Nealey, Integration of Density Multiplication in the Formation of Device-Oriented Structures by Directed Assembly of Block Copolymer–Homopolymer Blends, Adv. Funct. Mater., 2010, 20, 1251–1257 CrossRef CAS.
- J. K. W. Yang, Y. S. Jung, J.-B. Chang, R. A. Mickiewicz, A. Alexander Katz, C. A. Ross and K. K. Berggren, Complex self-assembled patterns using sparse commensurate templates with locally varying motifs, Nat. Nanotechnol., 2010, 5, 256–260 CrossRef CAS PubMed.
- J.-B. Chang, H. K. Choi, A. F. Hannon, A. Alexander-Katz, C. A. Ross and K. K. Berggren, Design rules for self-assembled block copolymer patterns using tiled templates, Nat. Commun., 2014, 5, 3305 Search PubMed.
- H. Tsai, J. W. Pitera, H. Miyazoe, S. Bangsaruntip, S. U. Engelmann, C.-C. Liu, J. Y. Cheng, J. J. Bucchignano, D. P. Klaus, E. A. Joseph, D. P. Sanders, M. E. Colburn and M. A. Guillorn, Two-Dimensional Pattern Formation Using Graphoepitaxy of PS-b-PMMA Block Copolymers for Advanced FinFET Device and Circuit Fabrication, ACS Nano, 2014, 8, 5227–5232 CrossRef CAS PubMed.
- G. S. Doerk, J. Y. Cheng, G. Singh, C. T. Rettner, J. W. Pitera, S. Balakrishnan, N. Arellano and D. P. Sanders, Enabling complex nanoscale pattern customization using directed self-assembly, Nat. Commun., 2014, 5, 5805 CrossRef PubMed.
- J. Cheng, G. S. Doerk, C. T. Rettner, G. Singh, M. Tjio, H. Truong, N. Arellano, S. Balakrishnan, M. Brink, H. Tsai, C.-C. Liu, M. Guillorn and D. P. Sanders, Customization and design of directed self-assembly using hybrid prepatterns, Proc. SPIE, 2015, 9423, 942307–942308 CrossRef.
- J. K. Bosworth, C. T. Black and C. K. Ober, Selective Area Control of Self-Assembled Pattern Architecture Using a Lithographically Patternable Block Copolymer, ACS Nano, 2009, 3, 1761–1766 CrossRef CAS PubMed.
- J. G. Son, J.-B. Chang, K. K. Berggren and C. A. Ross, Assembly of Sub-10-nm Block Copolymer Patterns with Mixed Morphology and Period Using Electron Irradiation and Solvent Annealing, Nano Lett., 2011, 11, 5079–5084 CrossRef CAS PubMed.
- A. Stein, G. Wright, K. G. Yager, G. S. Doerk and C. T. Black, Selective directed self-assembly of coexisting morphologies using block copolymer blends, Nat. Commun., 2016, 7, 12366 CrossRef CAS PubMed.
- G. Liu, F. Detcheverry, A. Ramírez-Hernández, H. Yoshida, Y. Tada, J. J. de Pablo and P. F. Nealey, Nonbulk Complex Structures in Thin Films of Symmetric Block Copolymers on Chemically Nanopatterned Surfaces, Macromolecules, 2012, 45, 3986–3992 CrossRef CAS.
- K. G. A. Tavakkoli, K. W. Gotrik, A. F. Hannon, A. Alexander-Katz, C. A. Ross and K. K. Berggren, Templating Three-Dimensional Self-Assembled Structures in Bilayer Block Copolymer Films, Science, 2012, 336, 1294–1298 CrossRef CAS PubMed.
- Y. J. Choi, J. Y. Kim, J. E. Kim, J. H. Mun, S. K. Cha and S. O. Kim, Hierarchical Directed Self-Assembly of Diblock Copolymers for Modified Pattern Symmetry, Adv. Funct. Mater., 2016, 26, 6462–6470 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |