DOI:
10.1039/C7ME00050B
(Review Article)
Mol. Syst. Des. Eng., 2017,
2, 370-379
Targeted drug delivery using iRGD peptide for solid cancer treatment
Received
5th July 2017
, Accepted 16th August 2017
First published on 16th August 2017
Abstract
Many solid tumor types, such as pancreatic cancer, have a generally poor prognosis, in part because the delivery of a therapeutic regimen is prohibited by pathological abnormalities that block access to tumor vasculature, leading to poor bioavailability. The recent development of the tumor-penetrating iRGD peptide that is covalently conjugated on the nanocarriers' surface or co-administered with nanocarriers becomes a popular approach for tumor targeting. More importantly, scientists have unlocked an important tumor transcytosis mechanism by which drug-carrying nanoparticles directly access solid tumors (that seems to be independent to leaky vasculature), thereby allowing systemically injected nanocarriers to more abundantly distribute at the tumor site with improved efficacy. In this focused review, we summarize the design and implementation strategy for iRGD-mediated tumor targeting. This includes the working principle of such a peptide and discussion on a patient-specific iRGD effect in vivo, commensurate with the level of key biomarker (i.e. neuropilin-1) expression in tumor vasculature. This highlights the necessity to contemplate the use of a personalized approach when iRGD technology is used in the clinic.
Design, System, Application
Engineering design of drug delivery nanocarrier systems holds great promise for cancer treatment by reducing their side effects and improving their efficacy. In this review, we have summarized the design and implementation strategies for nanocarrier tumor targeting mediated by iRGD, a recently developed tumor penetration peptide. The iRGD technology has been implemented by two major approaches, the iRGD covalent coupled approach and the iRGD co-administration approach, both of which have shown promising enhancement in tumor targeting in different cancer models. More and more evidence showed that the iRGD effect relies on the level of key biomarker (i.e. neuropilin-1) expression in tumor vasculature by a transcytosis mechanism. This highlights the necessity to consider personalized nanomedicine when using iRGD technology, which will promise more success of drug delivery in clinical application.
|
1. Introduction
The development of various nano-enabled drug delivery systems offers the great promise of a fundamental paradigm shift in cancer treatment, both in basic pharmaceutical research and in clinical application.1–4 It has been shown that under certain pathological circumstances, including certain solid tumors, the endothelial lining integrity of tumor vasculature is affected, which becomes leakier than normal tissues.5–7 This interesting phenomenon serves as the rationale and working principle for passive tumor targeting, a.k.a. the enhanced permeability and retention (EPR) effect, by which nanoparticles tend to preferably distribute at certain tumor sites after systemic administration.5–7 While there are concerns that the tumor types and heterogeneity among patients could lead to non-negligible variation in the magnitude of the EPR effect, the implementation of this technology has led to FDA-approved nanoformulations that positively impact cancer management in patients.8–10 However, EPR effect-dependent nanoparticle egress may not be the dominant mechanism in certain tumor types in which large tumor fenestration is blocked, which can be exemplified by the high pericyte coverage on pancreatic tumor vasculature. Another popular particle design approach is active tumor targeting, which frequently refers to the use of ligand–receptor mediated binding affinity to enhance particle uptake.11,12 While the early stage of the implementation of active targeting often contains overblown claims of targeted nanoparticles acting as “magic bullets”, the reality is that the enhanced binding affinity relies on close proximity (<0.5 nm), which means that it can only occur after nanoparticle extravasation through the tumor vasculature.13 Moreover, it is also important to consider the complexity of active targeting in terms of scale-up particle synthesis, quality control, potential for clinical use, abundance and distribution of targeted receptors in individual patients, and the cost of the nanomedicine product. While the calculations remain highly controversial, a recent review paper states that only ∼0.7% of the administered nanoparticle dose is found to be delivered to a solid tumor irrespective of active or passive targeting.14,15 In order to advance nanomedicine into the clinic, it is necessary to further improve the efficiency of nanoparticle tumor targeting, including the development of an alternative and more effective approach for tumor targeting in vivo.
While most nanocarriers that are currently being tested in clinical trials rely on passive and/or active delivery, which depends on the presence of leaky tumor vasculature, there is a complementary approach that involves tumor vascular endothelial cells, which display a network of tubular vesicles (a.k.a. the vesiculo-vacuolar organelle or VVO) that control vascular access of small molecules, nutrition substances, particulates, and even intact cells.16–19 In fact, this complementary mechanism, a.k.a. transcytosis, is a unique type of transcellular transport in which various macromolecules (some of which are in the nano-size range) are delivered across the interior of cells. For example, endothelial cells in the blood–brain barrier are responsible for brain homeostasis by restricting the access of a variety of compounds, including therapeutic drugs, and yet enable the supply of necessary nutrients, some of which are delivered via endocytosis.20 It was also shown that receptor-mediated transcytosis is involved for peptidic signaling and regulatory molecules (e.g. insulin, leptin, interleukins) and nutrients (e.g. iron, LDL) in different biological processes.20 In the setting of solid tumors, Dr. Erkki Ruoslahti et al. has described an endocytic transcytosis pathway in tumor endothelial cells that can be therapeutically accessed by tumor-penetrating iRGD peptides (CRGD[K/R]GP[D/E]C).17,19 While we will discuss the working principle of iRGD below, it suffices to mention here that iRGD is capable of promoting the penetration and tumor cell entry of a range of therapeutics (e.g. free drug, macromolecules, liposomes, and Abraxane®) in multiple cancer types.17,19 In our recent study, we demonstrated this major transcytosis mechanism to complement the classic EPR effect in the almost uniformly fatal pancreatic ductal adenocarcinoma (PDAC).30 Different from particle egress through tumor fenestration, we experimentally demonstrated the use of the iRGD peptide to activate transcytosis at the tumor site, leading to enhanced drug access and efficacy. While there are published review papers discussing the use of different peptides for cancer diagnosis and therapy,21–24 a focused discussion on iRGD-mediated targeting is necessary because it may enhance tumor targeting in an EPR effect independent fashion, and therefore useful to treat “non-leaky” tumor types. In this focused review, we will summarize the design and implementation strategy for iRGD-mediated tumor targeting, including our consideration of personalized nanomedicine when iRGD is used in the clinic.
2. iRGD-mediated tumor targeting
2.1 Discovery and working mechanism of iRGD in a tumor microenvironment
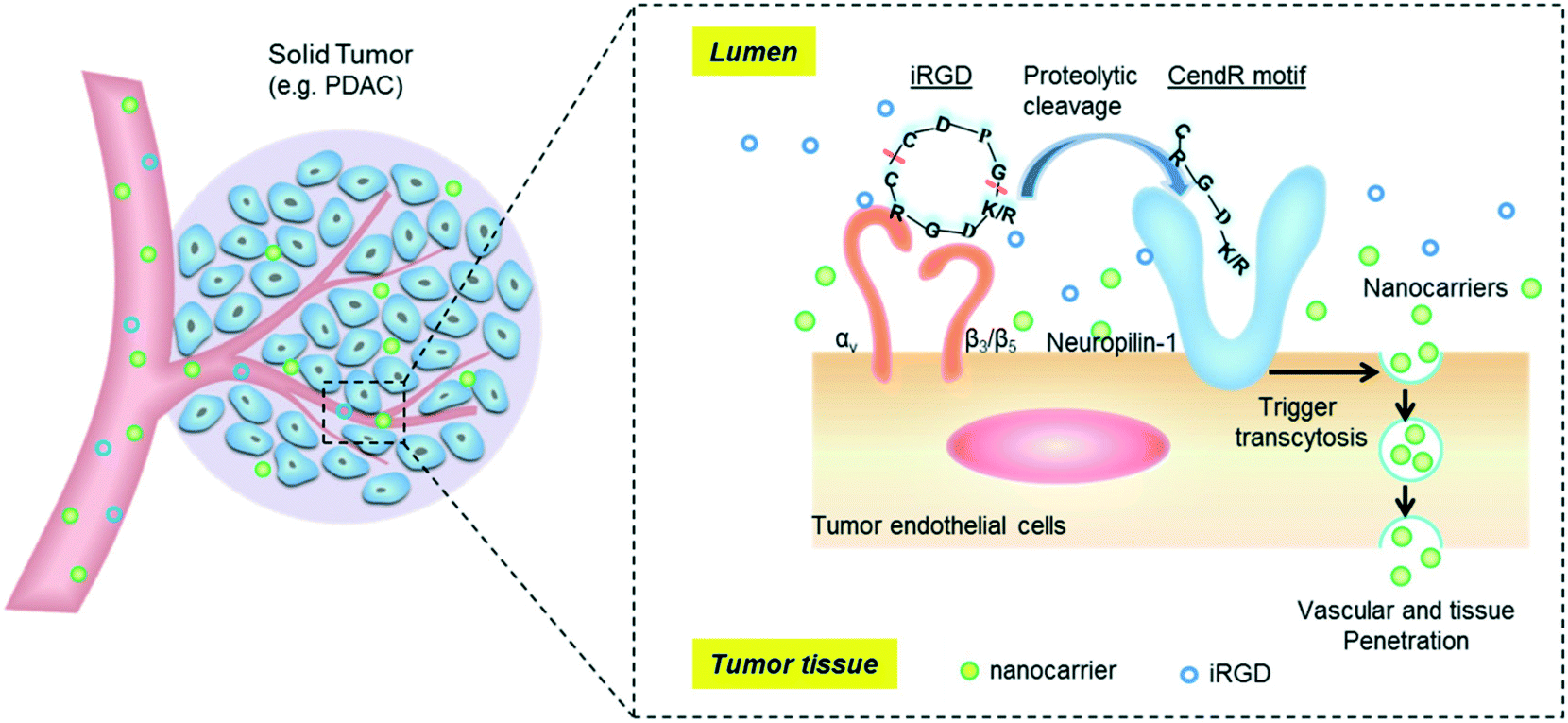
The tumor-penetrating peptide iRGD was developed by a phage screening method to identify peptides that selectively recognize tumor blood vessels in human prostate cancer animal models.17 This was achieved through the use of a cyclic CX7C (C, cysteine; X, any amino acid) peptide library displayed on T7 phage for three rounds of ex vivo phage display selection, followed by one round of in vivo selection. The combined ex vivo and in vivo experiments led to a new phage pool with 200–400× higher binding affinity to tumor-derived cell suspensions as compared to the average affinity of the original library.17 Three peptide sequences that contained the RGD motif (e.g. CRGDKGPDC, CRGDRGPDC, and CRGDKGPEC) became the most dominant peptides in the new pool. CRGDKGPDC, which binds to PPC-1 human prostate cancer cells at 4 °C and can be efficiently internalized by cancer cells at 37 °C, was named internalizing RGD peptide, a.k.a. “iRGD” (see the typical structure of the iRGD peptide in Fig. 1). More generally, cyclic 9-amino peptides with the sequences (CRGD[K/R]GP[D/E]C) are considered iRGD peptides. The working mechanism of the iRGD-mediated transport pathway is illustrated in Fig. 2.17,19,25–27 The peptide contains two critical sequence motifs, namely an integrin-binding RGD motif and a neuropilin-1 (NRP-1) binding motif. The RGD motif mediates the first binding of iRGD to αvβ3 or αvβ5 integrins, which are preferentially overexpressed in endothelial cells of tumor blood vessels and tumor cells.28,29 The binding of the cyclic peptide to the integrins is followed by a proteolytic cleavage and release of the C-terminal RXXR/K sequence, also dubbed the C-terminal rule (CendR) motif, which interacts with the NRP-1 receptor. NRP-1 binding leads to triggering of an endocytic transcytosis and trans-tissue transport pathway that can assist drug and nanoparticle delivery, including small drug molecules, monoclonal antibodies (e.g., trastuzumab), and nanoparticles (e.g., Abraxane®, doxorubicin liposomes and silicasomes).19,25,26,30 Since literature results show that the iRGD technology has been implemented by covalent coupled approach or co-administration approach, we have summarized these literature data in the following section.
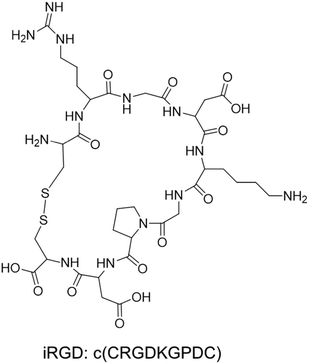 |
| | Fig. 1 Typical chemical structure of the cyclic iRGD peptide. | |
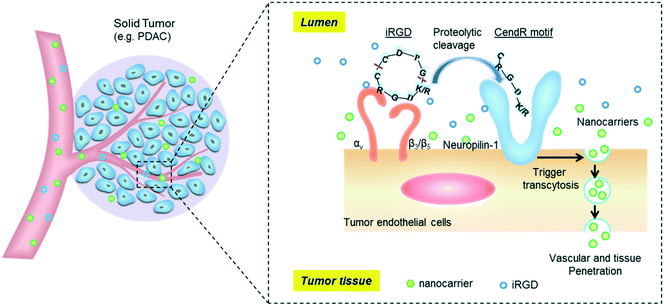 |
| | Fig. 2 Schematic of the iRGD-activated transcytosis mechanism for nanocarrier delivery in solid tumors. | |
2.2 Use of covalently conjugated iRGD to improve nanocarriers' tumor targeting
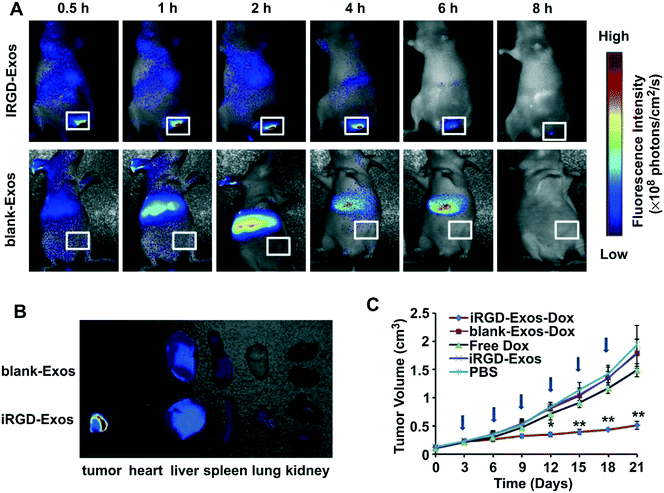
The effectiveness of iRGD-conjugated nanocarriers has been demonstrated in multiple cases that involve a variety of nanoformulations such as liposomes,31–36 polymer nanoparticles,37–43 polymer nanogels,44 polymersomes,45 nanocapsules,46 exosomes,47 protein nanoparticles,48 and inorganic nanoparticles (e.g., iron oxide nanoworms, porous silicon nanoparticles and mesoporous silica nanoparticles).49–53 A detailed summary of studies of iRGD-conjugated nanocarriers is listed in Table 1. Depending on the chemical composition, the conjugation reaction frequently involves the use of maleimide–thiol reaction,17,31–36,38,40–42,44–46,50,52,54 Michael addition of the acryloyl–amine reaction,39 alkyne–azide click reaction49,51 or amidation of the carboxyl–amine reaction,43,53 as demonstrated in Fig. 3. This includes the demonstration of tumor targeting effects which were achieved by iRGD conjugation to lipid micelles, Abraxane® (albumin/paclitaxel nanocomplex) and iron oxide nanoworms.17 In this study, the pristine iRGD peptide was modified with an extra cysteine residue and was conjugated to the nanocarriers through the maleimide–thiol reaction. In the lipid micelle, the iRGD-modified lipid was first prepared by coupling the iRGD to 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-maleimide (polyethylene glycol)2000 (DSPE-PEG2000), followed by mixing with other lipids that were further used in micelle preparation. For Abraxane® and iron oxide nanoworms, the iRGD was conjugated to the particle surface through a sulfo-SMCC (sulfosuccinimidyl 4-[N-maleimidomethyl]cyclohexane-1-carboxylate) cross-linker. The maleimide end of the sulfo-SMCC linker selectively reacted with the cysteine sulfhydryl group, whereas the succinimidyl end reacted with the primary amine group on the nanocarrier surface. In the subsequent animal studies using orthotopic human prostate, pancreatic and breast cancer models, the authors demonstrated that iRGD conjugation remarkably enhanced the penetration of nanoparticles into the tumor site, multiple hundreds of μm away from the tumor vasculature as compared to that of the control particles which primarily distributed in the peri-blood vessel region (Fig. 4). Since then, iRGD surface conjugation has become a popular approach to improve the nanocarriers' tumor targeting effect. Taking liposomal formulations for example, the effect of iRGD conjugation has been intensively studied in the past few years.31–36 The most commonly used conjugation reaction is the conjugation of iRGD to DSPE-PEG2000 to make a lipid film, followed by a rehydration process. The iRGD-conjugated lipid can be readily made by reacting the cysteine-modified iRGD with the commercially available DSPE-PEG2000–maleimide through the maleimide–thiol reaction.17,31 For example, through the iRGD conjugation to 230 nm doxorubicin-loaded liposomes, the authors successfully demonstrated facilitated cellular uptake of liposomes and an ∼2-fold enhanced antitumor effect in a subcutaneous breast cancer (4T1) model.31 Another example was testing the iRGD-conjugated doxorubicin liposome in melanoma in which the authors found an ∼57.5% improved antitumor efficacy and an ∼30% prolonged animal survival (43.5 days vs. 33 days) compared with the non-conjugated liposomes in the highly aggressive B16-F10 skin cancer model.32 In order to achieve the optimal therapeutic outcome, Dai et al. systemically dissected and studied the effect of iRGD density (1 mol% to 10 mol%) on chemo-delivery liposomes. This allowed the authors to show that the liposome with 5 mol% iRGD achieved the best cellular uptake and anticancer effect.33 Besides the classic cyclic iRGD, Liu et al. conjugated the CRGDK peptide, the CendR motif in iRGD, to an irinotecan-loaded peptide-crosslinked liposome.34 The data showed that the liposomes exhibited high targeting efficiency in cells with abundant NRP-1 expression, such as HT-29 colon cancer cells, as compared to the cancer type of low NRP-1 expression, such as MCF-7 breast cells. As a result, the use of the CRGDK peptide-conjugated liposome led to improved tumor targeting, penetration and antitumor effect compared to the non-conjugated liposomes in the HT-29 tumor in mice. In addition to the pristine iRGD peptide, iRGD derivatives were reported in the literature.35 Song et al. developed a “tadpole”-like peptide (nRGD) that contained iRGD and a tumor-associated macrophage (TAM) targeting sequence (alanine–alanine–asparagine).35 It was demonstrated that conjugating the nRGD to the liposomal carrier could dramatically improve antitumor efficacy, which is attributed to the specific interaction with tumor vascular endothelial cells capable of efficient tumor penetration, together with the capacity to deplete the TAMs in the breast tumor microenvironment. Interestingly, the iRGD peptide can also be used to improve the endogenous nanosubject for tumor targeting. In this regard, an exosome was used as a nanocarrier for doxorubicin encapsulation.47 Instead of chemical conjugation, iRGD was integrated into the exosome carrier via a fusion process by plasmid transfection. The iRGD-containing exosomes showed an ∼3-fold uptake increase in cultured MDA-MB-231 breast cancer cells and a 2–5-fold killing effect compared to the non-conjugated exosomes in vitro. Intravenous injection of iRGD-modified exosomes achieved a higher tumor distribution and antitumor effect compared to the non-conjugated exosomes in the orthotopic MDA-MB-231 cancer model in mice (Fig. 5).
Table 1 iRGD-mediated nanocarrier targeting through conjugation. As a general trend, the representative studies summarized in this table showed that iRGD-conjugated nanoparticles can be more effectively delivered at the tumor site. This is usually accompanied by improved antitumor efficacy in different cancer types
| NP type |
Conjugation chemistry |
Size |
Zeta potential |
Cancer type |
Ref. |
| Lipid micelles |
Maleimide–thiol reaction |
15–25 nm |
n/a |
Orthotopic human prostate (PC-3, PPC-1 and 22Rv1), pancreatic (MIA PaCa-2) and breast (BT474) cancers |
17
|
| Abraxane (albumin embedded paclitaxel) |
130 nm |
| Iron oxide nanoworms |
80 × 30 nm |
| Paclitaxel-loaded PCL–PVP polymer nanoparticles |
Thiazolidine ring |
40–50 nm |
−5–−10 mV |
Subcutaneous murine hepatic H22 tumor |
37
|
| siRNA-loaded PLGA–PLL–PEG polymer nanoparticles |
Maleimide–thiol reaction |
∼150 nm |
n/a |
Subcutaneous human non-small-cell lung cancer (A549) |
38
|
| Doxorubicin-loaded crosslinked multilayer liposome |
Maleimide–thiol reaction |
∼230 nm |
n/a |
Subcutaneous murine breast cancer (4T1) |
31
|
| Doxorubicin-loaded gold nanocluster-conjugated polymer nanogel |
Maleimide–thiol reaction |
∼182.4 nm |
−19.43 mV |
Murine melanoma B16 cells |
44
|
| Doxorubicin-loaded liposome |
Maleimide–thiol reaction |
∼90 nm |
−14.86 mV |
Subcutaneous murine melanoma B16-F10 tumor |
32
|
| Paclitaxel and survivin shRNA co-loaded polymer nanoparticles |
Acryloyl–amine reaction |
141–160 nm |
+30 mV |
Subcutaneous human non-small-cell lung cancer (A549/T) |
39
|
| Doxorubicin-loaded exosome |
Fused on exosome by plasmid transfection |
∼97 nm |
n/a |
Orthotopic human breast MDA-MB-231 tumor |
47
|
| Sorafenib-loaded porous silicon nanoparticles |
Azide–alkyne cycloaddition |
∼188.8 nm |
+11.2 mV |
Human endothelial EA.hy926 cells |
49
|
| Doxorubicin-loaded liposome |
Maleimide–thiol reaction |
∼95 nm |
−1.59 mV |
Subcutaneous murine melanoma B16 tumor |
33
|
| Magnetic core–shell nanoparticles (MCNPs) |
Maleimide–thiol reaction |
46.8 nm |
+15.78 mV |
Human glioblastoma (U87vIII) and breast cancer (MDA-MB-231, MCF-7) cells |
50
|
| Subcutaneous esophageal KYSE tumor |
| Doxorubicin-loaded chitosan-co-PLA/DPPE polymer nanoparticles |
Maleimide–thiol reaction |
229.9 nm |
−12.8 mV |
Human endothelial HUVEC and breast carcinoma MB-MDA-231 cells |
40
|
| Murine breast carcinoma 4T1 cells |
| Doxorubicin-conjugated PAMAM dendrimer |
Maleimide–thiol reaction |
∼22 nm |
+2.45 mV |
Orthotopic rat C6 glioma tumor in mouse |
54
|
| Irinotecan-loaded peptide-crosslinked liposome |
Maleimide–thiol reaction |
∼72 nm |
+5.7 mV |
Subcutaneous human colon HT-29 tumor |
34
|
| OSU03012-loaded protein nanocages |
Fusion of iRGD to protein using an E. coli protein expression system |
12–14 nm |
n/a |
Human pancreatic (AsPC-1, MIA PaCa-2 and Suit-2), colon (HT-29), and breast (MCF-7) cancer cells |
48
|
| Sorafenib-loaded porous silicon nanoparticles |
Azide–alkyne cycloaddition click chemistry |
202.8 nm |
−23.7 mV |
Subcutaneous human prostate PC3-MM2 cancer tumor |
51
|
| Iron oxide nanoworms |
Maleimide–thiol reaction |
∼70 × 30 nm |
n/a |
Metastasis human breast 231BR tumor and murine breast 4T1-BR5 tumor |
52
|
| Doxorubicin and sorafenib co-loaded lipid–polymer hybrid nanoparticles |
Maleimide–thiol reaction |
∼126 nm |
−21.4 nm |
Subcutaneous human hepatocellular Hep G2 tumor |
41
|
| Paclitaxel-loaded core–shell nanocapsules |
Maleimide–thiol reaction |
∼196.3 nm |
−28.63 nm |
Subcutaneous murine hepatoma H22 tumor |
46
|
| Doxorubicin-loaded liposomes |
Maleimide–thiol reaction |
∼150–170 nm |
−11.4–−13.6 mV |
Orthotopic murine breast 4T1 tumor |
35
|
| Paclitaxel-loaded polymersomes |
Maleimide–thiol reaction |
∼233 nm |
−2.7 mV |
Peritoneal or subcutaneous human gastric (MKN-45P) and murine colon (CT26) tumors |
45
|
| Vandetanib-loaded PEG–PLGA nanoparticles |
Maleimide–thiol reaction |
39.8 nm |
n/a |
Subcutaneous human hepatocellular BEL-7402 tumor |
42
|
| Indocyanine green (ICG)-loaded liposomes |
Maleimide–thiol reaction |
115.9 nm |
−34.21 mV |
Subcutaneous murine breast 4T1 tumor |
36
|
| Antiangiogenic (combretastatin A4) and chemotherapeutic (doxorubicin) co-loaded mesoporous silica nanoparticles (MSNs) |
Amide reaction |
∼70 nm |
∼−7 mV |
Subcutaneous human cervical Hela tumor |
53
|
| Survivin siRNA-loaded polymer nanoparticles |
Amide reaction |
60–90 nm |
+3–+9 mV |
Subcutaneous human prostate PC-3 tumor |
43
|
| Lipid-coated mesoporous silica nanoparticles (silicasome) |
Maleimide–thiol reaction |
∼130 nm |
∼−10 mV |
Orthotopic murine pancreatic KPC-derived tumor |
30
|
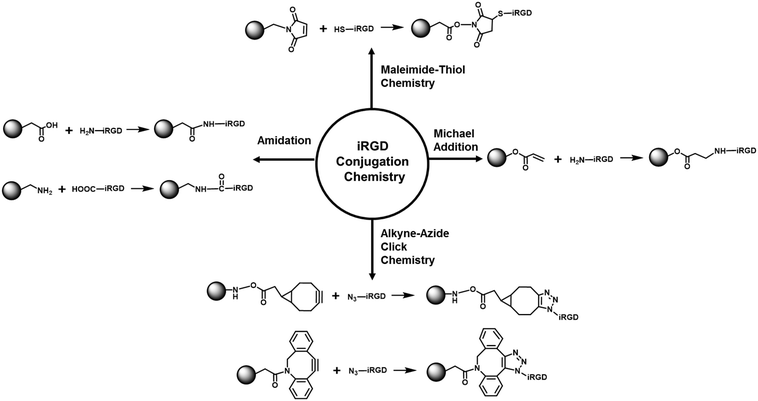 |
| | Fig. 3 Summary of the major bioconjugation reactions for covalent iRGD attachment on the nanocarrier surface. | |
 |
| | Fig. 4
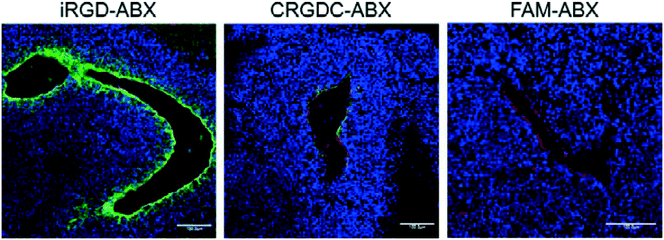
In vivo tumor penetration of Abraxane conjugates with or without iRGD conjugation. Confocal microscopy images of 22Rv1 orthotopic tumors from mice injected with the indicated Abraxane conjugates at a paclitaxel equivalent of 3 mg kg−1 after 3 h. iRGD-conjugated Abraxane showed more abundant accumulation and deeper penetration far away from the blood vessels compared to control peptide CRGDC-conjugated or non-conjugated Abraxane. Green, Abraxane; red, CD31; blue, DAPI. Scale bars = 100 μm. Adapted with permission from ref. 17. | |
 |
| | Fig. 5
In vivo tumor targeting ability of iRGD-containing exosomes (iRGD-Exos) and antitumor activity of Dox-loaded iRGD-Exos (iRGD-Exos–Dox) in MDA-MB-231 tumor-bearing mice. (A) In vivo fluorescence imaging of mice after being given a single intravenous injection of DiR-labeled blank-Exos or iRGD-Exos. Maximal fluorescence was detected at the tumor sites (white boxes) at 2 h. Two hours after injection of iRGD-Exos, no fluorescence was associated with the tumors of blank-Exos-treated mice at any time point. (B) Ex vivo fluorescence imaging of major organs from tumor-bearing mice, 2 h after intravenous injection with DiR-labeled blank-Exos or iRGD-Exos. (C) Tumor growth rate of mice treated by different reagents (PBS, iRGD-Exos, free Dox (3 mg kg−1), or an equivalent amount of Dox incorporated into blank-Exos (blank-Exos–Dox) or iRGD-Exos (iRGD-Exos–Dox)) every other day for a total of 6 injections (arrows). **, p < 0.01; *, p < 0.05. Adapted with permission from ref. 47. | |
In the case of polymeric nanoparticles, iRGD was frequently conjugated to the polymer chain during the polymer synthesis through different conjugation reactions, as we summarized in Fig. 3.37–43 Zhu et al. conjugated iRGD to paclitaxel-loaded PCL–PVP polymer nanoparticles. While iRGD surface modification moderately reduced particle circulation time in the blood stream, the authors demonstrated improved tumor targeting and antitumor efficacy in a subcutaneous hepatic H22 tumor.37 Another example was demonstrated by Shen et al., who conjugated the iRGD to paclitaxel/survivin shRNA co-loaded polymer nanoparticles through acryloyl–amine reaction.39 While there was no significant difference between the nanoparticle cellular uptakes with or without iRGD in vitro, the use of iRGD conjugation was advantageous in terms of tumor targeting and antitumor effect improvement in an A549/T lung cancer model in nude mice. Similar successes were demonstrated by conjugating iRGD to doxorubicin/sorafenib lipid–polymer hybrid nanoparticles or anti-angiogenesis agent vandetanib laden PEG–PLGA nanoparticles, which were tested in Hep G2 liver cancer or BEL-7402 liver cancer, respectively.41,42 Moreover, the iRGD-conjugated particles were also robustly tested in a gene delivery system in different cancer types, such as lung and prostate cancers.38,39,43 One example was to use iRGD modification to improve the systemic delivery of siRNA that targets survivin. It was shown that the iRGD-conjugated nanoparticles improved the siRNA tumor content by ∼3-fold, which led to an ∼3-fold greater knockdown effect in a PC3 prostate cancer xenograft model.43
Recently, the iRGD conjugation was also practiced in the emerging inorganic nanocarriers, such as magnetic core–shell nanoparticles (MCNPs), porous silicon nanoparticles, mesoporous silica nanoparticles (MSNPs), and iron oxide nanoworms.49–53 Please note that in a certain stroma-rich cancer type, such as a Kras-mutated orthotopic pancreatic cancer model, the iRGD conjugation effect is less prominent compared to other cancer types.30 Our interpretation is that in addition to the impenetrable pancreatic tumor stroma, the NRP-1 receptor abundance at the tumor vascular site may be the bottleneck that limits the access of conjugated particles to pass through the tumor site (see section 2.3).
Interestingly, the iRGD-conjugated nanocarriers also showed effectiveness when administered intraperitoneally for gastric and colon cancers.45 In this regard, the intraperitoneally administered iRGD-conjugated paclitaxel-loaded polymersomes showed improved accumulation, penetration and antitumor efficacy compared to the non-conjugated polymersomes.
2.3 Use of co-administrated iRGD to improve nanocarriers' tumor targeting via an activated transcytosis mechanism
While one can practice iRGD technology via the conjugation approach, recent advances indicate the possibility of using iRGD plus pharmaceutical products (including nanomedicine) by a co-administration approach, a new, but maybe a more effective way to use iRGD.19 We interpret this as sufficient NRP-1 receptor density initiates transmembrane uptake, while the receptor abundance at the tumor vascular site may be more limiting to the number of conjugated particles that dock and are allowed through.19,25 Remarkably, the iRGD effect can not only improve the tumor targeting of iRGD-conjugated nanocarriers, but can also be effective in a co-administration mode for the nanocarriers that do not couple to the peptide. The peptide can activate a bulk transport system that sweeps along nanocarriers present in the blood.19,30,54–61 Sugahara et al. first reported that without chemical conjugation to drugs, intravenous co-administration of iRGD boosted the vascular and tissue permeability in a tumor-specific and NRP-1-dependent manner.19 Researchers showed that free iRGD increased the tumor access of different drugs, including nanoformulations (e.g. nab-paclitaxel and liposomal doxorubicin). With the co-administered iRGD, these pharmaceutical products more efficiently penetrate into the deep tumor tissue, leading to significantly enhanced antitumor efficacy in orthotopic human breast tumor (BT474) and/or human prostate tumor (22Rv1) mouse models.19 Since then, the co-administration approach has been proven in multiple cancer types such as glioblastoma, non-small-cell lung cancer, breast carcinoma, melanoma and pancreatic cancer by different research groups, including our own (Table 2). For example, Agemy et al. found that intravenous co-injection of the iRGD into tumor tissue resulted in a markedly increased amount of proapoptotic peptide-coated iron oxide nanoworms, with dramatically prolonged mice survival in an orthotopic murine glioblastoma mouse model.55 Another example was co-administration of free iRGD with the paclitaxel-loaded PEG–PLA nanoparticles, demonstrating that iRGD significantly improved the nanoparticle access across the blood–brain tumor barrier and accumulation in glioma parenchyma, which led to longer survival in an orthotopic C6 glioma tumor model.57 Furthermore, Sun et al. showed that co-administration with iRGD was able to greatly corroborate the accumulation of paclitaxel-loaded red-blood-cell-mimetic nanoparticles in tumor parenchyma, and allowed the nanoparticles to diffuse far away from the blood vessels, with significantly improved tumor growth suppression and decreased lung metastasis compared to PTX-loaded polymer NPs alone in a breast 4T1 cancer model.61
Table 2 iRGD-mediated nanocarriers targeting through the co-administration mode. As a general trend, co-administration of nanocarriers plus non-conjugated iRGD improves drug delivery and antitumor efficacy in different cancer types, such as breast, prostate and pancreatic cancers. The effect of free iRGD is achieved by an activated transcytosis process, which mechanistically differs from the classic EPR effect, which requires enlarged fenestration in the tumor vasculature
| NP type |
Dose/treatment procedure |
Size |
Zeta potential |
Cancer type |
Ref. |
| Nab-paclitaxel nanoparticles |
4 μmol kg−1/after NP injection |
∼130 nm |
n/a |
Orthotopic human breast tumor (BT474) |
19
|
| Doxorubicin liposomes |
∼120 nm |
n/a |
Orthotopic human prostate tumor (22Rv1) |
| Proapoptotic peptide-coated iron oxide nanoworms |
4 mmol kg−1/mixed with NPs |
80–100 × 30 nm |
n/a |
Orthotopic murine glioblastoma 005 tumor |
55
|
| Cisplatin-loaded mPEG-b-PLG copolymer nanoparticles |
4 mg kg−1/mixed with NPs |
10–52 nm |
∼−7.82 mV |
Subcutaneous human non-small-cell lung cancer (A549) |
56
|
| Paclitaxel-loaded PEG–PLA polymer nanoparticles |
4 μmol kg−1/after NP injection |
∼131.3 nm |
∼−31.43 mV |
Orthotopic human C6 glioma tumor |
57
|
| Iron oxide nanoparticles |
n/a |
n/a |
n/a |
Human pancreatic cancer PANC-1 cells |
58
|
| n/a |
| Doxorubicin-conjugated PAMAM dendrimer |
4 μmol kg−1/mixed with NPs |
∼22 nm |
∼+2.45 mV |
Orthotopic rat C6 glioma tumor in mouse |
54
|
| Doxorubicin-conjugated AuNP modified on gelatin nanoparticles |
4 μmol kg−1/mixed with NPs |
∼131 nm |
∼−10.3 mV |
Subcutaneous murine breast carcinoma 4T1 cells |
59
|
| Doxorubicin-loaded liposome |
4 μmol kg−1/mixed with NPs |
∼90 nm |
∼−14.86 mV |
Subcutaneous murine melanoma B16-F10 tumor |
60
|
| Paclitaxel-loaded red-blood-cell-mimetic nanoparticles composed of a polymeric core and a RBC vesicle shell |
4 μmol kg−1/mixed with NPs |
∼147.9 nm |
∼−16.1 mV |
Orthotopic murine breast 4T1 tumor |
61
|
| Irinotecan-loaded lipid-coated mesoporous silica nanoparticles (silicasome) |
8 μmol kg−1/mixed with NPs |
∼130 nm |
∼−10 mV |
Orthotopic murine pancreatic KPC-derived tumor |
30
|
| Subcutaneous pancreatic cancer patient-derived tumor |
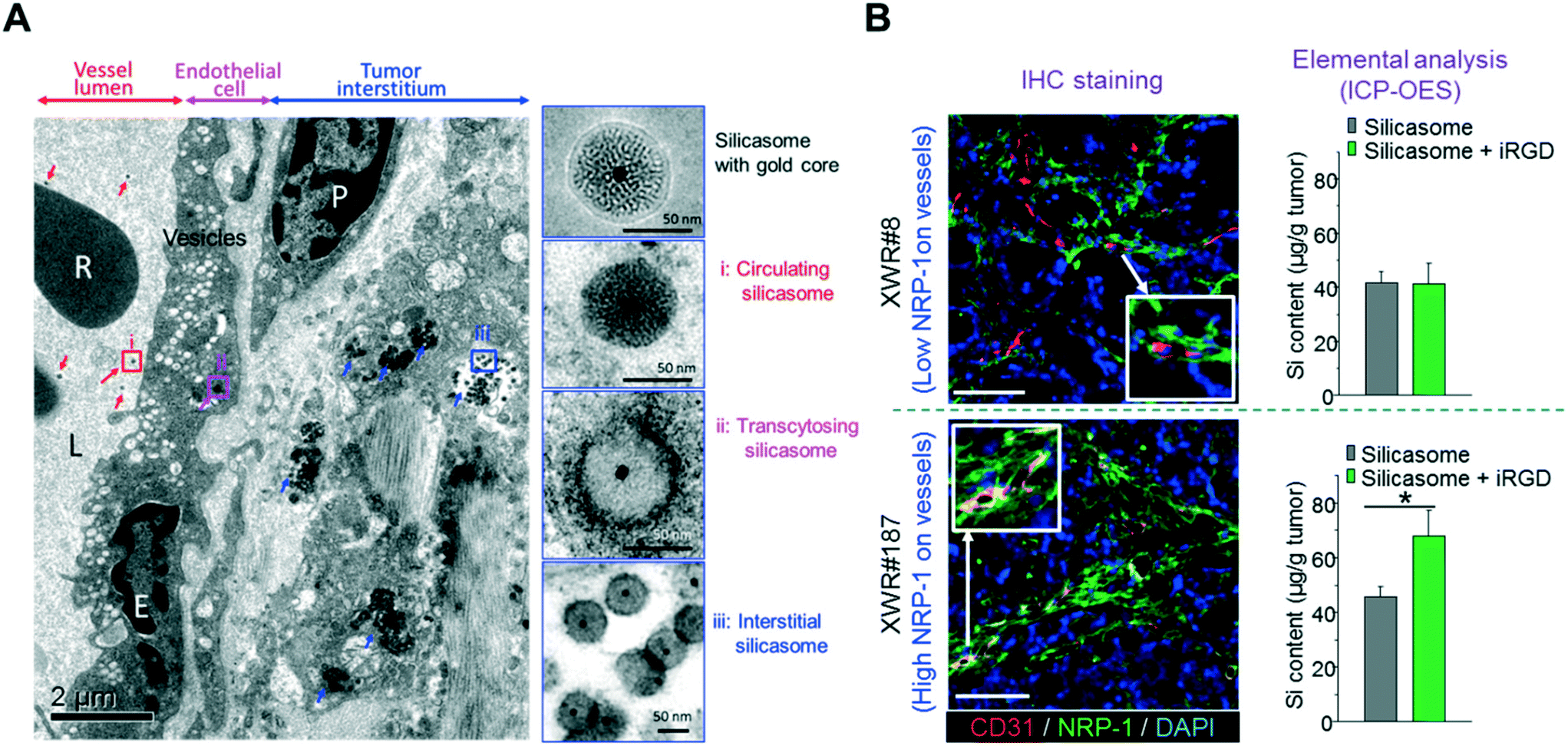
Recently, we showed that the anticancer activity of an irinotecan-loaded silicasome nanocarrier (lipid bilayer coated MSNP62,63) can be significantly improved by the co-administration of an unconjugated iRGD peptide (that does not require the covalent iRGD conjugation to the carrier).30 Through optical imaging and HPLC, we showed that the silicasome plus free iRGD increased the nanocarrier and payload abundance at orthotopic KPC tumors sites (2–4-fold), which is effective enough to introduce killing enhancement and survival prolonging, as well as metastasis inhibition. In this particular study, we have demonstrated that due to the electron density, it is not possible to obtain high resolution ultrastructure images of silica at the tumor site, but it has been demonstrated that the entrapment of a gold core in the silicasome can clearly show the transcytosis and lodging of our drug-laden silicasome in the tumor vasculature, interstitial tissues and cancer cells in the orthotopic pancreatic tumor site (Fig. 6A).30 More importantly, similar improved iRGD-mediated silicasome tumor accumulation was achieved in the patient-derived tumor with high NRP-1 expression in tumor blood vessels but not in the patient-derived tumor with low NRP-1 expression (Fig. 6B).30
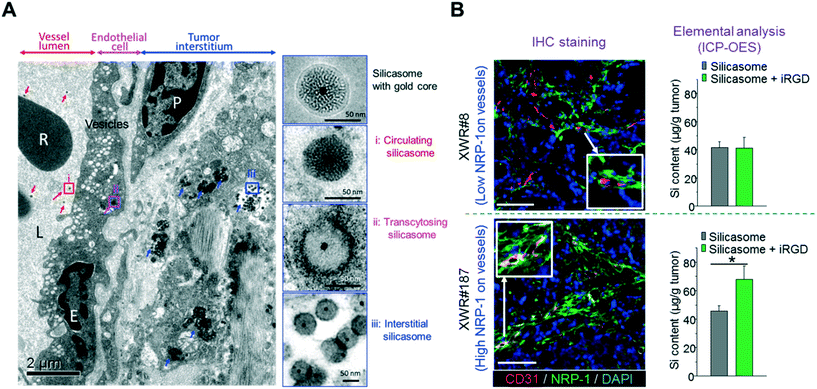 |
| | Fig. 6 (A) Ultrastructural TEM visualization of silicasome transcytosis mediated by iRGD co-administration in orthotopic pancreatic tumors. The electron micrograph shows Au-labeled silicasomes (i) in the lumen of a tumor blood vessel (red arrows), (ii) transported in the endothelial vesicles (pink arrow), and (iii) deposited in the tumor interstitium (blue arrows). High-magnification images of regions 1 through 3 are provided in the panels on the right. E, endothelial cell; P, pericyte. Scale bar: 2 μm (left panel); 50 nm (right panels). (B) iRGD-induced silicasome tumor accumulation in patient-derived xenografts. A pair of tumors (XWR#8 and XWR#187) with different levels of NRP-1 expression was selected for a biodistribution study in the absence and presence of iRGD co-administration. Multicolor IHC staining (left panel, green fluorescent antibody for NRP-1; red fluorescent antibody for CD31) demonstrated the relatively higher abundance of NRP-1 expression and higher extent of overlap with blood vessel endothelial cells for model XWR#187 than those of model XWR#8. ICP-OES measurement (right panel) showed that the uptake of silicasomes in tumors was significantly improved by iRGD co-administration in model XWR#187 but not in model XWR#8. Data represent mean ± SD. *p < 0.05, 2-tailed Student's t test. Adapted with permission from ref. 30. | |
3. Perspective and conclusion
While both the conjugation approach and co-administration approach have been used to improve tumor accumulation, only a few studies provided side-by-side comparisons in the same study. Sugahara et al. showed that the tumor accumulation of iRGD-conjugated Abraxane is slightly less than that of Abraxane co-administered with iRGD in breast and prostate cancers. The authors didn't obtain a statistical significance between the two groups.19 Recently, we showed that the co-administration approach is approximately 2.5× more effective than the conjugation approach in Kras-mutated or patient derived pancreatic cancer bearing mice, receiving IV injected silica-based nanocarriers.30 In our opinion, there are obvious advantages for the co-administration strategy from the pharmaceutical activity and nano-manufacturing perspectives. The co-administration may address a major limitation of peptide-conjugated nanocarriers, which rely on the available number of NRP-1 receptors, that is, the transport of the conjugated nanocarriers may be limited by the relatively small and finite number of target receptors on the vasculature, but separate injection of the free peptide can trigger bulk transfer of nanocarriers through the bystander (in greater number) at the tumor site.19,25 Moreover, introducing a covalently conjugated peptide may add complexity to the surface properties of nanocarriers that may lead to an undesired impact in the complicated in vivo system.64,65 It is possible that peptide conjugation, directly or indirectly, leads to particle opsonization and increased uptake by the reticuloendothelial system.66,67 The faster uptake by RES organs will result in shorter blood circulation time, ultimately leading to lower tumor accumulation.68,69 From the perspective of translational feasibility, the use of the free peptide is more practical and less expensive for clinical use compared to a conjugation process that inevitably enhances the cost and complexity of the nanocarrier.
Recently, there has been high-level coverage on the reproducibility project that aims to duplicate major experimental results in the field of cancer. One of the projects chosen, based on a prominent iRGD effect, was the duplication of the results of iRGD peptide co-administration for enhancing chemotherapy efficacy in a prostate cancer model.70 In spite of the fact that multiple international laboratories and our own study have validated this effect, a report showed that the authors executing that study failed to reproduce the iRGD co-administration results.70–72 While there can be many explanations for this discrepancy, some important possibilities include the verification of the biological activity of the iRGD peptide, as well as a demonstration of the actual presence of the NRP-1 receptor in the tumor model in order to obtain a valid comparison. In fact, our patient-derived model data suggested that the enhancing effect of the iRGD peptide could vary depending on the level of NRP-1 expression which is different from patient to patient and from one tumor model to another. All things considered, it is necessary to contemplate a personalized nano-therapeutic approach, especially in regard to pancreatic cancer (but also for other cancer types), to enhance the efficacy of cancer drugs through iRGD co-administration.
Conflicts of interest
IP developed by XL, HM and colleagues was licensed to Westwood Biosciences by The Regents of UC. HM is the co-founder in Westwood Biosciences Inc.
Acknowledgements
We would like to acknowledge the financial support from the U.S. Public Health Service Grant 1U01CA198846.
References
- D. Peer, J. M. Karp, S. Hong, O. C. Farokhzad, R. Margalit and R. Langer, Nat. Nanotechnol., 2007, 2, 751–760 CrossRef CAS PubMed.
- M. E. Davis, Z. Chen and D. M. Shin, Nat. Rev. Drug Discovery, 2008, 7, 771–782 CrossRef CAS PubMed.
- J. Wu and Z. Li, Chin. Sci. Bull., 2013, 58, 4515–4518 CrossRef.
- M. Ma, H. Chen and J. Shi, Sci. Bull., 2015, 60, 1170–1183 CrossRef CAS.
- F. Yuan, M. Dellian, D. Fukumura, M. Leunig, D. A. Berk, V. P. Torchilin and R. K. Jain, Cancer Res., 1995, 55, 3752–3756 CAS.
- H. Maeda, J. Wu, T. Sawa, Y. Matsumura and K. Hori, J. Controlled Release, 2000, 65, 271–284 CrossRef CAS PubMed.
- J. Fang, H. Nakamura and H. Maeda, Adv. Drug Delivery Rev., 2011, 63, 136–151 CrossRef CAS PubMed.
- A. Gabizon, R. Catane, B. Uziely, B. Kaufman, T. Safra, R. Cohen, F. Martin, A. Huang and Y. Barenholz, Cancer Res., 1994, 54, 987–992 CAS.
- D. D. Von Hoff, T. Ervin, F. P. Arena, E. G. Chiorean, J. Infante, M. Moore, T. Seay, S. A. Tjulandin, W. W. Ma, M. N. Saleh, M. Harris, M. Reni, S. Dowden, D. Laheru, N. Bahary, R. K. Ramanathan, J. Tabernero, M. Hidalgo, D. Goldstein, E. Van Cutsem, X. Wei, J. Iglesias and M. F. Renschler, N. Engl. J. Med., 2013, 369, 1691–1703 CrossRef CAS PubMed.
- A. Wang-Gillam, C.-P. Li, G. Bodoky, A. Dean, Y.-S. Shan, G. Jameson, T. Macarulla, K.-H. Lee, D. Cunningham, J. F. Blanc, R. A. Hubner, C.-F. Chiu, G. Schwartsmann, J. T. Siveke, F. Braiteh, V. Moyo, B. Belanger, N. Dhindsa, E. Bayever, D. D. Von Hoff and L.-T. Chen, Lancet, 2016, 387, 545–557 CrossRef CAS.
- J. D. Byrne, T. Betancourt and L. Brannon-Peppas, Adv. Drug Delivery Rev., 2008, 60, 1615–1626 CrossRef CAS PubMed.
- F. Danhier, O. Feron and V. Préat, J. Controlled Release, 2010, 148, 135–146 CrossRef CAS PubMed.
- Y. H. Bae and K. Park, J. Controlled Release, 2011, 153, 198–205 CrossRef CAS PubMed.
- S. Wilhelm, A. J. Tavares, Q. Dai, S. Ohta, J. Audet, H. F. Dvorak and W. C. W. Chan, Nat. Rev. Mater., 2016, 1, 16014 CrossRef CAS.
- S. E. McNeil, Nat. Rev. Mater., 2016, 1, 16073 CrossRef.
- D. Feng, J. A. Nagy, J. Hipp, H. F. Dvorak and A. M. Dvorak, J. Exp. Med., 1996, 183, 1981–1986 CrossRef CAS PubMed.
- K. N. Sugahara, T. Teesalu, P. P. Karmali, V. R. Kotamraju, L. Agemy, O. M. Girard, D. Hanahan, R. F. Mattrey and E. Ruoslahti, Cancer Cell, 2009, 16, 510–520 CrossRef CAS PubMed.
- E. Ruoslahti, S. N. Bhatia and M. J. Sailor, J. Cell Biol., 2010, 188, 759–768 CrossRef CAS PubMed.
- K. N. Sugahara, T. Teesalu, P. P. Karmali, V. R. Kotamraju, L. Agemy, D. R. Greenwald and E. Ruoslahti, Science, 2010, 328, 1031–1035 CrossRef CAS PubMed.
- J. Georgieva, D. Hoekstra and I. Zuhorn, Pharmaceutics, 2014, 6, 557–583 CrossRef CAS PubMed.
- S. Raha, T. Paunesku and G. Woloschak, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2011, 3, 269–281 CrossRef CAS PubMed.
- Z. Li and C. Cho, J. Transl. Med., 2012, 10, S1 CrossRef PubMed.
- J. Thundimadathil, J. Amino Acids, 2012, 2012, 1–13 CrossRef PubMed.
- R. J. Boohaker, M. W. Lee, P. Vishnubhotla, J. M. Perez and A. R. Khaled, Curr. Med. Chem., 2012, 19, 3794–3804 CrossRef CAS PubMed.
- E. Ruoslahti, Adv. Mater., 2012, 24, 3747–3756 CrossRef CAS PubMed.
- E. Ruoslahti, Adv. Drug Delivery Rev., 2017, 110–111, 3–12 CrossRef CAS PubMed.
- E. Ruoslahti, J. Clin. Invest., 2017, 127, 1622–1624 CrossRef PubMed.
- E. Ruoslahti and M. Pierschbacher, Science, 1987, 238, 491–497 CAS.
- D. Hanahan and R. A. Weinberg, Cell, 2000, 100, 57–70 CrossRef CAS PubMed.
- X. Liu, P. Lin, I. Perrett, J. Lin, Y.-P. Liao, C. H. Chang, J. Jiang, N. Wu, T. Donahue, Z. Wainberg, A. E. Nel and H. Meng, J. Clin. Invest., 2017, 127, 2007–2018 Search PubMed.
- Y. Liu, M. Ji, M. K. Wong, K.-I. Joo and P. Wang, BioMed Res. Int., 2013, 2013, 1–11 Search PubMed.
- X. Zhang, W.-Q. Yu, L.-M. Luo Zhang, D. Li Song, Ren Du, W.-L. Lu Huang and Q. Zhang, Int. J. Nanomed., 2013, 8, 2473–2485 CrossRef PubMed.
- W. Dai, Y. Fan, H. Zhang, X. Wang, Q. Zhang and X. Wang, Drug Delivery, 2015, 22, 10–20 CrossRef CAS PubMed.
- Y. Liu, D. Zhang, Z.-Y. Qiao, G.-B. Qi, X.-J. Liang, X.-G. Chen and H. Wang, Adv. Mater., 2015, 27, 5034–5042 CrossRef CAS PubMed.
- X. Song, Z. Wan, T. Chen, Y. Fu, K. Jiang, X. Yi, H. Ke, J. Dong, L. Yang, L. Li, X. Sun, T. Gong and Z. Zhang, Biomaterials, 2016, 108, 44–56 CrossRef CAS PubMed.
- F. Yan, H. Wu, H. Liu, Z. Deng, H. Liu, W. Duan, X. Liu and H. Zheng, J. Controlled Release, 2016, 224, 217–228 CrossRef CAS PubMed.
- Z. Zhu, C. Xie, Q. Liu, X. Zhen, X. Zheng, W. Wu, R. Li, Y. Ding, X. Jiang and B. Liu, Biomaterials, 2011, 32, 9525–9535 CrossRef CAS PubMed.
- J. Zhou, T. R. Patel, M. Fu, J. P. Bertram and W. M. Saltzman, Biomaterials, 2012, 33, 583–591 CrossRef CAS PubMed.
- J. Shen, Q. Meng, H. Sui, Q. Yin, Z. Zhang, H. Yu and Y. Li, Mol. Pharmaceutics, 2014, 11, 2579–2591 CrossRef CAS PubMed.
- X. Nie, J. Zhang, Q. Xu, X. Liu, Y. Li, Y. Wu and C. Chen, J. Mater. Chem. B, 2014, 2, 3232–3242 RSC.
- J. Zhang, J. Hu, H. F. Chan, M. Skibba, G. Liang and M. Chen, Nanomedicine, 2016, 12, 1303–1311 CrossRef CAS PubMed.
- J. Wang, H. Wang, J. Li, Z. Liu, H. Xie, X. Wei, D. Lu, R. Zhuang, X. Xu and S. Zheng, ACS Appl. Mater. Interfaces, 2016, 8, 19228–19237 CAS.
- X. Xu, J. Wu, Y. Liu, M. Yu, L. Zhao, X. Zhu, S. Bhasin, Q. Li, E. Ha, J. Shi and O. C. Farokhzad, Angew. Chem., Int. Ed., 2016, 55, 7091–7094 CrossRef CAS PubMed.
- S. Su, H. Wang, X. Liu, Y. Wu and G. Nie, Biomaterials, 2013, 34, 3523–3533 CrossRef CAS PubMed.
- L. Simón-Gracia, H. Hunt, P. Scodeller, J. Gaitzsch, V. R. Kotamraju, K. N. Sugahara, O. Tammik, E. Ruoslahti, G. Battaglia and T. Teesalu, Biomaterials, 2016, 104, 247–257 CrossRef PubMed.
- Z. Jin, Y. Lv, H. Cao, J. Yao, J. Zhou, W. He and L. Yin, Sci. Rep., 2016, 6, 27559 CrossRef CAS PubMed.
- Y. Tian, S. Li, J. Song, T. Ji, M. Zhu, G. J. Anderson, J. Wei and G. Nie, Biomaterials, 2014, 35, 2383–2390 CrossRef CAS PubMed.
- M. Murata, S. Narahara, T. Kawano, N. Hamano, J. S. Piao, J.-H. Kang, K. Ohuchida, T. Murakami and M. Hashizume, Mol. Pharmaceutics, 2015, 12, 1422–1430 CrossRef CAS PubMed.
- C.-F. Wang, E. M. Mäkilä, M. H. Kaasalainen, D. Liu, M. P. Sarparanta, A. J. Airaksinen, J. J. Salonen, J. T. Hirvonen and H. A. Santos, Biomaterials, 2014, 35, 1257–1266 CrossRef CAS PubMed.
- B. P. Shah, N. Pasquale, G. De, T. Tan, J. Ma and K.-B. Lee, ACS Nano, 2014, 8, 9379–9387 CrossRef CAS PubMed.
- C.-F. Wang, M. P. Sarparanta, E. M. Mäkilä, M. L. K. Hyvönen, P. M. Laakkonen, J. J. Salonen, J. T. Hirvonen, A. J. Airaksinen and H. A. Santos, Biomaterials, 2015, 48, 108–118 CrossRef CAS PubMed.
- A. M. Hamilton, S. Aidoudi-Ahmed, S. Sharma, V. R. Kotamraju, P. J. Foster, K. N. Sugahara, E. Ruoslahti and B. K. Rutt, J. Mol. Med., 2015, 93, 991–1001 CrossRef CAS PubMed.
- X. Li, M. Wu, L. Pan and J. Shi, Int. J. Nanomed., 2016, 11, 93–105 CAS.
- K. Wang, X. Zhang, Y. Liu, C. Liu, B. Jiang and Y. Jiang, Biomaterials, 2014, 35, 8735–8747 CrossRef CAS PubMed.
- L. Agemy, D. Friedmann-Morvinski, V. R. Kotamraju, L. Roth, K. N. Sugahara, O. M. Girard, R. F. Mattrey, I. M. Verma and E. Ruoslahti, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 17450–17455 CrossRef CAS PubMed.
- W. Song, M. Li, Z. Tang, Q. Li, Y. Yang, H. Liu, T. Duan, H. Hong and X. Chen, Macromol. Biosci., 2012, 12, 1514–1523 CrossRef CAS PubMed.
- G. Gu, X. Gao, Q. Hu, T. Kang, Z. Liu, M. Jiang, D. Miao, Q. Song, L. Yao, Y. Tu, Z. Pang, H. Chen, X. Jiang and J. Chen, Biomaterials, 2013, 34, 5138–5148 CrossRef CAS PubMed.
- H. D. Zuo, W. W. Yao, T. W. Chen, J. Zhu, J. J. Zhang, Y. Pu, G. Liu and X. M. Zhang, BioMed Res. Int., 2014, 2014, 1–8 Search PubMed.
- X. Cun, J. Chen, S. Ruan, L. Zhang, J. Wan, Q. He and H. Gao, ACS Appl. Mater. Interfaces, 2015, 7, 27458–27466 CAS.
- W.-Q. Zhang, K.-F. Yu, T. Zhong, L.-M. Luo, R. Du, W. Ren, D. Huang, P. Song, D. Li, Y. Zhao, C. Wang and X. Zhang, J. Drug Targeting, 2015, 23, 897–909 CrossRef CAS PubMed.
- J. Su, H. Sun, Q. Meng, Q. Yin, S. Tang, P. Zhang, Y. Chen, Z. Zhang, H. Yu and Y. Li, Adv. Funct. Mater., 2016, 26, 1243–1252 CrossRef CAS.
- H. Meng, M. Wang, H. Liu, X. Liu, A. Situ, B. Wu, Z. Ji, C. H. Chang and A. E. Nel, ACS Nano, 2015, 9, 3540–3557 CrossRef CAS PubMed.
- X. Liu, A. Situ, Y. Kang, K. R. Villabroza, Y. Liao, C. H. Chang, T. Donahue, A. E. Nel and H. Meng, ACS Nano, 2016, 10, 2702–2715 CrossRef CAS PubMed.
- X. Liu, H. Li, Q. Jin and J. Ji, Small, 2014, 10, 4230–4242 CAS.
- F. Zhao, H. Meng, L. Yan, B. Wang and Y. Zhao, Sci. Bull., 2015, 60, 3–20 CrossRef CAS.
- X. Liu, Q. Jin, Y. Ji and J. Ji, J. Mater. Chem., 2012, 22, 1916–1927 RSC.
- X. Liu, H. Zhu, Q. Jin, W. Zhou, V. L. Colvin and J. Ji, Adv. Healthcare Mater., 2013, 2, 352–360 CrossRef CAS PubMed.
- X. Liu, Y. Chen, H. Li, N. Huang, Q. Jin, K. Ren and J. Ji, ACS Nano, 2013, 7, 6244–6257 CrossRef CAS PubMed.
- X. Liu, H. Li, Y. Chen, Q. Jin, K. Ren and J. Ji, Adv. Healthcare Mater., 2014, 3, 1439–1447 CrossRef CAS PubMed.
- C. Mantis, I. Kandela and F. Aird, Reproducibility Project: Cancer Biology, eLife, 2017, 6, e17584 Search PubMed.
- J. Kaiser, Science, 2017, 355, 234–235 CrossRef CAS PubMed.
- M. Baker and E. Dolgin, Nature, 2017, 541, 269–270 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Jinhong
Jiang
a,
Ying
Ji
a,
Jianqin
Lu
a,
Ryan
Chan
a and
Huan
Meng
*ab
a,
Jinhong
Jiang
a,
Ying
Ji
a,
Jianqin
Lu
a,
Ryan
Chan
a and
Huan
Meng
*ab