
Insights from molecular dynamics simulations for computational protein design
Matthew Carter
Childers
 and
Valerie
Daggett
*
and
Valerie
Daggett
*
Department of Bioengineering, University of Washington, Seattle, WA 98195-5013, USA. E-mail: daggett@uw.edu; Fax: +1 206 685 3300; Tel: +1 206 685 7420
First published on 9th January 2017
Abstract
A grand challenge in the field of structural biology is to design and engineer proteins that exhibit targeted functions. Although much success on this front has been achieved, design success rates remain low, an ever-present reminder of our limited understanding of the relationship between amino acid sequences and the structures they adopt. In addition to experimental techniques and rational design strategies, computational methods have been employed to aid in the design and engineering of proteins. Molecular dynamics (MD) is one such method that simulates the motions of proteins according to classical dynamics. Here, we review how insights into protein dynamics derived from MD simulations have influenced the design of proteins. One of the greatest strengths of MD is its capacity to reveal information beyond what is available in the static structures deposited in the Protein Data Bank. In this regard simulations can be used to directly guide protein design by providing atomistic details of the dynamic molecular interactions contributing to protein stability and function. MD simulations can also be used as a virtual screening tool to rank, select, identify, and assess potential designs. MD is uniquely poised to inform protein design efforts where the application requires realistic models of protein dynamics and atomic level descriptions of the relationship between dynamics and function. Here, we review cases where MD simulations were used to modulate protein stability and protein function by providing information regarding the conformation(s), conformational transitions, interactions, and dynamics that govern stability and function. In addition, we discuss cases where conformations from protein folding/unfolding simulations have been exploited for protein design, yielding novel outcomes that could not be obtained from static structures.
Matthew received his B.Sc. in Biomedical Engineering from the School of Engineering and Applied Science at the University of Virginia in 2013. Afterwards, he began pursuing a Ph.D. in Bioengineering at the University of Washington under the mentorship of Dr. Valerie Daggett. |
Valerie Daggett received her Ph.D. in Pharmaceutical Chemistry from the University of California, San Francisco with Drs. Irwin Kuntz and Peter Kollman. Afterwards, she completed a postdoctoral fellowship with Dr. Michael Levitt at Stanford University. She joined the faculty at the University of Washington in 1993 where she is a Professor of Bioengineering, Biochemistry, Chemical Engineering, and Biomedical and Health Informatics. |
Design, System, ApplicationKnowledge of the relationship between protein sequence, structure, dynamics, and function has resulted in the design of novel proteins and the optimization of existing ones. Dynamics often underlie the molecular mechanisms of protein stability and function. To this end, methods for accurately simulating the dynamics of proteins represent opportunities to improve protein designs. On such method, molecular dynamics (MD), uses physics-based energy functions and explicit representations of atomic systems to model protein dynamics. Such simulations can guide protein design efforts in several ways. First, by providing atomistic details of the molecular interactions that contribute to protein stability or protein function, simulations can directly guide the rational design of proteins. Second, by monitoring specific behaviors over the course of many simulations, MD can evaluate designed proteins to identify promising candidates for further study. In addition MD can provide information about target structures or properties unobtainable from static native structures. Finally, MD can be used to assess and screen computational designs. The explicit inclusion of protein dynamics in design strategies has the capacity to improve protein design while simultaneously contributing to our general understanding of the relationship between sequence, structure, dynamics, and function. |
1. Introduction
Protein design is the creation of proteins either from scratch (de novo design) or through the modification of pre-existing structures (protein redesign). Protein design stands in contrast to experimental techniques, such as directed evolution, that generate new proteins by mimicking natural processes. Instead, protein design relies on a combination of chemical and biological intuition; known relationships between sequence, structure, and function; and computational resources to craft proteins. Protein design serves three main purposes. First, it can lead to the generation of proteins that have real-world applications, such as the design of more efficient enzymes to catalyze reactions, or the design of biotherapeutics. Second, protein design serves as a test of our understanding of protein chemistry and protein folding. Third, in the act of designing new proteins, we gain new insights into the determinants of protein structure and function.Early rationally designed proteins were pioneered in the 1970's by Gutte and coworkers, and include truncated bovine ribonucleases.1,2 These designs relied primarily on chemical and biological intuition. Later, as more protein crystal structures became available, computational predictions of secondary structure propensity3 paved the way for the design of elements of protein structure4 and topologically simple proteins.5–7 Deciphering relationships such as sequence composition, hydrophobic–hydrophilic patterning, and geometric constraints allowed for the design of more complex structures such as Felix8 and Betabellin.9 Richardson and Richardson provide an excellent review of the early days of protein design.10 The 1990s brought increasingly faster computers, enabling the development of more computational tools for protein design, such as side chain rotamer libraries.11,12 Over the next decade as components of the protein design toolbox were developed, refined, and integrated, we saw the first de novo design,13 the design of a fold not seen in nature,14 and the design of enzymes to catalyze reactions.15
In recent years, routine access to high performance computing resources, accompanied by refined protein design methodologies, has allowed for the design of increasingly sophisticated proteins with diverse topologies and functions. Other layers of complexity have been added to protein design strategies, including the dynamical nature of proteins, their binding partners, and the interplay between proteins and their environments. Static representations of proteins often mask the critical relationship between dynamics and function. Use of rotamer libraries was an early example of incorporating dynamics into design: allowing residues to sample multiple rotameric states allows for a greater number of potential sequences.
Protein dynamics are present over different spatial and temporal scales: from local fluctuations around equilibrium conformations to large-scale conformational changes upon binding. In most cases, dynamics are intimately connected to protein function. These functional dynamics range from large-scale conformational changes, such as the power stroke mechanism of myosin in muscle contraction16 to small-scale rearrangements of active site conformations mediated by a network of residues in cyclophilin A.17,18 Therefore it is beneficial to incorporate models of protein dynamics and flexibility in designs. When considering more sophisticated designs, especially designs where function is a target, flexibility and dynamics become even more important.
One way to integrate dynamics into protein design is to capitalize on insights from molecular dynamics (MD) simulations. Molecular dynamics uses a physics-based potential energy function, or force field, to simulate protein dynamics as a function of time according to classical Newtonian mechanics.19 While biophysical techniques, such as NMR spectroscopy, can yield insights into protein dynamics,20 MD is unparalleled with regard to the spatial and temporal resolution that it can provide, allowing us to see proteins in action, at least at fast time scales.21,22 MD has the power not only to identify functionally relevant conformations, which may be ‘hidden’ to experimental techniques, but it can also provide the details of transitions between these conformations.
No experimental technique is without error or limitation, and molecular simulations are no exception.23 There are three primary sources of error in MD simulations that dictate the extent to which a simulation correctly samples from a thermodynamic ensemble. The first source of error is the accuracy of the force field, an equation used to derive the forces interacting on all of the atoms in a system.24 Although force fields have been parameterized empirically, no force field can be truly accurate. However recent force field validations of model proteins have shown that contemporary force fields can reproduce experimental observables25–31 and the studies below demonstrate successful MD-derived predictions validated through protein design. The second main source of error is the extent of conformational sampling obtained from the simulation, or the extent to which simulations sample the heterogeneous distribution of conformational states, which is due to constraints on computing time and methods that limit the flexibility and sampling of the protein, as described previously.32 Determining the appropriate length of a simulation is a trade off between economy (shorter simulations) and sampling (longer simulations), constrained by the timescale(s) of the dynamics in question.33,34 However, longer simulations are not always better. Numerical errors such as round-off errors and truncation errors propagate as the simulation is extended as a result of numerical integration.19,35,36 Specialized integration algorithms and other computational methods for performing simulations have been developed to mitigate numerical error as much as possible.37,38 Although many of these approaches merely mask the problems associated with the use of physically unrealistic procedures and scaling of properties that yield discontinuous pathways.32,39
As evidenced by the validation of long simulations of BPTI40 and ubiquitin41 the combined errors from force field accuracy, conformational sampling, and numerical integration do not necessarily corrupt the simulated dynamics, but they don't necessarily provide more information beyond what is obtained in shorter simulations. Overall, advances in force field development,26–28 software development,42 parallelization schemes,43 and high performance computing resources47 have paved the way for simulations into the millisecond time scale and simulations are getting longer all of the time. Experiment is the ultimate arbiter of the accuracy and reliability of MD simulations. A thorough discussion of error and validation of MD simulations is beyond the context of this review; however, readers are referred to other theoretical and practical discussions of simulation validation.44–46 As simulated timescales approach those of experiment and biological relevance, the community can better assess the correspondence of simulation and experiment, and the incorporation of MD-derived predictions in protein design represents another important way to validate the methods.
The validity of protein dynamics derived from MD simulations can be assessed through its use in protein design and MD can inform protein design both directly and indirectly. Direct applications of MD include: the use of insights gained from MD to inform rational design and to generate testable hypotheses that are confirmed through design. Indirect applications include the use of MD: to rank, screen, and evaluate designs; to obtain general insights into protein dynamics; and to explain relationships between protein dynamics and the molecular mechanisms of protein stabilization. Once the mechanisms are well understood, they can be applied to other designs. Here, we review studies in which conventional MD simulations have been used either directly or indirectly in protein design. We cover three broad areas in which MD simulations have been employed: modulating protein stability, engineering functional regions, and engineering folding/unfolding pathways (Fig. 1).
 | ||
| Fig. 1 Applications of molecular dynamics for protein design. Molecular dynamics simulations can be used to (A) design stable protein variants, (B) engineer functional regions, or (C) provide insights from protein unfolding/folding pathways. | ||
2. Modulating protein stability
The use of natural proteins in biomedical and industrial applications is often limited due to the strict determinants of protein stability.47–49 Protein stability is governed by the net sum of forces that determine whether a protein will maintain a native, folded conformation or unfold.50 Technological applications of proteins may require exposure of the proteins to harsh environments or that the proteins remain stable for extended periods of time. Evolutionary pressure has driven natural proteins toward optimal activity, not stability per se, in their native environments. Thus, there have been many examples of proteins that have been mutated leading to increased stability, sometimes at the expense of function.51–56 Given that protein stability is not necessarily optimized for native conditions, it is unlikely that proteins are optimized for medical and industrial applications.57 For example, in the food industry, reactions involving enzymes are often carried out at high temperature to increase the reaction rate and reduce the potential for contamination.58 However, at elevated temperatures, proteins are susceptible to thermal inactivation due to unfolding or irreversible structural damage.59 Just as evolutionary pressure has given rise to proteins with optimal function in their native environments, it is possible to design protein variants that remain stable in alternative environments. Theoretical, experimental, and computational studies have identified strategies for designing stable protein variants such as: reducing mobility in flexible regions, introduction of disulfide bonds,60 introduction of salt bridges,61 and chemical modification of protein structures.62 However these design strategies are not guaranteed to always work, and they may lead to unexpected results.56,63 Instead, judicious selection of design strategies is necessary.Proteins are inherently dynamic molecules that sample various conformational states beyond those presented in the crystal structures in the Protein Data Bank (PDB).64 The ensembles of conformations proteins explore, the transitions between these conformational substates, and the response of these dynamic fluctuations to environmental conditions are coupled to protein stability. In this regard, MD simulations can provide access to atomic details of protein dynamics to aid in the design of stable protein variants.
One of the most common ways to stabilize proteins is to introduce a disulfide bond in the hope of locking down the structure. Increases in protein stability due to the introduction of a disulfide bond have been attributed to lowered entropy of the unfolded state,65 enthalpic changes to the unfolded state,66 and kinetic stabilization of the folded state.67 However the introduction of a disulfide can reduce protein stability due to poor packing and flexibility68,69 or stabilization of the denatured state.60 Thus, it is crucial to identify the optimal sites for the introduction of disulfide bonds and consider the effects of the disulfide bond on nonnative states. One early example focused on the incorporation of a disulfide bond into haloalkane dehalogenase (DhlA) from Xanthobacter autotrophicus, which catalyzes the hydrolytic dehalogenation of small, halogenated alkanes. Pikkemaat et al. used MD simulations of DhlA to identify flexible regions in DhlA that could be stabilized by a disulfide bond.70 Simulations performed at 298 K revealed a flexible region between residues 180 and 210. This region was classified as inflexible by the crystallographic β-factors. Pikkemaat et al.70 then used the SSBOND71 program to identify potential sites for the introduction of a disulfide bond. By cross-referencing sites identified by SSBOND with flexible regions identified from MD simulations, the authors selected residue positions 16 and 201 for the incorporation of a disulfide. Experimental studies followed. The residual activities of the WT protein and designed variants were monitored as a function of temperature and urea. Analogous to the melting temperature, Tm, denaturation transition values were defined as the temperature at which residual activity decreased to 50% of the initial value. The denaturation transition temperatures for the WT, oxidized mutant, and reduced mutant were, respectively, 47.5 °C, 52.7 °C, and 39.0 °C. The design was successful, leading to a variant with an increased activity-temperature profile, but introduction of the (reduced) Cys residues themselves was destabilizing. This study demonstrates the capacity of MD to aid in the rational selection of positions for the introduction of disulfide bonds into proteins. This approach improves on the prediction of amino acid pairs for the introduction of a disulfide bond by factoring in protein flexibility and conformational heterogeneity within the native state and has been used recently by others.72
2.1. Stability in harsh environments
The most common method for designing a stable protein variant using data obtained from MD simulations is known as the rigidification of flexible sites.73 MD is a convenient way to find such flexible sites, as mentioned above. Simulations at elevated temperatures, initially performed by Daggett and Levitt, were first used to study the mechanisms of protein folding by simulating unfolding pathways,74,75 after ensuring that the force field and methods were valid at high temperature.39,76 This approach was subsequently validated through extensive comparisons with experiment, including φ-values, pioneered by Fersht and coworkers.77,78 High temperature unfolding simulations methods have since been employed to design thermostable proteins. The method utilizes MD simulations to directly identify regions of the protein that are susceptible to unfolding by performing simulations at elevated temperatures (Fig. 2). Unstable regions are then redesigned through rational design or other computational algorithms, and MD is typically used to test the designs (Fig. 2, indirect application).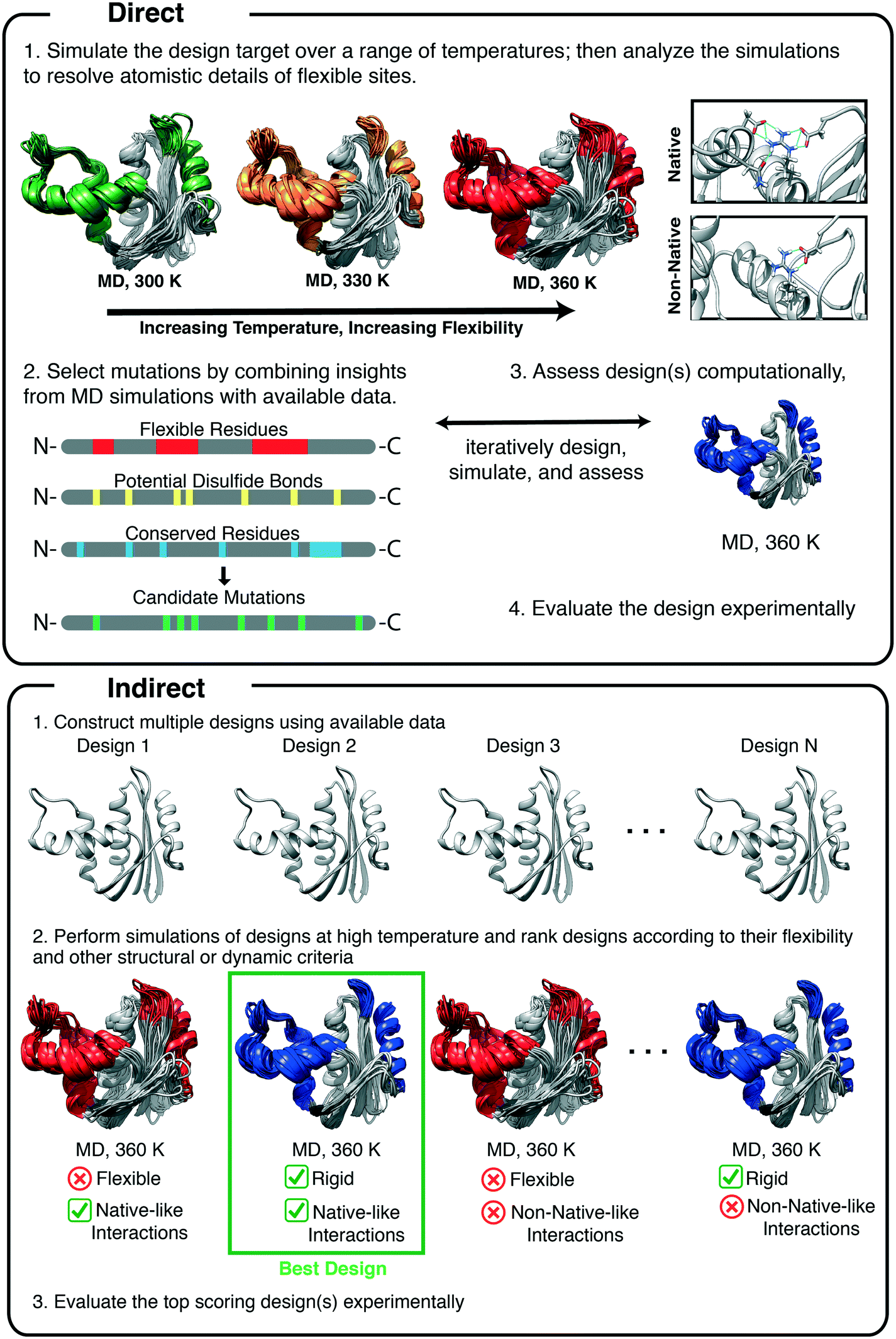 | ||
| Fig. 2 Overview of approaches to rigidify flexible regions of proteins. The rigidification of flexible sites is a common strategy for designing thermostable variants with MD simulations. In the direct application of the method, simulations are performed at various temperatures to identify regions of the protein that become flexible at high temperature. Interactions that underlie the increased flexibility at high temperature are identified by scrutinizing the trajectories. Then, proteins are designed (rationally or using other techniques) and simulated at high temperature to confirm that the designed variants are less flexible. In the indirect application of the method, high temperature simulations of multiple designs are performed and the top performing designs are selected according to their satisfaction of pre-determined criteria such as the retention of native-like contacts or their flexibility at high temperature. | ||
These methods have been applied in several studies of xylanases, which are a class of enzymes that degrade the polysaccharide β-1,4-xylan into xylose.79 They have numerous industrial applications in the pulp, paper, and food industries;80 however, they are inactivated at high temperature. Xylanase variants that are stable at high temperature are desired as as industrial reactions involving xylanases are often performed at elevated temperatures, which allows reactions to proceed quickly and prevents contamination by microbes.48 Xylanases are grouped into families based on their primary and tertiary structures.81 Family 11 xylanases have a single α-helix and two extended beta sheets, forming a jellyroll fold, and they have been the subject of numerous studies of protein thermostabilization (Fig. 3).
 | ||
| Fig. 3 Molecular dynamics derived insights for designing stable xylanase variants. (A) The structure of wild type xylanase from Bacillus circulans is shown, colored according to the four structural regions that resemble a hand. The catalytic residues are shown as balls and sticks. Xylanase has been the subject of numerous protein design studies. (B) [Direct use] molecular dynamics simulations may be used to directly inform rational design efforts by predicting regions of the protein that become unstructured under destabilizing conditions, such as high temperature or low pH. The final design of Yoo and coworkers is shown in which MD simulations were used to inform the redesign of a flexible loop.82,84 (C) [Indirect use] MD simulations may be used to generate rules for protein design. For example, the designed structure of Alponti et al. is shown, in which MD simulations of a thermophilic and mesophilic xylanase were compared to understand structural determinants of xylanase thermostabilization.92 (D) [Indirect use] MD simulations may also be used to rationalize the mechanism of protein stabilization and in so doing, provide insights for optimizing designs. For example, shown is the final design of Fonseca-Maldonaldo et al. in which MD simulations of various glycosylation states of xylanase were used to rationalize the relationship between glycosylation and xylanase stability. | ||
To design a thermostable variant of GH11 xylanase from Bacillus circulans, Yoo and coworkers first redesigned a flexible surface site and then refined their design using MD simulations.82 In the first generation design, a flexible surface cavity was identified by the FRODA83 webserver. Stabilization of this flexible cavity with three mutations (F48Y/T50V/T147L) resulted in a 15-fold increase in thermostability, as measured by t1/2 of thermal inactivation in functional assays, and 1.3-fold increase in catalytic efficiency.82 In a follow-up study, Yoo and coworkers used MD simulations to improve the design by optimizing interactions made by the previously mutated residues84 (Fig. 3B). MD simulations performed at 300 and 330 K showed that residues N52 and N151 became flexible at high temperature. The RosettaDesign85 server was used to predict stabilizing mutations for the flexible residues (N52 and N151) and others that line the redesigned cavity. Five mutations were predicted: I51V, N52Y, T143A, I144V, and T145D to improve stability. Experimental evaluation of single mutant designs revealed that the N52Y mutant had higher residual activity and a longer half-life than the first generation design. The F48Y/T50V/N52Y/T147L quadruple mutant had even higher catalytic efficiency, melting temperature, and resistance to heat inactivation. The increase in thermostability caused by the N52Y mutation was attributed to the formation of a hydrophobic cluster and an aromatic–aromatic interaction between N52Y and W185. Because residues N52 and W185 are conserved in xylanases from multiple species, the authors suggest that the N52Y mutation could be adapted to stabilize homologous xylanases. MD simulations were critical in the identification of the N52Y mutation. Although coarse-grain models of protein flexibility can yield valuable insights, MD provides more detailed descriptions of protein flexibility as well as the details of the interactions that underlie said flexibility.
Another strategy for designing thermostable proteins incorporates insights from the properties of proteins derived from thermophilic relatives. In a survey of the PDB, Argos et al. investigated thermophile/mesophile counterparts from 16 protein folds and concluded that thermophilic proteins are stabilized by an increased number of hydrogen bonds, salt bridges, and surface charges.86 MD simulations may also be used to infer general principles for protein design. One such application is to compare and contrast the dynamics and stabilities of homologous proteins from mesophilic and thermophilic organisms. Studies from our lab87 and others88,89 have suggested that thermophilic proteins dynamically adapt to function at higher temperatures due in part to the increased number of polar residues and salt bridges relative to their mesophilic counterparts. Although broad comparisons of thermophilic and mesophilic counterparts can yield general insights into protein design, the true challenge is to decipher the optimal residues to mutate that will improve protein stability without adversely affecting the structure, dynamics, or function of the particular protein in question.
Once stabilizing mechanisms of thermophilic proteins have been identified, it is possible to stabilize their mesophilic counterparts by grafting thermophilic sequences into mesophilic scaffolds. For example, Vieira and Degreve performed MD simulations of a thermophilic xylanase from Thermomyces lanuginosus (TLX) and a mesophilic xylanase from Bacillus circulans (BCX).90 They used MD simulations to compare residue flexibility and salt bridge occupancy between TLX and BCX at temperatures between 25° and 70 °C. TLX has a larger number of intramolecular interactions among residues in the ‘fingers’ domain as well as more favorable interactions between side chains and solvent. Building off of the results from Vieira et al.,90,91 Alponti et al. grafted charges from a thermophilic GH11 xylanase into a mesophilic scaffold92 (Fig. 3C). In total, five mutations were made in the ‘fingers’ domain: S22E, S27E, N32D, N54E, N181R. These five single amino acid mutants were used in an experimental combinatorial library, yielding the S22E/N32D mutant, which displayed higher stability than the WT and single mutant designs. The S22E/N32D mutant had 1.9 fold higher specific activity (WT: 25.0 U mg−1vs. S22E/N32D: 46.9 U mg−1 where U = activity units), with a t1/2 of thermal inactivation 6.3 times that of WT (WT: 13.3 min vs. S22E/N32D: 84.8 min). Comparisons of thermophilic and mesophilic homologue dynamics also led to the design of a thermostable variant of nitrile hydratase (NHase).93 By grafting thermophilic sequences into a mesophilic homologue, the resulting designed protein had enhanced thermal stability relative to WT.
Successful design of more stable proteins is not limited to enzymes. Repeat proteins, proteins with modular scaffolds, consist of repeated elements of a fundamental building block of structure. Ankyrin repeat proteins consist of repeats of a 33-residue helix–loop–helix–loop motif.94,95 The development of highly stable variants of repeat proteins was accomplished using the consensus design approach,96 by which homologous sequences were compared to determine the optimal residues at each position in the repeat unit.97,98 To improve the thermostability of designed ankyrin repeats, Caflisch and co-workers combined equilibrium unfolding experiments and high temperature MD simulations to investigate the effects of the number of repeats on stability.99 They modeled ankyrin repeats of the form NIxC, where Ix refers to the number of internal consensus repeats, and N and C refer to the N- and C-terminal capping repeats, respectively. Native state simulations (300 K) of NI1C, NI2C, and NI3C showed that a network of salt bridges formed at inter-repeat interfaces stabilized the proteins. High temperature unfolding simulations (400 K) showed that NI1C and NI2C unfolded while NI3C retained a native-like conformation, in agreement with experiments showing a correlation between thermal stability and the number of repeat units. Additional unfolding experiments showed that NI4 is more stable than NI4C and has two-state folding characteristics, but is less soluble and more prone to aggregate. This suggested an opportunity to redesign the internal I:C interface to strengthen inter-repeat interactions while simultaneously providing the aggregation-disfavoring properties of the cap. In total, six redesigns of NI1C were made, each with between two and 11 mutations grouped into three categories: mutations to redesign the hydrophobic interface, mutations to facilitate hydrogen bond formation, or mutations to elongate the second helix of the C-terminal. Subsequent high temperature MD simulations of the designs predicted that the hydrophobic mutations did not significantly contribute to protein stabilization while the hydrophilic and helix elongation mutations did. Thermal unfolding experiments confirmed that the introductions of hydrogen bond-promoting mutations and helix-elongating mutations stabilized the protein, as evidenced by an increase in the Tm from 60 °C for WT to 77 °C for one of the designs. MD simulations were essential in the identification of residues at the interface where mutation would be most beneficial.
To design proteins for increased stability in other environments, the general strategy remains the same; however, instead of performing simulations at elevated temperatures, the simulation conditions are set to mimic the environment where enhanced stability is desired. For example, to design pH stable protein variants, simulations are performed at different pH levels by modifying the protonation states of titratable residues. For example, armadillo repeat proteins were designed using a consensus method, but NMR revealed unfavorable interactions at neutral pH that were absent at high pH. A pH stable variant was designed by mutating Lys residues to Gln based on MD simulations.100
Mutations involving titratable side chains can modulate pH-dependent protein stability.101 Penicillin acylase from Escherichia coli and other members of the Ntn-hydrolase superfamily102 cleave amide bonds.103,104 Members of this family have diverse pH stability profiles and E. coli penicillin acylase becomes inactive at acidic or alkaline pH.105 Suplatov et al. combined sequence analysis with constant pH MD simulations to design a pH stable variant suitable for industrial applications.105 At neutral pH two residues in the B1 domain, Glu482 and Asp484, share a proton and coordinate a hydrogen bond network that stabilizes the active site. At alkaline pH, when both residues are negatively charged the hydrogen bond network collapsed. The authors attributed the pH-mediated inactivation of the enzyme to the loss of the active site conformation. They then took this a step further and aligned the sequence of penicillin acylase from E. coli to penicillin acylase from Alcaligenes faecalis, which does not become inactive at alkaline pH. A potential mutation, D484N, was suggested by cross-referencing the MD-derived mechanism of instability at alkaline pH with potential mutations identified by inferring mutability from the sequence alignment. Simulations of the D484N of E. coli protein at neutral pH were comparable to those of the WT but the mutant was stabilized at alkaline pH, as predicted. Experimental follow-up confirmed that the inactivation rate decreased 9 fold for the mutant relative to WT.
Simulations of nonnative environments can also be combined with elevated temperature simulations. For example, to engineer a pH stable variant of the sweet protein MNEI, Leone and Picone performed MD simulations at native and elevated temperature and for various protonation states of Glu23.106 When E23 was protonated, the simulations remained stable over a range of temperatures. Protonated E23 contributed to overall stability through formation of a hydrogen bond network that was disrupted when it was deprotonated. A pH-stable variant was designed by mutating E23 to Q, and the melting temperature increased by ∼10 °C at both neutral and alkaline pH without altering stability at low pH. With careful modeling of the desired conditions and the selection of appropriate controls, MD is able to simulate many environments. Once potential design sites have been identified from MD simulations, it is possible to combine the results with a long list of established protein design strategies, including performing simulations at high temperature.
2.2. Post-translational modifications and non-standard amino acids
Post-translational modifications (PTMs) and non-standard amino acids represent another route to stabilize proteins and regulate function.107 Of the over 25 distinct PTMs that have been experimentally identified, phosphorylation, acetylation, and N-linked glycosylation are the most frequent, and extensive databases allow for routine access to statistics and properties of PTMs.108–110 It is possible to introduce or remove PTMs into proteins through the modulation of sequence motifs (e.g. N-X-T/S for N-linked glycosylation).More than half of all proteins are predicted to be glycosylated.111 Protein glycoengineering is accomplished by the introduction or removal of glycosylation sequences at specific sites in proteins.112,113 Glycosylation can be incorporated into protein design to confer advantageous properties, such as increases in in vivo half-life and stability, burial of hydrophobic batches, decreases in aggregation, and increasing solubility.110 In pharmaceutical formulations of interferon-β, solvent-exposed hydrophobic residues drive the formation of protein aggregates, rendering the protein non-functional. The majority of designed proteins have focused on making mutations to favor a specific conformation or function, i.e. positive design; however, in some cases it is necessary to make designs that prevent access to particular interactions, conformational states or functions, i.e. negative design.114,115 To reduce the aggregation propensity of interferon-β, Samoudi et al. used negative design to create hyper-glycosylated variants of interferon-β. MD simulations were used to evaluate the designs by comparing the structure, dynamics, residue flexibility, and solvent flexibility of the designed glycosylation sites to WT.116 Glycosylation sites were selected by combining web server predictions, a literature search, and structure-based design from available crystal structures. By comparing the flexibility of designed, glycosylated proteins with wild type (WT), two models were selected for experimental characterization, which confirmed hyper-glycosylation and retention of function. Negative design can also be used to avoid conformations and functions such as off-pathway folding intermediates, conformational substates of the native ensemble, protein–protein interactions, protein–ligand interactions, aggregation-prone states, and pathological states such as amyloid. High temperature MD simulations have also been used to generate reference structures for ‘negative states’.117
In an example of the use of MD in this area, Fonseca-Maldonaldo and Ward investigated the relationship between the pattern of glycosylation and xylanase thermostability by systematically modulating the pattern of glycosylation118 (Fig. 3D). GH11 xylanase from Bacillus subtilis has 6 putative glycosylation sites (N8, N20, N25, N29, N141 and N181) and in WT, 4 sites were glycosylated (N20, N25, N141 and N181) when expressed in Pichia pastoris.118 Fonseca-Maldonaldo and Ward systematically mutated Asn residues to Gln in order to generate mutants with different patterns of glycosylation. By correlating glycan–protein, glycan–glycan, and protein–protein interactions observed in silico to experimental measures of stability, they showed that both the pattern and extent of glycosylation contributed to protein stability. In general, glycosylation improved stability; however, the number and pattern of glycosylation sites finely tunes protein stability. For example, the most stable protein in this study had three of five glycosylation sites occupied. Additionally, removal of some glycosylation sites allowed previously unoccupied sites (8 and 29) to become occupied. MD simulations were also used to explain why more glycosylation does not always increase stability, as well as the molecular mechanisms behind glycosylation and stability.
Another technique to expand the repertoire in the computational design of proteins is to use non-standard amino acids. Non-standard amino acids are amino acids other than the twenty common amino acids coded for by DNA. The ability to incorporate non-standard amino acids, such as D-amino acids, and PTMs into designed proteins greatly increases protein design space and offers alternative strategies for designing stable proteins. One such opportunity is to use D-amino acids to stabilize unfavorable φ/ψ backbone dihedral angles. The left-handed α-helical (αL) region of φ/ψ space is rarely populated in ‘normal’ globular proteins, and is generally an unfavorable conformation for non-Gly L-amino acids.119 In contrast to L-amino acids, D-amino acids have greater preference for αL conformations and the right-handed α-helical (αR) region of φ/ψ space is disfavored.120 Rodriguez-Granillo et al. hypothesized that mutation of a C-terminal helix capping Gly residue to a polar D-amino acid could stabilize the α-helix of Trp-cage version 5b.121 A D-amino acid in the αL conformation at a C-terminal helix capping position additionally allows for helix stabilization through the formation of a hydrogen bond between the side chain of the D-amino acid and the backbone of the helix. They first mutated G10 to bulkier L-amino acids, G10A and G10Q, at the C-terminal end of the α-helix and found that these mutations were destabilizing by MD. Next, they focused on D-amino acid mutations at this position G10a, G10n, and G10q (where lowercase is used for D-amino acids). They found that the extent of fluctuations and hydrogen bond occupancies at the mutation site correlated with stability. However, they also found that the D-Ala mutation did not increase stability; thus, the magnitude of gains in stability is, not surprisingly, dependent on both the backbone chirality and side chain properties of the amino acid. In addition to their use in helix capping, D-amino acids have also been incorporated into the design of stable β-turns,122 α-sheet peptides to inhibit amyloid formation,123,124 design of novel protein folds,125,126 stabilization of large proteins,127 and stabilizing β-hairpins.128
2.3. Tradeoffs between stability and function in design
Candida antartica lipase B catalyzes a variety of organic reactions,129 and it is used in numerous industrial applications due to its enantioselectivity and wide range of substrates. One need for a thermostable variant of C. antartica lipase B is in biodiesel production. To aid in the design of thermostable variants of C. antartica Lipase B, Park et al. performed MD simulations at four different temperatures: 300, 330, 360, and 400 K (ref. 130) to determine vulnerable regions of the structure. Park et al. used the rigidification of flexible sites method to design thermostable variants. The designs were tested experimentally by determining their specific activities and residual activities. A T158S mutant had 2× the specific activity of WT whereas the A251E mutant had 0.5× the specific activity of WT. The WT, T158S, and A251E designs showed 55%, 44%, and 75% residual activity after 4–5 hours, respectively. The results of their designs showed an important tradeoff. The mutant most susceptible to thermal denaturation (T158S) had the highest specific activity under native conditions, while the design with the lowest specific activity (A251E) retained its activity for longer periods of time at elevated temperatures. Similar negative correlations between protein stability and function have been observed for mutants of dihydrofolate reductase,51 the Hesx-1 DNA binding domain,52,131 prolactin,53,132 troponin C,54 and a M2 transmembrane proton channel.56 However the relationship between protein thermodynamic stability, function, and flexibility is complex: positive correlations between stability and function have been reported for human fibroblast growth factor 1133 and calbindin D9K.134,135 These conflicting results suggest that systems respond to mutations in distinct ways. This highlights an important design consideration: often multiple objectives must be balanced. Importantly, designs that increase protein stability should also avoid changes that disrupt the native fold, dynamics, or functional regions of the design.3. Engineering functional regions into proteins
Functional sites in proteins include active sites, ligand binding sites, and protein–protein interaction sites. Proteins depend upon coordinated dynamics of these sites to perform their functions. It is often desirable to engineer these regions of proteins in order to optimize the activity of natural proteins or to introduce new functionality into existing protein scaffolds.3.1. Designing binding sites
The design of protein–protein and protein–ligand binding sites is challenging due to the large surface areas involved, their conformational flexibility, and the interplay of solvent and protein dynamics at the sites.136,137 There are two overarching design strategies for engineering binding sites. The first is to engineer the binding site for enhanced protein:ligand or protein:protein interactions by mutating residues that are directly involved in the interaction. The second strategy is to alter the conformational diversity of the binding site by mutating residues that drive the stabilization of alternate conformational states. By resolving details of the interactions that favor or disfavor binding and by identifying residues that regulate binding site geometry, MD simulations can provide a wealth of details to guide design.Given its dynamic nature the 13-residue peptide compstatin has been the subject of numerous MD design efforts. This peptide inhibits the cleavage of human C3138 and has potential therapeutic use as a treatment for unregulated complement activation.139 It is active against C3 from several primate species, but not in lower mammals,140 such that rodents are poor disease models as human compstatin does not interact with rat C3. To create a viable rat model for evaluating compstatin as a therapeutic, Tamamis et al. capitalized on MD simulations to design a ‘transgenic’ rat C3 protein that binds human compstatin and retains its other functions.141 The crystal structure of the complex between C3c and an acetylated double mutant of compstatin138 (Ac-Val4Trp/His9Ala, henceforth ‘W4A9’), which is more active than WT compstatin, served as the starting conformation for MD simulations. The static structure of the complex sheds little light on the improved activity of W4A9 against compstatin or why human compstatin fails to inhibit cleavage of C3 in lower mammals. MD simulations were performed of the complex between W4A9 and truncated human C3c, the complex of W4A9 and truncated rat C3c, and free human C3c,142 resulting in four specific regions in human C3c with strong interactions with W4A9. In the rat C3c:W4A9 complex, local conformational changes proximal to the compstatin binding site distorted the structure of the binding site and eliminated or reduced intermolecular interactions between rat C3c and W4A9. Two transgenic designs were created in which the positions mostly likely to introduce W4A9 binding competency in rat C3 were selected on the basis of interactions that were lost or destabilized in the rat:W4A9 simulation. In this example, it was important to balance two design constraints. First the binding region was humanized to promote compstatin:C3 interactions; second, the structural scaffold was preserved to retain rat-like characteristics. Subsequent MD simulations of the humanized designs showed increased interactions between rat C3 and the W4A9 compound. Insights from this paper also aided in the design of compstatin analogues by the same group.143 This work demonstrates how MD simulations can be used to engineer a binding site to interact with a nonnative ligand and highlights the fact that the goals of a designed protein must be balanced against other objectives, such as the retention of other interactions in the designed protein.
Cytochrome b5 (cyt b5) is a heme-containing protein that participates in electron transfer reactions and can interact with multiple proteins. MD simulations of cyt b5 demonstrated that collective motions of surface residues of cyt b5 resulted in the transient exposure of a hydrophobic cleft on the surface of the protein144 (Fig. 4). While short by today's standards, this study was the longest simulation at the time and it was cited as the first time a simulation had significantly deviated from the starting position and returned,145 and prior to this deviations led to irrecoverable changes and there was the fear that numerical errors would spoil long simulations. Furthermore, with respect to the substates sampled, both the open form, discovered by MD, and the closed form, observed in crystal and NMR structures, are consistent with solution NMR data. Based on the proximity of the cleft to residues implicated in protein–protein recognition, it was proposed that cleft dynamics regulate protein recognition as well as access to the heme through the cleft. In a follow up study, Storch et al. then engineered cytochrome b5 to alter cleft dynamics.146 They created two designs to inhibit cleft formation. In the first design a single mutation, S18D, introduced a salt bridge across the site of cleft formation. In the second design, a disulfide bond was introduced at positions 18 and 47, across the cleft. In this way the formation of the cleft, and subsequent access to the heme, could be controlled by reducing the disulfide bond or breaking the salt bridge. Subsequent MD simulations confirmed that the designs prevented the opening and closing of the cleft (Fig. 4). Follow-up experimental studies employing a variety of techniques, most notably NMR, verified the formation of the cleft as well as the effects of the mutations designed to rationally control cleft formation.147 This study demonstrates the power of MD simulations to reveal functionally relevant conformations that are excursions from the static, averaged experimental structures, as well as the molecular motions that control exchange between alternate conformations.
 | ||
| Fig. 4 Discovery of functional region in cytochrome b5 by MD and subsequent computational design to regulate function. Cyclic opening and closing of a dynamic cleft providing access to the heme for electron transfer identified through MD simulations. Space-filling representations of cytochrome b5 showing designed salt bridge to close the cleft (S18D) and introduction of disulfide bond (S18C/R47C). The buried hydrophobic residues that intermittently became exposed upon cleft formation are colored red. The heme group is colored blue. | ||
Later, de Groot and coworkers used a similar approach to shift the conformational equilibrium of ubiquitin.148 Ubiquitin populates both an open and closed state and dynamically fluctuates between the two via a pincer-like motion. Ubiquitin's many binding partners preferentially interact with one of the two states. Janssen and coworkers engineered ubiquitin to skew the conformational preferences such that one state was more populated than the other to obtain selective binding.138 To do this MD simulations of unbound ubiquitin were performed to identify the most promising mutations to alter the populations of the two states. Umbrella sampling simulations were subsequently used to construct free energy profiles of the pincer mode, and 11 out of 15 proposed mutations shifted the conformational equilibrium towards one state over another. They then evaluated binding properties experimentally and confirmed their computational predictions.
3.2. Designing enzyme active sites
Enzymes are attractive catalysts because of their efficiency, selectivity, specificity, and biodegradability.52 However, there is a limited number of naturally occurring enzymes, which can limit the feasibility of using natural enzymes as catalysts in technological applications. Computational design seeks to overcome this limitation by engineering enzyme efficiency, stabilizing naturally occurring enzymes, and designing enzymes to catalyze arbitrary chemical reactions.P450 TxtE from Streptomyces scabies is an enzyme from the cytochrome P450 superfamily that regioselectively catalyzes aromatic nitration.149 These enzymes rely on dynamic rearrangements of the B/C and F/G loops, which seal the active site upon substrate binding, to regulate the high reactivities of reaction intermediates. Arnold and coworkers obtained crystal structures of P450 TxtE that revealed a functional role of the B/C loop; however, they were unable to determine the interactions made by the F/G loop, as it is disordered in the crystal structure.149 Computational approaches were used to model the structure and dynamics of the disordered F/G loop.150 First, the F/G loop was constructed via homology modeling followed by short MD simulations. Snapshots were taken from the trajectories at regular intervals and subjected to geometry optimization. Of these snapshots, the Top 100 energetically favorable structures were used as starting points for production MD.
The transition from a disordered state to an ordered closed state occurs on long time scales. To circumvent this computational bottleneck and to avoid biasing the results via enhanced sampling schemes, Arnold and coworkers used multiple rounds of adaptive sampling to exhaustively simulate the open-to-closed transition (Fig. 5). The combined production runs were used to construct coarse-grained and fine-grained Markov models of the transition. The coarse-grained model encompassed 9 unique conformational states, i.e. macrostates. The fine-grained model contained many microstates whose conformations differed by small amplitude changes (such as side chain rearrangements).
 | ||
| Fig. 5 Identification of the opening/closing transition of the P450 TxtE F/G loop and engineering of regioselective variants. P450 TxtE from Streptomyces scabies adopts both closed (a) and open (c) conformations of the F/G loop. Molecular dynamics simulations revealed the mechanism of conformational exchange between these two states, schematically illustrated in (b). In the open conformation, the binding pocket is exposed to solvent, which enables substrate (L-Trp) binding and product (4NT) release (f). In the closed conformation, water molecules (W1–3) and residues G58, Y175, and Y89 form a network of interactions that stabilize and orient substrate in the binding pocket (d). In the transition state, identified by MD, H176 and Y89 form transient interactions with substrate, which suggested that mutation of H176 would shift the conformational equilibrium between open and closed states when substrate is bound (e). Figures reproduced from Dodani et al. 2016.150 Our panels a and c correspond to Fig. 1b, panel b is Fig. 2c, and panels 5d–f correspond to Fig. 2a, b and d. | ||
The Markov state models were used to organize the trajectories into discrete conformational states and to identify the connectivity of the conformational states, resulting in a predicted transition mechanism of the F/G loop from an open, disordered conformation to a closed, ordered conformation (Fig. 5). A single conformational state was observed in all transitions and had characteristics of both the bound and unbound macrostates. This conformational state was defined as the transition state of the closing and opening of the F/G loop. In the closed state, Y175 forms an interaction with Y89; in the transition state H176 interacts with Y89 as opposed to Y175. These results suggested that mutation of H176 could be used to stabilize the closed state of the FG loop in the presence of substrate. Two mutations, H176Y and H176F, were tested experimentally and resulted in 15- and 8-fold higher binding affinities, respectively, and produced 5-nitro-L-tryptophan (5NT) instead of 4-nitro-L-tryptophan (4NT). Additional mutations at these positions modulated the production of 5NT and 4NT. The WT produces 4NT. The WT protein (H176) produced 4NT; designs with N, G, S, or M at position 176 produced a mix of 4NT and 5NT; and designs with F, Y, or W at position 176 produced 5NT. Simulations of the His176 mutations with aromatic residues (F, Y, or W) also demonstrated a stabilization of the closed conformation of the F/G loop. Crystal structures of designs with an aromatic mutation at position 176 had fully resolved F/G loops in the closed state. For some proteins, such as TxtE, simulations on the microsecond–millisecond timescale may be necessary to resolve the functional dynamics. For other proteins simulations on the nanosecond–microsecond timescale are sufficient. It may be difficult to know a priori the time scale necessary to capture functional dynamics, but in general the processes of interest are quite fast with long waiting times determining the timescale. In such situations performing many independent short trials can increase the odds of observing conformational changes beyond the simulation timescale while still maintaining unbiased dynamics.151
The Kemp elimination reaction, i.e. the deprotonation of carbon, is a well-characterized reaction for which no known naturally occurring enzyme exists.152 Several groups have designed novel enzymes to catalyze the reaction.153–156 Building off of the successes of other designed Kemp eliminases, Mayo and coworkers combined the “inside out” design method of Zanghellini et al.157 with MD simulations to iteratively design a Kemp eliminase.158 Xylanase from Thermoascus aurantiacus was used as the scaffold in the first generation design, HG-1. Seven mutations were made to the scaffold to create the Kemp eliminase. Designs in the active site were inspired by prior Kemp eliminase designs from Röthlisberger et al.153 and the catalytic antibody 34E4.154,159 Although HG-1 did fold to the designed structure, it did not demonstrate any Kemp eliminase activity. To understand why the design showed no activity, Mayo and coworkers determined the crystal structure of HG-1 and performed MD simulations. The crystal structure showed that two of the active site residues were rotated out of the active site, which prevented substrate binding and exposed the active site to solvent. MD simulations performed in the presence and absence of substrate showed that the active site was too exposed to solvent and that the active site residues had a high degree of flexibility. Furthermore, the simulations predicted that the active site explored conformations that were inconsistent with the initial design and incompatible with catalytic function. In the second-generation design, HG-2, Mayo and coworkers redesigned the active site to increase its size, hydrophobicity, and burial in the protein. MD simulations of HG-2 suggested that it should be active, although two distinct conformations of the active site were populated during the simulation. Subsequent experiments confirmed that their design was active, with a catalytic efficiency comparable to that of the best designs of Röthlisberger et al.153 Additional analysis of the HG-2 simulations suggested that the alternate binding mode was more populated than the designed conformation and had a greater potential for catalytic activity. In the third generation design, HG-3, an additional mutation (S265T) was made to decrease the flexibility of active site residues, to reduce the conformational heterogeneity of the active site, and to promote the alternate conformation. Simulations of HG-3 confirmed a loss of conformational heterogeneity in the active site. Experimental evaluation of HG-3 showed higher catalytic efficiency than any of the previous designs, demonstrating the added value of incorporating MD with other design methods (Fig. 5).
MD simulations have also been used to understand the shortcomings of designs to catalyze the retro-aldol reaction. Jiang et al. created de novo designs to catalyze the multi-step retro-aldol reaction.160 In that study, 32 out of 72 reported designs showed retro-aldolase activity; however, the best performing designs from that study had rate enhancements 2–3 orders of magnitude lower than catalytic antibodies.15,161 To investigate whether the geometric criteria for retro-aldolase activity were maintained, Ruscio et al.162 performed MD simulations of RA22 from Jiang et al.160 in complex with substrate. The simulations revealed that two substrate orientations were possible and that two geometric criteria, corresponding to the nucleophilic attack and proton abstraction steps in the retro-aldol reaction, were satisfied only ∼50% and ∼25% of the time during simulations, and thus only rarely were both criteria met simultaneously. Ruscio et al. suggested that explicit exclusion of protein and ligand flexibility during the design process concealed the inability of the designs to maintain productive active site geometry. They attributed the low rate enhancements of the designs to the inability of the active site to maintain the designed geometry. Later, Baker and coworkers refined another retro-aldol design, RA34, using directed evolution.163 The refined RA34.6 variant had higher catalytic efficiency, but structural studies revealed some persistent design flaws. Namely, the flaws were increased conformational heterogeneity of catalytic lysine residue and heterogeneity of the substrate within the binding pocket. These shortcomings are quite similar to those suggested by the MD simulations of Ruscio et al.162
3.3. Screening and ranking computational designs
Natural enzymes are usually orders of magnitude more efficient than even the best designed proteins. Furthermore, designed enzymes with high sequence identity can have drastically different catalytic properties. A challenge in protein design is to successfully distinguish active from inactive designs and to rank active designs. By providing atomic details of active site conformations and dynamics, MD simulations have the capacity to aid in the screening and ranking of enzyme designs.164 Simulations of potential designs are screened according to pre-determined metrics, e.g. the stability of active site conformations, and top performing designs are chosen for refinement or experimental characterization. For example, Houk and coworkers were not able to differentiate between active and inactive Kemp eliminase designs using quantum mechanics (QM) cluster models and full enzyme QM/MM and PM3/PDDG/MC calculations.165 The major problem was that these approaches cannot model changes in protein structure and solvent accessibility accurately. They then performed MD simulations of Kemp eliminase (KE) with and without substrate, and tested whether the active site geometry was preserved in 23 KE designs using cathepsin K and catalytic antibody 34E4 as references. Designs were predicted to be inactive if active site geometries deviated from the designed conformation. The activity of 20 out of 23 designs was predicted correctly. In another example, Bjelic et al. used QM and MD simulations to design a protein to catalyze the slow, multistep Morita–Baylis–Hillman (MBH) reaction.166 The MD simulations predicted designs with stable active site conformations, which were subsequently confirmed experimentally. Privett et al.158 also used MD simulations to predict whether active site “recapitulation” designs would be active using similar criteria to those used in Kiss et al.165 MD simulations successfully predicted the activities of five out of six designs. These studies demonstrate the utility of MD for evaluating designs as a filter for the design process.4. Design insights from unfolding/folding pathways
Protein folding is a complex process by which an unfolded polypeptide chain adopts a well-defined, compact structure. Protein folding begins at the denatured state, which can contain varying degrees of dynamic residual secondary and/or tertiary structure. The polypeptide chain proceeds through a series of intermediate states before arriving at a native, folded state. The relationship between sequence and structure is complex, as highlighted by the heteromorphic proteins GA and GB, which have high sequence identity (77–98%), but distinct topologies.167,168 The solution to this conundrum came through MD-derived predictions that interactions in the denatured state determined whether the α-helical GA fold or β-grasp fold of GB would be adopted.169 These MD predictions were later confirmed by experiment.170 Thus, atomic details of protein folding that are often hidden or obscured by experimental techniques can be revealed by MD simulations. Conventional MD simulations of ab initio protein folding with full atomic resolution remain computationally intense, even for very small proteins. As an alternative to ab initio folding simulations, less computationally intensive techniques, such as high temperature MD, have been developed to sample protein unfolding/folding pathways74,75 and microscopic reversibility has been demonstrated.171,172 Predictions about the folding/unfolding mechanism of a protein, whether they are obtained from experiment or simulation, can be tested through protein design. Once such mechanisms have been characterized, protein design may then be used to alter the kinetics and thermodynamics of folding pathways.4.1. Designing fast folding protein variants
The first design based on a simulated protein unfolding/folding pathway focused on the transition state, paving the way for the design of faster folding proteins. This study entailed high temperature simulations to obtain the unfolding pathway of chymotrypsin inhibitor 2 (CI2).173 The transition state of unfolding/folding was predicted by conformational clustering and subsequently validated by experiment.78,174,175 For a high temperature unfolding simulation, the putative transition state (TS) ensemble of folding/unfolding can be identified using conformational clustering.171,176 In this technique, the Cα root mean squared deviation is calculated between all structures in the trajectory and placed into a N × N matrix. Then the matrix is reduced to a 3 × N dimensional matrix using a dimensionality reduction algorithm. This set of points is then viewed as a 3D trajectory with sequential points connected in time and with the trajectory clustered by conformational similarity. The TS is predicted to be the when the protein leaves the native cluster (Fig. 6). Analysis of the MD-derived TS using this method revealed two regions of CI2 with unfavorable nonnative interactions.175 We reasoned that mutations that minimize these unfavorable interactions in the TS would accelerate folding provided they did not also stabilize the native or denatured states. In this way, the MD simulations were extremely useful because the interactions were evaluated in all WT states and the designed mutations were also simulated prior to experimental validation. The first region identified for redesign was at the C-terminus of the α-helix. In the native state, D23 forms a salt bridge with K2. In the TS, the unpaired D23 made unfavorable interactions with the helix dipole, specifically the unpaired backbone carbonyl group at the C-terminus of the α-helix. The D23A mutation was made to relieve charge repulsion and made use of a helix-promoting residue. The second region was the protease-binding loop, which became distorted in the TS, resulting in charge repulsion between the three Arg residues that are normally involved in interactions with the main chain in the native state (Fig. 6). A multiple sequence alignment between CI2 and 18 homologous proteins showed that both R46 and R48 are highly conserved, but that Phe was observed at position 48 in two homologues and Trp was observed at position 48 in one homologue. The R48F mutation was selected and predicted to increase the folding rate by removal of unfavorable electrostatic interactions. Experimental studies supported the predictions and both mutants folded more rapidly than WT, and R48F yielded that fast folding CI2 variant with nearly two orders of magnitude improvement in the rate of folding (Fig. 6). MD was critical to achieve this result since knowledge of both native and nonnative interactions of the TS and denatured state was necessary. | ||
| Fig. 6 Thermal denaturation simulations to map unfolding pathways and characterize transition state structures for the rate-determining step of chymotrypsin inhibitor 2 (CI2). Top: Conformational clustering to identify putative TS regions and comparison of the identified structural ensembles with experiment (right). Center: Snapshots from different MD conformational ensembles are displayed highlighting the loss of structure and increased conformational sampling upon unfolding. Bottom: Interactions were identified in the MD-derived TS structures that could be improved upon with mutation while now affecting those same interactions in the native and denatured states (structures shown in stereo). Designs were made and tested in silico and the predictions were validated by experiment. The goal of creating a faster folding version of CI2 was realized through the designed mutations and the targeted interactions would not have been evident from static experimental structures or native state MD simulations. The images in the top panel are reproduced from Li and Daggett.173 | ||
Later, Piana et al. used a similar approach to design a faster folding FiP35 WW domain.177 An ab initio folding simulation of the FiP35 WW domain40 was used to construct the folding pathway of the FiP35 WW domain. They capitalized on knowledge of residual structure in the unfolded state, rate limiting steps to hairpin formation, and structure of the transition state ensemble to design a faster folding variant, which they subsequently validated experimentally.
Myoglobin is heme binding protein and a member of the globin fold family and has 8 α-helices arranged in two hydrophobic cores: ABGH (formed by helices A, B, G, and H) and CDEF (formed by helices C, D, E and F). In the absence of the heme prosthetic group, apomyoglobin (ApoMb) adopts a fold similar to the native structure of holo-myoglobin.178 The CDEF domain contains more local contacts and it was expected to fold prior to the ABGH domain. Despite these predictions, stopped flow refolding experiments showed that in the absence of heme, the ABGH domain forms rapidly, followed by formation of CDEF.179–181 This unexpected behavior was attributed to the stabilization of the CDEF hydrophobic core by heme. Gruebele and coworkers increased the speed of folding of the CDEF domain by using MD simulations to design apomyoglobin variants with well-packed hydrophobic cores in the absence of heme.182 Two initial mutants were introduced that were obtained from the literature that lead to greater stability, secondary structure content, and faster folding of CDEF: H64F and P88A.183,184 Three additional mutations were made that reduced cavity volume and increased hydrophobic interactions in the heme cavity: L89W, V68W, I107M. In total, five variants were simulated and experimentally tested. The heme cavity in the WT protein has a void volume of 638 Å3. MD simulations showed that as the number of hydrophobic mutations increased, so did the reduction in cavity volume, up to 21%. These mutations also increased the size of the side chain residue interaction network connecting the C, D, and E α-helices. The ApoMb-3, 4, and 5 designs had ∼20% greater helix content than WT, as monitored by CD. All ApoMb designs had melting temperatures approximately 20 °C higher. Finally, stopped flow refolding measurements showed that two of the mutants refolded four times faster than WT.
4.2. Stabilizing nonnative and intermediate states of proteins
MD also proved to be instrumental in characterizing the folding/unfolding pathway of the engrailed homeodomain (EnHD). This simple 3-helix bundle protein is particularly interesting because it is an ultrafast unfolding and folding protein, allowing the time scale of the process to be probed directly by MD at experimentally accessible temperatures in a collaborative study between the Daggett and Fersht groups. In temperature-jump experiments, EnHD folds to an intermediate state in ≤1.5 μs and the transition from I to N takes ∼15 μs. The relaxation kinetics were followed to 338 K and extrapolated to 348–373 K.185 The thermal unfolding of EnHD was also investigated at a variety of temperatures and the time taken to reach the TS in the simulations is in agreement with the unfolding times determined experimentally (Fig. 7), and the simulations were done as predictions 3 years prior.186 The TS of EnHD contains native-like secondary structure and a partially packed hydrophobic core. The calculated and experimental Φ-values for the TS, using the method described above for CI2, are in good agreement (R = 0.85, Fig. 7), and again the MD preceded the experimental study.187,188 The simulated unfolding process is independent of temperature, and essentially the same TSs are obtained at 348, 373, and 498 K.189,190 Higher temperature accelerates the process without changing the overall pathway. By MD, from the TS, reorientation of the helices, expansion and disruption of the helix docking leads to the intermediate. This intermediate has a high helical content and few tertiary contacts (Fig. 7).185–187 Five years later the structure was determined directly by NMR,191 and it is in remarkable agreement with the MD prediction (Fig. 7). | ||
| Fig. 7 Experimental rates for unfolding and folding of the engrailed homeodomain (EnHD), along with MD-predicted TS times. Comparison of calculated S-values and experimental Φ-values, which both reflect the extent of structure for each residue probed, S-values through direct structure in the MD ensemble and Φ-values through inferred structure from ratios of free energies upon mutation. MD-predicted intermediate structure and NMR-derived intermediate structure determined several years later. | ||
The EnHD work set the stage for further design efforts. We have also investigated other members of the EnHD family. There is a change from 3-state to 2-state kinetics of folding across the homeodomain superfamily of proteins as the mechanism slides from framework to nucleation-condensation. The tendency for framework folding in this family correlates with inherent helical propensity. The cellular myeloblastic protein (c-Myb) falls in the mechanistic transition region. In-depth analysis of the MD trajectories showed that folding can be attributed to both of these mechanisms in different regions of the protein.188 Experimentally, however, c-Myb folds by apparent two-state kinetics, but the MD simulations predict that the kinetics hide a high-energy intermediate.188 The primary distinction between c-Myb and other members of the homeodomain family is that c-Myb has 5 (rather than 4) residues in the loop connecting the second and third helices. A mutant with a deletion of P174 was predicted to increase the helical propensity of helix 2 and to stabilize the turn connecting H2 and H3. In simulations, deletion of P174 allowed for the formation of native like contacts in the helix–turn–helix motif. The intermediate state was more populated in the mutant than in the WT simulations. Urea induced unfolding of c-Myb fit a two-state folding transition. Urea induced unfolding of c-Myb ΔP174 deviated from a two-state folding transition. Additional urea induced unfolding of c-Myb ΔP174/L155A showed evidence for the buildup of an intermediate during folding. We stabilized this folding intermediate discovered by MD by deleting a residue (P174) in the loop between its second and third helices, and the mutant intermediate is long-lived in simulations. Later equilibrium and kinetic experiments demonstrated that folding of the ΔP174 mutant is indeed 3-state.192 Their results showed that MD simulations can identify intermediate states that may be ‘invisible’ to experimental techniques.
Tenascin C is found in the extracellular matrix and regulates cell–matrix interactions through the transduction of mechanical forces to biological signals. Application of a stretching force induces unfolding of the fibronectin type III domain of Tenascin-C (TNfn3), resulting in lengthening of Tenascin-C. Atomic force microscopy (AFM) and steered MD simulations193,194 showed that force-induced unfolding of TNfn3 results in a complex unfolding pathway with two highly populated intermediate states. One strategy for modulating the mechanical stability of proteins is to incorporate a bi-His metal chelation site into the protein.195 Based on MD-derived forced-unfolding intermediates of TNfn3, Zhuang et al. engineered a bi-His metal chelation site into TNfn3 that allows the mechanical stability of Tenascin-C to be modulated, and it may be used to regulate the activity of ECM proteins in vivo.196 Two intermediate states are populated along the forced-unfolding trajectory, I1 and I2, and AFM suggested that the rate-limiting unfolding transition state lies between the intermediates. A hydrogen bond between the backbone atoms of residues 6 and 23 was associated with the transition from I1 to I2 in the simulations and a metal chelation site was incorporated to increase the energy barrier of the transition state for force-induced unfolding. Both chemical unfolding experiments and AFM showed increased TNfn3 stability in the presence of Ni2+, which coordinates with the designed bi-His residues. Thus, the mechanical stability of mutants that incorporate a designed chelation site can be tuned by the addition or removal of Ni2+. This approach may be extended to other protein where modulation of mechanical signal transduction is desired.
4.3. Probing misfolded states of proteins
Unfolding simulations, like those discussed above, can also be used to characterize vulnerabilities in structures associated with disease, such as the consequences of single nucleotide polymorphisms197 and misfolding diseases. With respect to misfolding diseases, this is something of a misnomer. Instead of a competition between misfolding and folding correctly to the native state, these amyloidogenic proteins fold correctly but then partially unfold, often in response to low pH, and misfold into aggregation prone amyloidogenic intermediate states. The process of amyloidogenesis is characterized by the conversion of soluble, native conformations into toxic soluble oligomers and finally mature fibrils.198,199 There are now over 40 different human amyloid diseases.200 Characterization of the conformational conversions during amyloidogenesis has not been possible using traditional structure determination methods due to dynamics, aggregation, and heterogeneity of the species. To this end, MD simulations can be applied to flesh out the molecular aspects of the process, typically triggering the process by lowering the pH.In particular, as it was becoming clear that experimental methods were not able to provide detailed accounts of the conformational changes involved in amyloidogenesis, MD were employed in an effort to map the process. The first such account focused on the prion protein and the pH-induced changes in the structure in MD are in good agreement with experiment.201,202 Some altered β-sheet-like structure was observed in the resulting putative amyloidogenic intermediate states and more extensive simulations with other amyloidogenic proteins with diverse topologies showed that they, too, adopt α-sheet structure, including transthyretin,203–206 lysozyme,207 β2-microglobulin,208 and polyglutamine.209,210 Interestingly, α-sheet structure was modeled by Pauling and Corey in 1951,211 but they, rightly, dismissed it in favor of β-sheet structure for native folded proteins. α-sheets are similar to β-sheets except that instead of being formed by successive residues in the β-region of (ϕ,ψ) space, they alternate between αL and αR conformations, which produce an extended sheet with the main chain carbonyls aligned on one side of the strand and the amide NH groups on the other (Fig. 8).
 | ||
| Fig. 8 Schematic highlighting conversion of a β-strand in the amyloidogenic protein transthyretin at low pH to an α-strand with alignment of the carbonyl oxygens in red. Small α-sheet hairpins were designed to be complementary to the structure discovered through MD. Binding of the de novo designed α-sheet hairpin peptides inhibits aggregation and neutralizes toxicity associated with the soluble oligomers. Furthermore many amyloidogenic proteins and peptides adopt the α-sheet structure in MD and the same peptide designs cross react and inhibit aggregation in many amyloid disease systems. | ||
Based on the MD, we proposed the α-sheet hypothesis, which posits that α-sheet structure is shared by unrelated amyloidogenic proteins and is linked to toxicity and harbored by the toxic oligomers.204,212 As such, α-sheet structure represents a novel target for neutralizing toxicity and inhibiting fibril formation by employing the molecular dynamics-generated structures for rational structure-based design to create structurally complementary inhibitors (Fig. 8). To this end, anti-α-sheet peptides, which are themselves α-sheets, were designed in silico, tested by MD and the highest-ranking designs were synthesized and tested experimentally. The designs make use of alternating D- and L-amino acids to help lock in the α-sheet structure, making use of conformational propensity125,213–215 and rotamer libraries216–218 for D- and L-amino acids from our Dynameomics project,219,220 which contains simulations of representatives of essentially all known protein folds and multiple host–guest peptide systems. This MD-derived Dynameomics database is another way in which MD can contribute to design efforts, as it contains 105 more structures than the Protein Data Bank and represents a rich source of information regarding dynamic protein structures in solution.
If the α-sheet hypothesis is correct, then an α-sheet design should prevent aggregation in multiple amyloid systems. This is found to be the case, the designs α-sheet inhibitors preferentially bind the toxic oligomeric species and inhibit aggregation of Aβ (associated with Alzheimer's disease), transthyretin (systemic amyloid disease) and amylin (type 2 diabetes).123,124 Furthermore, all α-sheet designs investigated to date inhibit amyloidogenesis independent of sequence such that the stability of the α-sheet structure determines inhibitory potency not the sequence per se.124 These inhibitors are unique in their mechanism of action through selective binding to the toxic oligomers, which supports the idea that the toxic species formed during amyloidogenesis adopt α-sheet and that unrelated amyloidogenic proteins and peptides funnel through this structure despite having different sequences and native structures. MD has been crucial to these design efforts, as without the MD the α-sheet structure would not have been discovered and this new family of cross-reacting amyloid inhibitors represent the only compounds that specifically target the toxic oligomeric species.
MD simulations have also been used to modulate the aggregation propensity of β2-microglobulin to form amyloid.221,222 In particular, Fogolari et al.222 investigated the behavior of β2-microglobulin monomers under amyloidogenic conditions and suggested that W60 and other neighboring hydrophobic residues play a role in the early stages of aggregation by providing a hydrophobic interface for intermolecular association.222 Interestingly, this region formed α-sheet structure in the earlier simulations discussed above,222 and Fogolari et al. also noted that D59/W60 formed the α-sheet repeating unit in their simulations. Mutation of W60 to Gly (W60G) resulted in a variant that is less prone to aggregation.223 More recently, Camilloni et al. performed extensive replica averaged metadynamics simulations of WT and W60G β2-microglobulin to gain insights into the relationship between conformational flexibility and aggregation propensity. These simulations allowed for the design of variants with diverse aggregation kinetics.221
5. Conclusions
Protein dynamics underlie mechanisms of protein stability, function, and folding. Because of this relationship, consideration of protein dynamics provides unique perspectives for protein design. Molecular dynamics simulations can provide critical insights into the relationship between protein dynamics and function, thereby leading to novel designs unobtainable through the use of static native structures alone. Furthermore, MD is unparalleled in its ability to provide atomic details of distinct nonnative conformational states as well as the transitions between these states that may be inaccessible by other techniques. Dynamics can be incorporated into different points in the protein design cycle. For example, simulations of wild type proteins performed prior to protein design can directly aid in the rational design of protein variants. After proteins have been designed, simulations can be used to evaluate and systematically rank them. A common strength of MD for protein design is to identify the optimal residues for making mutations. MD simulations can also help filter through candidate mutations derived from other computational design tools. Finally, there is a high degree of synergy between MD and other techniques: it can provide atomic details of mechanisms identified by experiment. In turn, experiment can confirm or reject predictions made by MD. As computational protein design matures, our understanding of the complex interplay between structure, dynamics, and function improves and as does our ability to use this information for applications in industry and medicine.Acknowledgements
We are grateful for financial support provided by the National Institutes of Health (GMS 95808).References
- B. A. Gutte, Synthetic 70-amino acid residue analog of ribonuclease S-protein with enzymic activity, J. Biol. Chem., 1975, 250, 889–904 CAS.
- B. Gutte, Study of RNase A mechanism and folding by means of synthetic 63-residue analogs, J. Biol. Chem., 1977, 252, 663–670 CAS.
- M. Levitt and J. Greer, Automatic identification of secondary structure in globular proteins, J. Mol. Biol., 1977, 114, 181–239 CrossRef CAS PubMed.
- E. T. Kaiser and F. J. Kézdy, Secondary structures of proteins and peptides in amphiphilic environments, Proc. Natl. Acad. Sci. U. S. A., 1983, 80, 1137–1143 CrossRef CAS.
- R. Moser, et al., Expression of the synthetic gene of an artificial DDT-binding polypeptide in Escherichia coli, Protein Eng., Des. Sel., 1987, 1, 339–343 CrossRef CAS.
- R. Moser, R. M. Thomas and B. Gutte, An artificial crystalline DDT-binding polypeptide, FEBS Lett., 1983, 157, 247–251 CrossRef CAS.
- B. Gutte, M. Däumigen and E. Wittschieber, Design, synthesis and characterisation of a 34-residue polypeptide that interacts with nucleic acids, Nature, 1979, 281, 650–655 CrossRef CAS PubMed.
- M. Hecht, J. Richardson, D. Richardson and R. Ogden, De novo design, expression, and characterization of Felix: a four-helix bundle protein of native-like sequence, Science, 1990, 249, 973 Search PubMed.
- B. Erickson, et al., in Computer Graphics and Molecular Modeling, ed. R. Fletterick and M. Zoller, Cold Spring Harbor Laboratory, 1986, pp. 53–57 Search PubMed.
- J. S. Richardson and D. C. Richardson, The de novo design of protein structures, Trends Biochem. Sci., 1989, 14, 304–309 CrossRef CAS PubMed.
- R. L. Dunbrack and M. Karplus, Backbone-dependent rotamer library for proteins. Application to side-chain prediction, J. Mol. Biol., 1993, 230, 543–574 CrossRef CAS PubMed.
- C. Wilson, L. M. Gregoret and D. A. Agard, Modeling side-chain conformation for homologous proteins using an energy-based rotamer search, J. Mol. Biol., 1993, 229, 996–1006 CrossRef CAS PubMed.
- B. I. Dahiyat and S. L. Mayo, De novo protein design: fully automated sequence selection, Science, 1997, 278, 82–87 CrossRef CAS PubMed.
- B. Kuhlman, et al., Design of a novel globular protein fold with atomic-level accuracy, Science, 2003, 302, 1364–1368 CrossRef CAS PubMed.
- R. Sterner, R. Merkl and F. M. Raushel, Computational Design of Enzymes, Chem. Biol., 2008, 15, 421–423 CrossRef CAS PubMed.
- S. Sivaramakrishnan, E. Ashley, L. Leinwand and J. A. Spudich, Insights into human beta-cardiac myosin function from single molecule and single cell studies, J. Cardiovasc. Transl. Res., 2009, 2, 426–440 CrossRef PubMed.
- E. Z. Eisenmesser, et al., Intrinsic dynamics of an enzyme underlies catalysis, Nature, 2005, 438, 117–121 CrossRef CAS PubMed.
- E. Z. Eisenmesser, D. A. Bosco, M. Akke and D. Kern, Enzyme dynamics during catalysis, Science, 2002, 295, 1520–1523 CrossRef CAS PubMed.
- M. P. Allen and D. J. Tildesley, Computer Simulations of Liquids, Clarendon Press, 1987 Search PubMed.
- A. G. Palmer, NMR probes of molecular dynamics: Overview and comparison with other techniques, Annu. Rev. Biophys. Biomol. Struct., 2001, 30, 129–155 CrossRef CAS PubMed.
- H. van den Bedem and J. S. Fraser, Integrative, dynamic structural biology at atomic resolution--it's about time, Nat. Methods, 2015, 12, 307–318 CrossRef CAS PubMed.
- R. O. Dror, M. Ø. Jensen, D. W. Borhani and D. E. Shaw, Exploring atomic resolution physiology on a femtosecond to millisecond timescale using molecular dynamics simulations, J. Gen. Physiol., 2010, 135, 555–562 CrossRef CAS PubMed.
- T. D. Romo and A. Grossfield, Unknown unknowns: the challenge of systematic and statistical error in molecular dynamics simulations, Biophys. J., 2014, 106, 1553–1554 CrossRef CAS PubMed.
- L. Monticelli and D. P. Tieleman, in Methods in Molecular Biology, 2013, vol. 924, pp. 197–213 Search PubMed.
- E. A. Cino, W.-Y. Choy and M. Karttunen, Comparison of Secondary Structure Formation Using 10 Different Force Fields in Microsecond Molecular Dynamics Simulations, J. Chem. Theory Comput., 2012, 8, 2725–2740 CrossRef CAS PubMed.
- D. J. Price and C. L. Brooks, Modern protein force fields behave comparably in molecular dynamics simulations, J. Comput. Chem., 2002, 23, 1045–1057 CrossRef CAS PubMed.
- K. Lindorff-Larsen, et al., Systematic validation of protein force fields against experimental data, PLoS One, 2012, 7, e32131 CAS.
- K. A. Beauchamp, Y.-S. Lin, R. Das and V. S. Pande, Are Protein Force Fields Getting Better? A Systematic Benchmark on 524 Diverse NMR Measurements, J. Chem. Theory Comput., 2012, 8, 1409–1414 CrossRef CAS PubMed.
- O. F. Lange, D. van der Spoel and B. L. de Groot, Scrutinizing molecular mechanics force fields on the submicrosecond timescale with NMR data, Biophys. J., 2010, 99, 647–655 CrossRef CAS PubMed.
- Y. Gu, D.-W. W. Li and R. Brüschweiler, NMR Order Parameter Determination from Long Molecular Dynamics Trajectories for Objective Comparison with Experiment, J. Chem. Theory Comput., 2014, 10, 2599–2607 CrossRef CAS PubMed.
- E. S. O'Brien, A. J. Wand and K. A. Sharp, On the ability of molecular dynamics force fields to recapitulate NMR derived protein side chain order parameters, Protein Sci., 2016, 25, 1156–1160 CrossRef PubMed.
- D. A. C. Beck, R. S. Armen and V. Daggett, Cutoff size need not strongly influence molecular dynamics results on solvated polypeptides, Biochemistry, 2005, 44, 609–616 CrossRef CAS PubMed.
- G. Hernández, J. S. Anderson and D. M. Lemaster, Experimentally assessing molecular dynamics sampling of the protein native state conformational distribution, Biophys. Chem., 2012, 163–164, 21–34 CrossRef PubMed.
- K. C. Cunha, et al., Assessing protein conformational sampling and structural stability via de novo design and molecular dynamics simulations, Biopolymers, 2015, 103, 351–361 CrossRef CAS PubMed.
- S. D. Bond and B. J. Leimkuhler, Molecular dynamics and the accuracy of numerically computed averages, Acta Numer., 2007, 16, 1–65 CrossRef.
- D. C. C. Rapaport, The Art of Molecular Dynamics Simulation, Cambridge University Press, 2nd edn, 2004 Search PubMed.
- R. D. Skeel, in The Graduate Student's Guide to Numerical Analysis '98, 1999, vol. 26, pp. 119–176 Search PubMed.
- M. E. Tuckerman and G. J. Martyna, Understanding Modern Molecular Dynamics: Techniques and Applications, J. Phys. Chem. B, 2000, 104, 159–178 CrossRef CAS.
- M. Levitt, M. Hirshberg, R. Sharon, K. E. Laidig and V. Daggett, Calibration and Testing of a Water Model for Simulation of the Molecular Dynamics of Proteins and Nucleic Acids in Solution, J. Phys. Chem. B, 1997, 101, 5051–5061 CrossRef CAS.
- D. E. Shaw, et al., Atomic-level characterization of the structural dynamics of proteins, Science, 2010, 330, 341–346 CrossRef CAS PubMed.
- K. Lindorff-Larsen, P. Maragakis, S. Piana and D. E. Shaw, Picosecond to Millisecond Structural Dynamics in Human Ubiquitin, J. Phys. Chem. B, 2016, 120, 8313–8320 CrossRef CAS PubMed.
- M. J. Abraham, et al., GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers, SoftwareX, 2015, 1, 19–25 CrossRef.
- V. S. Pande, et al., Atomistic protein folding simulations on the submillisecond time scale using worldwide distributed computing, Biopolymers, 2003, 68, 91–109 CrossRef CAS PubMed.
- W. F. van Gunsteren, J. Dolenc and A. E. Mark, Molecular simulation as an aid to experimentalists, Curr. Opin. Struct. Biol., 2008, 18, 149–153 CrossRef CAS PubMed.
- W. F. van Gunsteren and A. E. Mark, Validation of molecular dynamics simulation, J. Chem. Phys., 1998, 108, 6109 CrossRef CAS.
- V. Daggett, Molecular Dynamics Simulations of the Protein Unfolding/Folding Reaction, Acc. Chem. Res., 2002, 35, 422–429 CrossRef CAS PubMed.
- G. D. Haki and S. K. Rakshit, Developments in industrially important thermostable enzymes: a review, Bioresour. Technol., 2003, 89, 17–34 CrossRef CAS PubMed.
- J.-M. Choi, S.-S. Han and H.-S. Kim, Industrial applications of enzyme biocatalysis: Current status and future aspects, Biotechnol. Adv., 2015, 33, 1443–1454 CrossRef CAS PubMed.
- C. J. Wilson, Rational protein design: developing next-generation biological therapeutics and nanobiotechnological tools, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2015, 7, 330–341 CrossRef CAS PubMed.
- C. N. Pace, B. A. Shirley, M. McNutt and K. Gajiwala, Forces contributing to the conformational stability of proteins, FASEB J., 1996, 10, 75–83 CAS.
- A. Yokota, H. Takahashi, T. Takenawa and M. Arai, Probing the roles of conserved arginine-44 of Escherichia coli dihydrofolate reductase in its function and stability by systematic sequence perturbation analysis, Biochem. Biophys. Res. Commun., 2010, 391, 1703–1707 CrossRef CAS PubMed.
- M. Torrado, et al., Role of conserved salt bridges in homeodomain stability and DNA binding, J. Biol. Chem., 2009, 284, 23765–23779 CrossRef CAS PubMed.
- J.-B. Jomain, et al., Structural and thermodynamic bases for the design of pure prolactin receptor antagonists: X-ray structure of Del1-9-G129R-hPRL, J. Biol. Chem., 2007, 282, 33118–33131 CrossRef CAS PubMed.
- R. S. Fredricksen and C. A. Swenson, Relationship between stability and function for isolated domains of troponin C, Biochemistry, 1996, 35, 14012–14026 CrossRef CAS PubMed.
- A. L. Stouffer, V. Nanda, J. D. Lear and W. F. DeGrado, Sequence determinants of a transmembrane proton channel: an inverse relationship between stability and function, J. Mol. Biol., 2005, 347, 169–179 CrossRef CAS PubMed.
- M. Masazumi, G. Signor and B. W. Matthews, Substantial increase of protein stability by multiple disulphide bonds, Nature, 1989, 342, 291–293 CrossRef PubMed.
- T. Sikosek and H. S. Chan, Biophysics of protein evolution and evolutionary protein biophysics, J. R. Soc., Interface, 2014, 11, 20140419 CrossRef PubMed.
- S. Kapoor, A. Rafiq and S. Sharma, Protein Engineering and Its Applications in Food Industry, Crit. Rev. Food Sci. Nutr., 2015, 11 DOI:10.1080/10408398.2014.1000481.
- K. Teilum, J. G. Olsen and B. B. Kragelund, Protein stability, flexibility and function, Biochim. Biophys. Acta, Proteins Proteomics, 2011, 1814, 969–976 CrossRef CAS PubMed.
- J. Clarke, A. M. Hounslow, C. J. Bond, A. R. Fersht and V. Daggett, The effects of disulfide bonds on the denatured state of barnase, Protein Sci., 2000, 9, 2394–2404 CrossRef CAS PubMed.
- S. S. Pendley, Y. B. Yu and T. E. Cheatham, Molecular dynamics guided study of salt bridge length dependence in both fluorinated and non-fluorinated parallel dimeric coiled-coils, Proteins, 2009, 74, 612–629 CrossRef CAS PubMed.
- S.-H. H. Chao, et al., Two structural scenarios for protein stabilization by PEG, J. Phys. Chem. B, 2014, 118, 8388–8395 CrossRef CAS PubMed.
- S. Xiao, et al., Rational modification of protein stability by targeting surface sites leads to complicated results, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 11337–11342 CrossRef CAS PubMed.
- H. M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov and P. E. Bourne, The Protein Data Bank, Nucleic Acids Res., 2000, 28, 235–242 CrossRef CAS PubMed.
- P. J. Flory, Statistical Thermodynamics of Semi-Flexible Chain Molecules, Proc. R. Soc. A, 1956, 234, 60–73 CrossRef CAS.
- A. J. Doig and D. H. Williams, Is the hydrophobic effect stabilizing or destabilizing in proteins?: The contribution of disulphide bonds to protein stability, J. Mol. Biol., 1991, 217, 389–398 CrossRef CAS PubMed.
- J. Clarke and A. R. Fersht, Engineered disulfide bonds as probes of the folding pathway of barnase: increasing the stability of proteins against the rate of denaturation, Biochemistry, 1993, 32, 4322–4329 CrossRef CAS PubMed.
- C. M. Johnson, M. Oliveberg, J. Clarke and A. R. Fersht, Thermodynamics of denaturation of mutants of barnase with disulfide crosslinks, J. Mol. Biol., 1997, 268, 198–208 CrossRef CAS PubMed.
- S. F. Betz, Disulfide bonds and the stability of globular proteins, Protein Sci., 1993, 2, 1551–1558 CrossRef CAS PubMed.
- M. G. Pikkemaat, A. B. M. Linssen, H. J. C. Berendsen and D. B. Janssen, Molecular dynamics simulations as a tool for improving protein stability, Protein Eng., 2002, 15, 185–192 CrossRef CAS PubMed.
- B. Hazes and B. W. Dijkstra, Model building of disulfide bonds in proteins with known three-dimensional structure, Protein Eng., 1988, 2, 119–125 CrossRef CAS PubMed.
- C. Niu, L. Zhu, X. Xu and Q. Li, Rational Design of Disulfide Bonds Increases Thermostability of a Mesophilic 1,3-1,4-β-Glucanase from Bacillus terquilensis, PLoS One, 2016, 11, e0154036 Search PubMed.
- H. Yu and H. Huang, Engineering proteins for thermostability through rigidifying flexible sites, Biotechnol. Adv., 2014, 32, 308–315 CrossRef CAS PubMed.
- V. Daggett and M. Levitt, A model of the molten globule state from molecular dynamics simulations, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 5142–5146 CrossRef CAS.
- V. Daggett and M. Levitt, Protein unfolding pathways explored through molecular dynamics simulations, J. Mol. Biol., 1993, 232, 600–619 CrossRef CAS PubMed.
- M. Levitt, M. Hirshberg, R. Sharon and V. Daggett, Potential Energy Function and Parameters for Simulations of the Molecular Dynamics of Proteins and Nucleic Acids in Solution, Comput. Phys. Commun., 1995, 91, 215–231 CrossRef CAS.
- A. Matouschek, J. T. Kellis Jr, L. Serrano and A. R. Fersht, Mapping the transition state and pathway of protein folding by protein engineering, Nature, 1989, 340, 122–126 CrossRef CAS PubMed.
- V. Daggett, A. Li, L. S. Itzhaki, D. E. Otzen and A. R. Fersht, Structure of the transition state for folding of a protein derived from experiment and simulation, J. Mol. Biol., 1996, 257, 430–440 CrossRef CAS PubMed.
- N. Kulkarni, A. Shendye and M. Rao, Molecular and biotechnological aspects of xylanases, FEMS Microbiol. Rev., 1999, 23, 411–456 CrossRef CAS PubMed.
- Q. Beg, M. Kapoor, L. Mahajan and G. S. Hoondal, Microbial xylanases and their industrial applications: a review, Appl. Microbiol. Biotechnol., 2001, 56, 326–338 CrossRef CAS PubMed.
- T. Collins, C. Gerday and G. Feller, Xylanases, xylanase families and extremophilic xylanases, FEMS Microbiol. Rev., 2005, 29, 3–23 CrossRef CAS PubMed.
- J. C. Joo, S. Pohkrel, S. P. Pack and Y. J. Yoo, Thermostabilization of Bacillus circulans xylanase via computational design of a flexible surface cavity, J. Biotechnol., 2010, 146, 31–39 CrossRef CAS PubMed.
- S. Wells, S. Menor, B. Hespenheide and M. F. Thorpe, Constrained geometric simulation of diffusive motion in proteins, Phys. Biol., 2005, 2, S127–S136 CrossRef CAS PubMed.
- J. C. Joo, S. P. Pack, Y. H. Kim and Y. J. Yoo, Thermostabilization of Bacillus circulans xylanase: Computational optimization of unstable residues based on thermal fluctuation analysis, J. Biotechnol., 2011, 151, 56–65 CrossRef CAS PubMed.
- Y. Liu and B. Kuhlman, RosettaDesign server for protein design, Nucleic Acids Res., 2006, 34, W235–W238 CrossRef CAS PubMed.
- G. Vogt, S. Woell and P. Argos, Protein thermal stability, hydrogen bonds, and ion pairs, J. Mol. Biol., 1997, 269, 631–643 CrossRef CAS PubMed.
- E. D. Merkley, W. W. Parson and V. Daggett, Temperature dependence of the flexibility of thermophilic and mesophilic flavoenzymes of the nitroreductase fold, Protein Eng., Des. Sel., 2010, 23, 327–336 CrossRef CAS PubMed.
- A. Szilágyi and P. Závodszky, Structural differences between mesophilic, moderately thermophilic and extremely thermophilic protein subunits: results of a comprehensive survey, Structure, 2000, 8, 493–504 CrossRef.
- K. Yokota, K. Satou and S.-Y. Ohki, Comparative analysis of protein thermostability: differences in amino acid content and substitution at the surfaces and in the core regions of thermophilic and mesophilic proteins, Sci. Technol. Adv. Mater., 2006, 7, 255–262 CrossRef CAS.
- D. S. Vieira and L. Degreve, An insight into the thermostability of a pair of xylanases: the role of hydrogen bonds, Mol. Phys., 2009, 107, 59–69 CrossRef CAS.
- D. S. Vieira, L. Degrève and R. J. Ward, Characterization of temperature dependent and substrate-binding cleft movements in Bacillus circulans family 11 xylanase: a molecular dynamics investigation, Biochim. Biophys. Acta, 2009, 1790, 1301–1306 CrossRef CAS PubMed.
- J. S. Alponti, R. Fonseca Maldonado and R. J. Ward, Thermostabilization of Bacillus subtilis GH11 xylanase by surface charge engineering, Int. J. Biol. Macromol., 2016, 87, 522–528 CrossRef CAS PubMed.
- J. Chen, H. Yu, C. Liu, J. Liu and Z. Shen, Improving stability of nitrile hydratase by bridging the salt-bridges in specific thermal-sensitive regions, J. Biotechnol., 2012, 164, 354–362 CrossRef CAS PubMed.
- J. Li, A. Mahajan and M. D. Tsai, Ankyrin repeat: A unique motif mediating protein–protein interactions, Biochemistry, 2006, 45, 15168–15178 CrossRef CAS PubMed.
- L. K. Mosavi, T. J. Cammett, D. C. Desrosiers and Z.-Y. Peng, The ankyrin repeat as molecular architecture for protein recognition, Protein Sci., 2004, 13, 1435–1448 CrossRef CAS PubMed.
- B. T. Porebski and A. M. Buckle, Consensus protein design, Protein Eng., Des. Sel., 2016, 29, 245–251 CrossRef CAS PubMed.
- H. K. Binz, M. T. Stumpp, P. Forrer, P. Amstutz and A. Plückthun, Designing repeat proteins: Well-expressed, soluble and stable proteins from combinatorial libraries of consensus ankyrin repeat proteins, J. Mol. Biol., 2003, 332, 489–503 CrossRef CAS PubMed.
- P. Forrer, M. T. Stumpp, H. K. Binz and A. Plückthun, A novel strategy to design binding molecules harnessing the modular nature of repeat proteins, FEBS Lett., 2003, 539, 2–6 CrossRef CAS PubMed.
- G. Interlandi, S. K. Wetzel, G. Settanni, A. Plückthun and A. Caflisch, Characterization and further stabilization of designed ankyrin repeat proteins by combining molecular dynamics simulations and experiments, J. Mol. Biol., 2008, 375, 837–854 CrossRef CAS PubMed.
- P. Alfarano, et al., Optimization of designed armadillo repeat proteins by molecular dynamics simulations and NMR spectroscopy, Protein Sci., 2012, 21, 1298–1314 CrossRef CAS PubMed.
- A.-S. Yang and B. Honig, On the pH Dependence of Protein Stability, J. Mol. Biol., 1993, 231, 459–474 CrossRef CAS PubMed.
- C. Oinonen and J. Rouvinen, Structural comparison of Ntn-hydrolases, Protein Sci., 2000, 9, 2329–2337 CrossRef CAS PubMed.
- M. Arroyo, I. de la Mata, C. Acebal and M. P. Castillón, Biotechnological applications of penicillin acylases: state-of-the-art, Appl. Microbiol. Biotechnol., 2003, 60, 507–514 CrossRef CAS PubMed.
- D. T. Guranda, T. S. Volovik and V. K. Svedas, pH Stability of penicillin acylase from Escherichia coli, Biochemistry, 2004, 69, 1386–1390 CAS.
- D. Suplatov, et al., Computational design of a pH stable enzyme: understanding molecular mechanism of penicillin acylase's adaptation to alkaline conditions, PLoS One, 2014, 9, e100643 Search PubMed.
- S. Leone, et al., Molecular Dynamics Driven Design of pH-Stabilized Mutants of MNEI, a Sweet Protein, PLoS One, 2016, 11, e0158372 Search PubMed.
- M. Strumillo and P. Beltrao, Towards the computational design of protein post-translational regulation, Bioorg. Med. Chem., 2015, 23, 2877–2882 CrossRef CAS PubMed.
- K.-Y. Huang, et al., dbPTM 2016: 10-year anniversary of a resource for post-translational modification of proteins, Nucleic Acids Res., 2016, 44, D435–D446 CrossRef PubMed.
- G. A. Khoury, et al., Proteome-wide post-translational modification statistics: frequency analysis and curation of the swiss-prot database, Sci. Rep., 2011, 1, 4–8 Search PubMed.
- R. J. Solá and K. Griebenow, Effects of glycosylation on the stability of protein pharmaceuticals, J. Pharm. Sci., 2009, 98, 1223–1245 CrossRef PubMed.
- R. Apweiler, H. Hermjakob and N. Sharon, On the frequency of protein glycosylation, as deduced from analysis of the SWISS-PROT database, Biochim. Biophys. Acta, 1999, 1473, 4–8 CrossRef CAS.
- A. M. Sinclair and S. Elliott, Glycoengineering: The effect of glycosylation on the properties of therapeutic proteins, J. Pharm. Sci., 2005, 94, 1626–1635 CrossRef CAS PubMed.
- J. L. Baker, E. Çelik and M. P. DeLisa, Expanding the glycoengineering toolbox: the rise of bacterial N-linked protein glycosylation, Trends Biotechnol., 2013, 31, 313–323 CrossRef CAS PubMed.
- H. W. Hellinga, Rational protein design: combining theory and experiment, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 10015–10017 CrossRef CAS.
- I. N. Berezovsky, K. B. Zeldovich and E. I. Shakhnovich, Positive and negative design in stability and thermal adaptation of natural proteins, PLoS Comput. Biol., 2007, 3, e52 Search PubMed.
- M. Samoudi, et al., Rational design of hyper-glycosylated interferon beta analogs: A computational strategy for glycoengineering, J. Mol. Graphics Modell., 2015, 56, 31–42 CrossRef CAS PubMed.
- J. A. Davey, A. M. Damry, C. K. Euler, N. K. Goto and R. A. Chica, Prediction of Stable Globular Proteins Using Negative Design with Non-native Backbone Ensembles, Structure, 2015, 23, 2011–2021 CrossRef CAS PubMed.
- R. Fonseca-Maldonado, et al., Engineering the pattern of protein glycosylation modulates the thermostability of a GH11 xylanase, J. Biol. Chem., 2013, 288, 25522–25534 CrossRef CAS PubMed.
- G. N. Ramachandran, C. Ramakrishnan and V. Sasisekharan, Stereochemistry of polypeptide chain configurations, J. Mol. Biol., 1963, 7, 95–99 CrossRef CAS PubMed.
- C.-L. Towse, G. Hopping, I. Vulovic and V. Daggett, Nature versus design: the conformational propensities of D-amino acids and the importance of side chain chirality, Protein Eng., Des. Sel., 2014, 27, 447–455 CrossRef CAS PubMed.
- A. Rodriguez-Granillo, S. Annavarapu, L. Zhang, R. L. Koder and V. Nanda, Computational design of thermostabilizing D-amino acid substitutions, J. Am. Chem. Soc., 2011, 133, 18750–18759 CrossRef CAS PubMed.
- B. Imperiali, R. A. Moats, S. L. Fisher and T. J. Prins, A conformational study of peptides with the general structure Ac-L-Xaa-Pro-D-Xaa-L-Xaa-NH2: spectroscopic evidence for a peptide with significant beta-turn character in water and in dimethyl sulfoxide, J. Am. Chem. Soc., 1992, 114, 3182–3188 CrossRef CAS.
- G. Hopping, et al., Designed α-sheet peptides inhibit amyloid formation by targeting toxic oligomers, eLife, 2014, 3, e01681 Search PubMed.
- J. Kellock, G. Hopping, B. Caughey and V. Daggett, Peptides Composed of Alternating L- and D-Amino Acids Inhibit Amyloidogenesis in Three Distinct Amyloid Systems Independent of Sequence, J. Mol. Biol., 2016, 428, 2317–2328 CrossRef CAS PubMed.
- S. Rana, B. Kundu and S. Durani, Stereospecific peptide folds. A rationally designed molecular bracelet, Chem. Commun., 2004, 2462–2463 RSC.
- S. Rana, B. Kundu and S. Durani, A small peptide stereochemically customized as a globular fold with a molecular cleft, Chem. Commun., 2005, 207–209 RSC.
- F. I. Valiyaveetil, M. Sekedat, R. Mackinnon and T. W. Muir, Glycine as a D-amino acid surrogate in the K(+)-selectivity filter, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 17045–17049 CrossRef CAS PubMed.
- K. M. Makwana and R. Mahalakshmi, Capping β-hairpin with N-terminal d-amino acid stabilizes peptide scaffold, Biopolymers, 2016, 106, 260–266 CrossRef CAS PubMed.
- E. M. Anderson, K. M. Larsson and O. Kirk, One biocatalyst--many applications: the use of Candida antarctica B-lipase in organic synthesis, Biocatal. Biotransform., 1998, 16, 181–204 CrossRef CAS.
- H. J. Park, K. Park, Y. H. Kim and Y. J. Yoo, Computational approach for designing thermostable Candida antarctica lipase B by molecular dynamics simulation, J. Biotechnol., 2014, 192(Pt A), 66–70 CrossRef CAS PubMed.
- I. de la Mata, et al., The impact of R53C mutation on the three-dimensional structure, stability, and DNA-binding properties of the human Hesx-1 homeodomain, ChemBioChem, 2002, 3, 726–740 CrossRef CAS PubMed.
- S. Kinet, S. Bernichtein, P. A. Kelly, J. A. Martial and V. Goffin, Biological properties of human prolactin analogs depend not only on global hormone affinity, but also on the relative affinities of both receptor binding sites, J. Biol. Chem., 1999, 274, 26033–26043 CrossRef CAS PubMed.
- M. Zakrzewska, D. Krowarsch, A. Wiedlocha, S. Olsnes and J. Otlewski, Highly stable mutants of human fibroblast growth factor-1 exhibit prolonged biological action, J. Mol. Biol., 2005, 352, 860–875 CrossRef CAS PubMed.
- K. Julenius, E. Thulin, S. Linse and B. E. Finn, Hydrophobic core substitutions in calbindin D9k: effects on stability and structure, Biochemistry, 1998, 37, 8915–8925 CrossRef CAS PubMed.
- B. B. Kragelund, et al., Hydrophobic core substitutions in calbindin D9k: effects on Ca2+ binding and dissociation, Biochemistry, 1998, 37, 8926–8937 CrossRef CAS PubMed.
- A. Morin, J. Meiler and L. S. Mizoue, Computational design of protein-ligand interfaces: potential in therapeutic development, Trends Biotechnol., 2011, 29, 159–166 CrossRef CAS PubMed.
- S. Liang, et al., Exploring the molecular design of protein interaction sites with molecular dynamics simulations and free energy calculations, Biochemistry, 2009, 48, 399–414 CrossRef CAS PubMed.
- B. J. C. Janssen, E. F. Halff, J. D. Lambris and P. Gros, Structure of compstatin in complex with complement component C3c reveals a new mechanism of complement inhibition, J. Biol. Chem., 2007, 282, 29241–29247 CrossRef CAS PubMed.
- D. Ricklin and J. D. Lambris, Compstatin: a complement inhibitor on its way to clinical application, Adv. Exp. Med. Biol., 2008, 632, 273–292 CrossRef CAS PubMed.
- A. Sahu, D. Morikis and J. D. Lambris, Compstatin, a peptide inhibitor of complement, exhibits species-specific binding to complement component C3, Mol. Immunol., 2003, 39, 557–566 CrossRef CAS PubMed.
- P. Tamamis, et al., Design of a modified mouse protein with ligand binding properties of its human analog by molecular dynamics simulations: The case of C3 inhibition by compstatin, Proteins: Struct., Funct., Bioinf., 2011, 79, 3166–3179 CrossRef CAS PubMed.
- P. Tamamis, D. Morikis, C. A. Floudas and G. Archontis, Species specificity of the complement inhibitor compstatin investigated by all-atom molecular dynamics simulations, Proteins: Struct., Funct., Bioinf., 2010, 78, 2655–2667 CAS.
- P. Tamamis, et al., Molecular Dynamics in Drug Design: New Generations of Compstatin Analogs, Chem. Biol. Drug Des., 2012, 79, 703–718 CAS.
- E. M. Storch and V. Daggett, Molecular dynamics simulation of cytochrome b5: implications for protein–protein recognition, Biochemistry, 1995, 34, 9682–9693 CrossRef CAS PubMed.
- J. Moult, Comparison of database potentials and molecular mechanics force fields, Curr. Opin. Struct. Biol., 1997, 7, 194–199 CrossRef CAS PubMed.
- E. M. Storch, V. Daggett and W. M. Atkins, Engineering out motion: introduction of a de novo disulfide bond and a salt bridge designed to close a dynamic cleft on the surface of cytochrome b5, Biochemistry, 1999, 38, 5054–5064 CrossRef CAS PubMed.
- E. M. Storch, J. S. Grinstead, A. P. Campbell, V. Daggett and W. M. Atkins, Engineering Out Motion: A Surface Disulfide Bond Alters the Mobility of Trp 22 in Cytochrome b5 as Probed by Time-Resolved Fluorescence and 1H-NMR Experiments, Biochemistry, 1999, 38, 5065–5075 CrossRef CAS PubMed.
- S. Michielssens, et al., A designed conformational shift to control protein binding specificity, Angew. Chem., Int. Ed., 2014, 53, 10367–10371 CrossRef CAS PubMed.
- S. C. Dodani, et al., Structural, functional, and spectroscopic characterization of the substrate scope of the novel nitrating cytochrome P450 TxtE, ChemBioChem, 2014, 15, 2259–2267 CrossRef CAS PubMed.
- S. C. Dodani, et al., Discovery of a regioselectivity switch in nitrating P450s guided by molecular dynamics simulations and Markov models, Nat. Chem., 2016, 8, 419–425 CrossRef CAS PubMed.
- D. A. C. Beck and V. Daggett, A one-dimensional reaction-coordinate for identification of transition states from explicit solvent Pfold-like calculations, Biophys. J., 2007, 93, 3382–3391 CrossRef CAS PubMed.
- D. S. Kemp and M. L. Casey, Physical Organic Chemistry of Benzisoxazoles. II. Linearity of the Brönsted Free Energy Relationship for the Base-Catalyzed Decomposition of Benzisoxazoles, J. Am. Chem. Soc., 1973, 95, 6670–6680 CrossRef CAS.
- D. Röthlisberger, et al., Kemp elimination catalysts by computational enzyme design, Nature, 2008, 453, 190–195 CrossRef PubMed.
- S. N. Thorn, R. G. Daniels, M. T. Auditor and D. Hilvert, Large rate accelerations in antibody catalysis by strategic use of haptenic charge, Nature, 1995, 373, 228–230 CrossRef CAS PubMed.
- F. Hollfelder, A. J. Kirby and D. S. Tawfik, Off-the-shelf proteins that rival tailor-made antibodies as catalysts, Nature, 1996, 383, 60–62 CrossRef CAS PubMed.
- I. V. Korendovych, et al., Design of a switchable eliminase, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 6823–6827 CrossRef CAS PubMed.
- A. Zanghellini, et al., New algorithms and an in silico benchmark for computational enzyme design, Protein Sci., 2006, 15, 2785–2794 CrossRef CAS PubMed.
- H. K. Privett, et al., Iterative approach to computational enzyme design, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 3790–3795 CrossRef CAS PubMed.
- E. W. Debler, et al., Structural origins of efficient proton abstraction from carbon by a catalytic antibody, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 4984–4989 CrossRef CAS PubMed.
- L. Jiang, et al., De novo computational design of retro-aldol enzymes, Science, 2008, 319, 1387–1391 CrossRef CAS PubMed.
- B. List, C. F. Barbas and R. A. Lerner, Aldol sensors for the rapid generation of tunable fluorescence by antibody catalysis, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 15351–15355 CrossRef CAS.
- J. Z. Ruscio, J. E. Kohn, K. A. Ball and T. Head-Gordon, The influence of protein dynamics on the success of computational enzyme design, J. Am. Chem. Soc., 2009, 131, 14111–14115 CrossRef CAS PubMed.
- L. Wang, et al., Structural analyses of covalent enzyme-substrate analog complexes reveal strengths and limitations of de novo enzyme design, J. Mol. Biol., 2012, 415, 615–625 CrossRef CAS PubMed.
- G. Kiss, V. S. Pande and K. N. Houk, Molecular dynamics simulations for the ranking, evaluation, and refinement of computationally designed proteins, Methods Enzymol., 2013, 523, 145–170 CAS.
- G. Kiss, D. Röthlisberger, D. Baker and K. N. Houk, Evaluation and ranking of enzyme designs, Protein Sci., 2010, 19, 1760–1773 CrossRef CAS PubMed.
- S. Bjelic, et al., Computational design of enone-binding proteins with catalytic activity for the Morita-Baylis-Hillman reaction, ACS Chem. Biol., 2013, 8, 749–757 CrossRef CAS PubMed.
- P. A. Alexander, D. A. Rozak, J. Orban and P. N. Bryan, Directed Evolution of Highly Homologous Proteins with Different Folds by Phage Display: Implications for the Protein Folding Code, Biochemistry, 2005, 44, 14045–14054 CrossRef CAS PubMed.
- Y. He, D. C. Yeh, P. Alexander, P. N. Bryan and J. Orban, Solution NMR structures of IgG binding domains with artificially evolved high levels of sequence identity but different folds, Biochemistry, 2005, 44, 14055–14061 CrossRef CAS PubMed.
- K. A. Scott and V. Daggett, Folding mechanisms of proteins with high sequence identity but different folds, Biochemistry, 2007, 46, 1545–1556 CrossRef CAS PubMed.
- A. Morrone, M. E. McCully, P. N. Bryan, M. Brunori, S. Gianni, V. Daggett and C. Travaglini-Allocatelli, The denatured state dictates the topology of two proteins with almost identical sequence but different native structure and function, J. Biol. Chem., 2011, 286, 3863–3872 CrossRef CAS PubMed.
- R. Day and V. Daggett, Direct observation of microscopic reversibility in protein folding, J. Mol. Biol., 2007, 366, 677–686 CrossRef CAS PubMed.
- M. E. McCully, D. A. C. Beck and V. Daggett, Microscopic reversibility of protein folding in molecular dynamics simulations of the engrailed homeodomain, Biochemistry, 2008, 47, 4079–7089 CrossRef PubMed.
- A. Li and V. Daggett, Characterization of the transition state of protein unfolding by use of molecular dynamics: chymotrypsin inhibitor 2, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 10430–10434 CrossRef CAS.
- A. Li and V. Daggett, Identification and characterization of the unfolding transition state of chymotrypsin inhibitor 2 by molecular dynamics simulations, J. Mol. Biol., 1996, 257, 412–429 CrossRef CAS PubMed.
- A. G. Ladurner, L. S. Itzhaki, V. Daggett and A. R. Fersht, Synergy between simulation and experiment in describing the energy landscape of protein folding, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 8473–8478 CrossRef CAS.
- M. Levitt, Molecular dynamics of native protein, II. Analysis and nature of motion, J. Mol. Biol., 1983, 168, 621–657 CrossRef CAS PubMed.
- S. Piana, et al., Computational design and experimental testing of the fastest-folding β-sheet protein, J. Mol. Biol., 2011, 405, 43–48 CrossRef CAS PubMed.
- M. J. Cocco and J. T. Lecomte, Characterization of hydrophobic cores in apomyoglobin: a proton NMR spectroscopy study, Biochemistry, 1990, 29, 11067–11072 CrossRef CAS PubMed.
- P. A. Jennings and P. E. Wright, Formation of a molten globule intermediate early in the kinetic folding pathway of apomyoglobin, Science, 1993, 262, 892–896 CAS.
- R. M. Ballew, J. Sabelko and M. Gruebele, Direct observation of fast protein folding: the initial collapse of apomyoglobin, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 5759–5764 CrossRef CAS.
- R. M. Ballew, J. Sabelko and M. Gruebele, Observation of distinct nanosecond and microsecond protein folding events, Nat. Struct. Biol., 1996, 3, 923–926 CrossRef CAS PubMed.
- J. S. Goodman, S. H. Chao, T. V. Pogorelov and M. Gruebele, Filling up the heme pocket stabilizes apomyoglobin and speeds up its folding, J. Phys. Chem. B, 2014, 118, 6511–6518 CrossRef CAS PubMed.
- C. Garcia, C. Nishimura, S. Cavagnero, H. J. Dyson and P. E. Wright, Changes in the apomyoglobin folding pathway caused by mutation of the distal histidine residue, Biochemistry, 2000, 39, 11227–11237 CrossRef CAS PubMed.
- P. Picotti, et al., Modulation of the structural integrity of helix F in apomyoglobin by single amino acid replacements, Protein Sci., 2004, 13, 1572–1585 CrossRef CAS PubMed.
- U. Mayor, C. M. Johnson, J. G. Grossmann, S. Sato, G. S. Jas, S. M. V. Freund, N. R. Guydosh, D. O. V. Alonso, V. Daggett and A. R. Fersht, The Complete Folding Pathway of a Protein from Nanoseconds to Microseconds, Nature, 2003, 421, 863–867 CrossRef CAS PubMed.
- U. Mayor, C. M. Johnson, V. Daggett and A. R. Fersht, Protein folding and unfolding in microseconds to nanoseconds by experiment and simulation, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 13518–13522 CrossRef CAS PubMed.
- M. L. DeMarco, D. O. V. Alonso and V. Daggett, Diffusing and colliding: The atomic level folding/unfolding pathway of a small helical protein, J. Mol. Biol., 2004, 341, 1109–1124 CrossRef CAS PubMed.
- S. Gianni, N. R. Guydosh, F. Khan, T. D. Caldas, U. Mayor, G. W. N. White, M. L. DeMarco, V. Daggett and A. R. Fersht, Unifying features in protein-folding mechanisms, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 13286–13291 CrossRef CAS PubMed.
- M. McCully, D. A. C. Beck and V. Daggett, Microscopic reversibility in single-molecule folding/unfolding of a protein at its Tm, Biochemistry, 2008, 47, 7079–7089 CrossRef CAS PubMed.
- T. D. Sharpe, A. L. Jonsson, T. J. Rutherford, V. Daggett and A. R. Fersht, The role of the turn in β-hairpin formation during WW domain folding, Protein Sci., 2007, 16, 2233–2239 CrossRef CAS PubMed.
- T. L. Religa, J. S. Markson, U. Mayor, S. M. V. Freund and A. R. Fersht, Solution structure of a protein denatured state and folding intermediate, Nature, 2005, 437, 1053–1056 CrossRef CAS PubMed.
- G. W. N. White, S. Gianni, J. G. Grossman, P. Jemth, A. R. Fersht and V. Daggett, Simulation and Experiment Conspire to reveal Cryptic Intermediates and the Slide from the Nucleation-Condensation to Framework Mechanism of Folding, J. Mol. Biol., 2005, 350, 757–775 CrossRef CAS PubMed.
- Q. Peng, et al., Mechanical design of the third FnIII domain of tenascin-C, J. Mol. Biol., 2009, 386, 1327–1342 CrossRef CAS PubMed.
- S. P. Ng, et al., Mechanical unfolding of TNfn3: the unfolding pathway of a fnIII domain probed by protein engineering, AFM and MD simulation, J. Mol. Biol., 2005, 350, 776–789 CrossRef CAS PubMed.
- Y. Cao, T. Yoo and H. Li, Single molecule force spectroscopy reveals engineered metal chelation is a general approach to enhance mechanical stability of proteins, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 11152–11157 CrossRef CAS PubMed.
- S. Zhuang, Q. Peng, Y. Cao and H. Li, Modulating the Mechanical Stability of Extracellular Matrix Protein Tenascin-C in a Controlled and Reversible Fashion, J. Mol. Biol., 2009, 390, 820–829 CrossRef CAS PubMed.
- K. Rutherford and V. Daggett, Polymorphisms and Disease: Hotspots of Inactivation in Methyltransferases, Trends Biochem. Sci., 2010, 35, 531–538 CrossRef CAS PubMed.
- F. Chiti and C. M. Dobson, Protein misfolding, functional amyloid, and human disease, Annu. Rev. Biochem., 2006, 75, 333–366 CrossRef CAS PubMed.
- C. Haass and D. J. Selkoe, Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid beta-peptide, Nat. Rev. Mol. Cell Biol., 2007, 8, 101–112 CrossRef CAS PubMed.
- J. D. Sipe, et al., Amyloid fibril proteins and amyloidosis: chemical identification and clinical classification International Society of Amyloidosis 2016 Nomenclature Guidelines, Amyloid, 2016, 23, 209–213 CrossRef CAS PubMed.
- D. O. V. Alonso, S. DeArmond, F. Cohen and V. Daggett, Mapping the Early Steps in the Conversion of the Prion Protein, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 2985–2989 CrossRef CAS PubMed.
- D. O. V. Alons, C. An and V. Daggett, Simulations of Biomolecules: Characterization of the Early Steps in the pH-Induced Conformational Conversion of the Hamster, Bovine, and Human Forms of the Prion Protein, Philos. Trans. R. Soc., A, 2002, 360, 1165–1178 CrossRef PubMed.
- R. S. Armen, D. O. V. Alonso and V. Daggett, Anatomy of an amyloidogenic intermediate: Conversion of β-sheet to α-sheet structure in transthyretin at acidic pH, Structure, 2004, 12, 1847–1863 CrossRef CAS PubMed.
- R. S. Armen, M. L. DeMarco, D. O. V. Alonso and V. Daggett, Pauling and Corey's alpha-pleated sheet structure may define the prefibrillar amyloidogenic intermediate in amyloid disease, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 11622–11627 CrossRef CAS PubMed.
- R. E. Steward, R. S. Armen and V. Daggett, Different disease-causing mutations in transthyretin trigger the same conformational conversion, Protein Eng., Des. Sel., 2008, 21, 187–195 CrossRef CAS PubMed.
- M. Yang, M. Lei, B. Yordanov and S. Huo, Peptide plane can flip in two opposite directions: Implication in amyloid formation of transthyretin, J. Phys. Chem. B., 2006, 110, 5829–5833 CrossRef CAS PubMed.
- S. L. Kazmirski and V. Daggett, Non-native interactions in protein folding intermediates: molecular dynamics simulations of hen lysozyme, J. Mol. Biol., 1998, 284, 793–806 CrossRef CAS PubMed.
- R. S. Armen and V. Daggett, Characterization of two distinct beta2-microglobulin unfolding intermediates that may lead to amyloid fibrils of different morphology, Biochemistry, 2005, 44, 16098–16107 CrossRef CAS PubMed.
- R. S. Armen, B. M. Bernard, R. Day, D. O. V. Alonso and V. Daggett, Characterization of a possible amyloidogenic precursor in glutamine-repeat neurodegenerative diseases, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 13433–13438 CrossRef CAS PubMed.
- V. Babin, C. Roland and C. Sagui, The α-sheet: A missing-in-action secondary structure?, Proteins, 2011, 79, 937–946 CrossRef CAS PubMed.
- L. Pauling and R. B. Corey, The pleated sheet, a new layer configuration of polypeptide chains, Proc. Natl. Acad. Sci. U. S. A., 1951, 37, 251–256 CrossRef CAS.
- V. Daggett, α-sheet: The toxic conformer in amyloid diseases?, Acc. Chem. Res., 2006, 39, 594–602 CrossRef CAS PubMed.
- D. A. C. Beck, D. O. V. Alonso, D. Inoyama and V. Daggett, The intrinsic conformational propensities of the twenty naturally occurring amino acids and reflection of these propensities in proteins, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 12259–12264 CrossRef CAS PubMed.
- C.-L. Towse, J. Vymetal, J. Vondrasek and V. Daggett, Insights into unfolded proteins from the intrinsic φ/ψ propensities of the AAXAA host-guest series, Biophys. J., 2016, 110, 348–361 CrossRef CAS PubMed.
- C.-L. Towse, G. Hopping, I. Vulovic and V. Daggett, Nature versus design: The conformational propensities of D-amino acids and the importance of side chain chirality, Protein Eng., Des. Sel., 2014, 27, 447–455 CrossRef CAS PubMed.
- M. C. Childers, C.-L. Towse and V. Daggett, The effect of chirality and steric hindrance on intrinsic backbone conformational propensities: Tools for protein design, Protein Eng., Des. Sel., 2016, 29, 271–280 CrossRef CAS PubMed.
- C.-L. Towse, S. J. Rysavy, I. M. Vulovic and V. Daggett, New Dynamic Rotamer Libraries: Data-Driven Analysis of Side Chain Conformational Propensities, Structure, 2016, 24, 187–199 CrossRef CAS PubMed.
- M. C. Childers, C. L. Towse and V. Daggett, Molecular dynamics-derived rotamer libraries for D-amino acids, 2016, submitted for publication.
- D. A. C. Beck, A. L. Jonsson, D. Schaeffer, K. A. Scott, R. Day, R. D. Toofanny, D. O. V. Alonso and V. Daggett, Dynameomics: Mass annotation of protein dynamics and unfolding in water by high-throughput atomistic molecular dynamics simulations, Protein Eng., Des. Sel., 2008, 21, 353–368 CrossRef CAS PubMed.
- M. W. Van der Kamp, P. C. Anderson, D. A. C. Beck, N. C. Benson, A. L. Jonsson, E. D. Merkley, R. D. Schaeffer, A. D. Scouras, A. Simms, R. D. Toofanny and V. Daggett, Dynameomics: A comprehensive database of protein dynamics, Structure, 2010, 18, 423–435 CrossRef CAS PubMed.
- C. Camilloni, et al., Rational design of mutations that change the aggregation rate of a protein while maintaining its native structure and stability, Sci. Rep., 2016, 6, 25559 CrossRef CAS PubMed.
- F. Fogolari, et al., Molecular dynamics simulation suggests possible interaction patterns at early steps of beta2-microglobulin aggregation, Biophys. J., 2007, 92, 1673–1681 CrossRef CAS PubMed.
- G. Esposito, et al., The Controlling Roles of Trp60 and Trp95 in β2-Microglobulin Function, Folding and Amyloid Aggregation Properties, J. Mol. Biol., 2008, 378, 885–895 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |