
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDevelopment of NMR and thermal shift assays for the evaluation of Mycobacterium tuberculosis isocitrate lyase inhibitors†
Ram Prasad
Bhusal
 a,
Krunal
Patel
a,
Brooke X. C.
Kwai‡
a,
Anne
Swartjes‡
ab,
Ghader
Bashiri
cd,
Jóhannes
Reynisson
a,
Jonathan
Sperry
*a and
Ivanhoe K. H.
Leung
*a
a,
Krunal
Patel
a,
Brooke X. C.
Kwai‡
a,
Anne
Swartjes‡
ab,
Ghader
Bashiri
cd,
Jóhannes
Reynisson
a,
Jonathan
Sperry
*a and
Ivanhoe K. H.
Leung
*a
aSchool of Chemical Sciences, The University of Auckland, Private Bag 92019, Victoria Street West, Auckland 1142, New Zealand. E-mail: j.sperry@auckland.ac.nz (JS); i.leung@auckland.ac.nz (IKHL)
bInstitute for Molecules and Materials, Radboud University, Heyendaalseweg 135, 6525 AJ, Nijmegen, The Netherlands
cSchool of Biological Sciences, The University of Auckland, Private Bag 92019, Victoria Street West, Auckland 1142, New Zealand
dMaurice Wilkins Centre for Molecular Biodiscovery, The University of Auckland, Private Bag 92019, Victoria Street West, Auckland 1142, New Zealand
First published on 17th October 2017
Abstract
The enzymes isocitrate lyase (ICL) isoforms 1 and 2 are essential for Mycobacterium tuberculosis survival within macrophages during latent tuberculosis (TB). As such, ICLs are attractive therapeutic targets for the treatment of tuberculosis. However, there are few biophysical assays that are available for accurate kinetic and inhibition studies of ICL in vitro. Herein we report the development of a combined NMR spectroscopy and thermal shift assay to study ICL inhibitors for both screening and inhibition constant (IC50) measurement. Operating this new assay in tandem with virtual high-throughput screening has led to the discovery of several new ICL1 inhibitors.
Introduction
Tuberculosis (TB) is a high burden infectious disease that is caused by Mycobacterium tuberculosis.1,2 In 2015, there were over 10 million new TB cases and around 1.8 million TB-related deaths.3 TB has a long latency period; once a human is infected with M. tuberculosis, the bacteria may stay inactive within macrophages for many years leading to a syndrome that is known as latent TB.4–6 The environment inside macrophages is relatively hypoxic and lacking in external nutrients. In order to survive under these conditions, M. tuberculosis is able to simultaneously catabolise different carbon sources, including fatty acids and cholesterol that are available in relative abundance inside macrophages.7–9 The enzymes isocitrate lyase (ICL) isoforms 1 and 2 play essential roles in this metabolic adaptation.10 ICLs are key enzymes in both the M. tuberculosis glyoxylate and methylcitrate cycles. In the glyoxylate cycle, ICLs catalyse the conversion of the tricarboxylic acid (TCA) cycle intermediate isocitrate into glyoxylate and succinate (Fig. 1a), thus bypassing the two decarboxylation steps in the TCA cycle and preserving these carbons for gluconeogenesis.11,12 In the methylcitrate cycle, ICLs catalyse the conversion of methylisocitrate, an intermediate of the propionate degradation pathway, to pyruvate and succinate. Propionate, which is toxic to the bacteria, is generated by β-oxidation of odd chain fatty acids and cholesterol that M. tuberculosis may utilise as carbon sources.12,13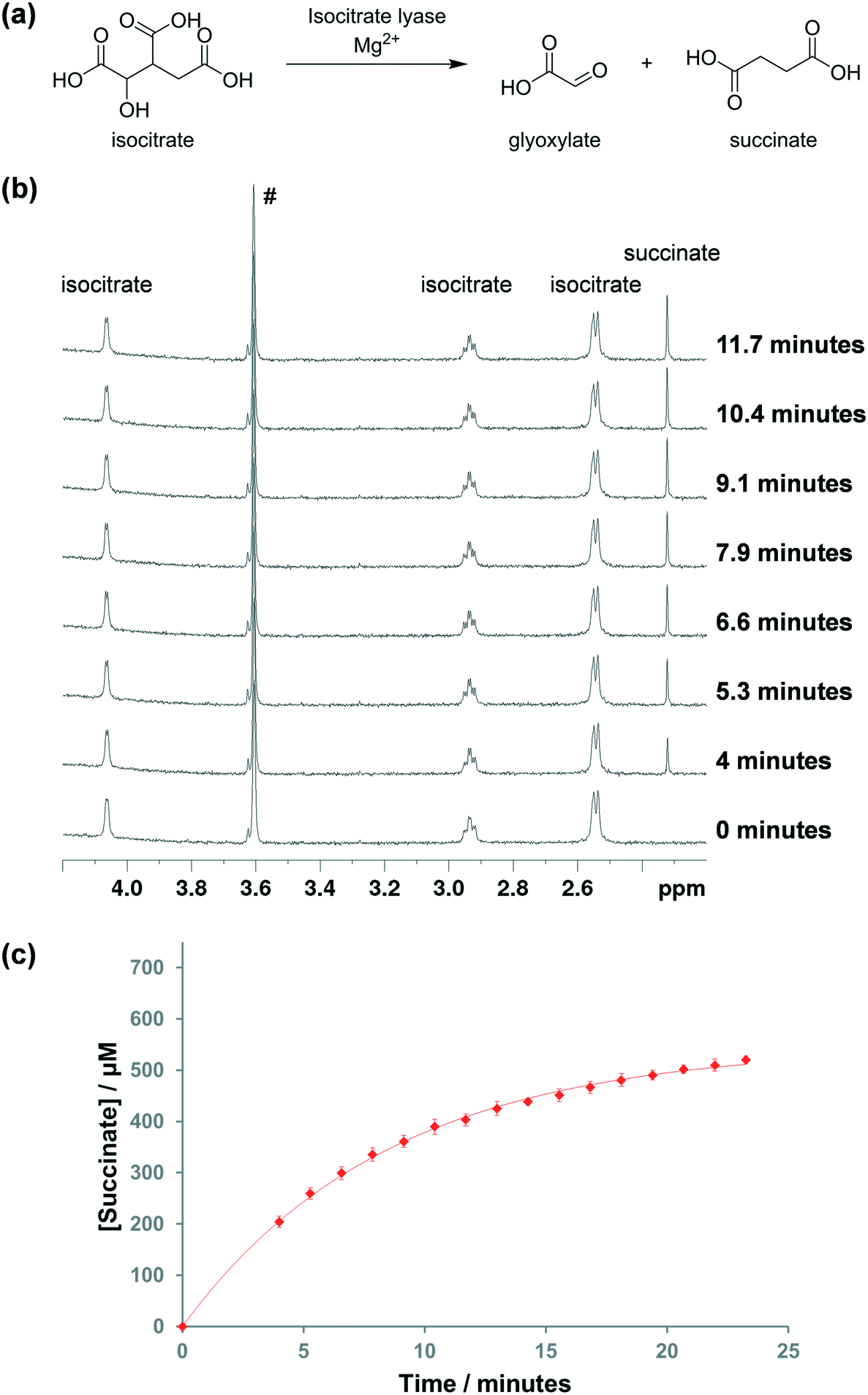 | ||
| Fig. 1 (a) Isocitrate lyase catalyses the conversion of isocitrate to glyoxylate and succinate; (b) 1H NMR spectroscopy to monitor ICL1-catalysed turnover of isocitrate into succinate; (c) corresponding plot of the isocitrate turnover data. The curve was added to aid visualisation. Sample contained 190 nM ICL1, 1 mM DL-isocitrate, 5 mM MgCl2 and 50 mM Tris/Tris-D11 (pH 7.5) in 90% H2O and 10% D2O. The hashtag (#) indicates Tris/Tris-D11 peak. The errors shown are the standard deviation from three separate measurements. | ||
Given the pivotal roles ICLs play in the survival of M. tuberculosis inside macrophages, ICLs are attractive inhibition targets for the treatment of latent TB.14–16 We recently instigated a research programme aimed at identifying new inhibitors of ICLs, but it was readily apparent that few accurate biophysical assays are available to study ICL kinetics and inhibition in vitro. Most ICL assays rely on ultraviolet/visible (UV/vis) spectrophotometry to determine the amount of glyoxylate that is formed as a result of ICL-catalysed reactions. For example, one method uses lactate dehydrogenase (LDH) to catalyse the reduction of glyoxylate to glycolate, during which NADH (a cosubstrate of LDH) is oxidised to NAD+. The decrease in NADH concentration is then measured by UV/vis spectrophotometry using a tetrazolium/formazan dye.17 Another method relies on the reaction phenylhydrazine with glyoxylate to form a hydrazone, which is subsequently analysed by UV/vis spectrophotometry.18 In addition to kinetic assays, the use of native non-denaturing mass spectrometry and intrinsic protein fluorescence to monitor ICL-inhibitor binding interactions have also been reported.19 However, these assays have several drawbacks. Auto-oxidation of NADH to NAD+ may affect the accuracy of the LDH-coupled assay.20 In addition, this method is not suitable for measuring the methylisocitrate lyase activity of ICLs because LDH cannot take pyruvate as substrate.20 For the phenylhydrazine-coupled assay, the accuracy of the assay may be compromised by the rate of the glyoxylate-phenylhydrazone complex formation, which is pH dependent and gets slower above pH 7.21,22 Phenylhydrazine is also unstable at pH 7 and above, with the breakdown products may lead to a slow increase in the UV absorption, thus affecting the accuracy of the measurements.21,22
1H nuclear magnetic resonance (NMR) spectroscopy is an established technique for the study of enzyme kinetics that has been used to characterise different enzyme systems including (but not limited to) carbohydrate-processing enzymes,23,24 enzymes related to antibiotic resistance25 and oxygenases.26,271H NMR spectroscopy enables the direct monitoring of reaction kinetics in real time and accurate, quantitative information can be obtained by following changes in the peak area of the resonances associated with the substrate and/or reaction product(s). In contrast, thermal shift assay is a simple and high-throughput method that can be used to study protein–ligand binding interactions by measuring the melting temperature of a protein by the use of a fluorescence dye that is sensitive to changes in hydrophobic environment.28–30 When a protein unfolds, it exposes its hydrophobic core. This enables the dye to bind to the exposed hydrophobic regions, which lead to fluorescence. Ligand binding may stabilise or destabilise the protein towards thermal denaturation, leading to a positive or negative shift in the protein's melting temperature.31,32 We reasoned that a strategy that combined 1H NMR spectroscopy and a thermal shift assay would provide a good way to accurately monitor ICL kinetics and inhibition. By using M. tuberculosis ICL1 as a model system, we have optimised the experimental conditions and demonstrated the feasibility of applying a joint NMR and thermal shift strategy for inhibitor screening, inhibition constant (IC50) measurement and for elucidating the modes of action of ICL inhibitors. Finally, this technique is exemplified by work in tandem with virtual high-throughput screening, which subsequently led to the discovery of several novel ICL1 inhibitors. ICL2 was not used in this study because it was reported to be unstable in vitro17 and it possesses relatively low catalytic activity when compared to ICL1.12
Results and discussion
ICL1 enzyme kinetics by 1H NMR
We first tested the use of 1H NMR spectroscopy to monitor the ICL1-catalysed turnover of isocitrate to succinate and glyoxylate. DL-Isocitrate, which is available commercially, was used as the substrate. MgCl2 was added to the reaction mixture as it was previously shown to be important for ICL1 activity.171H spectra were recorded at ∼1.3 minute intervals. Upon addition of the enzyme, the peaks corresponding to isocitrate dropped in intensity, which was accompanied by the appearance of a new singlet peak at 2.3 ppm, corresponding to succinate (Fig. 1b). Integration of the isocitrate and succinate peaks showed that the reaction appeared to slow down when ∼50% of the isocitrate was consumed (Fig. 1c). As the isocitrate was a racemic mixture, this result infers that ICL1 has a preference for one enantiomer, which is in agreement with a previous study that showed D-isocitrate is the preferred substrate of the enzyme.33Divalent metals play important role in the activity of ICL1. Previous studies showed that Mg2+ (and to a lesser extent, Mn2+) are required for optimal ICL1 activity.17,34 In order to confirm the concentration of divalent magnesium that is required for optimal activity of the enzyme, the reaction was run using different concentrations of MgCl2. Under our reaction conditions, 500 μM of MgCl2 was required for the optimal activity (Fig. S1†). At least 500 μM of MgCl2 was used in all subsequent kinetic and inhibition assays.
The kinetic parameters for ICL1 with DL-isocitrate were then evaluated by 1H NMR. The Michaelis constant (KM) was found to be 290 ± 10 μM and the catalytic constant (kcat) was determined to be 4.3 ± 0.1 s−1 (Fig. S2†). These values were similar to those obtained by Gould et al. using the aforementioned LDH assay, which were 190 μM and 5.24 s−1 respectively (Table 1).12 The slight discrepancy between the two measurements is likely due to differences in the reaction conditions. Overall, this validated the accuracy of our 1H NMR assay to study ICL1 kinetics.
| K M/μM | k cat/s−1 | |
|---|---|---|
| This study | 290 ± 10 | 4.3 ± 0.1 |
| Gould et al.12 | 190 | 5.24 |
We then tested the use of 1H NMR spectroscopy to monitor the ICL1-catalysed turnover of methylisocitrate to pyruvate and succinate (Fig. S3a†). (2S,3R)-2-Methylisocitrate was used as substrate. In the presence of ICL1 and Mg2+, two new singlet signals at ∼2.3 ppm, one corresponded to succinate and the other corresponded to pyruvate, were found to increase in intensity over time (Fig. S3b†). This was coupled with a drop in intensity of the methylisocitrate signals. We then repeated the experiments at different methylisocitrate concentrations. Interestingly, substrate inhibition was observed when methylisocitrate concentration exceeded 1 mM (Fig. S4†). Substrate inhibition was not observed when isocitrate was used as substrate. Further investigations are required to fully understand the biological significance of these observations. Overall, our results showed that 1H NMR spectroscopy is a versatile and informative tool to study ICL1 kinetics that allows the use of different substrates and enables kinetic information to readily be measured and quantified.
Inhibition studies of ICL1 by 1H NMR
We then applied our new NMR-based assay to study ICL1 inhibition. Initially, we chose four known ICL inhibitors, including three so-called first generation inhibitors itaconic acid,35 3-nitropropionate36 and 3-bromopyruvate.37 Itaconic acid and 3-nitropropionate are noncovalent inhibitors of ICL1 whereas 3-bromopyruvate inhibits ICL1 in a covalent manner. Methyl 4-(4-methoxyphenyl)-4-oxobut-2-enoate, an inhibitor that was discovered last year by Liu et al. using high-throughput screening, was also evaluated (Table 2).38| Inhibitor | Structure | IC50/μM |
|---|---|---|
| 3-Nitropropionate |

|
14.7 ± 1.8 |
| 3-Bromopyruvate |
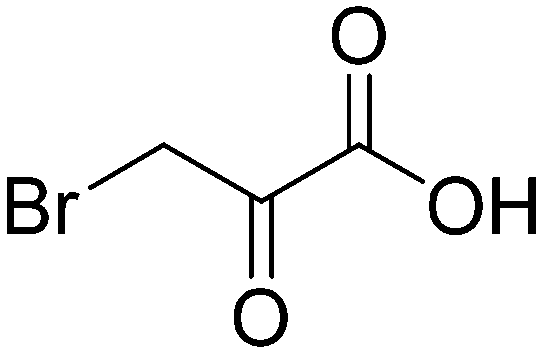
|
17.5 ± 1.0 |
| Itaconic acid |

|
29.4 ± 4.1 |
| Methyl-4-(4-methoxyphenyl)-4-oxobut-2-enoate |

|
250 ± 7 |
| Compound 29 |

|
>100 |
| Compound 38 |
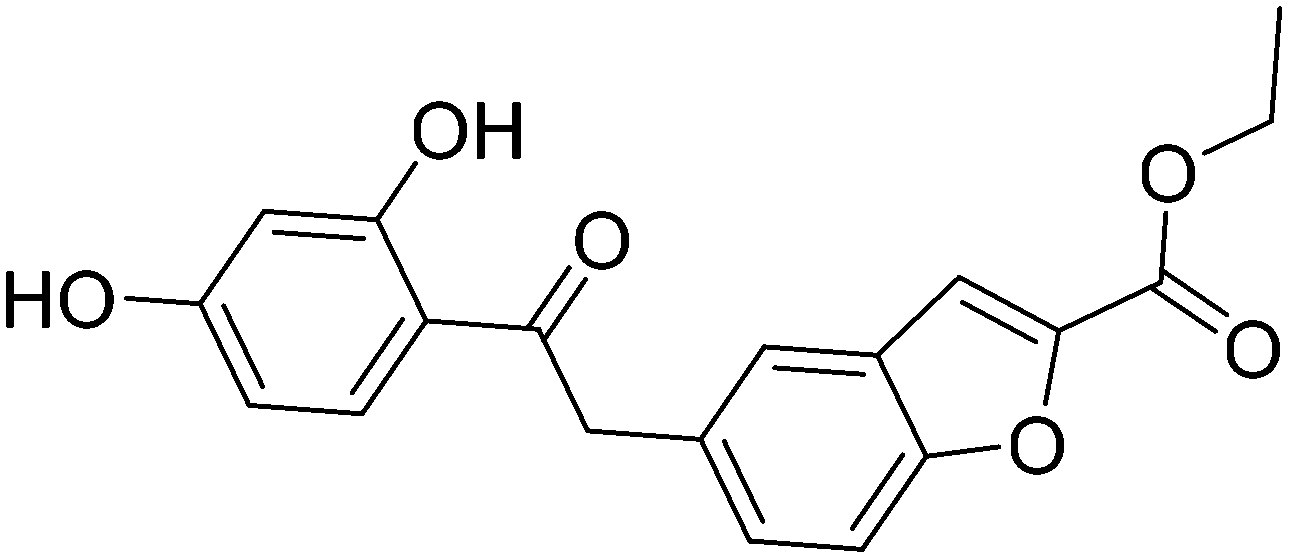
|
>100 |
Single concentration inhibition experiments were first conducted (Fig. S5†). In agreement with previous studies,17 our 1H NMR assay showed that 3-nitropropionate was the most potent inhibitor amongst 3-nitropropionate, 3-bromopyruvate and itaconic acid. Under our assay condition, methyl 4-(4-methoxyphenyl)-4-oxobut-2-enoate was the weakest of the four tested. We then repeated our measurements at different inhibitor concentrations in order to obtain quantitative inhibition information (IC50; Table 2 and Fig. S6–S9†). The IC50 values of 3-nitropropionate, 3-bromopyruvate, itaconic acid and methyl 4-(4-methoxyphenyl)-4-oxobut-2-enoate were found to be 14.7 ± 1.8 μM, 17.5 ± 1.0 μM, 29.4 ± 4.1 μM and 250 ± 7 μM respectively. The reported IC50 value for methyl 4-(4-methoxyphenyl)-4-oxobut-2-enoate was 30.9 μM.38 The slight discrepancy in our measured and reported IC50 values for methyl 4-(4-methoxyphenyl)-4-oxobut-2-enoate is likely due to the different reaction conditions and assays used in the two studies. Overall, our results show that 1H NMR is a useful tool to study ICL1 inhibition in vitro, enabling a rapid evaluation of inhibitor strength as well as providing quantitative information such as IC50.
Thermal shift assay to study ICL1-inhibitor interactions
Although 1H NMR spectroscopy was found to be a useful method to study ICL1 inhibition, it is relatively low throughput and labour intensive. A high throughput assay is needed to facilitate the efficient screening and development of new ICL inhibitors. Thermal shift assays are a widely-used method to study protein–ligand interactions.28–30 The principle of a thermal shift assay is based on the premise that ligand binding can stabilise or destabilise protein to thermal denaturing, and therefore lead to a shift in the protein's melting temperature.First, the melting temperature of ICL1 was measured. As MgCl2 is important for the activity of the enzyme, a saturating concentration of 1 mM was used. The melting temperature of ICL1 in the presence of MgCl2 was found to be 43.0 °C. Next, the melting temperatures of ICL1 in the presence of a saturating concentration (1 mM) of the aforementioned inhibitors and MgCl2 were measured. Addition of 3-bromopyruvate or itaconic acid were found to stabilise ICL1, with positive shifts to melting temperatures of 52.5 °C and 53.3 °C respectively. Interestingly, 3-nitropropionate and methyl 4-(4-methoxyphenyl)-4-oxobut-2-enoate were found to destabilise the protein, with negative thermal shifts to 40.9 °C and 37.6 °C respectively (Fig. S10†).
A negative thermal shift upon ligand binding has been previously observed for other protein systems.31,32 A positive thermal shift may be observed if the ligand induces the protein to adapt a more stable ‘closed’ conformation, whilst negative thermal shift may be observed if the ligand keeps the protein in a less stable ‘open’ conformation. Previous structural studies by Sharma et al. showed that ICL1 may undergo a two-step conformation change upon substrate binding (Fig. S11†).39 Indeed, a crystal structure of ICL1 in the presence of both 3-nitropropionate and glyoxylate was found to adapt a ‘closed’ conformation (PDB id: 1F8I). 3-Nitropropionate is a structural analogue of succinate.36 We reasoned that the binding of 3-nitropropionate on its own may keep ICL1 in the open conformation in order to allow glyoxylate to bind. However, in the presence of both 3-nitropropionate and glyoxylate, the protein can then undergo conformational change to the ‘closed’ conformation, as suggested by the crystal structure, to catalyse the reverse reaction. This proposal is consistent with the mechanism suggested by Sharma et al.39 It should also be noted that binding of 3-nitropropionate or methyl 4-(4-methoxyphenyl)-4-oxobut-2-enoate may induce a change in the oligomeric state of ICL1, which exists as a tetramer in solution.39 However, based on evidence from X-ray crystallography39 and molecular docking,38 both compounds are not known to bind at the oligomerisation interface between the ICL1 monomers, or interfere with the residues that were previously identified as important for the protein's oligomerisation.40
Application of the combined 1H NMR and thermal shift assays with virtual high-throughput screening
Virtual high-throughput screening is a cost-effective and efficient strategy to identify chemical structures that are potentially important for binding to a target protein.41–43 Obviously, the hits identified by virtual high-throughput screening needed to be verified experimentally. In order to test the applicability of the 1H NMR and thermal shift assays and to identify new inhibitors of ICL1, a virtual screen was conducted. Using the crystal structure of ICL1 (PDB ID: 1F8I, resolution 2.25 Å),39 a screen was performed with the InterBioScreen Ltd natural product collection.44 9050 compounds were screened and four different scoring functions, including GoldScore (GS),45 ChemScore (CS),46,47 Piecewise Linear Potential (ChemPLP)48 and Astex Statistical Potential (ASP),49 were used. Ten docking runs were allowed for each compound with virtual screening setting (30%). Based on the scores, ligands with predicted low GS (<45), CS (<20), ChemPLP (<45), ASP (<20) as well as those with no hydrogen bonding (HB = 0) were eliminated, which resulted in 840 compounds. These compounds were screened again with high search efficiency (100%) and fifty docking runs. Candidates with low GS (<38), CS (<17), ChemPLP (<38), ASP (<17) as well as those with predicted limited hydrogen bonding (HB < 1) were eliminated, resulting in 205 candidates. For both rounds of screening the cut-off values of the scores were determined based on the scores of the known inhibitors itaconic acid, 3-nitropropionate and 3-bromopyruvate. Furthermore, only compounds with predicted hydrogen bonding were taken forward since hydrogen bond is important not only for the affinity but also the specificity of the ligand binding.50 These candidates were then visually inspected for consensus of the best predicted configuration of the ligands between the scoring functions. Ligands that showed plausible configurations, i.e., not strained, lipophilic moieties not pointing into the water environment resulting in an entropy penalty, were taken forward. Furthermore, compounds that did not contain undesirable moieties that are linked to general cell toxicity (e.g. thiourea and aliphatic ketones) and chemical reactivity (e.g. Michael acceptors and imines), were chosen.51,52 This screening methodology has been successfully applied previously to find active ligands for various biomolecular systems.53–56 In total, 41 compounds were selected for experimental testing (Fig. S12 and Table S1†).We then applied the 1H NMR and thermal shift assays to verify the hits obtained from the virtual screen. First, we tested the compounds using the thermal shift assay. Out of the 41 compounds, 19 induced a shift of more than 0.5 °C (positive or negative) in the melting temperature of ICL (Fig. S13†). This was followed with 1H NMR-based single concentration inhibition experiment to test the 19 hits. The result showed that two molecules significantly inhibited ICL1 (compounds 29 and 38, Fig. S14†). The IC50 of the molecules were both >100 μM (Fig. S15 and S16† and Table 2). Molecular modelling suggests compounds 29 and 38 both occupy the substrate and Mg2+ binding sites. The reason for the relatively low IC50 values is due to the removal of the Mg2+ ion from the binding pocket upon inhibitor binding. Mg2+ sits within a cavity that is predicted to be occupied by aliphatic moieties of the inhibitors. Thus, the inhibitors need to displace the magnesium ion to bind efficiently, which would require a considerable energy expenditure due to the saturation concentration (5 mM) of the ion in the experimental setup. The main stream molecular descriptors (molecular weight, log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P, hydrogen bond donors/acceptors, polar surface area and rotatable bonds see Table S2†) for compounds 29 and 38 were calculated and they conform to drug-like chemical space except logP and numbers of hydrogen bond donors, which are in lead-like chemical space (for the definition of chemical space see ref. 57). Furthermore, the molecular weight for the hits is in the low to mid 300s, making them excellent starting points for chemical modification and further development. Finally, nine close structural derivatives were purchased to generate a structural activity (SAR) profile (Fig. S17†), but none showed any activity. In general, docking to the binding site showed that these compounds are too bulky to fit into it. Overall, our results show that combining 1H NMR and thermal shift assays is an effective strategy for screening potential ICL1 inhibitors.
P, hydrogen bond donors/acceptors, polar surface area and rotatable bonds see Table S2†) for compounds 29 and 38 were calculated and they conform to drug-like chemical space except logP and numbers of hydrogen bond donors, which are in lead-like chemical space (for the definition of chemical space see ref. 57). Furthermore, the molecular weight for the hits is in the low to mid 300s, making them excellent starting points for chemical modification and further development. Finally, nine close structural derivatives were purchased to generate a structural activity (SAR) profile (Fig. S17†), but none showed any activity. In general, docking to the binding site showed that these compounds are too bulky to fit into it. Overall, our results show that combining 1H NMR and thermal shift assays is an effective strategy for screening potential ICL1 inhibitors.
Conclusions
ICL isoforms 1 and 2 are important enzymes for the survival of M. tuberculosis in macrophages, enabling the bacteria to utilise fatty acids and cholesterol as carbon sources. ICLs are attractive inhibition targets for the treatment of latent TB. By using ICL1 as a model system, we have demonstrated the general applicability of a combined 1H NMR and thermal shift assays to screen for and evaluate ICL inhibitors. Both methods presented herein are relatively simple to carry out. In contrast to current fluorescence-based assays that rely on enzyme or chemically coupled reactions, the NMR assay described herein enables a direct observation of substrate consumption and product formation and is therefore less prone to errors. One minor drawback of the NMR assay is the low throughput associated with monitoring reactions in real time. Typically, around 15 to 20 minutes of measurement time (six to eleven 1H experiments) were needed to obtain an initial rate. This equates to around seven hours of total measurement time to obtain a full kinetic analysis or a complete IC50 curve with, for example, six concentration points in triplicate. In contrast, the thermal shift assay is a high-throughput method enabling semi-automatic measurements using multi-well plates. Ligand binding to ICL1 was easily identified through a change in the protein's melting temperature. Interestingly, we observed both positive (i.e. stabilising) and negative (i.e. destabilising) thermal shifts of ICL1 with four known inhibitors, likely due to the inhibitors keeping ICL1 in either the open or closed conformations. We have demonstrated the utility of these assays by validating a small library of compounds that were obtained by a virtual high-throughput screen. This ultimately led to the discovery of two novel ICL1 inhibitors that are the subject of ongoing medicinal chemistry studies in our laboratories.Experimental section
Materials
Unless otherwise stated, all chemicals were purchased from Sigma-Aldrich/Merck, Thermo Fisher Scientific, Environmental Control Products (ECP), AK Scientific, Global Science – a VWR/Bio-Strategy Company and Bio-Rad. Tris-D11 and D2O were from Cambridge Isotope Laboratories or Cortecnet. Restriction enzymes BasI-HF and T4 DNA polymerase were obtained from New England Biolabs. Competent cells XL10-Gold and BL21 (DE3) were obtained from Agilent. The Bio-Rad Precision Plus Protein Kaleidoscope Prestained Protein Standards were used for sodium dodecylsulphate polyacrylamide gel electrophoresis (SDS-PAGE). Methyl 4-(4-methoxyphenyl)-4-oxobut-2-enoate was obtained from Enamine. Compounds from virtual screening were obtained from InterBioScreen.Cloning of ICL1
Synthetic gene (gBlocks) encoding M. tuberculosis ICL1 (Table S3†) were obtained from Integrated DNA Technologies. The pNIC28-Bsa4 vector was a gift from Opher Gileadi (Addgene plasmid #26103).58 In order to clone onto the pNIC28-Bsa4 vector, the tacttccaatccatg sequence was added to the 5′ end and taacagtaaaggtggata was added to the 3′ end of the DNA sequence encoding M. tuberculosis ICL1 (Table S3†) when designing the synthetic gene. The synthetic gene and the pNIC28-Bsa4 vector were prepared and cloned using the procedure reported by Gileadi et al. with XL10-Gold.59 The recombinant plasmid was confirmed by DNA sequencing (DNA Sequencing Centre, The University of Auckland). The correct plasmid were then used to transform BL21 (DE3) competent cells for protein expression.Production and purification of ICL1
The recombinant plasmid was first transformed in E. coli BL21(DE3). Starter culture was incubated overnight at 37 °C with shaking in 2YT media. The starter culture was then diluted with fresh 2YT media, which was then incubated at 37 °C with shaking until OD600 of 0.6. Isopropyl-β-D-thiogalactopyranoside (IPTG; 200 μM final concentration) was then added and further incubated at 18 °C with shaking for a further 16 to 20 hours. Cells were harvested by centrifugation. Cell pellets were obtained by centrifugation and resuspended in 50 mM HEPES buffer pH 7.8 with 5 mM imidazole and 500 mM NaCl. The cells were lysed on ice by sonication (4 × 20 seconds) burst at 60% amplitude with 40 seconds rest in between. The protein was purified by 5 mL His-trap column eluting with Tris-HCl buffer pH 7.8 with 500 mM imidazole and followed by gel filtration using 50 mM Tris-HCl buffer pH 7.5.Thermal shift assay
Thermal shift assay was carried out using a BioRad MyiQ real time PCR instrument. The assay was carried out using 20 μM ICL1, 1 mM compounds and 1 mM MgCl2 in 50 mM Tris-HCl pH 7.5. Protein unfolding was monitored by measuring the fluorescence of the SYPRO Orange dye. The dye stock (5000× concentrate) was first diluted in 50 mM Tris-HCl (pH 7.5) to a 200× concentrate before diluting by 5 times into the sample. Temperature was increased from 25 to 95 °C at 1 °C increment every 60 seconds. All measurements were performed in triplicate. For determination of protein melting temperature values, melting curve for each data set was analysed by SigmaPlot 13 (USA) and fitted with the Sigmoid, 3 parameter model.NMR experiments
NMR experiments were conducted at a 1H frequency of 500 MHz using a Bruker Avance III HD spectrometer equipped with a BBFO probe. Experiments were conducted at 300 K. Standard 5 mm NMR tubes (Wilmad) using a sample volume of 500 μL were used in all experiments. The pulse tip-angle calibration using the single-pulse nutation method (Bruker pulsecal routine) was undertaken for each sample.60 All measurements were performed in triplicate.Time course experiments were monitored by standard Bruker 1H experiments with water suppression by excitation sculpting. Unless otherwise stated, the number of transients was 16, and the relaxation delay was 2 seconds. The lag time between addition of enzyme and the end of the first experiment was usually 4 minutes. Initial rates were calculated by linear fitting using Excel 2013 (Microsoft) for data points up to 20% turnover. Kinetic parameters were obtained using the Hanes plot. Linear fitting was done using Excel 2013 (Microsoft). All NMR samples contained 190 nM ICL1, 1 mM DL-isocitrate and 5 mM MgCl2 buffered with 50 mM Tris-D11 (pH 7.5) in 90% H2O and 10% D2O. For kinetic parameter measurements, the isocitrate concentrations ranged from 50 μM to 750 μM. For single concentration inhibition assay, 100 μM inhibitors was used. For IC50 measurements, varying concentrations of inhibitors were used. IC50 values were obtained by SigmaPlot 13 and fitted with the Sigmoid, 3 parameter model.
Virtual high throughput screening
The compounds were docked to the crystal structure of ICL I (PDB ID: 1F8I),39 which was obtained from the Protein Data Bank (PDB).61,62 The Scigress version FJ 2.6 program (Scigress: Version FJ 2.6; Fijitsu Limited, 2008–2016) was used to prepare the crystal structure for docking, i.e., hydrogen atoms added, the co-crystallised succinic acid and glyoxylic acid were removed from protein, the magnesium ion as well as crystallographic water molecules. The Scigress software suite was also used to transfer the structures from 2D to 3D followed by structural optimisation using the MM2 force field.63 The centre of the binding was defined on the co-crystallised ligand with coordinates (x = 5.931, y = 56.950, z = 83.843) with 10 Å radius. For the initial screen 30% search efficiency was used (virtual screen) with ten runs per compound. For the second phase (re-dock) 100% efficiency was used in conjunction with fifty docking runs. The GoldScore,45 ChemScore,46,47 ChemPLP48 and ASP49 scoring functions were implemented to validate the predicted binding modes and relative energies of the ligands using GOLD v5.2 software suite. The InterBioScreen Ltd natural product collection was used for the screening.44 The robustness of the protocol was tested by re-docking the co-crystallised ligand (succinic acid) with these results: RMSD (root-mean-square deviation) GS – 1.750 Å, CS – 0.929 Å, PLP – 0.747 Å and ASP – 1.882 Å, verifying the validity of the procedure. The QikProp v3.21 (QikProp v3.2, Schrödinger, New York, 3.2, 2009) software package was used to calculate the molecular descriptors of the compounds. The reliability of the prediction power of QikProp is established for the molecular descriptors used in this study.64Synthesis of (2S,3R)-2-methylisocitrate
(2S,3R)-2-Methylisocitrate was prepared according to the procedure reported by Darley et al.65Conflicts of interest
The authors declare no competing interests.Acknowledgements
We thank the University of Auckland for a Doctoral Scholarship (R. P. B) and the Maurice and Phyllis Paykel Trust for funding. G. B. is supported by a Sir Charles Hercus Fellowship awarded through the Health Research Council of New Zealand. We thank Professor Bernard Golding (Newcastle University, UK) for his advice when preparing (2S,3R)-2-methylisocitrate. We thank Dr M. Schmitz for maintenance of the NMR facility and Ms K. Boxen for the DNA sequencing service.References
- M. Pai, M. A. Behr, D. Dowdy, K. Dheda, M. Divangahi, C. C. Boehme, A. Ginsberg, S. Swaminathan, M. Spigelman, H. Getahun, D. Menzies and M. Raviglione, Tuberculosis, Nat. Rev. Dis. Primers, 2016, 27, 16076 CrossRef PubMed.
- A. Zumla, M. Raviglione, R. Hafner and C. F. von Reyn, Tuberculosis, N. Engl. J. Med., 2013, 368, 745–755 CrossRef CAS PubMed.
- World Health Organization and Regional Office for South-East Asia, Bending the curve – ending TB: Annual report 2017, New Delhi, 2017 Search PubMed.
- H. Getahun, A. Matteelli, R. E. Chaisson and M. Raviglione, Latent Mycobacterium tuberculosis infection, N. Engl. J. Med., 2015, 372, 2127–2135 CrossRef CAS PubMed.
- H. Esmail, C. E. Barry, D. B. Young and R. J. Wilkinson, The ongoing challenge of latent tuberculosis, Philos. Trans. R. Soc., B, 2014, 369, 20130437 CrossRef CAS PubMed.
- P. L. Lin and J. L. Flynn, Understanding latent tuberculosis: a moving target, J. Immunol., 2010, 185, 15–22 CrossRef CAS PubMed.
- L. P. S. de Carvalho, S. M. Fischer, J. Marrero, C. Nathan, S. Ehrt and K. Y. Rhee, Metabolomics of Mycobacterium tuberculosis reveals compartmentalized co-catabolism of carbon substrates, Chem. Biol., 2010, 17, 1122–1131 CrossRef CAS PubMed.
- D. G. Russell, B. C. VanderVen, W. Lee, R. B. Abramovitch, M.-J. Kim, S. Homolka, S. Niemann and K. H. Rohde, Mycobacterium tuberculosis wears what it eats, Cell Host Microbe, 2010, 8, 68–76 CAS.
- D. Schnappinger, S. Ehrt, M. I. Voskuil, Y. Liu, J. A. Mangan, I. M. Monahan, G. Dolganov, B. Efron, P. D. Butcher, C. Nathan and G. K. Schoolnik, Transcriptional adaptation of Mycobacterium tuberculosis within macrophages: insights into the phagosomal environment, J. Exp. Med., 2003, 198, 693–704 CrossRef CAS PubMed.
- E. J. Muñoz-Elías and J. D. McKinney, Mycobacterium tuberculosis isocitrate lyases 1 and 2 are jointly required for in vivo growth and virulence, Nat. Med., 2005, 11, 638–644 CrossRef PubMed.
- J. D. McKinney, K. Höner zu Bentrup, E. J. Muñoz-Elías, A. Miczak, B. Chen, W. T. Chan, D. Swenson, J. C. Sacchettini, W. R. Jacobs Jr and D. G. Russell, Persistence of Mycobacterium tuberculosis in macrophages and mice requires the glyoxylate shunt enzyme isocitrate lyase, Nature, 2000, 406, 735–738 CrossRef CAS PubMed.
- T. A. Gould, H. Van De Langemheen, E. J. Muñoz-Elías, J. D. McKinney and J. C. Sacchettini, Dual role of isocitrate lyase 1 in the glyoxylate and methylcitrate cycles in Mycobacterium tuberculosis, Mol. Microbiol., 2006, 61, 940–947 CrossRef CAS PubMed.
- H. Eoh and K. Y. Rhee, Methylcitrate cycle defines the bactericidal essentiality of isocitrate lyase for survival of Mycobacterium tuberculosis on fatty acids, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 4976–4981 CrossRef CAS PubMed.
- R. P. Bhusal, G. Bashiri, B. X. C. Kwai, J. Sperry and I. K. H. Leung, Targeting isocitrate lyase for the treatment of latent tuberculosis, Drug Discovery Today, 2017, 22, 1008–1016 CrossRef CAS PubMed.
- M. Krátký and J. Vinšová, Advances in mycobacterial isocitrate lyase targeting and inhibitors, Curr. Med. Chem., 2012, 19, 6126–6137 CrossRef.
- Y. V. Lee, H. A. Wahab and Y. S. Choong, Potential inhibitors for isocitrate lyase of Mycobacterium tuberculosis and non-M. tuberculosis: a summary, BioMed Res. Int., 2015, 2015, 895453 Search PubMed.
- K. Höner Zu Bentrup, A. Miczak, D. L. Swenson and D. G. Russell, Characterization of activity and expression of isocitrate lyase in Mycobacterium avium and Mycobacterium tuberculosis, J. Bacteriol., 1999, 181, 7161–7167 Search PubMed.
- G. H. Dixon and H. L. Kornberg, Assay methods for key enzymes of the glyoxylate cycle, Proc. Biochem. Soc., 1959, 72, 3p Search PubMed.
- T. V. Pham, A. S. Murkin, M. M. Moynihan, L. Harris, P. C. Tyler, N. Shetty, J. C. Sacchettini, H.-l. Huang and T. D. Meek, Mechanism-based inactivator of isocitrate lyases 1 and 2 from Mycobacterium tuberculosis, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 7617–7622 CrossRef CAS PubMed.
- H. K. Chenault and G. M. Whitesides, Lactate dehydrogenase-catalyzed regeneration of NAD from NADH for use in enzyme-catalyzed synthesis, Bioorg. Chem., 1989, 17, 400–409 CrossRef CAS.
- P. Vanni, E. Giachetti, G. Pinzauti and B. A. Mcfadden, Comparative structure, function and regulation of isocitrate lyase, an important assimilatory enzyme, Comp. Biochem. Physiol., 1990, 95B, 431–458 CrossRef CAS.
- N. S. T. Lui and O. A. Roels, An improved method for determining glyoxylic acid, Anal. Biochem., 1970, 38, 202–209 CrossRef CAS PubMed.
- C. Tyl, S. Felsinger and L. Brecker, In situ proton NMR of glycosidase catalyzed hydrolysis and reverse hydrolysis, J. Mol. Catal. B: Enzym., 2004, 28, 55–63 CrossRef CAS.
- P. Spangenberg, V. Chiffoleau-Giraud, C. André, M. Dion and C. Rabiller, Probing the transferase activity of glycosidases by means of in situ NMR spectroscopy, Tetrahedron: Asymmetry, 1999, 10, 2905–2912 CrossRef CAS.
- S. S. van Berkel, J. E. Nettleship, I. K. H. Leung, J. Brem, H. Choi, D. I. Stuart, T. D. W. Claridge, M. A. McDonough, R. J. Owens, J. Ren and C. J. Schofield, Binding of (5S)-penicilloic acid to penicillin binding protein 3, ACS Chem. Biol., 2013, 8, 2112–2116 CrossRef CAS PubMed.
- I. K. H. Leung, T. J. Krojer, G. T. Kochan, L. Henry, F. von Delft, T. D. W. Claridge, U. Oppermann, M. A. McDonough and C. J. Schofield, Structural and mechanistic studies on γ-butyrobetaine hydroxylase, Chem. Biol., 2010, 17, 1316–1324 CrossRef CAS PubMed.
- N. M. Mbenza, P. G. Vadakkedath, D. J. McGillivray and I. K. H. Leung, NMR studies of the non-haem Fe(II) and 2-oxoglutarate-dependent oxygenases, J. Inorg. Biochem. DOI:10.1016/j.jinorgbio.2017.08.032.
- M. W. Pantoliano, E. C. Petrella, J. D. Kwasnoski, V. S. Lobanov, J. Myslik, E. Graf, T. Carver, E. Asel, B. A. Springer, P. Lane and F. R. Salemme, High-density miniaturized thermal shift assays as a general strategy for drug discovery, J. Biomol. Screening, 2001, 6, 429–440 CrossRef CAS PubMed.
- R. Zhang and F. Monsma, Fluorescence-based thermal shift assays, Curr. Opin. Drug Discovery Dev., 2010, 13, 389–402 CAS.
- M.-C. Lo, A. Aulabaugh, G. Jin, R. Cowling, J. Bard, M. Malamas and G. Ellestad, Evaluation of fluorescence-based thermal shift assays for hit identification in drug discovery, Anal. Biochem., 2004, 332, 153–159 CrossRef CAS PubMed.
- D. Matulis, J. K. Kranz, F. R. Salemme and M. J. Todd, Thermodynamic stability of carbonic anhydrase: measurements of binding affinity and stoichiometry using ThermoFluor, Biochemistry, 2005, 44, 5258–5266 CrossRef CAS PubMed.
- P. Cimmperman, L. Baranauskiene, S. Jachimoviciūte, J. Jachno, J. Torresan, V. Michailoviene, J. Matuliene, J. Sereikaite, V. Bumelis and D. Matulis, A quantitative model of thermal stabilization and destabilization of proteins by ligands, Biophys. J., 2008, 95, 3222–3231 CrossRef CAS PubMed.
- R. A. Smith and I. C. Gunsalus, Isocitritase: enzyme properties and reaction equilibrium, J. Biol. Chem., 1957, 229, 305–319 CAS.
- E. Giachetti and P. Vanni, Effect of Mg2+ and Mn2+ on isocitrate lyase, a non-essentially metal-ion-activated enzyme. A graphical approach for the discrimination of the model for activation, Biochem. J., 1991, 276, 223–230 CrossRef CAS PubMed.
- B. A. McFadden and S. Purohit, Itaconate, an isocitrate lyase directed inhibitor in Pseudomonas indigofera, J. Bacteriol., 1977, 131, 136–144 CAS.
- J. V. Schloss and W. W. Cleland, Inhibition of isocitrate lyase by 3-nitropropionate, a reaction-intermediate analogue, Biochemistry, 1982, 21, 4420–4427 CrossRef CAS PubMed.
- Y. H. Ko and B. A. McFadden, Alkylation of isocitrate lyase from Escherichia coli by 3-bromopyruvate, Arch. Biochem. Biophys., 1990, 278, 373–380 CrossRef CAS PubMed.
- Y. Liu, S. Zhou, Q. Deng, X. Li, J. Meng, Y. Guan, C. Li and C. Xiao, Identification of a novel inhibitor of isocitrate lyase as a potent antitubercular agent against both active and non-replicating Mycobacterium tuberculosis, Tuberculosis, 2016, 97, 38–46 CrossRef CAS PubMed.
- V. Sharma, S. Sharma, K. Hoener zu Bentrup, J. D. McKinney, D. G. Russell, W. R. Jacobs Jr and J. C. Sacchettini, Structure of isocitrate lyase, a persistence factor of Mycobacterium tuberculosis, Nat. Struct. Biol., 2000, 7, 663–668 CrossRef CAS PubMed.
- H. Shukla, V. Kumar, A. K. Singh, N. Singh, M. Kashif, M. I. Siddiqi, M. Yasoda Krishnan and M. Sohail Akhtar, Insight into the structural flexibility and function of Mycobacterium tuberculosis isocitrate lyase, Biochimie, 2015, 110, 73–80 CrossRef CAS PubMed.
- D. B. Kitchen, H. Decornez, J. R. Furr and J. Bajorath, Docking and scoring in virtual screening for drug discovery: methods and applications, Nat. Rev. Drug Discovery, 2004, 3, 935–949 CrossRef CAS PubMed.
- A. Lavecchia and C. Di Giovanni, Virtual screening strategies in drug discovery: a critical review, Curr. Med. Chem., 2013, 20, 2839–2860 CrossRef CAS PubMed.
- E. Lionta, G. Spyrou, D. K. Vassilatis and Z. Cournia, Structure-based virtual screening for drug discovery: principles, applications and recent advances, Curr. Top. Med. Chem., 2014, 14, 1923–1938 CrossRef CAS PubMed.
- InterBioScreen Ltd., 121019 Moscow, P.O. Box 218, Russia, http://www.ibscreen.com (accessed August 16, 2017).
- G. Jones, P. Willett, R. C. Glen, A. R. Leach and R. Taylor, Development and validation of a genetic algorithm for flexible docking, J. Mol. Biol., 1997, 267, 727–748 CrossRef CAS PubMed.
- M. D. Eldridge, C. W. Murray, T. R. Auton, G. V. Paolini and R. P. Mee, Empirical scoring functions: I. The development of a fast empirical scoring function to estimate the binding affinity of ligands in receptor complexes, J. Comput.-Aided Mol. Des., 1997, 11, 425–445 CrossRef CAS PubMed.
- M. L. Verdonk, J. C. Cole, M. J. Hartshorn, C. W. Murray and R. D. Taylor, Improved protein–ligand docking using GOLD, Proteins, 2003, 52, 609–623 CrossRef CAS PubMed.
- O. Korb, T. Stutzle and T. E. Exner, Empirical scoring functions for advanced protein-ligand docking with PLANTS, J. Chem. Inf. Model., 2009, 49, 84–96 CrossRef CAS PubMed.
- W. Mooij and M. L. Verdonk, General and targeted statistical potentials for protein-ligand interactions, Proteins, 2005, 61, 272–287 CrossRef CAS PubMed.
- A. Fersht, Structure and mechanism in protein science: a guide to enzyme catalysis and protein folding, W. H. Freeman and Company, New York, 1999 Search PubMed.
- T. I. Oprea, C. Bologa and M. Olah, Compound selection for virtual screening, in Virtual screening in drug discovery, ed. J. Alvarez and B. K. Shoichet, Taylor & Francis, London, 2005, pp. 89–106 Search PubMed.
- P. Axerio-Cilies, I. P. Castañeda, A. Mirza and J. Reynisson, Investigation of the incidence of “undesirable” molecular moieties for high-throughput screening compound libraries in marketed drug compounds, Eur. J. Med. Chem., 2009, 44, 1128–1132 CrossRef CAS PubMed.
- J. Reynisson, C. O'Neill, J. Day, L. Patterson, E. McDonald, P. Workman, M. Katan and S. A. Eccles, The identification of novel PLC-gamma inhibitors using virtual high throughput screening, Bioorg. Med. Chem., 2009, 17, 3169–3176 CrossRef CAS PubMed.
- E. Robinson, E. Leung, A. M. Matuszek, N. Krogsgaard-Larsen, D. P. Furkert, M. A. Brimble, A. Richardson and J. Reynisson, Virtual screening for novel Atg5–Atg16 complex inhibitors for autophagy modulation, Med. Chem. Commun., 2015, 6, 239–246 RSC.
- T. Khomenko, A. Zakharenko, T. Odarchenko, H. J. Arabshahi, V. Sannikova, O. Zakharova, D. Korchagina, J. Reynisson, K. Volcho and N. Salakhutdinov, New inhibitors of tyrosyl-DNA phosphodiesterase I (Tdp 1) combining 7-hydroxycoumarin and monoterpenoid moieties, Bioorg. Med. Chem., 2016, 24, 5573–5581 CrossRef CAS PubMed.
- R. Huang, D. M. Ayine-Tora, M. N. Muhammad Rosdi, Y. Li, J. Reynisson and I. K. H. Leung, Virtual screening and biophysical studies lead to HSP90 inhibitors, Bioorg. Med. Chem. Lett., 2017, 27, 277–281 CrossRef CAS PubMed.
- F. Zhu, G. Logan and J. Reynisson, Wine Compounds as a Source for HTS Screening Collections. A Feasibility Study, Mol. Inf., 2012, 31, 847–855 CrossRef CAS PubMed.
- P. Savitsky, J. Bray, C. D. Cooper, B. D. Marsden, P. Mahajan, N. A. Burgess-Brown and O. Gileadi, High-throughput production of human proteins for crystallization: The SGC experience, J. Struct. Biol., 2010, 172, 3–13 CrossRef CAS PubMed.
- O. Gileadi, N. A. Burgess-Brown, S. M. Colebrook, G. Berridge, P. Savitsky, C. E. A. Smee, P. Loppnau, C. Johansson, E. Salah and N. H. Pantic, High throughput production of recombinant human proteins for crystallography, in Structural proteomics: high-throughput methods, ed. B. Kobe, M. Guss and T. Huber, Humana Press, New York, 2008, ch. 14, pp. 221–246 Search PubMed.
- P. S. C. Wu and G. Otting, Rapid pulse length determination in high-resolution NMR, J. Magn. Reson., 2005, 176, 115–119 CrossRef CAS PubMed.
- H. Berman, K. Henrick and H. Nakamura, Announcing the worldwide Protein Data Bank, Nat. Struct. Biol., 2003, 10, 980 CrossRef CAS PubMed.
- H. M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov and P. E. Bourne, The Protein Data Bank, Nucleic Acids Res., 2000, 28, 235–242 CrossRef CAS PubMed.
- N. L. Allinger, Conformational analysis. 130. MM2. A hydrocarbon force field utilizing V1 and V2 torsional terms, J. Am. Chem. Soc., 1977, 99, 8127–8134 CrossRef CAS.
- L. Ioakimidis, L. Thoukydidis, A. Mirza, S. Naeem and J. Reynisson, Benchmarking the reliability of QikProp. Correlation between experimental and predicted values, QSAR Comb. Sci., 2008, 27, 445–456 CAS.
- D. J. Darley, T. Selmer, W. Clegg, R. W. Harrington, W. Buckel and B. T. Golding, Stereocontrolled synthesis of (2R,3S)-2-methylisocitrate, a central intermediate in the methylcitrate cycle, Helv. Chim. Acta, 2003, 86, 3991–3999 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7md00456g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |