
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAntiproliferative activities of alkaloid-like compounds†
XiXi
Xu
 a,
Tristan
Rawling
a,
Ariane
Roseblade
b,
Roger
Bishop
c and
Alison T.
Ung
*a
a,
Tristan
Rawling
a,
Ariane
Roseblade
b,
Roger
Bishop
c and
Alison T.
Ung
*a
aSchool of Mathematical and Physical Sciences, University of Technology Sydney, Broadway, NSW 2007, Australia. E-mail: Alison.Ung@uts.edu.au; Tel: +61 2 9514 1881
bDiscipline of Pharmacy, Graduate School of Health, University of Technology Sydney, Broadway, NSW 2007, Australia
cSchool of Chemistry, University of New South Wales, UNSW, Sydney, NSW 2052, Australia
First published on 25th October 2017
Abstract
Tricyclic alkaloid-like compounds were synthesised in a few steps, via the bridging Ritter reaction. The compounds were evaluated for their antiproliferative activity against the MCF-7 and the aggressive MDA-MB-231 breast cancer cells. The anti-cancer activities of 2c were found to be selective towards the aggressive and more challenging to treat triple negative (MDA-MB-231) cell line while exhibiting no antiproliferative activities towards the MCF-7 cells at the highest concentration tested (50 μM). The IC50 of compound 2c was determined to be 7.9 μM for the MDA-MB-231 cell line. Furthermore, 2c arrested cell cycle at the G2/M phase and induced apoptosis in a dose-dependent manner. Besides in-house anti-cancer screening, compound 3 was selected for anti-cancer screening by the National Cancer Institute and was found to have broad anti-cancer activity with selectivity against particular leukaemia, colon, melanoma, and breast cancer cell lines. Cytotoxicities of compounds 2c and 3 were also tested against noncancerous mammalian cells (VERO cell line), and found to be selective towards cancerous cells. The facile synthetic route, unique chemical structures and the biological data make these alkaloid-like compounds worthwhile lead compounds for further anti-cancer drug development.
Introduction
Alkaloids have played a significant part in drug development. However, research in this area has since started to decline, and the early 1980s saw a move towards derivatisation of natural alkaloids, as well as the synthesis of non-natural alkaloid-like compounds using well known natural product scaffolds.1–4Alkaloids and their derivatives are to date the most successful anti-cancer drugs on the market. Despite tremendous research efforts made in anti-cancer drug development, cancer remains one of the leading causes of death in the modern world with an alarming rising mortality rate.
Multidrug resistance is one of the major contributing factors in the battle against cancer. Cancer cells can grow resistance either due to the nature and genetic background of the cancer cell or as a result of genetic changes following chemotherapy treatments. All of these factors make the pursuit of better anti-cancer agents a challenging task.5–7
The intramolecular variation of the standard Ritter reaction offers a diverse variety of heterocyclic products with an imine functional group.8–10 Also, there are a small number of cases involving the formation of a bridged bicyclic structure, known as the bridging Ritter product. This bridging cyclisation has been used previously by our research group to synthesise more complex alkaloid-like molecules.11,12
Our research team further explored the reactivity of the tricyclic imine scaffolds with dimethyl acetylenedicarboxylate (DMAD) to form a multicyclic molecule 3 that is highly reminiscent of the natural alkaloids, as shown in Scheme 1.
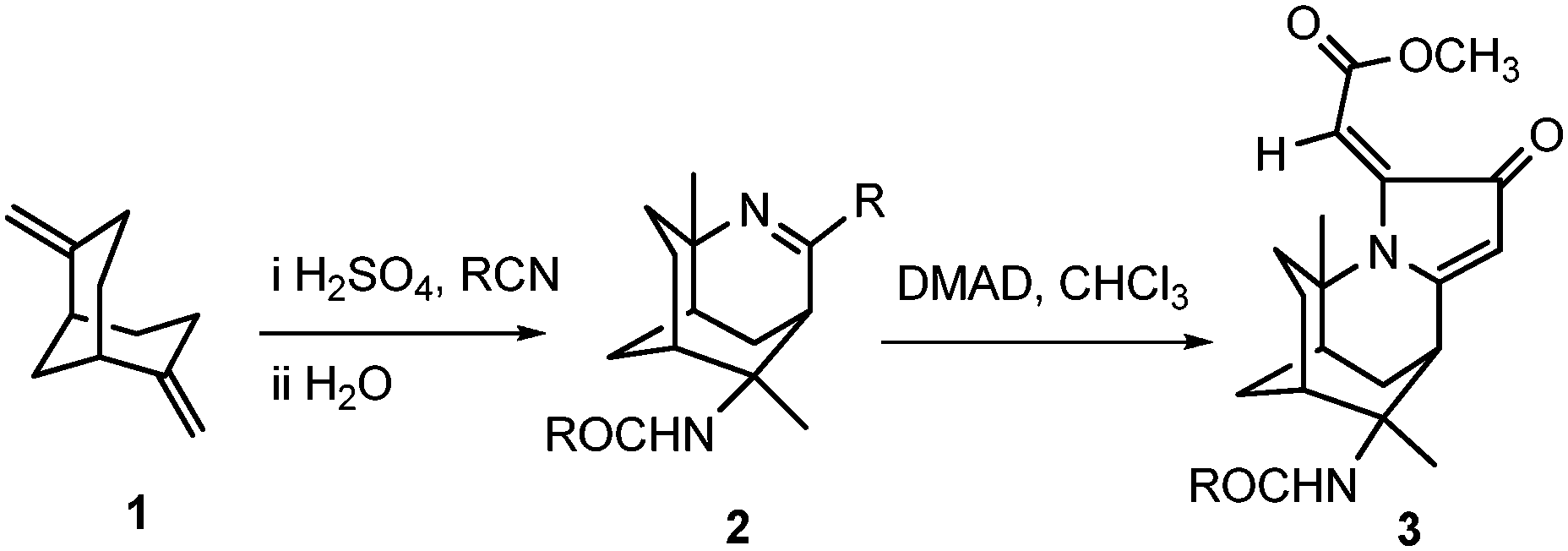 | ||
| Scheme 1 Two-step synthesis of 3 from 2,6-dimethylenebicyclo[3.3.1]nonane 1. | ||
The unique chemical scaffold and drug-like properties of 3-azatricyclo[5.3.1.04,9]undec-2-ene 2, would make it the ideal building block for drug discovery. Therefore, the imine 2 has been developed into a small library of novel and relatively complex alkaloid-like compounds.
Breast cancer is the most common cancer amongst women; it comprises distinct molecular subtypes that affect the patient's response to different pathway-targeted therapies. Breast cancers that express estrogen receptor (ER) and/or progesterone receptor (PR) can be treated with hormonal therapy.13 Triple-negative breast cancers lack these receptors, as well as the human epidermal growth factor receptor 2 (HER2), and are therefore unresponsive to targeted breast cancer drugs. Moreover, triple negative breast cancers are more aggressive and metastatic. Thus, in this paper, we report on the synthesis of alkaloid-like compounds and assess their cytotoxicities in-house against the MDA-MB-231 triple negative breast cancer cell line, as well as against a broad panel of cancers using the NCI screen.14
Results and discussion
Chemistry
As part of our ongoing research into alkaloid-like compounds as anti-cancer agents,15,16 we have synthesised a library of tricyclic compounds using the bridging Ritter reaction as the key reaction. Various nitriles were used, and subsequent reactions such as reduction, reductive alkylation, and DMAD cycloaddition were used to derivatise these small molecules to heterocyclic alkaloid-like compounds.17Bridged Ritter reaction with various nitriles
Our research group previously prepared the cyclic diene 1 as the precursor for generating the tricyclic imine scaffold.11,12,18,19 Tricyclic imines 2a–c were synthesised via the bridging Ritter reaction in benzene under strongly acidic conditions (Scheme 2). The nitriles were chosen to provide a variety of substituents at the C-2 position that alters the reactivity of the respective imine group. The reaction mechanism for the formation of the tricyclic imines was described in our previous publications.18,19 | ||
| Scheme 2 Synthesis of tricyclic imines (2a–c). | ||
The bulky and electron poor nature of ClCH2CN has resulted in the decrease in the percentage yield of 2c in comparison to 2a and 2b. This phenomenon has also been observed in our earlier work, where outcomes of the bridging products were significantly influenced by the nitriles.15
Reduction and reductive alkylation of the 3-azatricyclo[5.3.1.04,9]undec-2-ene system
Reduction of 2a–b was carried out under mild reduction conditions with NaBH(OAc)3 in dry methanol (Scheme 3). The hydride reduces the imine exclusively from the less hindered face of the tricyclic ring structure to yield diastereoisomers 6a and 6b in 71% and 97% yields, respectively. | ||
| Scheme 3 Reduction and reductive alkylation of tricyclic imines. | ||
The structures of 6a and 6b were confirmed by NMR analysis. The 1H NMR spectrum showed the newly formed H-2 peaks at 3.50 and 3.12 ppm for 6a and 6b respectively. The rest of the spectral features remained similar to those of their corresponding imines, with slight upfield shifts of H-3. The stereochemistry of H-2 was evident in the 1D NOESY NMR studies, where cross peaks were observed for H-2 and H-3. This stereoselectivity results from the Re face of the cyclic ring system being hindered by the bicyclic cyclohexane rings.
Compounds 6a and 6b were then subjected to reductive alkylation with 28% formaldehyde to yield compounds 7a and 7b in 49% and 26% yield, respectively. The experimental yields of 7a and 7b highlight the significance of inaccessibility of N-3 due to the steric hindrance created by the substituents at the C-2 and C-4 positions.
Structures of 7a and 7b were confirmed by NMR analysis. The 1H NMR spectrum showed the newly formed –NCH3 peaks at 2.29 and 2.42 ppm, respectively, while the 13C NMR spectrum confirmed the formation of two –NCH3 peaks at 35.97 and 39.30 ppm corresponding to the structures of 7a and 7b respectively.
The reduction of 2c using either NaBH(OAc)3 or NaBH4 provided an unidentified decomposed material as indicated by 1H NMR analysis of the crude mixtures. In an attempt to reduce 2c by hydrogenation in the presence of 5% Pd/C, we recovered the unexpected product 8 in 79% yield (Scheme 4). The formation of 8 can be explained via the palladium catalysed dehalogenation of α-haloamides.20,21 Compound 8 was then subjected to reduction using NaBH4 to give 9 in 72% yield. The structure of 9 was confirmed by NMR analysis; the 1H NMR spectrum showed the newly formed –CH peak at 3.61 ppm as a quintet. The splitting of this –CH peak further confirms that the imine has been reduced. The rest of the spectrum remained similar to that of the other reduced imines.
 | ||
| Scheme 4 Hydrogenation and reduction of compound 2c. | ||
Cycloaddition of DMAD to 3-azatricyclo[5.3.1.04,9]undec-2-ene and reduction of 3
Cyclic amide 3 was synthesised from a one-pot reflux in chloroform with acetylenedicarboxylate (DMAD) and tricyclic imine 2a in 40% yield, according to the method that was previously reported.18,22 It was then selectively reduced with NaBH4 in the presence of NiCl2·6H2O to yield novel compound 10 in 70% yield (Scheme 5). | ||
| Scheme 5 Derivatisation of tricyclic imine scaffold 2a. | ||
The 1H NMR spectrum of 10 showed the disappearance of the C-21 alkene proton peak of compound 3 at 5.86 ppm. A newly formed doublet of doublets at 2.91 and 2.81 ppm corresponded to the structure of 3 being reduced at the C-12 and C-21 positions. The 13C NMR spectrum further confirmed the presence of HC-12 and H2C-21 at 61.6 and 40.9 ppm, respectively. 2D NOESY measurements were carried out on 10 to ascertain the relative stereochemistry at C-12. There were significant cross-peaks between H-12 & H-15 and between H-13 & H-21. The lowest-energy conformation of 10 was calculated using Spartan 10 molecular modelling software (WAVEFUNCTION, INC). The calculation was carried out using the setup menu in Spartan, with the specification of equilibrium conformation, at ground states, semi-empirical model at AM1 level. The molecular modelling of compound 10 (Fig. 1) predicted the distances between H-12 & H-15, and between H-13 & H-21 to be 2.61 and 2.37 Å, respectively. This observation supports the (S)-configuration at C-12 which further indicates that the hydride attacked the C-12 position from the less hindered Si face of the heterocyclic ring.
 | ||
| Fig. 1 Molecular model of compound 10 (Spartan 10, generated structure), showing NOE correlations between H-12 and H-15, and H-13 and H-21. | ||
Biological evaluation
| Tumour type | Cell line | IC50 (μM) |
|---|---|---|
| a Concentration of compound 3 required inhibiting cell growth by 50%. | ||
| Leukemia | CCRF-CEM | 4.4 |
| HL-60 (TB) | 5.5 | |
| MOLT-4 | 2.4 | |
| RPMI-8226 | 4.1 | |
| Non-small cell lung | NCI-H522 | 2.0 |
| EKVX | 12.2 | |
| Colon cancer | COLO-205 | 7.8 |
| HT29 | 7.5 | |
| SW-620 | 2.8 | |
| CNS cancer | SF-539 | 3.3 |
| Melanoma | SK-MEL-28 | 1.8 |
| SK-MEL-5 | 6.3 | |
| MDA-MB-435 | 5.9 | |
| Ovarian cancer | OVCAR-3 | 11.8 |
| Renal cancer | CAKI-1 | 5.3 |
| RXF 393 | 6.7 | |
| DU-145 | 16.9 | |
| Prostate cancer | PC-3 | 15.7 |
| DU-145 | 16.9 | |
| Breast cancer | MCF-7 | 7.1 |
| MDA-MB-231/ATCC | 38.9 | |
| HS 578T | 10.9 | |
| T-47D | 22.7 | |
The dose response curves from the screening of compound 3 against leukaemia, breast cancer, colon and melanoma cells lines determined by NCI are presented in Fig. 2.
 | ||
| Fig. 2 Dose response curves of compound 3 against leukaemia, breast cancer, colon, and melanoma cell lines. | ||
Compound 3 potently inhibited the proliferation of leukaemia, colon, renal and melanoma cancer cell lines with IC50 values below 10 μM. Amongst the breast cancer cell lines, some variation in activity was observed. The proliferation of most of these cell lines was potently inhibited by 3. However, aggressive and highly metastatic MDA-MB-231 cell line was the least active (IC50 = 38.9 μM). This suggests that compound 3 is more active towards acute lymphoblastic leukaemia both T-cell and B-cell, as well as acute promyelocytic leukaemia, and less active towards chronic myelogenous leukaemia which observed a threefold increase in the IC50 values comparing to that of lymphoblastic and promyelocytic leukaemia (Table 1). The selectivity of compound 3 towards the lymphoblastic and promyelocytic leukaemia cell lines could be due to the similarities present in the immunohistochemistry of the CCRF-CEM, HL60, MOLT4, and RPMI-8226 cell lines such as the CD antigen expression patterns, and the expression of both TCR-αβ and TdT genes.23–25 Future investigation is needed to determine the connection in the observed activity across these cell lines from the broad screening.
Diversification of compound structures was achieved by selectively reducing the α,β-unsaturated system in compound 3 to yield compound 10. However, the removal of the double bond at C-21 resulted in decreased the cytotoxicities of compound 10 (IC50 > 50 μM) against MDA-MB-231 cells as shown in Fig. 3.
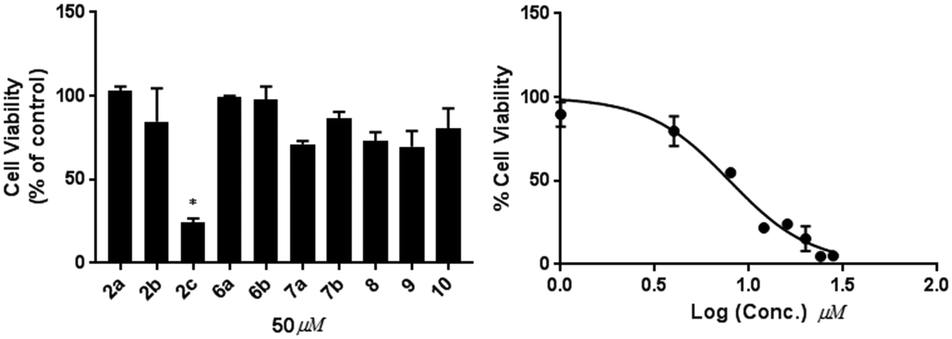 | ||
| Fig. 3 Effects of 2a–c, 6a–b, 7a–7b, 8, 9 and 10 (50 μM, 48 hours) on the proliferation of MDA-MB-231 breast cancer cells (left). * indicate p = 0.001. Dose response curve of 2c is shown on the right. | ||
Cell cycle analysis
Further investigation into the mechanism of action of these compounds was determined to explain for the observed selectivity. To investigate the primary mechanism of action for active compound 2c, we assessed the ability of 2c to induce cell cycle arrest and apoptosis in MDA-MB-231 cells using flow cytometry. Cell cycle studies revealed that compound 2c (20 μM, 24 hours) induced cell cycle arrest and altered the distribution of cells throughout the cell cycle (Fig. 4). Thus, treatment with 2c significantly decreased the population of MDA-MB-231 cells in G0/G1 by 20% (p < 0.0001), and increased the proportion of cells in the G2/M phase by 15% (p < 0.0001) comparing to that of control. These findings suggest that cell cycle arrest at the G2/M phase contributes to the antiproliferative activity of 2c. | ||
| Fig. 4 Effect of 2c on the cell cycle progression of MDA-MB-231 cells; cells were treated for 24 h with (A) DMSO (control), (B) 2c (20 μM), (C) percentage change from control. Data represent the mean ± SEM of at least 3 independent experiments. ** indicates p < 0.0001. | ||
Induction of apoptosis
The translocation of phospholipid phosphatidylserine (PS) to the outer leaflet of the cell plasma membrane is one of the hallmarks of apoptosis.26 This is followed by a loss of membrane integrity and eventual cell death. The ability of compound 2c to induce apoptosis in MDA-MB-231 cells was also assessed using flow cytometry. MDA-MB-231 cells were treated with 20 and 40 μM of 2c for 48 h and then stained with Annexin V-FITC (which binds to exposed PS) and PI. This allows for the determination of the proportion of cells of undergoing apoptosis at either early apoptosis (Annexin-positive, PI-negative), late apoptosis (Annexin-positive, PI-positive) or total apoptosis (early + late apoptosis) (Fig. 5). | ||
| Fig. 5 Effect of compound 2c on the induction of apoptosis in MDA-MB-231 cells; cells were treated with 20 and 40 μM concentrations of 2c for 48 h, stained with Annexin V-FITC and PI, and the apoptotic effect was assessed by flow cytometry. Representative results are shown, with quadrants indicating the proportion of cells that are necrotic: Q1, late apoptotic: Q2, early apoptotic: Q3, and viable: Q4. Images are representative of 3 independent experiments. | ||
MDA-MB-231 cells tested positive for Annexin V-FITC after treatment with 2c (20 and 40 μM, 48 hours) p = 0.0223 and p = 0.0047 respectively, which indicates the cells are in the early stage of apoptosis after treatment of the drug (Fig. 5). The population of cells that underwent early apoptosis after treatment with 2c (20 and 40 μM) was increased by 21% and 31% respectively, which suggests a dose-dependent increase in the apoptotic population.
Cytotoxicity of active of compound 2c against MCF-7
The selectivity of compound 2c was investigated by performing MTS cell proliferation assay in varying concentrations (10–50 μM) on the ER+ (MCF-7) breast cancer cell line Fig. 6. The effects of compounds 2c on the proliferation and viability of MCF-7 cells after 48 h treatment was determined. Compound 2c exhibited no antiproliferative activities towards the MCF-7 cells at the highest concentration tested (50 μM). | ||
| Fig. 6 Effects of 2c (10–50 μM, 48 hours) on the proliferation of MCF-7 breast cancer cells. | ||
Cytotoxicity of compounds 2c and 3 against VERO cells
To assess the selectivity of compounds 2c and 3 towards cancer cells, the cytotoxicities of these compounds were assessed against the noncancerous VERO cell line at 50 μg mL−1 (or 158 μM for 2c, and 139 μM for 3). In these assays, 2c and 3 reduced VERO cell viability by 84% and 77% of control, respectively. In contrast 2c reduced MDA-MB-231 cell viability to 90% of control at 20 μM, and in the case of 3 viability was reduced to 80% at 75 μM. These data clearly demonstrate the selectivity of 2c and 3 towards cancer cells over noncancerous cell lines.Conclusions
A small library of alkaloid-like compounds was synthesised and their anticancer activities were evaluated. Promising anti-cancer activity was established for compound 3 across multiple cancer types, and for compound 2c against the MDA-MB-231 breast cancer cell line, while showed no activity against MCF-7 at 50 μM. Furthermore, cell cycle and apoptosis studies revealed compound 2c promotes cell cycle arrest in the G2/M phase, as well as inducing apoptosis after 48 hours of treatment. The observed selectivity of 2c against MDA-MB-231 over noncancerous VERO cells warrants further development in these alkaloid-like compounds via bridging Ritter reaction.Experimental
General information
Reagents and analytical grade solvents were purchased from commercial sources. Compounds were purified by column chromatography using flash silica gel (60–80 μm). The purity of compounds was determined by 1H NMR and GC/MS. 1H and 13C NMR spectra were recorded on an Agilent 500 MHz spectrometer (500 MHz 1H, 125 MHz 13C) in deuterated chloroform (CDCl3) unless otherwise specified. NMR assignments were based on COSY, HSQC, NOESY and DEPT experiments. 1H and 13C NMR assignments are based on the numbering system used in the systematic name. Low-resolution mass spectra were obtained on an Agilent 6890GC fitted with 5% polysilphenylene, 95% polydimethylsiloxane column, and an Agilent 5973n MS (EI) spectrometer. High-resolution mass spectra were obtained on an Agilent 6510 Accurate-Mass Q-TOF mass spectrometer, equipped with an ESI source.General procedure for the synthesis of imines via bridging Ritter reaction
A mixture of 98% H2SO4 (2 mL) and nitrile (41.60 mmol, 8 mole equiv.) in 100 mL round bottomed flask fitted with a condenser and drying tube, was stirred in an ice bath. A solution of 2,6-dimethylenebicyclo[3.3.1]nonane 1 (0.77 g, 5.20 mmol) in benzene (5 mL) was added to the mixture drop-wise but rapidly. The reaction mixture was stirred at 0 °C for 30 min, then at room temperature overnight. Water was added to the mixture and stirring was continued for a further 30 min. The mixture was extracted with diethyl ether. The aqueous layer was washed with 1 M NaOH solution (18 mL) and extracted with chloroform. The organic layer was dried over Na2SO4 and solvent removed under reduced pressure. The crude product was purified by column chromatography ethyl acetate![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :hexane (4:6).
:1 ethyl acetate/methanol) 0.75; 1H NMR (500 MHz, CDCl3) δ 5.22 (s, 1H, NH-15), 3.36 (s, 1H, H-1), 2.36 (q, J = 7.5 Hz, 2H, CH2-18), 2.21–2.15 (m, 2H, CH2-14), 1.82 (d, J = 10.5 Hz, 1H, H-9), 1.77–1.68 (m, 3H, H-7, H-10a, H-5a), 1.64–1.48 (m, 4H, H-8a, H-10b, H-5b, H-6a), 1.46 (s, 3H, CH3-13), 1.27 (s, 3H, CH3-12), 1.32–1.22 (m, 2H, H-6b, H8b), 1.14 (t, J = 7.5 Hz, 6H, CH3-19, CH3-20); 13C NMR δ 174.8 (C
:hexane (4:6).
:1 ethyl acetate/methanol) 0.75; 1H NMR (500 MHz, CDCl3) δ 5.22 (s, 1H, NH-15), 3.36 (s, 1H, H-1), 2.36 (q, J = 7.5 Hz, 2H, CH2-18), 2.21–2.15 (m, 2H, CH2-14), 1.82 (d, J = 10.5 Hz, 1H, H-9), 1.77–1.68 (m, 3H, H-7, H-10a, H-5a), 1.64–1.48 (m, 4H, H-8a, H-10b, H-5b, H-6a), 1.46 (s, 3H, CH3-13), 1.27 (s, 3H, CH3-12), 1.32–1.22 (m, 2H, H-6b, H8b), 1.14 (t, J = 7.5 Hz, 6H, CH3-19, CH3-20); 13C NMR δ 174.8 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 173.1 (C-2), 62.1 (C-11/4), 61.0 (C-11/4), 40.7 (CH-1), 40.1 (CH-9), 37.7 (CH2-18), 35.7 (CH2-5), 34.9 (CH-7), 34.4 (CH3-12), 33.6 (CH2-14), 30.6 (CH2-8), 30.3 (CH2-10), 25.4 (CH3-13), 22.1 (CH2-6), 14.3 (CH3-19/20), 12.6 (CH3-19/20); FT-IR νmax (neat) 3440, 3249, 3190, 2925, 2110, 1650, 1554, 1449, 1371, 1354, 1112, 959, 941, 744 cm−1; HRMS (ESI): [M + H]+, found 277.2273, C17H29N2O required 277.2280.
:1 ethyl acetate/methanol) 0.76; 1H NMR (500 MHz, CDCl3) δ 6.14 (br s, 1H, NH-15), 4.19 (d, J = 11.50 Hz, 1H, H-18a), 4.12 (d, J = 11.5 Hz, 1H, H-18b), 4.00 (s, 2H, CH2-14), 3.48 (br s, 1H, H-1), 2.05 (d, J = 10.5 Hz, 1H, H-9), 1.85 (br s, 1H, H-7), 1.84–1.73 (m, 2H, H-10a, H-5a), 1.69 (d, J = 10.5 Hz, 1H, H-8a), 1.64–1.52 (m, 3H, H-6a, H-10b, H-5b), 1.50 (s, 3H, CH3-12), 1.38–1.36 (m, 1H, H-8b), 1.33 (s, 3H, CH3-13); 13C NMR δ 169.1 (CO), 165.0 (CN), 60.4 (C-11/4), 60.1 (C-11/4), 49.7 (CH2-18), 43.5 (CH2-14), 37.0 (CH-1), 36.6 (CH-9), 33.2 (CH2-10), 32.5 (CH-7), 31.6 (CH3-13), 28.3 (CH2-8), 27.8 (CH2-10), 22.7 (CH3-12), 19.6 (CH2-6); FT-IR νmax (neat) 3530, 3455, 3267, 3210, 2954, 2146, 1660, 1554, 1450, 1391, 1367, 1355, 1120, 944, 850, 722 cm−1; HRMS (ESI): [M + H]+, found 317.1195, C15H23Cl2N2O required 317.1187.
O), 173.1 (C-2), 62.1 (C-11/4), 61.0 (C-11/4), 40.7 (CH-1), 40.1 (CH-9), 37.7 (CH2-18), 35.7 (CH2-5), 34.9 (CH-7), 34.4 (CH3-12), 33.6 (CH2-14), 30.6 (CH2-8), 30.3 (CH2-10), 25.4 (CH3-13), 22.1 (CH2-6), 14.3 (CH3-19/20), 12.6 (CH3-19/20); FT-IR νmax (neat) 3440, 3249, 3190, 2925, 2110, 1650, 1554, 1449, 1371, 1354, 1112, 959, 941, 744 cm−1; HRMS (ESI): [M + H]+, found 277.2273, C17H29N2O required 277.2280.
:1 ethyl acetate/methanol) 0.76; 1H NMR (500 MHz, CDCl3) δ 6.14 (br s, 1H, NH-15), 4.19 (d, J = 11.50 Hz, 1H, H-18a), 4.12 (d, J = 11.5 Hz, 1H, H-18b), 4.00 (s, 2H, CH2-14), 3.48 (br s, 1H, H-1), 2.05 (d, J = 10.5 Hz, 1H, H-9), 1.85 (br s, 1H, H-7), 1.84–1.73 (m, 2H, H-10a, H-5a), 1.69 (d, J = 10.5 Hz, 1H, H-8a), 1.64–1.52 (m, 3H, H-6a, H-10b, H-5b), 1.50 (s, 3H, CH3-12), 1.38–1.36 (m, 1H, H-8b), 1.33 (s, 3H, CH3-13); 13C NMR δ 169.1 (CO), 165.0 (CN), 60.4 (C-11/4), 60.1 (C-11/4), 49.7 (CH2-18), 43.5 (CH2-14), 37.0 (CH-1), 36.6 (CH-9), 33.2 (CH2-10), 32.5 (CH-7), 31.6 (CH3-13), 28.3 (CH2-8), 27.8 (CH2-10), 22.7 (CH3-12), 19.6 (CH2-6); FT-IR νmax (neat) 3530, 3455, 3267, 3210, 2954, 2146, 1660, 1554, 1450, 1391, 1367, 1355, 1120, 944, 850, 722 cm−1; HRMS (ESI): [M + H]+, found 317.1195, C15H23Cl2N2O required 317.1187.
General procedure for hydride reduction of tricyclic imines
Imines 2a (1.81 mmol) was dissolved in dry MeOH (15 mL) and placed in a round bottom flask fitted with a drying tube, NaBH(OAc)3 (2.30 g, 10.86 mmol, 6 mole equiv.) was slowly added. The mixture was stirred overnight at room temperature. The reaction was quenched with a saturated solution of NaHCO3 (20 mL), extracted with CHCl3 (20 mL × 2), and washed with a saturated solution of NaCl (20 mL). The organic layer was dried with Na2CO3 and solvent was removed under reduced pressure to yield a viscous liquid which solidified upon standing over a day. The product was purified by column chromatography ethyl acetate:methanol (8:2) to afford the reduced compound 6a.
:2 ethyl acetate/methanol) 0.60; 1H NMR (500 MHz, CDCl3) δ 5.31 (s, 1H, NH-15), 3.50 (q, J = 7.5 Hz, 1H, H-2), 2.44 (br s, 1H, H-1), 2.09–2.06 (m, 1H, H-9), 1.95 (s, 3H, CH3-12), 1.83–1.59 (m, 7H, H-6a, H-6b, H-8a, H-8b, H-7, H-10a, H-5a), 1.58 (s, 3H, CH3-18), 1.50 (br d, J = 13.0 Hz, 1H, H-10b), 1.34 (d, J = 7.5 Hz, 3H, CH3-14), 1.17–1.12 (m, 1H, H-5b), 1.10 (s, 3H, CH3-13); 13C NMR δ 171.7 (CO), 63.9 (C-11/4), 56.8 (CH-2), 55.2 (C-11/4), 41.6 (CH-1), 38.8 (CH2-5), 37.5 (CH3-18), 37.1 (CH-9), 36.1 (CH-7), 34.3 (CH2-8), 30.5 (CH2-10), 28.1 (CH3-12), 27.8 (CH3-14), 26.3 (CH3-13), 22.8 (CH2-6); FT-IR νmax (neat) 3256, 3191, 2956, 2925, 2873, 1648, 1550, 1457, 1372, 1353, 1234, 1112, 1076, 940, 721 cm−1; HRMS (ESI): [M + H]+, found 251.2111, C15H27N2O required 251.2123.
:2 ethyl acetate/methanol) 0.62; 1H NMR (500 MHz, CDCl3) δ 5.29 (s, 1H, NH-15), 3.12 (quint, J = 5.5 Hz, 1H, H-2), 2.58 (br s, 1H, H-1), 2.16 (dq, J = 1.0, 7.5 Hz, 2H, CH2-14), 1.96 (dd, J = 1.0, 11.0 Hz, 1H, H-9), 1.98–1.82 (m, 1H, H-6a), 1.82–1.76 (m, 2H, H-5a, H-7), 1.72–1.65 (m, 2H, H-6b, H-10a), 1.58 (s, 3H, CH3-12), 1.60–1.55 (m, 3H, H-10b, CH2-18), 1.48 (t, J = 1.5 Hz, 1H, H-10b), 1.46 (br s, 1H, H-8a), 1.27 (s, 1H, H-8b), 1.14 (t, J = 7.5 Hz, 3H, CH3-20), 1.10 (s, 3H, CH3-13), 0.97 (t, J = 7.5 Hz, 3H, CH3-19); 13C NMR δ 172.63 (CO), 61.9 (CH-2), 61.0 (C-11/4), 37.3 (CH-1), 36.6 (CH2-18), 35.5 (CH-9), 34.6 (CH3-12), 34.3 (CH-7), 32.3 (CH2-5), 31.8 (CH2-14), 31.2 (CH2-8), 28.2 (CH2-10), 23.3 (CH3-13), 20.4 (CH2-6), 12.7 (CH3-19), 10.1 (CH3-20); FT-IR νmax (neat) 3286, 3260, 2985, 2883, 2673, 1659, 1549, 1432, 1377, 1320, 1247, 1102, 1060, 989, 756 cm−1; HRMS (ESI): [M + H]+, found 279.2436, C17H31N2O required 279.2436.
O), 165.7 (C-2), 62.6 (C-11/4), 61.4 (C-11/4), 45.9 (CH2-14), 42.8 (CH-1), 39.5 (CH-9), 35.6 (CH2-5), 34.4 (CH-7), 34.3 (CH3-13), 31.9 (CH3-18), 30.2 (CH2-8), 30.0 (CH2-10), 25.2 (CH3-12), 22.0 (CH2-6); FT-IR νmax (neat) 3554, 3420, 3228, 3210, 2983, 2150, 1665, 1595, 1450, 1351, 1331, 1311, 1125, 957, 880, 778 cm−1; HRMS (ESI): [M + H]+, found 283.1581, C15H24ClN2O required 283.1579.
:methanol (9.5:0.5) to yield amine 9 as an off white viscous liquid (0.035 g, 72%), Rf (9.5:0.5 ethyl acetate/methanol) 0.64; 1H NMR (500 MHz, CDCl3) δ 6.58 (br s, 1H, NH-15), 3.98 (s, 2H, H-14), 3.61 (dt, J = 7.0, 14.0 Hz, 1H, H-2), 2.45 (br s, 1H, CH-1), 2.18–2.12 (m, 1H, H-9), 1.86–1.76 (m, 4H, H-8a, H-7, H-6a, H-6b), 1.74–1.65 (m, 2H, H-8b, H-10a), 1.64–1.62 (m, 1H, H-5a), 1.60 (s, 3H, CH3-18), 1.56–1.50 (m, 1H, H-10b), 1.39 (d, J = 7.5 Hz, 3H, CH3-13), 1.34–1.27 (m, 1H, H-5b), 1.17 (s, 3H, CH3-12); 13C NMR δ 166.8 (CO), 64.2 (C-11/4), 56.6 (CH-2), 56.0 (C-11/4), 46.0 (CH2-14), 41.3 (CH-1), 37.9 (CH2-5), 36.9 (CH-9), 36.9 (CH3-12), 35.8 (CH-7), 33.7 (CH2-8), 30.1 (CH2-10), 27.5 (CH3-13), 26.1 (CH3-18), 22.7 (CH2-6); FT-IR νmax (neat) 3279, 3265, 2952, 2800, 2651, 1660, 1589, 1450, 1395, 1388, 1200, 1120, 1080, 989, 877, 756 cm−1; HRMS (ESI): [M + H]+, found 285.1715, C15H26ClN2O required 285.1734.
General conditions for reductive alkylation of 3-azatricyclo[5.3.1.04,9]undec-2-ene
Amine 6 (0.92 mmol) was dissolved in MeCN (20 mL), to which NaBH3CN (1.15 g, 18.4 mmol, 20 mole equiv.), 40% formaldehyde solution (2 mL) and a few drops of glacial acetic acid were added. The solution was stirred for 3 hours at room temperature. The solvent was removed under reduced pressure. The crude was dissolved in CHCl3 and washed with saturated NaHCO3, saturated NaCl, and dried with Na2SO4 to yield a pale red solid 7a.:1 ethyl acetate/methanol) 0.26; 1H NMR (500 MHz, CDCl3) δ 5.17 (s, 1H, NH-15), 3.05–3.01 (m, 1H, H-2), 2.47 (br 1H, H-1), 2.29 (s, 3H, NCH3), 2.01–1.98 (m, 1H, H-9), 1.93 (s, 3H, CH3-12), 1.90–1.64 (m, 7H, H-6a, H-6b, H-8a, H-8b, H-7, H-10a, H-5a), 1.58 (s, 3H, CH3-18), 1.49 (d, J = 4.0 Hz, 1H, H-10b), 1.30 (d, J = 7.5 Hz, 3H, CH3-14), 1.22–1.14 (m, 1H, H-5b), 1.11 (s, 3H, CH3-13); 13C NMR δ 171.7 (CO), 63.9 (C-11/4), 56.8 (CH-2), 55.2 (C-11/4), 41.6 (CH-1), 38.8 (CH2-5), 37.5 (CH3-18), 37.1 (CH-9), 36.1 (CH-7), 36.0 (NCH3), 34.3 (CH2-8), 30.5 (CH2-10), 28.1 (CH3-12), 27.8 (CH3-14), 26.3 (CH3-13), 22.8 (CH2-6); FT-IR νmax (neat) 3360, 3225, 2976, 2803, 2670, 1660, 1548, 1432, 1377, 1320, 1247, 1102, 1020, 982, 750 cm−1; HRMS (ESI): [M + H]+, found 265.2256, C16H29N2O required 265.2280.
:1 ethyl acetate/methanol) 0.30; 1H NMR (500 MHz, CDCl3) δ 5.27 (s, 1H, NH-15), 2.57 (br s, 1H, H-1), 2.38 (br s, 3H, NCH3), 2.15 (ddd, J = 3.0, 7.5, 10.5 Hz, 2H, CH2-14), 1.98–1.90 (m, 2H, H-6a, H-9), 1.84–1.76 (m, 3H, H-5a, H-7, H-8a), 1.74–1.68 (m, 2H, H-5b, H-10a), 1.66–1.60 (m, 4H, H-6b, CH2-18, H-8b), 1.59 (s, 3H, CH3-12), 1.48–1.42 (m, 1H, H-10b), 1.15 (br s, 3H, CH3-13), 1.13 (t, J = 7.5 Hz, 3H, CH3-20), 1.07 (t, J = 7.5 Hz, 3H, CH3-19); 13C NMR δ 175.9 (CO), 63.7 (C-11/4), 42.0 (CH-2), 40.0 (CH-1), 39.3 (NCH3), 39.3 (CH-9), 39.2 (CH-7), 34.1 (CH2-14), 33.8 (CH2-8), 31.6 (CH2-18), 30.3 (CH3-13), 29.7 (CH2-10), 25.1 (CH3-12), 23.9 (CH2-8), 16.7 (CH2-6), 15.3 (CH3-19), 12.7 (CH3-20); FT-IR νmax (neat) 3360, 3200, 2966, 2873, 2652, 1648, 1540, 1467, 1377, 1335, 1240, 1174, 1033, 960, 763 cm−1; HRMS (ESI): [M + H]+, found 293.2576, C18H32N2O required 293.2593.
:1 ethyl acetate/methanol) 0.53; 1H NMR (500 MHz, CDCl3) δ 5.48 (br s, 1H, NH-16), 5.09 (s, 1H, H-10), 3.95 (d, J = 2.0 Hz, H-7), 3.44 (s, 3H, OCH3), 2.91 (dd, J = 15.5, 3.5 Hz, 1H, H-21a), 2.81 (dd, J = 15.5, 6.5 Hz, 1H, H-21b), 2.14 (s, 1H, H-12), 2.07 (br s, 1H, H-3), 1.70–1.60 (m, 3H, H-6a, H-14a, H-4a), 1.98 (m, 1H, H-5), 1.96 (s, 3H, COCH3), 1.95–1.85 (m, 3H, H-13a, H-4b, H-6b), 1.54–1.30 (2H, H-14b, H-13b), 1.50 (s, 3H, CH3-15), 1.34 (s, 3H, CH3-19); 13C NMR δ 201.9 (C-11), 187.1 (C-9), 173.4 (C-17), 172.5 (C-22), 103.5 (CH-10), 62.9 (C-2/8), 62.6 (C-2/8), 61.6 (CH-12), 54.5 (OCH3), 40.9 (CH2-21), 39.6 (CH-3), 39.0 (CH-7), 38.0 (CH-5), 33.9 (CH3-15), 32.8 (CH2-13), 32.3 (CH2-4), 28.5 (CH2-6), 27.4 (CH3-25), 24.5 (CH3-19), 21.8 (CH2-14). HRMS (ESI): [M + H]+, found 361.2107, C20H29N2O4 required 361.2127.
Cell biology
Compound 3 was screened under the standard conditions of the NCI anticancer screening program, against 48 cell lines covering nine cancer types.14General reagents for cell culture
Penicillin and streptomycin, trypsin and fetal bovine serum were purchased from Thermo Fisher Scientific (Scoresby, Vic, Australia). Dulbecco's modified Eagle's medium (DMEM), phosphate-buffered saline (PBS) and DMSO were purchased from Sigma-Aldrich (Castle Hill, NSW, Australia). The CellTiter 96® AQueous One Solution Reagent was purchased from Promega (Alexandria, NSW, Australia).Cell culture and viability assays
Human MDA-MB-231 breast cancer cells were obtained as a gift from Prof. Michael Murray (University of Sydney) and grown at 37 °C in a humidified atmosphere of 5% CO2 in DMEM supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. Confluent cells (80–90%) were harvested using trypsin/EDTA after washing with PBS.For the MTS assay cells were seeded in 96-well flat-bottom plates at a density of 7 × 103 cells per well. Serum was removed after 24 hours, after which cells were treated with various concentrations of 2a–c, 6a–b and 10 (1–50 μM) in DMSO (maximum concentration 0.1%) for 48 hours; control cells received solvent alone. Cell viability was determined using the CellTiter 96® AQueous One Solution Cell Proliferation Assay (Promega, USA) as per the manufacturer's recommendation. MTS reagent was added to each well, and the absorbance of all wells was determined by measuring optical density at 550 nm (Infinite M1000Pro, Tecan) after 4 hours incubation at 37 °C. Blank-subtracted absorbance values were normalised to the DMSO control, which was arbitrarily assigned as 100%. IC50 values were defined as the drug concentration that prevented cell growth of more than 50% relative to the DMSO control and were determined using nonlinear regression analysis with Prism 7.0 (GraphPad Software, CA, USA). Each compound was tested in triplicates. Note: the MTS assay was repeated with human MCF-7 breast cancer cells obtained as a gift from Asso. Prof. Mary Bebawy (University of Technology Sydney).
Cell cycle analysis
Cell cycle distribution was evaluated in MDA-MB-231 cells that were seeded in 6-well flat bottom plates at a density of 5 × 105 cells per well. Serum was removed after 24 hours, after which the cells were treated with various concentrations of drugs (20–40 μM) in DMSO (maximum concentration 0.1%) for 24 hours; control wells only contained DMSO at 0.1%. Treated cells were then harvested and fixed overnight at 0 °C in 80% ethanol, centrifuged at 500 g for 10 minutes at 4 °C. Fixed cells were then washed twice with PBS and resuspended in 0.1 M PBS containing 0.1% (v/v) Triton X-100 and 0.1 mg mL−1 RNAse A. After staining with PI (50 μg mL−1), cells were incubated for 30 minutes in the dark at 37 °C and subjected to flow cytometry (BD LSRFortessa X-20), with 1 × 104 events, per sample acquired. Cell cycle distribution was calculated using Flowing Software version 2.5.1.Apoptosis/necrosis
Annexin V-FITC/PI staining was used to evaluate the extent of apoptosis in MDA-MB-231 cells as per manufacturer's recommendation (BD Bioscience, Sydney, NSW, Australia). Cells were seeded in 6-well plates at a density of 1 × 105 cells per well, 24 hours after serum removal the cells were treated with 2c (20–40 μM) for 48 hours. Control wells were treated with DMSO only at 0.1% (v/v). Treated cells were trypsinised and washed twice with cold PBS. The cells were then resuspended in binding buffer (0.1 mL), 5 μL of Annexin V-FITC reagent and PI (50 μg mL−1). The cells were incubated for 20 minutes in the dark at 0 °C and subjected to flow cytometry as described above. The percentages of viable (Annexin V−/PI−), early apoptotic (Annexin V+/PI−), late apoptotic (Annexin V+/PI+), and necrotic (Annexin V−/PI+) cells were calculated using Flowing Software version 2.5.1.Cytotoxicity against VERO cells
Cytotoxicity of compounds 2c and 3 against VERO cells (African Green Monkey kidney cell lines) was determined externally by Bioassay Laboratory, BIOTEC, Thailand,27 using the following procedure.28GFP-expressing Vero cell line was generated in-house by stably transfected the African green monkey kidney cell line (VERO, ATCC CCL-81) with plasmid carried gfp gene (pEGFP-N1, Clontech). The cell line was maintained in minimal essential medium (MEM) supplemented with 10% heat-inactivated fetal bovine serum, 1 mM sodium pyruvate, 2.2 g L−1 NaHCO3 and 0.8 mg mL−1 geneticin and incubated at 37 °C in a humidity incubator with 5% CO2. Before performing the assay, cells at a logarithmic growth were harvested and diluted to 3.3 × 104 cells per mL suspension in complete medium without geneticin.
Compounds 2c and 3 were assayed in 384-well plate in a quadruplicate. Briefly, test wells were added with 5 μL of samples (or positive or negative control agents) and followed with 45 μL of cell suspension addition. Positive and negative control agents were ellipticine, and 0.5% DMSO, respectively. The plate was then incubated at 37 °C in humidity incubator with 5% CO2 for 4 days. Fluorescence was measured in a bottom-reading mode with excitation and emission wavelengths of 485 and 535 nm. The fluorescent signal of day-4 was subtracted with that of day-0 before calculation. The percentage of cytotoxicity was calculated by the following equation:
| % Cytotoxicity = [1 − (FUT/FUC)] × 100 |
Statistical analysis
All measurements were performed at least in triplicates unless otherwise stated. Data from multiple treatments were analysed by one-way ANOVA in combination with Fisher's protected least significant difference test.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the University of Technology of Sydney. We like to thanks, Prof Michael Murray (University of Sydney) and Assoc. Prof Mary Bebawy (University of Technology Sydney) for generously providing as gifts the MDA-MB-231 and MCF-7 breast cancer cells, respectively. We like to thank NCI for carrying out broad screening compound 3.References
- O. Genilloud, Recent Pat. Anti-Infect. Drug Discovery, 2012, 7, 189 CrossRef CAS.
- V. Spanò, A. Attanzio, S. Cascioferro, A. Carbone, A. Montalbano, P. Barraja, L. Tesoriere, G. Cirrincione, P. Diana and B. Parrino, Mar. Drugs, 2016, 14, 226 CrossRef PubMed.
- A. Carbone, B. Parrino, G. Di Vita, A. Attanzio, V. Spanò, A. Montalbano, P. Barraja, L. Tesoriere, M. A. Livrea, P. Diana and G. Cirrincione, Mar. Drugs, 2015, 13, 460 CrossRef PubMed.
- B. Parrino, A. Attanzio, V. Spanò, S. Cascioferro, A. Montalbano, P. Barraja, L. Tesoriere, P. Diana, G. Cirrincione and A. Carbone, Eur. J. Med. Chem., 2017, 138, 371 CrossRef CAS PubMed.
- F. Kratz, I. A. Müller, C. Ryppa and A. Warnecke, ChemMedChem, 2008, 3, 20 CrossRef CAS PubMed.
- M. B. Sporn and K. T. Liby, Nat. Clin. Pract. Oncol., 2005, 2, 518 CrossRef CAS PubMed.
- G. Szakacs, J. K. Paterson, J. A. Ludwig, C. Booth-Genthe and M. M. Gottesman, Nat. Rev. Drug Discovery, 2006, 5, 219 CrossRef CAS PubMed.
- J. J. Ritter and F. X. Murphy, J. Am. Chem. Soc., 1952, 74, 763 CrossRef CAS.
- R. Bishop, in Comprehensive Organic Synthesis - Selectivity, Strategy and Efficiency in Modern Organic Chemistry, Volumes 1–9, ed. B. M. Trost, I. Fleming and E. Winterfeldt, Pergamon, Oxford, 1991, vol. 6, p. 261 Search PubMed.
- R. Bishop, in Comprehensive Organic Synthesis, ed. G. A. Molander and P. Knochel, Elsevier, Oxford, 2014, 2nd edn, vol. 6, p. 239 Search PubMed.
- R. Bishop, S. C. Hawkins and I. C. Ibana, J. Org. Chem., 1988, 53, 427 CrossRef CAS.
- I. C. C. Bong, A. T. Ung, D. C. Craig, M. L. Scudder and R. Bishop, Aust. J. Chem., 1989, 42, 1929 CrossRef CAS.
- C. K. Baumann and M. Castiglione-Gertsch, Drugs, 2007, 67, 2335 CrossRef CAS PubMed.
- National Cancer Institute, Division of Cancer Treatment and Diagnosis, https://dtp.cancer.gov/discovery_development/nci-60/methodology.htm, (accessed 10.03.17).
- A. T. Ung, S. G. Williams, A. Angeloski, J. Ashmore, U. Kuzhiumparambil, M. Bhadbhade and R. Bishop, Monatsh. Chem., 2014, 145, 983 CrossRef CAS.
- A. T. Ung, A. N. West, M. J. A. Phillips and S. G. Williams, Monatsh. Chem., 2016, 147, 1737 CrossRef CAS.
- C. Hemtasin, A. T. Ung, S. Kanokmedhakul, K. Kanokmedhakul, R. Bishop, T. Satraruji and D. Bishop, Monatsh. Chem., 2012, 143, 955 CrossRef CAS.
- A. T. Ung, R. Bishop, D. C. Craig, M. L. Scudder and J. Yunus, Aust. J. Chem., 1992, 45, 553 CrossRef CAS.
- R. Bishop and A. Landers, Aust. J. Chem., 1979, 32, 2675 CrossRef CAS.
- D. Kikelj, J. Kidrič, P. Pristovšek, S. Pečar, U. Urleb, A. Krbavčič and H. Hönig, Tetrahedron, 1992, 48, 5915 CrossRef CAS.
- T. Matsumura, M. Akiba, S. Arai, M. Nakagawa and A. Nishida, Tetrahedron Lett., 2007, 48, 1265 CrossRef CAS.
- Q. Lin, D. Djaidi, R. Bishop, D. C. Craig and M. L. Scudder, Aust. J. Chem., 1998, 51, 799 CrossRef CAS.
- L. Bullinger and P. J. Valk, J. Clin. Oncol., 2005, 23, 6296 CrossRef CAS PubMed.
- A. Andersson, P. Eden, D. Lindgren, J. Nilsson, C. Lassen, J. Heldrup, M. Fontes, A. Borg, F. Mitelman, B. Johansson, M. Hoglund and T. Fioretos, Leukemia, 2005, 19, 1042 CrossRef CAS PubMed.
- L. Belov, O. de la Vega, C. G. dos Remedios, S. P. Mulligan and R. I. Christopherson, Cancer Res., 2001, 61, 4483 CAS.
- S. Elmore, Toxicol. Pathol., 2007, 35, 495 CrossRef CAS PubMed.
- Bioassay Laboratory, National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Thailand Science Park, Phahonyothin Road, Khlong Nueng, Khlong Luang, Pathum Thani 12120 THAILAND, http://www.biotec.or.th/bioassay/index.php/bioassay-service, (accessed 14.10.17).
- L. Hunt, M. Jordan, M. De Jesus and F. M. Wurm, Biotechnol. Bioeng., 1999, 65, 201 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7md00435d |
| This journal is © The Royal Society of Chemistry 2017 |