
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Histone lysine methyltransferase structure activity relationships that allow for segregation of G9a inhibition and anti-Plasmodium activity†‡
Sandeep
Sundriyal
 a,
Patty B.
Chen
bcd,
Alexandra S.
Lubin
a,
Gregor A.
Lueg
a,
Fengling
Li
e,
Andrew J. P.
White
a,
Nicholas A.
Malmquist
bcd,
Masoud
Vedadi
ef,
Artur
Scherf
bcd and
Matthew J.
Fuchter
*a
a,
Patty B.
Chen
bcd,
Alexandra S.
Lubin
a,
Gregor A.
Lueg
a,
Fengling
Li
e,
Andrew J. P.
White
a,
Nicholas A.
Malmquist
bcd,
Masoud
Vedadi
ef,
Artur
Scherf
bcd and
Matthew J.
Fuchter
*a
aDepartment of Chemistry, Imperial College London, London SW7 2AZ, UK. E-mail: m.fuchter@imperial.ac.uk; Fax: +44 (0)2075945805; Tel: +44 (0)2075945815
bUnité Biologie des Interactions Hôte-Parasite, Département de Parasites et Insectes Vecteurs, Institut Pasteur, Paris 75015, France
cCNRS ERL 9195, Paris 75015, France
dINSERM Unit U1201, Paris 75015, France
eStructural Genomics Consortium, University of Toronto, Toronto, ON M5G 1L7, Canada
fDepartment of Pharmacology and Toxicology, University of Toronto, Toronto, ON M5S 1A8, Canada
First published on 15th March 2017
Abstract
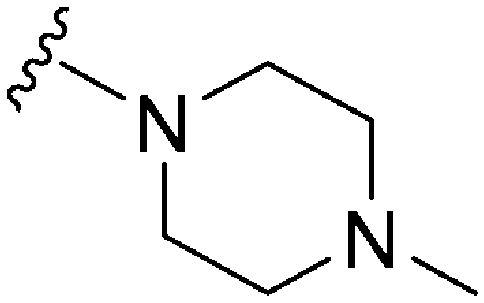
Plasmodium falciparum HKMTs (PfHKMTs) play a key role in controlling Plasmodium gene expression and represent exciting new anti-malarial epigenetic targets. Using an inhibitor series derived from the diaminoquinazoline HKMT inhibitory chemotype, we have previously identified compounds with highly promising antimalarial activity, including irreversible asexual cycle blood stage-independent cytotoxic activity at nM concentrations, oral efficacy in in vivo models of disease, and the unprecedented ability to reactivate dormant liver stage parasites (hypnozoites). However, future development of this series will need to address host versus parasite selectivity, where inhibitory activity against human G9a is removed from the lead compounds, while maintaining potent anti-Plasmodium activity. Herein, we report an extensive study of the SAR of this series against both G9a and P. falciparum. We have identified key SAR features which demonstrate that high parasite vs. G9a selectivity can be achieved by selecting appropriate substituents at position 2, 4 and 7 of the quinazoline ring. We have also, in turn, discovered that potent G9a inhibitors can be identified by employing a 6-carbon ‘Nle mimic’ at position 7. Together, this data suggests that while broadly similar, the G9a and potential PfHKMT target(s) binding pockets and/or binding modes of the diaminoquinazoline analogues exhibit clear and exploitable differences. Based on this, we believe this scaffold to have clear potential for development into a novel anti-malarial therapeutic.
Introduction
Despite a global reduction in malaria incidence and mortality rate over the past 15 years, this disease remains a major health burden, with over 200 million new cases reported in 2015.1 The majority of the malaria related deaths occur in sub-Saharan Africa, particularly amongst children under five and pregnant women. The emergence of multi-drug resistant strains of Plasmodium, the malaria causing parasite, have rendered many of the conventional antimalarial drugs ineffective.2,3 Artemisinin-based combination therapies (ACTs) have, in part, addressed this issue and are widely used as an effective treatment.4 However, recent reports of artemisinin resistant Plasmodium strains along the Thai–Cambodian border5,6 is of significant concern. Thus, there is an increasing demand for the discovery of novel classes of antimalarial drugs, with distinct modes of action, in order to tackle multi-drug resistant Plasmodium parasites.7Epigenetic regulation has been shown to affect gene expression throughout the life cycle of Plasmodium8,9 and thus, modulation of epigenetic targets in the malarial parasite presents a novel approach for antimalarial drug discovery. Indeed, inhibitors of malarial histone deacetylases (PfHDACs) have been shown to possess parasite killing activity and are currently being explored as a new class of antimalarial drugs.10–14 However, the poor selectivity, unfavourable pharmacokinetics and toxicity issues associated with the hydroxamic acid-based HDAC inhibitors poses a major challenge to their clinical development.15 Thus other potential epigenetic targets in Plasmodium are of significant interest.
Histone lysine methyltransferases (HKMTs) act as vital components of epigenetic regulation by serving as ‘writers’ that install methyl marks on histones and other proteins. Among various human HKMTs, G9a (EHMT2) is a well-studied enzyme, which catalyses the addition of one or two methyl groups to lysine 9 of histone H3 (H3K9me1 and H3K9me2).16 Like most of the other HKMTs, the active site of G9a resides in the SET (suppressor of variegation 3–9, enhancer of zeste and trithorax) domain, where the substrate peptide binds and is, in turn, methylated by an S-adenosyl methionine (SAM) cofactor. G9a has been shown to play key role in various physiological and pathophysiological processes such as mental health,17 cocaine addiction,18,19 differentiation and cancer20–22 and thus together with other HKMTs, it is under investigation in context of drug discovery.23–26
Plasmodium falciparum HKMTs (PfHKMTs) play key role in controlling Plasmodium gene expression through epigenetic pathways.9 Computational analysis predicts the presence of ten SET domain containing PfHKMTs,27 six of which were found to be essential in the asexual blood-stages of the parasite and thus may represent good drug targets.28,29 Moreover, knockout of PfSET2 (now renamed PfSETvs) was found to reverse the silencing of the var gene family, which is centrally involved in the immune evasion mechanism by which Plasmodium avoids the host antibody response.8,28 Despite this potential, production of enzymatically active PfHKMTs has proved to be challenging, with only a few successful reports in the literature,29,30 thus hindering the prospect of PfHKMT inhibitor discovery.
We have recently reported our initial attempts to validate the PfHKMTs as a novel approach for antimalarial therapy.31–34 In the absence of the full complement of purified essential PfHKMTs – required for target-based hit discovery and SAR – we used a phenotypically-led approach; examining the activity of an established HKMT chemotype for antimalarial activity. Specifically, a focused library of inhibitors exemplifying the diaminoquinazoline HKMT chemotype was explored. Diaminoquinazoline HKMT activity was initially identified through a high throughput screen, with BIX01294 (1, Fig. 1) identified as an inhibitor of human G9a.35 While a number of medicinal chemistry studies – most notably those of Jin and co-workers – have been reported that improve the activity, selectivity, cell permeability and in vivo activity of G9a probes derived from 1,36–41 it is becoming increasingly apparent that the HKMT activity of this chemotype is not limited to G9a. Indeed, by modifying the amino side chains of this scaffold, diaminoquinazoline inhibitors have been reported exhibiting human SETD842,43 and EZH226 activity (Fig. 1). Given this broad HKMT activity, it would seem that ‘repurposing’ the diaminoquinazoline scaffold as inhibitors of the homologous PfHKMTs (for a comparison of the homology of select P. falciparum SET domains to human proteins, see Table S1‡ in Malmquist et al.31) is a valid approach to progress these exciting new drug targets.
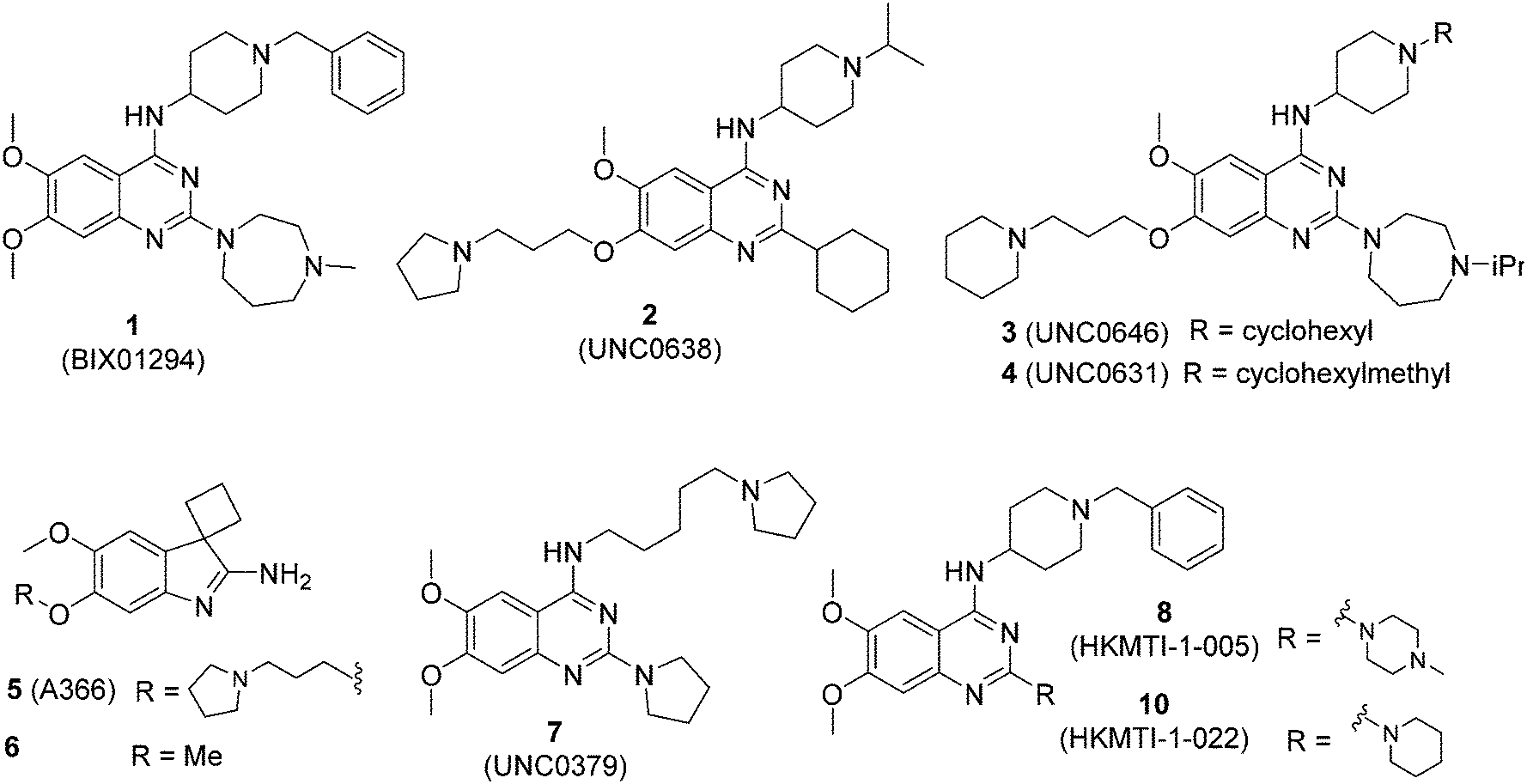 | ||
| Fig. 1 Representative examples of G9a (1–6), SET8 (7) and dual G9a/EZH2 (8, 10) inhibitors. | ||
In our initial studies, 1 (Fig. 1) and a related analogue TM2-115 (60, Table 3) were found to exhibit rapid and irreversible asexual cycle blood stage-independent cytotoxic activity at nM concentrations, comparable potency against resistant strains (including artemisinin) and clinical isolates of P. falciparum and P. vivax, and oral efficacy in in vivo mouse models of P. berghei and P. falciparum infection.31,32 Highly promising effects were also observed for other life cycle stages, with mature gametocyte progression to gamete formation inhibited at submicromolar concentrations,32 and the unprecedented ability to reactivate dormant liver stages (hypnozoites) in a novel in vitro model system.34 A dose-dependent reduction in histone methylation (H3K4 and, to a lesser extent H3K9) was observed in parasites upon treatment (Western analysis), suggesting on-target PfHKMT activity,31 and that the broad ranging effects of these compounds is likely due to their target. A preliminary SAR study on our diaminoquinazoline series revealed that some pharmacophoric features might be conserved for both parasite-killing and G9a inhibition, thereby suggesting potential similarities between G9a and the yet unidentified PfHKMT target(s) responsible for the anti-parasitic activity.33 However, future development of this series will need to address host versus parasite selectivity; where inhibitory activity against human G9a is removed from the lead compounds, while maintaining potent anti-Plasmodium activity. Hence, we set out to identify regions around the scaffold that can be fine-tuned to improve the parasite-killing to G9a inhibition ratio. Herein, we report an extensive study of the SAR of this series against both G9a and P. falciparum. To provide a more complete picture of the underlying SAR, some of the analogues included (mainly in the ESI‡), and their anti-Plasmodium activities, were previously reported by us.33 However, G9a inhibition for such analogues is reported here for the first time. Such cases are clearly marked in Tables 1–4. Important and previously unidentified trends are determined for activity against both targets and, critically, we elucidate features of this scaffold that allow for high parasite versus host selectivity. We believe this study further cements the potential of this scaffold as a candidate for development into greatly needed novel therapies to control malaria.
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| R2 | Pf3D7 IC50 (nM)* | G9a IC50 (nM)* | G9a/Pf3D7 | HepG2 IC50 (nM) | HepG2/Pf3D7 | clog![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P P |
TPSA | |
| a Parasite-killing activity reported earlier.33 b IC50 reported using enzyme-coupled SAH detection (ECSD) assay.36,37 c IC50 reported using chemiluminescence-based oxygen tunnelling (CLOT) assay.36,37 ND = not determined; *IC50 determination experiments were performed either in duplicates or triplicates. | ||||||||
| 1 BIX01294 |
|
43a | 67 (110b/290c) | 1.6 | 4800 | 111.6 | 3.86 | 65.99 |
| 8 |
|
18a | 101 | 5.6 | 5500 | 305.5 | 3.48 | 65.99 |
| 9 |
|
29a | 332 | 11.4 | 3800 | 131 | 5.10 | 62.75 |
| 10 |
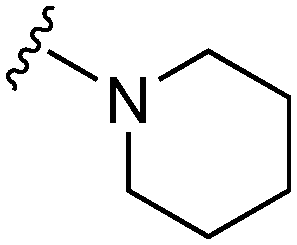
|
23a | 472 | 20.5 | 4700 | 201.7 | 4.71 | 62.75 |
| 11 |
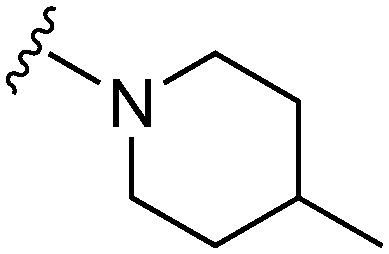
|
76a | 1326 | 17.4 | 5400 | 71.0 | 4.96 | 62.75 |
| 12 |
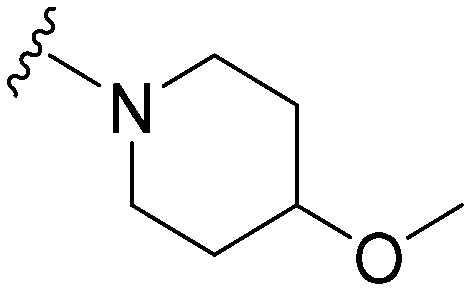
|
38a | 576 | 15.2 | 10100 |
265.8 | 4.34 | 71.98 |
| 13 |
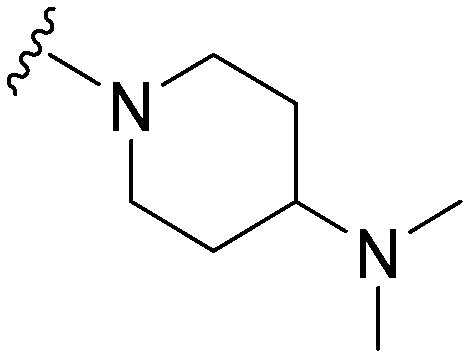
|
37a | 506 | 13.7 | 5900 | 159.5 | 4.25 | 65.99 |
| 14 |
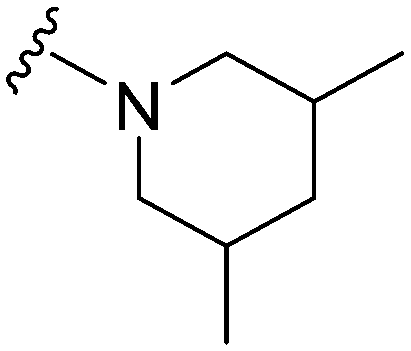
|
67a | ∼10000 |
∼149.3 | 3600 | 53.7 | 5.21 | 62.75 |
| 15 |
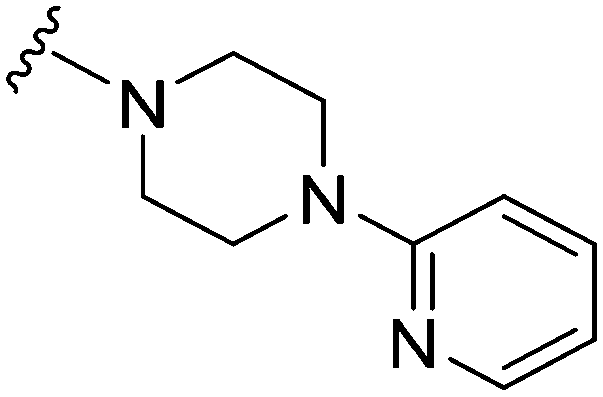
|
26a | 3190 | 122.7 | 2600 | 100 | 4.44 | 78.88 |
| 16 |
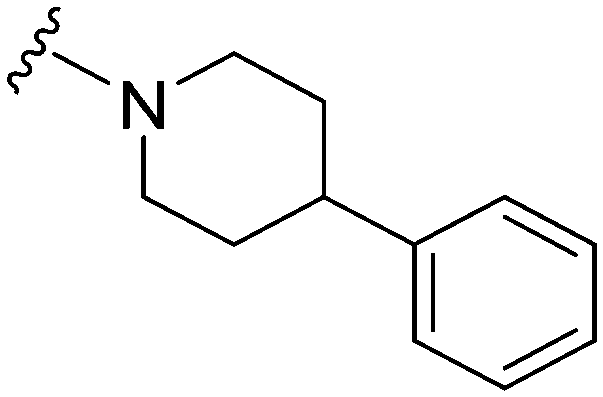
|
72a | >10000 |
>138.9 | 6100 | 84.7 | 6.11 | 62.75 |
| 17 |
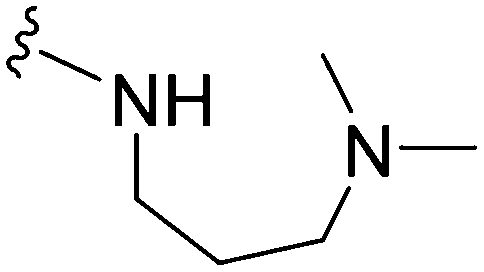
|
>1000 | 123 | <0.1 | ND | ND | 4.09 | 74.78 |
| Pyrimethamine | 33 | ND | ND | ND | ND | |||
| Chloroquine | 8 | ND | ND | ND | ND | |||
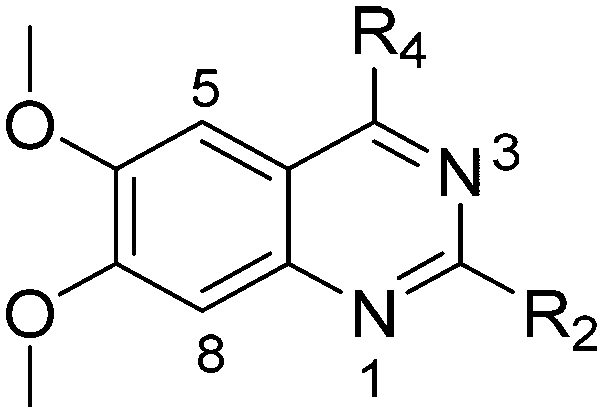
Chemistry
Molecules 8–59 (Tables 1 and 2) were synthesized following a well-established two-step synthetic procedure (Scheme 1).36–38,41 Hence, 6,7-dimethoxy-2,4-dichloroquinazoline was treated with various commercially available or synthesized (see ESI‡) N-substituted-4-piperidylamines, in order to obtain 4-substituted quinazoline derivatives. These intermediates were subsequently heated with a second amine nucleophile to access the desired diaminoquinazolines analogues.|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| ID | R4 | R2 | Pf3D7 IC50 (nM)* | G9a IC50 (nM)* | G9a/Pf3D7 | HepG2 IC50 (nM) | HepG2/Pf3D7 | clogP |
TPSA |
| a Parasite-killing activity reported earlier.33 b IC50 reported using enzyme-coupled SAH detection (ECSD) assay.36,37 c IC50 reported using chemiluminescence-based oxygen tunnelling (CLOT) assay.36,37 ND = not determined; NT = not tested; *IC50 determination experiments were performed either in duplicates or triplicates. | |||||||||
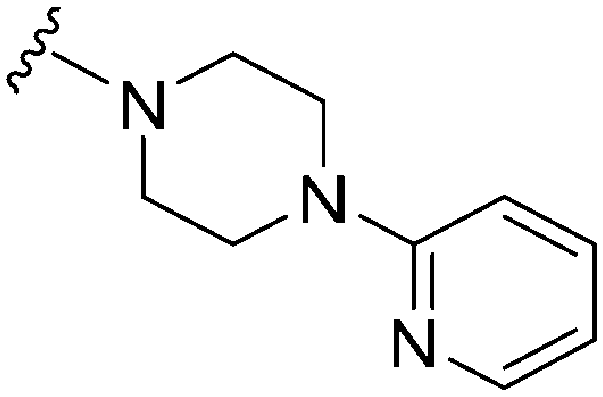
| 18 |
|
|
74 | 116 | 1.6 | 3100 | 41.9 | 3.78 | 65.99 |
| 19 |
|
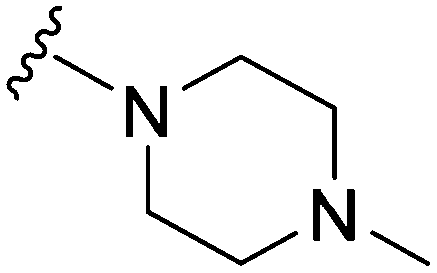
|
69 | 78 | 1.1 | 3300 | 47.8 | 3.78 | 65.99 |
| 20 |
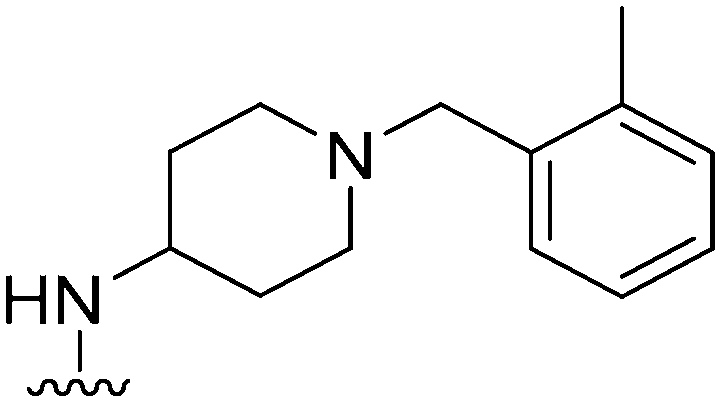
|
|
80 | 169 | 2.1 | 4100 | 51.2 | 3.78 | 65.99 |
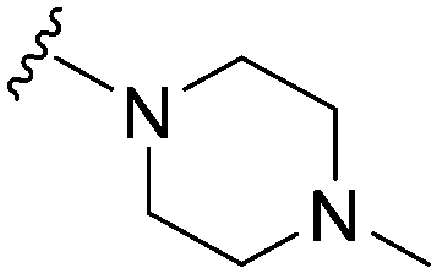
| 21 |
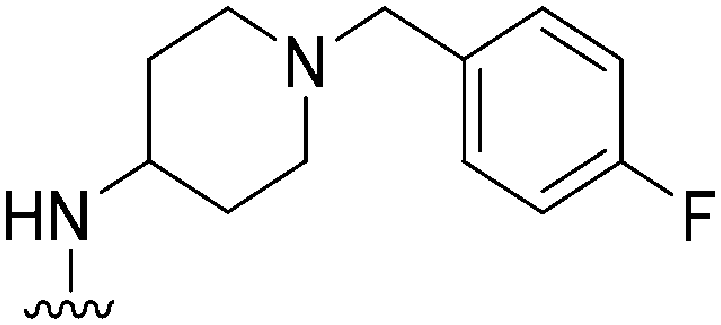
|
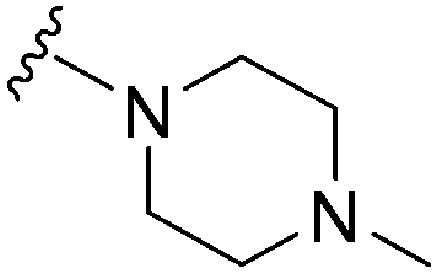
|
NT | 112 | — | ND | ND | 3.61 | 65.99 |
| 22 |
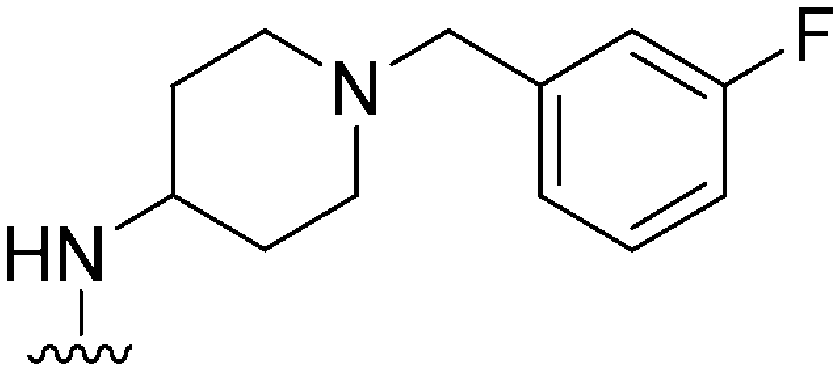
|
|
148 | 132 | 0.9 | ND | ND | 3.61 | 65.99 |
| 23 |
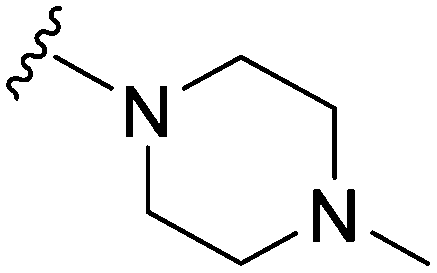
|
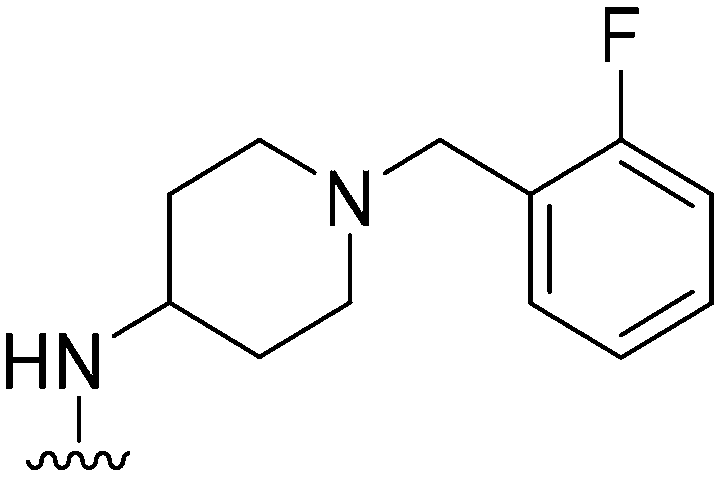
|
137 | 116 | 0.8 | ND | ND | 3.61 | 65.99 |
| 24 |
|
|
237 | 128 | 0.5 | ND | ND | 3.48 | 75.22 |
| 25 |
|
|
77 | 869 | 11.3 | 2800 | 36.4 | 5.02 | 62.75 |
| 26 |
|
|
77 | 690 | 9.0 | 2700 | 35.1 | 5.02 | 62.75 |
| 27 |
|
|
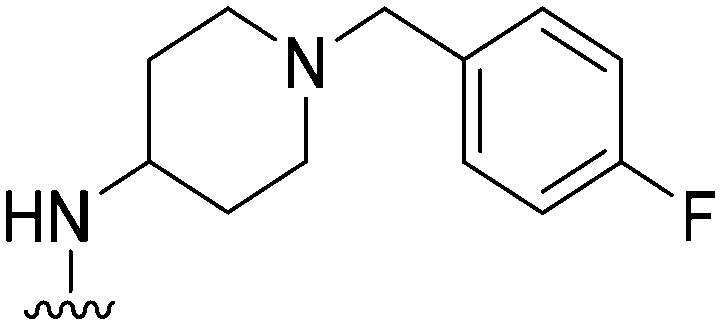
78 | ∼1000 | ∼12.8 | 2900 | 37.2 | 5.02 | 62.75 |
| 28 |
|
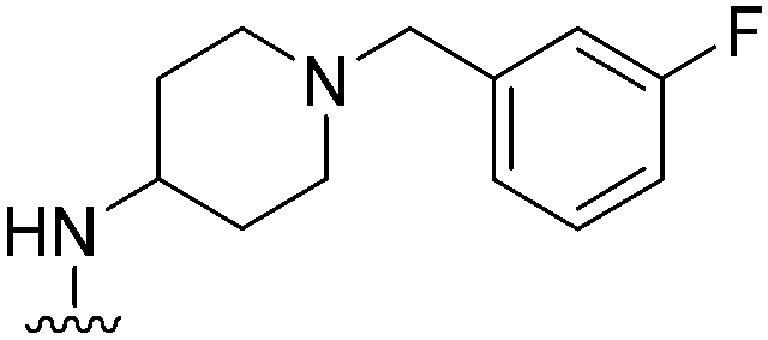
|
191 | 1214 | 6.4 | ND | ND | 4.85 | 62.75 |
| 29 |
|
|
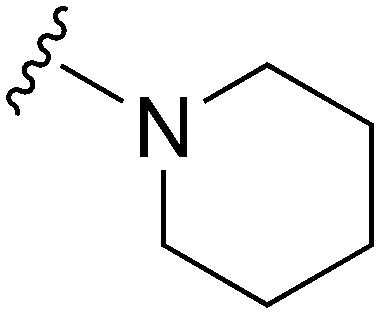
NT | ∼1000 | — | ND | ND | 4.85 | 62.75 |
| 30 |
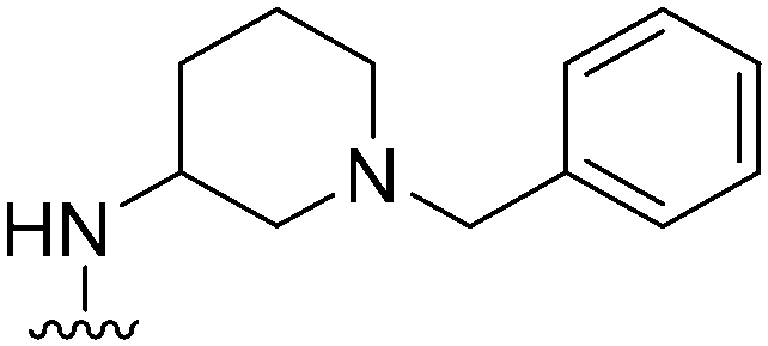
|
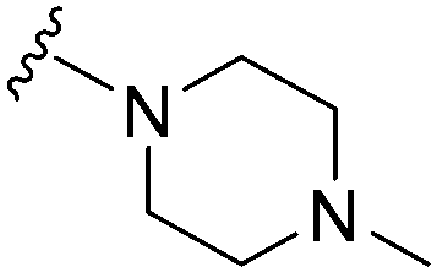
|
197a | ∼1000 | ∼5.1 | 10500 |
53.3 | 3.48 | 65.99 |
| 31 |
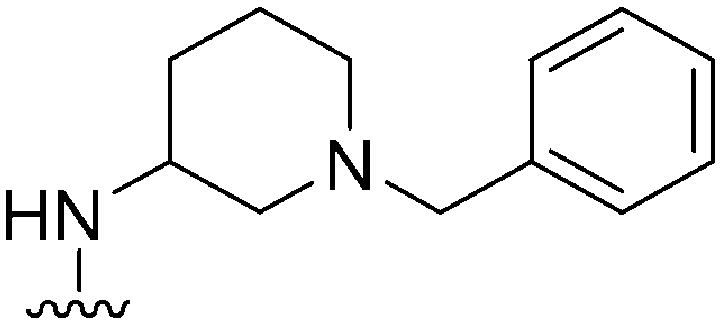
|
|
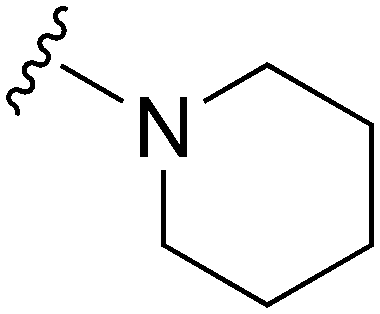
178a | ∼1000 | ∼5.6 | 6300 | 35.4 | 4.71 | 62.75 |
| 32 |
|
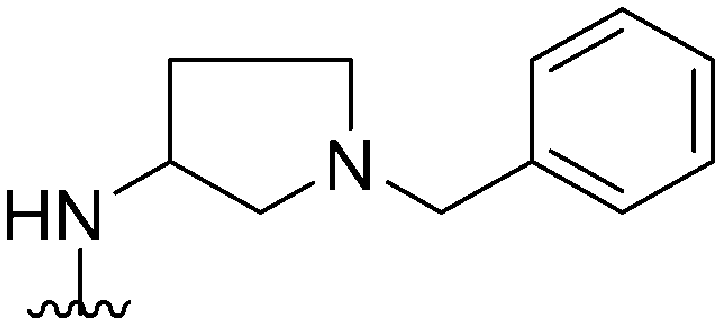
|
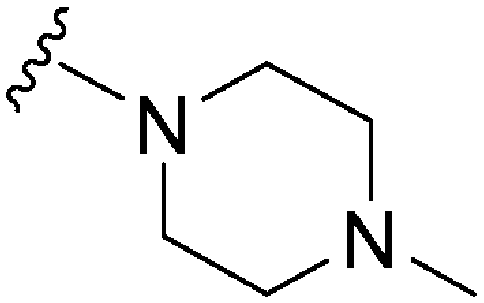
330a | 344 | 1.0 | ND | ND | 3.09 | 65.99 |
| 33 |
|
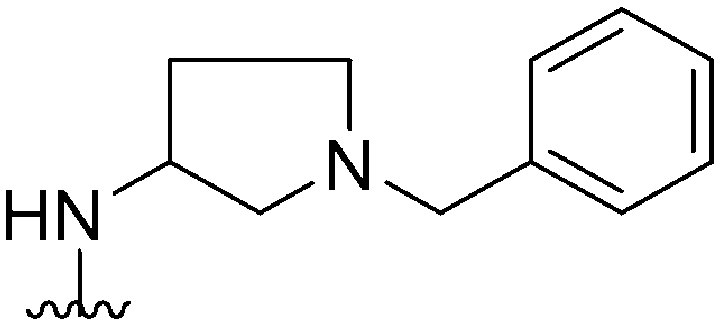
|
94a | 830 | 8.8 | 6300 | 67 | 4.32 | 62.75 |
| 34 |
|
|
93a | ∼10000 |
∼107.5 | 5600 | 60.2 | 4.06 | 78.88 |
| 35 |
|
|
247a | >10000 |
>40.5 | ND | ND | 4.08 | 95.95 |
| 36 |
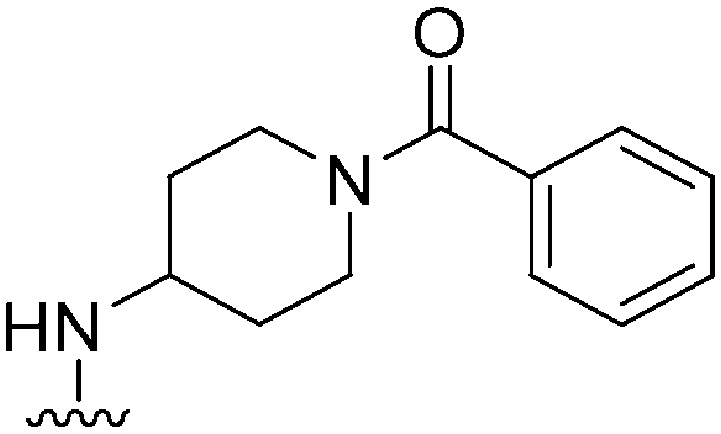
|
|
369a | >10000 |
>27.1 | ND | ND | 4.35 | 79.82 |
| 37 |
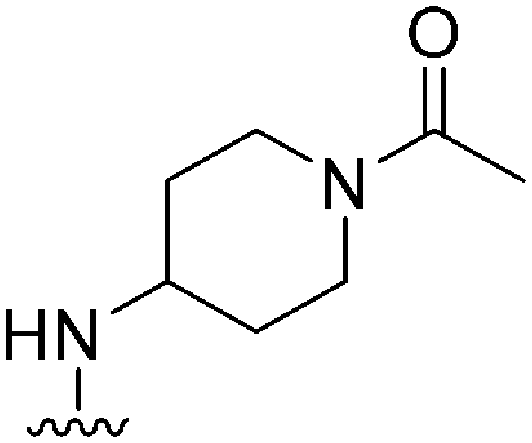
|
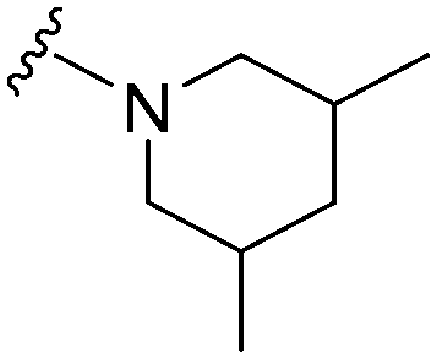
|
107a | >1000 | >9.3 | 17800 |
166.3 | 3.55 | 79.82 |
| 38 |
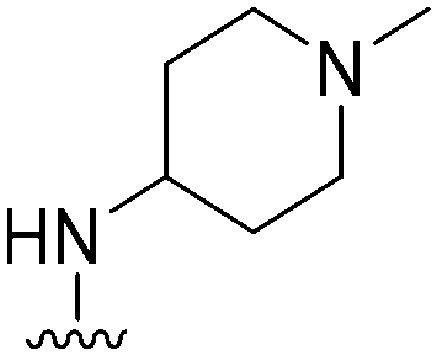
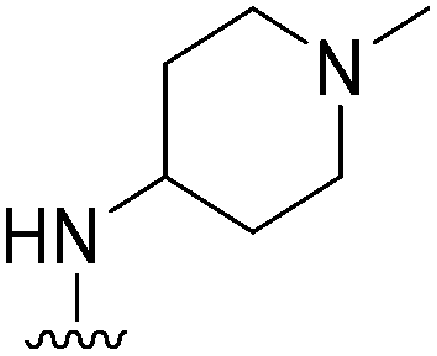
|
|
>300a | 330b/230c | — | ND | ND | 2.30 | 65.99 |
| 39 |
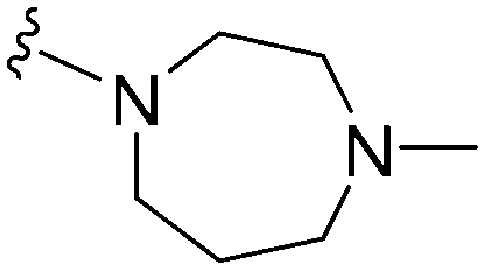
|
|
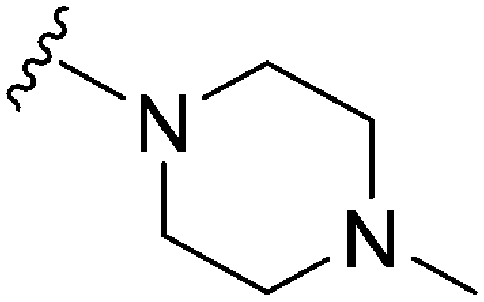
>300a | 680b/200c | — | ND | ND | 1.90 | 65.99 |
| 40 |
|
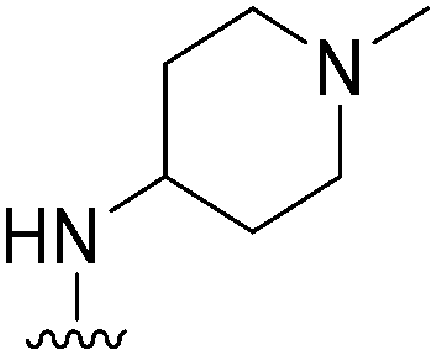
|
37a | 591 | 16.0 | 14200 |
383.8 | 3.14 | 62.75 |
| 41 |
|
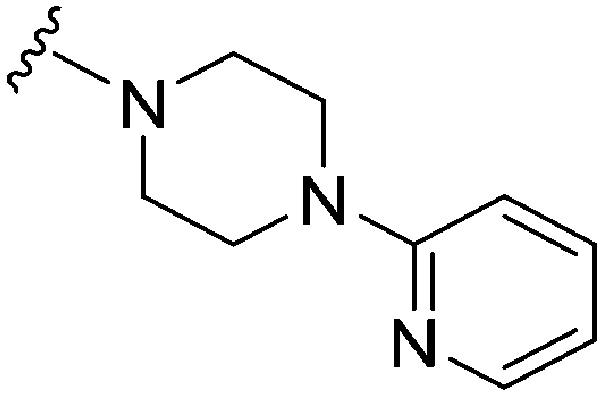
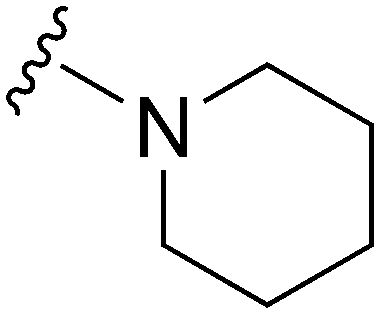
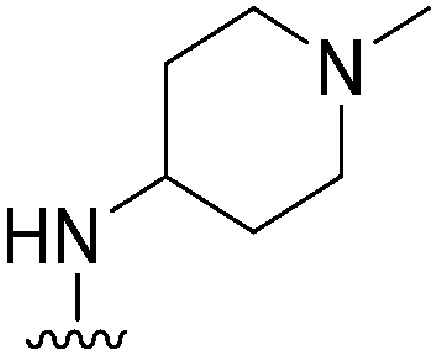
|
56a | >1000 | >17.9 | 5500 | 98.2 | 2.88 | 78.88 |
| 42 |
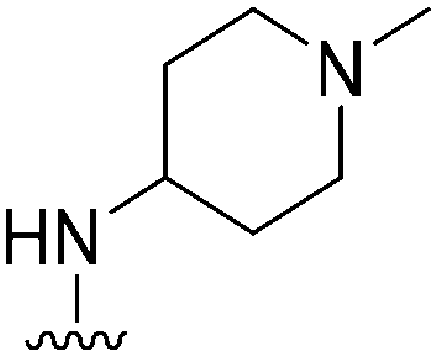
|
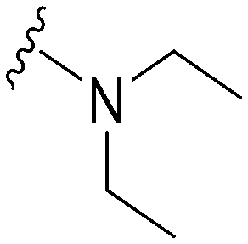
|
28a | 910b/6500c | — | >10000 |
>357 | 3.00 | 62.75 |
| 43 |
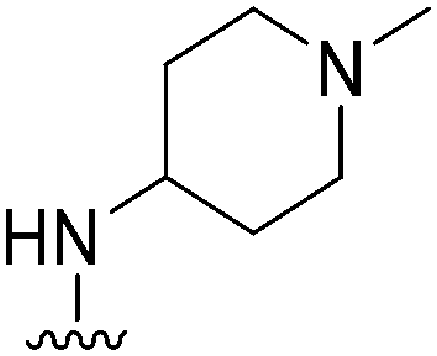
|
|
34a | 1100b/900c | — | >10000 |
>294 | 2.22 | 62.75 |
| 44 |
|
|
174 | 10 | 0.06 | ND | ND | 4.25 | 65.99 |
| 45 |
|
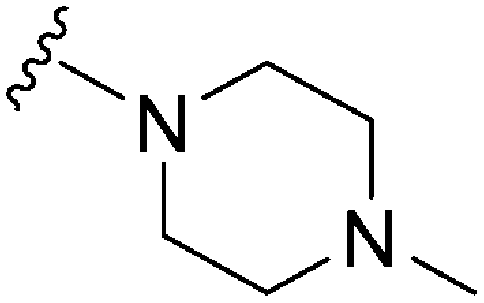
|
130 | 25 | 0.19 | ND | ND | 3.86 | 65.99 |
| 46 |
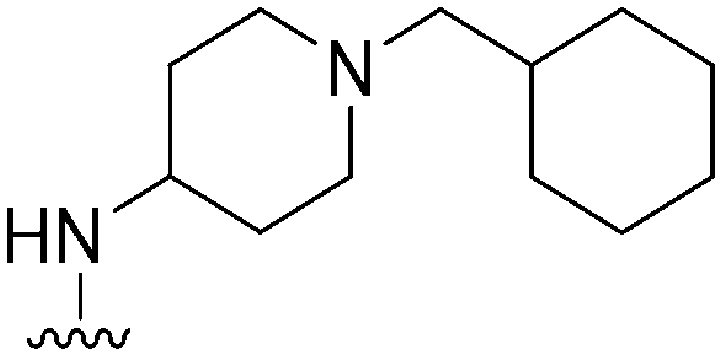
|
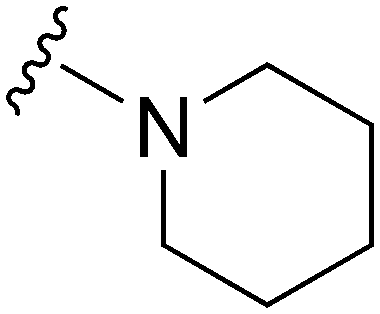
|
144 | 295 | 2.0 | ND | ND | 5.09 | 62.75 |
| 47 |
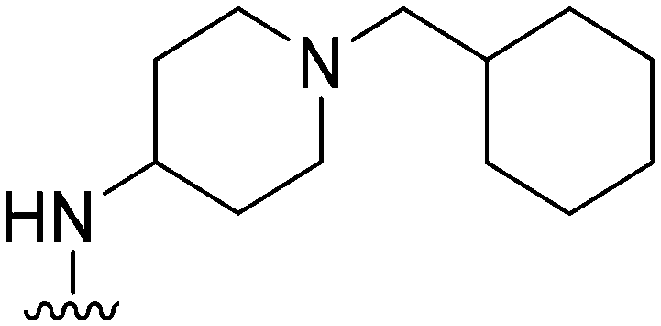
|
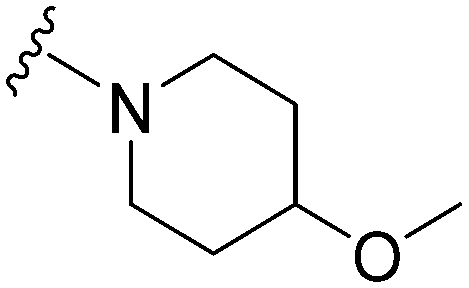
|
47 | 185 | 3.9 | ND | ND | 4.72 | 71.98 |
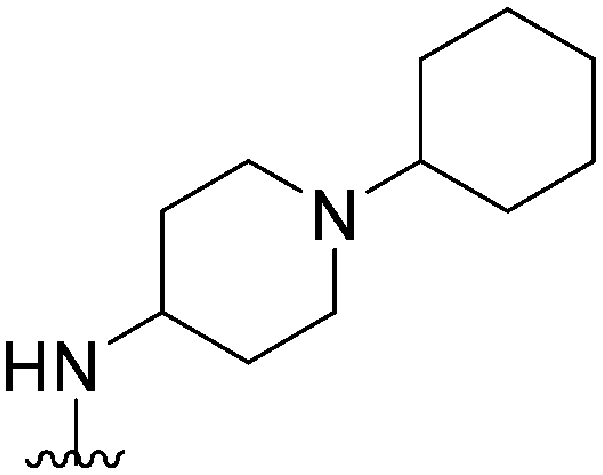
| 48 |
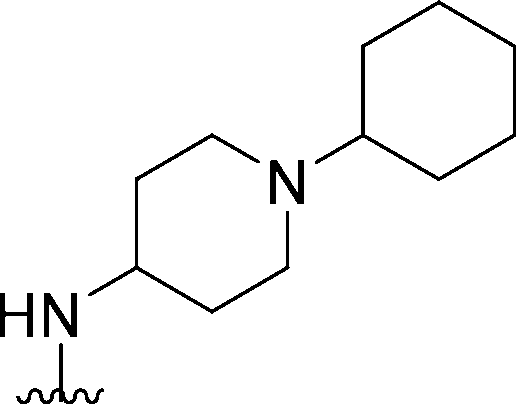
|
|
205 | 55 | 0.27 | ND | ND | 4.00 | 65.99 |
| 49 |
|
|
222 | 49 | 0.22 | ND | ND | 3.61 | 65.99 |
| 50 |
|
|
108 | NT | — | ND | ND | 4.85 | 62.75 |
| 51 |
|
|
470 | 55 | 0.12 | ND | ND | 3.07 | 65.99 |
| 52 |
|
|
280 | 123 | 0.44 | ND | ND | 2.68 | 65.99 |
| 53 |
|
|
126 | NT | — | ND | ND | 3.55 | 71.98 |
| 54 |
|
|
71 | 319 | 4.5 | ND | ND | 3.92 | 62.75 |
| 55 |
|
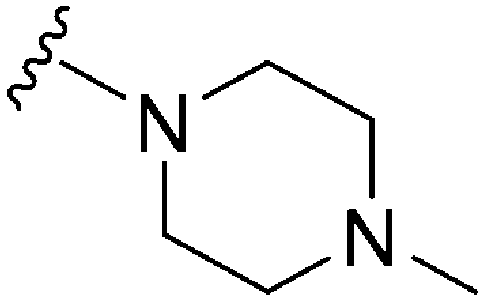
|
215 | 6438 | 29.9 | ND | ND | 3.14 | 62.75 |
| 56 |
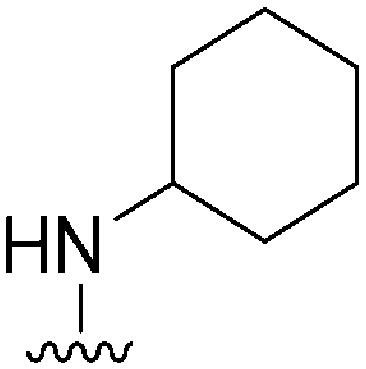
|
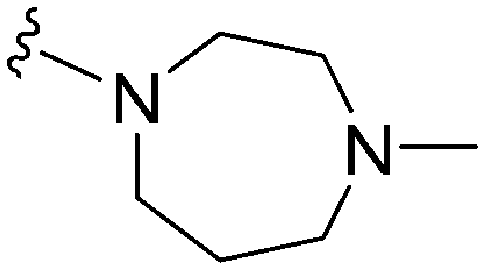
|
107 | 3877 | 36.2 | ND | ND | 3.53 | 62.75 |
| 57 |
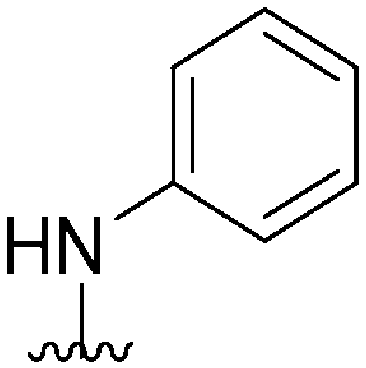
|
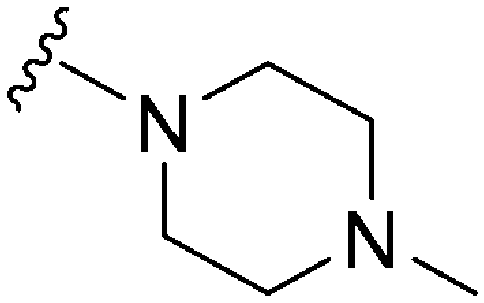
|
570a | >10000 |
>17.5 | ND | ND | 3.14 | 62.75 |
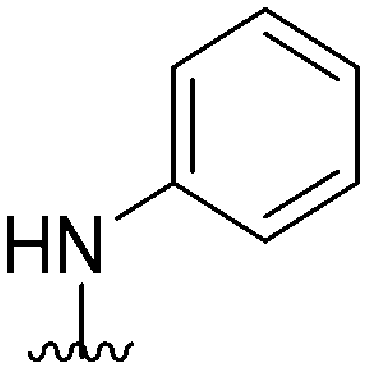
| 58 |
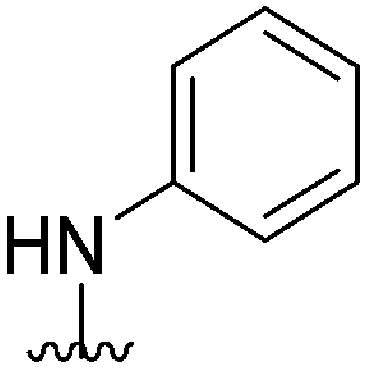
|
|
161a | >10000 |
>62.1 | ND | ND | 4.38 | 59.51 |
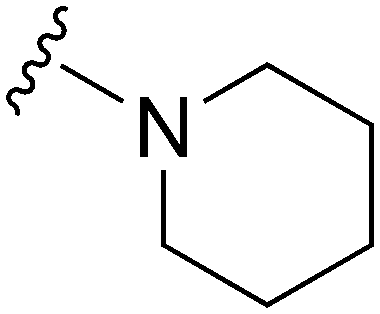
| 59 |
|
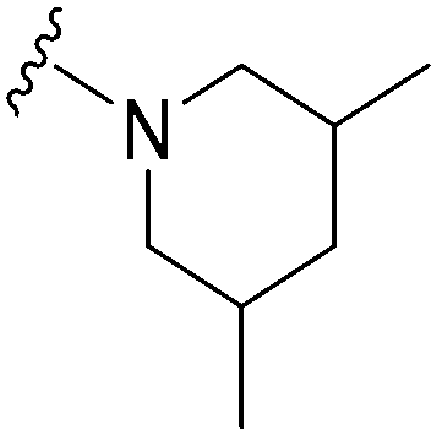
|
77a | >10000 |
>129.9 | 12200 |
158.4 | 4.87 | 59.51 |
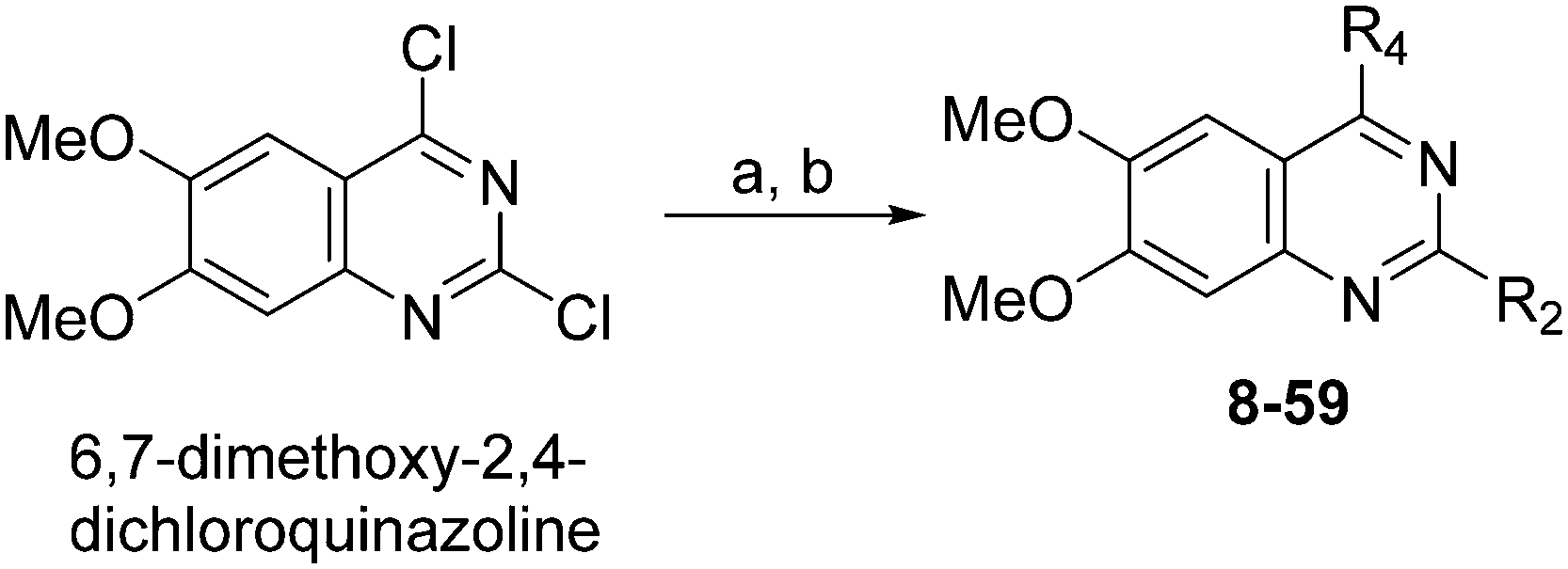 | ||
| Scheme 1 Synthesis of diaminoquinazolines in Tables 1 and 2. Reagents and conditions: (a) various amines, Et3N (or DIEA), THF (or DMF), RT, 18–24 h; (b) various amines (5–10 equiv.), microwave, toluene (or neat), 130–185 °C, 30–50 min or i-PrOH, 4 M HCl/dioxane, microwave, 160 °C, 15 min. | ||
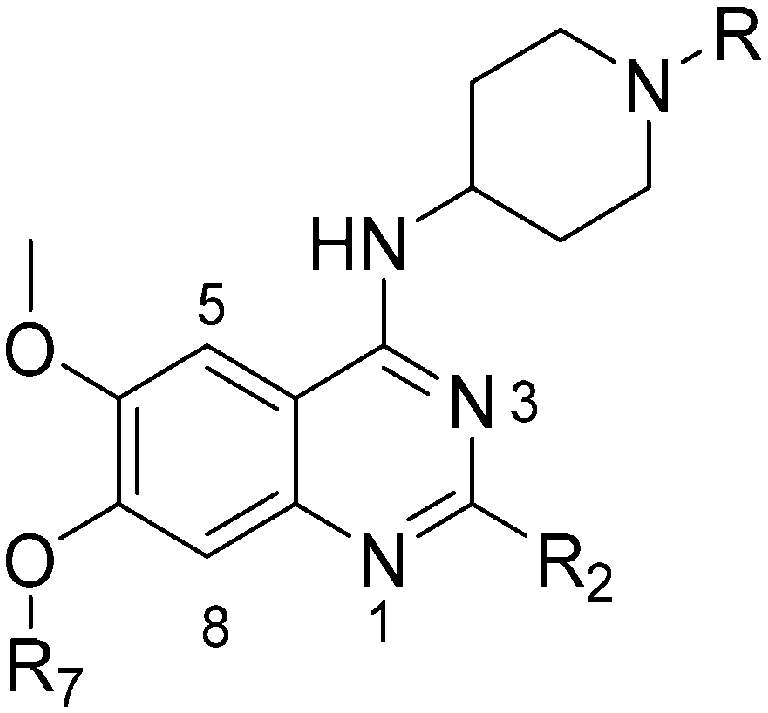
Analogues with benzyl or alkoxy substituents at position 7 (Table 3) were synthesized following a previously described methodology,36–38 with slight modifications, as shown in Scheme 2. The phenolic oxygen of the commercially available 4-hydroxy-3-methoxybenzoate (92) was first benzylated to give 93. Nitration of 93 gave nitro compound 94 which was further reduced to obtain aniline 95. Conversion of 95 into a urea intermediate, followed by base-mediated ring closure yielded quinazolinedione 96 that was subsequently heated with phosphorous oxychloride to obtain the 2,4-dichloroquinazoline derivative 97. Subsequent displacement of the chloride atoms from position 4 and 2, analogously to that described in Scheme 1, yielded final compounds 60–63 possessing 7-OBn substituents. Alternatively, 98 was debenzylated under acidic conditions to yield intermediates 99 possessing a free phenol group at position 7. The phenol oxygen was either alkylated using a selection of alkyl halides or else treated with primary alcohols under Mitsunobu conditions to give substituted 2-chloroquinazolines 100. Finally, substitution of the chloride at position 2 of 100 with a second amine yielded the desired analogues 68–70 and 73–84. The synthesis of analogues 64–67, 71, 72 (Table 3) has been reported earlier by Jin et al. using similar synthetic scheme.36–38
|
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| R | R7 | R2 | Pf3D7 IC50 (nM)* | G9a IC50 (nM)* | G9a/Pf3D7 | HepG2 IC50 (nM) | HepG2/Pf3D7 | clogP |
TPSA | |
| a Parasite-killing activity reported earlier.33 b IC50 reported using enzyme-coupled SAH detection (ECSD) assay.36,37 c IC50 reported using chemiluminescence-based oxygen tunnelling (CLOT) assay.36,37 ND = not determined; *IC50 determination experiments were performed either in duplicates or triplicates. | ||||||||||
| 60 TM2-115 | –Me | –Bn |
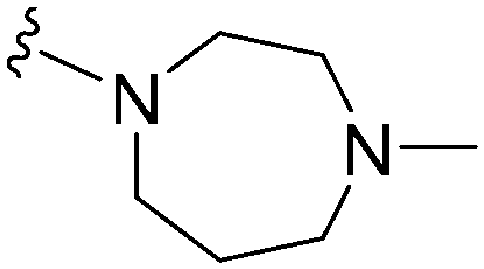
|
43a | >1000 | 23.2 | 4700 | 110.1 | 3.86 | 65.99 |
| 61 | –Bn | –Bn |
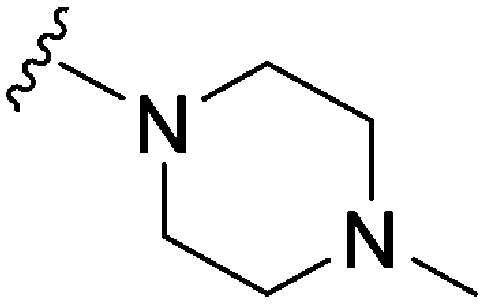
|
82 | ∼10000 |
122 | 2900 | 35.4 | 5.05 | 65.99 |
| 62 | –Bn | –Bn |
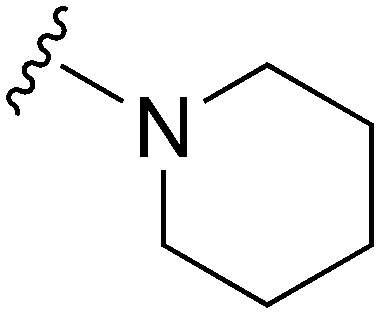
|
57 | >50000 |
877.2 | 2900 | 50.9 | 6.28 | 62.75 |
| 63 | –Me | –Bn |
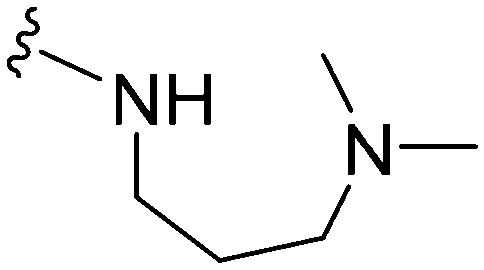
|
>1000 | >1000 | — | ND | ND | 4.09 | 74.78 |
| 64 | –Me |
|
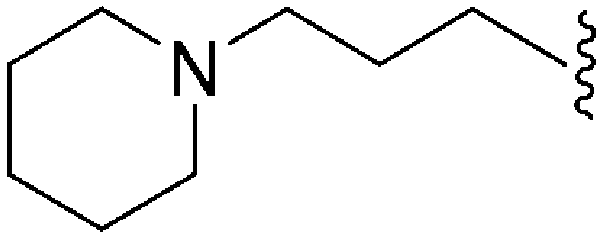
|
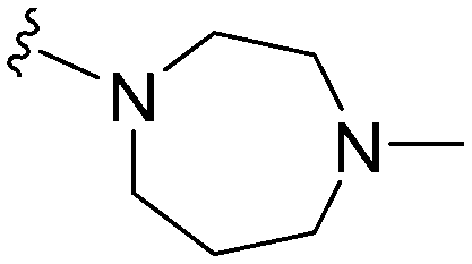
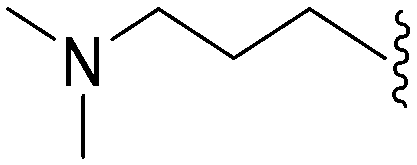
>2000a | 25b/20c | — | ND | ND | 3.54 | 69.23 |
| 65 | –Me |
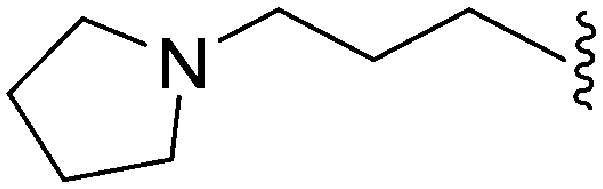
|
|
>2000a | 8b | — | ND | ND | 3.15 | 69.23 |
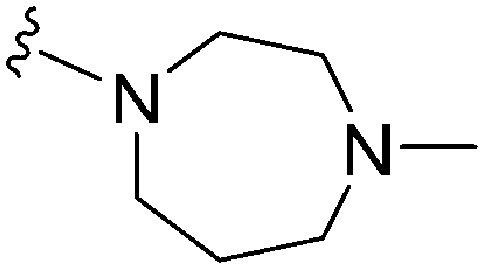
| 66 (UNC0224) | –Me |
|
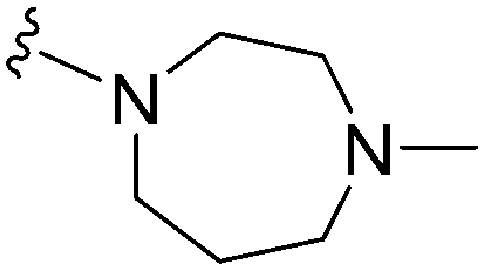
|
>2000a | 43b/57c | — | ND | ND | 2.62 | 69.23 |
| 67 | –Me |
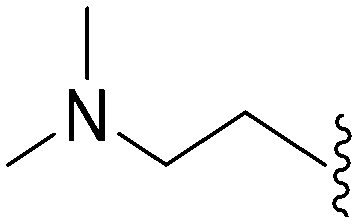
|
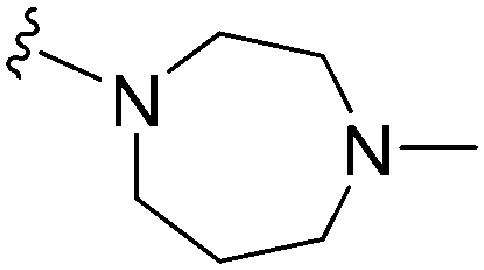
|
>2000a | 110b/120c | — | ND | ND | 2.23 | 69.23 |
| 68 | –iPr |
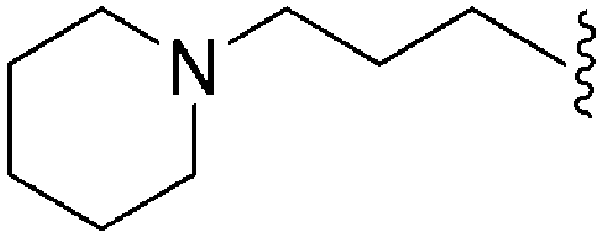
|
|
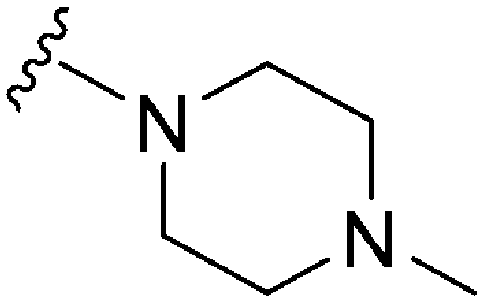
1120 | 4 | 0.004 | ND | ND | 3.93 | 69.23 |
| 69 | –Bn |
|
|
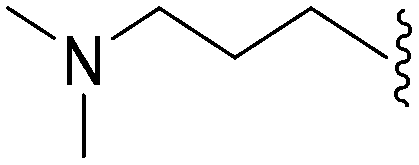
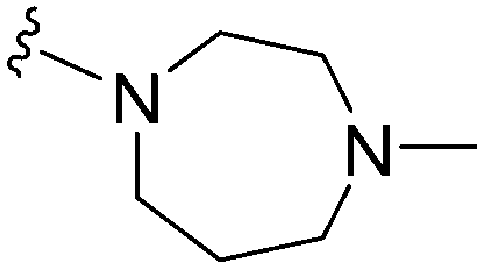
632 | <3 | >0.005 | ND | ND | 4.19 | 69.23 |
| 70 | –Bn |
|
|
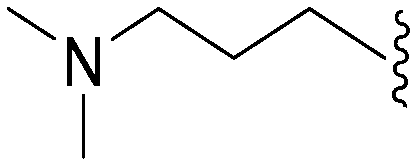
449 | 8 | 0.018 | ND | ND | 3.80 | 69.23 |
| 71 | –Me |
|
|
319a | >10000c |
— | ND | ND | 3.09 | 75.22 |
| 72 | –Me |

|
|
150a | 3400b/5200c | — | ND | ND | 4.10 | 65.99 |
| 73 | –Bn |
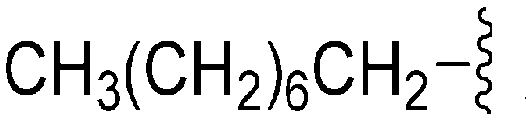
|
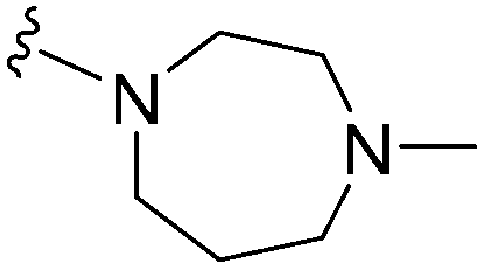
|
268 | 211 | 0.79 | ND | ND | 6.60 | 65.99 |
| 74 | –Bn |
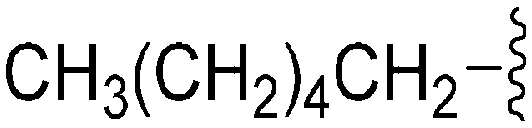
|
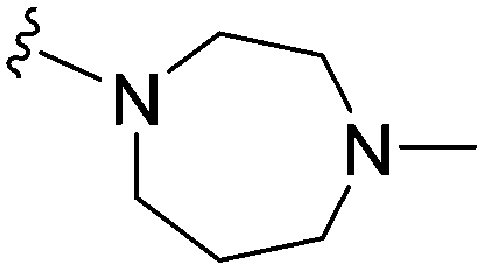
|
235 | 39 | 0.17 | ND | ND | 5.82 | 65.99 |
| 75 | –Bn |
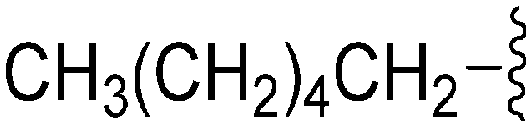
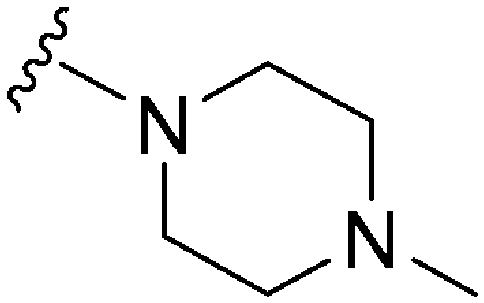
|
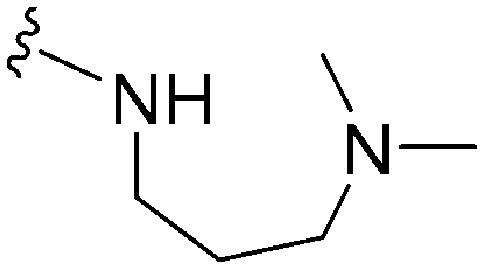
|
248 | 39 | 0.16 | ND | ND | 6.04 | 74.78 |
| 76 | –Bn |
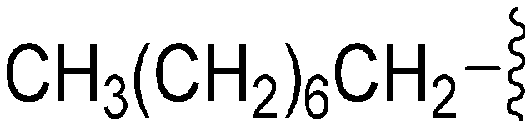
|
|
242 | 1018 | 4.2 | ND | ND | 6.21 | 65.99 |
| 77 | –Bn |
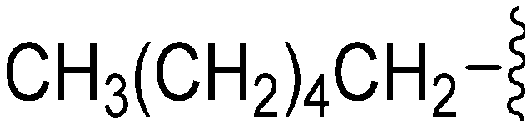
|
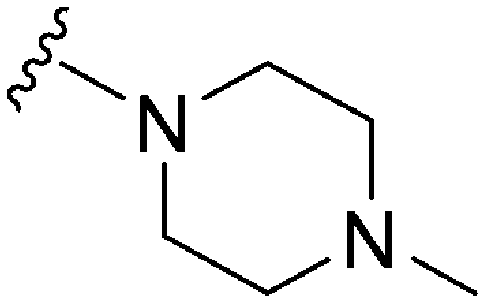
|
231 | 114 | 0.5 | ND | ND | 5.43 | 65.99 |
| 78 | –Bn |
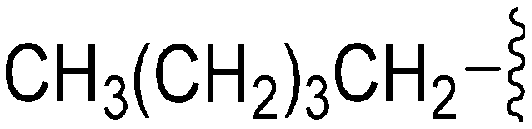
|
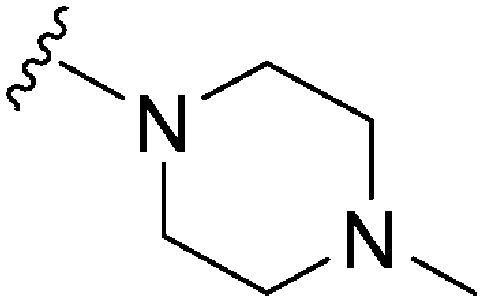
|
173 | ∼1000 | ∼5.8 | ND | ND | 5.04 | 65.99 |
| 79 | –Bn |
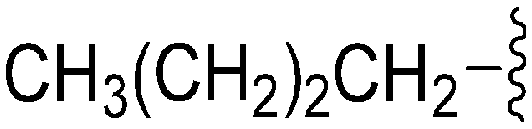
|
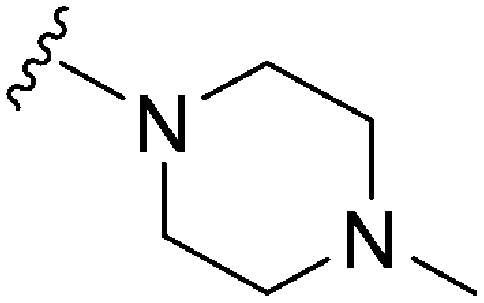
|
62 | 683 | 11.0 | 2900 | 46.8 | 4.65 | 65.99 |
| 80 | –iPr |
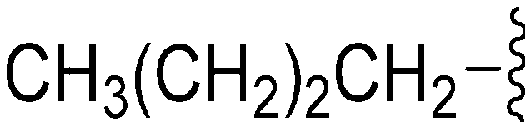
|
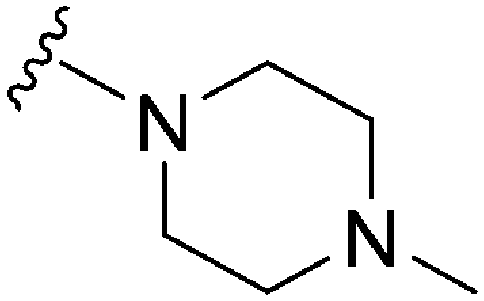
|
56 | 843 | 15.0 | 4000 | 71.4 | 3.85 | 65.99 |
| 81 | –Bn |
|
|
61 | 5344 | 87.6 | 1800 | 29.5 | 4.89 | 65.99 |
| 82 | –iPr |
|
|
37 | 2034 | 55.0 | 2400 | 64.9 | 4.10 | 65.99 |
| 83 | –Bn |
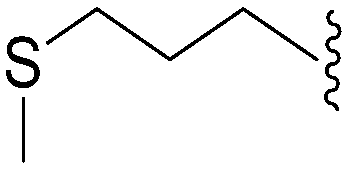
|
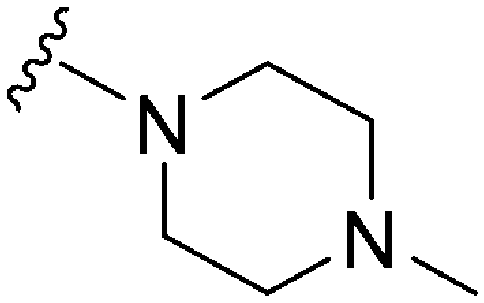
|
162 | 223 | 1.4 | ND | ND | 4.60 | 65.99 |
| 84 | –Bn |
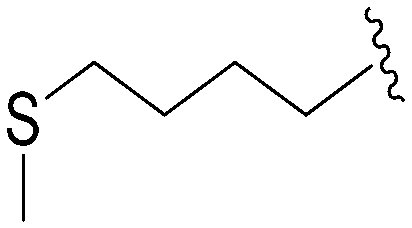
|
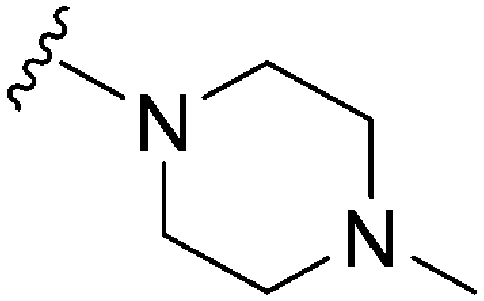
|
241 | 575 | 2.4 | ND | ND | 4.99 | 65.99 |
 | ||
| Scheme 2 Synthesis of diaminoquinazolines in Table 3. Reagents and conditions: (i) benzyl bromide, K2CO3, DMF, 0 °C – RT, 18 h; (ii) HNO3, Ac2O, RT, 18 h; (iii) Fe, NH4Cl, i-PrOH/H2O, reflux, 18 h; (iv) a) NaOCN, H2O/AcOH, RT, 18 h; b) NaOH, MeOH, reflux, 6 h; (v) POCl3, DIEA, acetonitrile, reflux, 6 h; (vi) various amines, Et3N (or DIEA), THF (or DMF), RT, 18–24 h; (vii) various amines (5–10 equiv.), microwave, toluene (or neat), 130–185 °C, 30–50 min or i-PrOH, 4 M HCl/dioxane, microwave, 160 °C, 15 min; (viii) TFA, reflux, 3 h; (ix) K2CO3 (or Cs2CO3), DMF, alkyl halides, 3–18 h 80 °C or PPh3, DIAD, THF, 20 h, RT. | ||
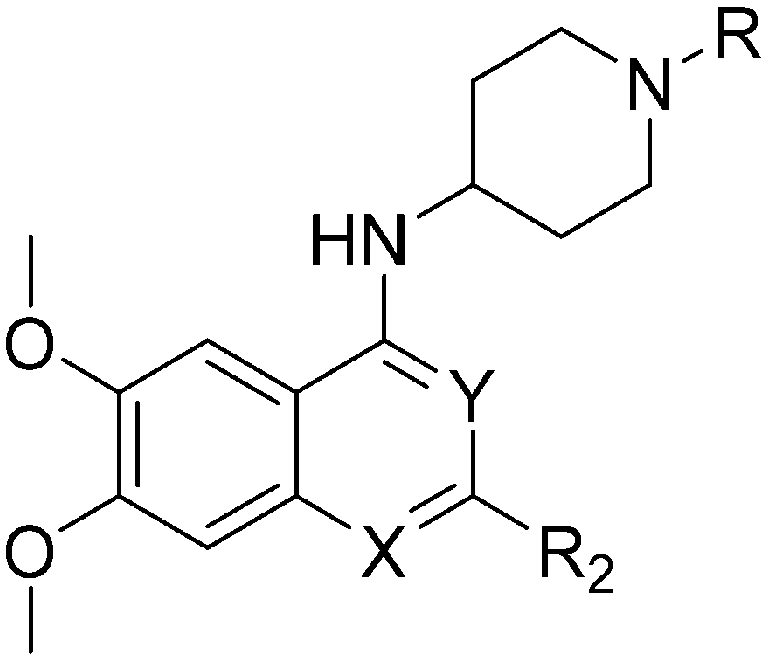
For the synthesis of diaminoisoquinoline and diaminoquinoline analogues 85–91 (Table 4) a route analogous to the synthesis of diaminoquinazolines was envisaged, where the desired amines could be installed late stage. This strategy required the synthesis of the corresponding 1,3-dichloro-6,7-dimethoxyisoquinoline (107) and 2,4-dichloro-6,7-dimethoxyquinoline (110) intermediates, as shown in Schemes 3 and 4, respectively.
|
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| ID | R | X | Y | R2 | Pf3D7 IC50 (nM)* | G9a IC50 (nM)* | G9a/Pf3D7 | HepG2 IC50 (nM) | HepG2/Pf3D7 | SlogP |
TPSA |
| ND = not determined; NT = not tested; *IC50 determination experiments were performed either in duplicates or triplicates. | |||||||||||
| 85 | –Bn | C | N |
|
955 | NT | — | ND | ND | 4.08 | 53.1 |
| 86 | –Bn | C | N |
|
1087 | >50000 |
>46.00 | ND | ND | 4.94 | 59.09 |
| 87 | –Bn | C | N |
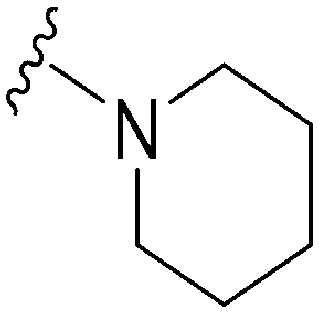
|
1367 | >25000 |
>18.3 | ND | ND | 5.32 | 49.86 |
| 88 | –Bn | N | C |
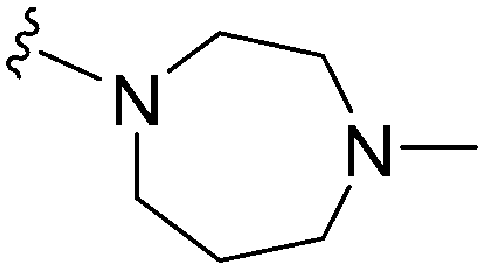
|
97 | 13 | 0.13 | 5600 | 57.7 | 4.47 | 53.1 |
| 89 | –Bn | N | C |
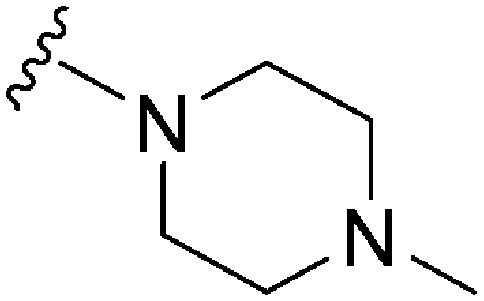
|
46 | 21 | 0.46 | 3400 | 73.9 | 4.08 | 53.1 |
| 90 | –Bn | N | C |
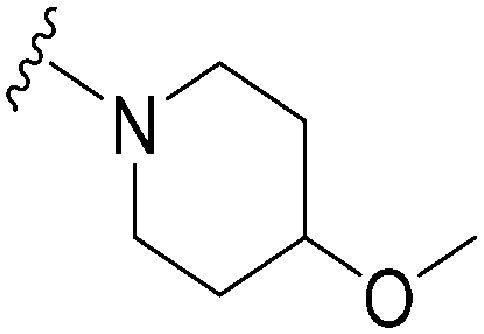
|
49 | 119 | 2.4 | 3600 | 73.5 | 4.94 | 59.09 |
| 91 | –Me | N | C |
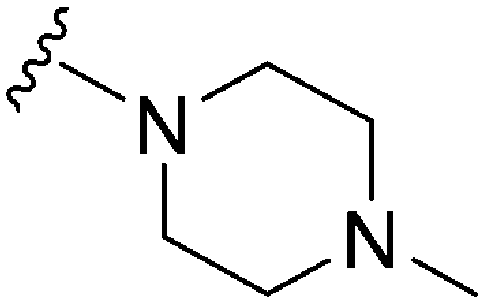
|
203 | 30 | 0.15 | 13500 |
66.5 | 2.51 | 53.1 |
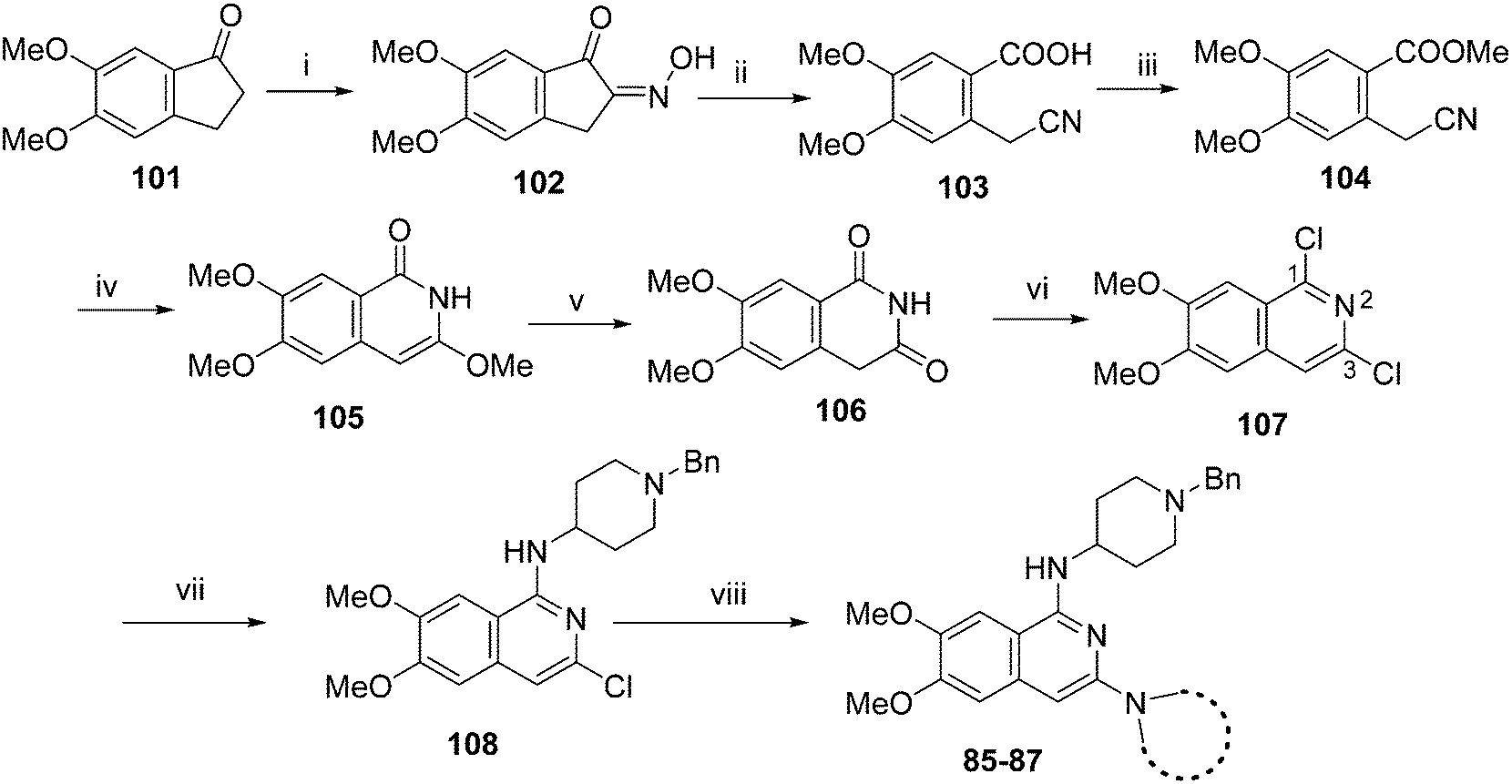 | ||
| Scheme 3 Synthesis of isoquinoline analouges in Table 4. Reagents and conditions: (i) butyl nitrite, HCl (cat.), MeOH, 40 °C, 15 min; (ii) a) NaOH, 50 °C, TosCl b) 80 °C, 15 min; (iii) K2CO3, MeI, DMF, 2 h; (iv) NaH, MeOH (anhyd), 80 °C, 3 h; (v) 3 M HCl, MeOH, 100 °C, 1 h; (vi) PhP(O)Cl2, 160 °C, 3 h, sealed tube; (vii) 180 °C, 1,2-dichlorobenzene, 1-benzyl-4-piperidylamine, microwave, 2 h; (viii) (SPhos) palladium(II) phenethylamine chloride, K-tOBu, various cyclic amines THF, 90 °C, 5–8 h. | ||
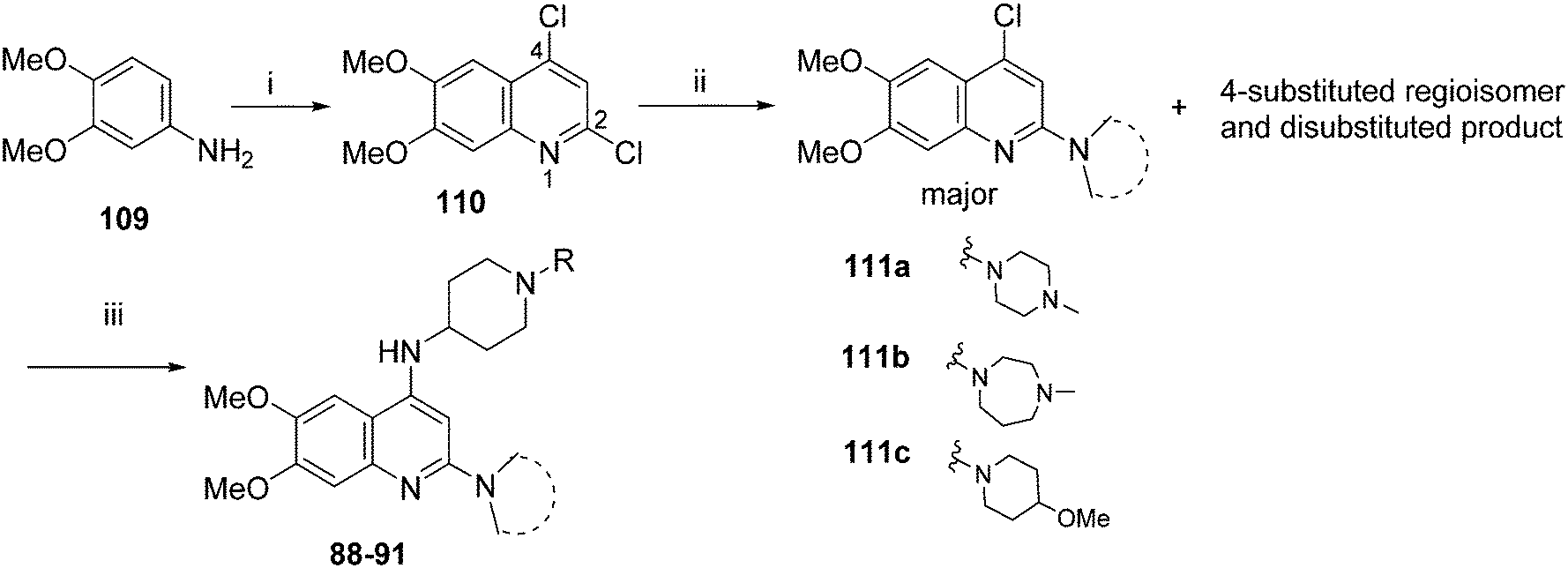 | ||
| Scheme 4 Synthesis of quinoline analogues in Table 4. Reagents and conditions: (i) malonic acid, POCl3, reflux, 3 h; (ii) THF, DIEA, various amines, 120 °C, microwave, 48 h; (iii) Pd-PEPPSI-iPr, Li-tOBu (1 M in THF), 1-benzyl-4-piperidylamine or 1-methyl-4-piperidylamine, THF, 100 °C, 18 h. | ||
Following a reported procedure, 5,6-dimethoxy-1-indanone (101) was treated with butylnitrite under acidic conditions to yield keto-oxime 102 (Scheme 3).44,45 Compound 102 was tosylated and ring-opened in a one pot46 procedure to give the benzoic acid derivative 103 that was esterified to obtain 104. Subsequent, addition of methoxide to the nitrile moiety of 104, followed by cyclisation resulted in the isoquinolin-1-one derivative 105 that was further hydrolysed under acidic conditions to obtain dihydroisoquinoline-1,3-dione (106). Attempts to convert 106 to the key intermediate 1,3-dichloro-6,7-dimethoxyisoquinoline (107) using widely-used chlorinating reagents, such as phosphorous oxychloride, phosphorous pentachloride or thionyl chloride, all failed with little/no conversion of the starting material. Finally, the use of dichlorophenylphosphine oxide47 at high temperature was identified as a suitable method to obtain 107 in good yield.
It has been previously reported that conversion of 1,3-dichloroisoquinoline (with no methoxy groups in position 6 and 7) to diaminoisoquinolines is possible under thermal conditions, albeit in low yield.48 Notably, the first substitution reaction (with ammonia or a primary amine) was reported to occur at the more reactive position 1 at 100 °C, while temperatures up to 220 °C were used for the second substitution reaction (using a secondary amine) at position 3. However, in case of 107, which contains methoxy groups in positions 6 and 7, a higher temperature was required for the first substitution reaction. Hence, treatment of 107 with 1-benzyl-4-piperidylamine at 180 °C resulted in the substituted isoquinoline analogue 108. Prolonged reaction time or the use of higher temperatures resulted in significant degradation of the starting material. Attempts to substitute position 3 of 108 with either ammonia or a secondary amine at temperatures of up to 220 °C proved fruitless, resulting in either no conversion, or degradation of 108 into a complex intractable mixture. Given this, a Buchwald–Hartwig C–N coupling reaction was investigated as an alternative approach. While a comparable coupling reaction to mediate C–N bond formation at position 3 was not found in the literature, one example was identified which described the coupling between an aromatic amine and position 1 of a 1-chloro-3-aminoisoquinoline using palladium acetate and BINAP under microwave irradiation.46 Based on the precedent of successful coupling between heteroaromatic halides and cyclic secondary amines using Pd-PEPPSI-iPr,49–51 we initially surveyed this catalyst for coupling of 108 and cyclic amines. However, low conversion and an intractable mixture of components was obtained under the standard coupling conditions. Hence, dialkylbiaryl phosphane based palladium catalysts were employed which are known for their high stability52 and broad applicability53 especially for arylchloride substrates.54 The use of a SPhos based palladium catalyst in particular resulted in excellent reaction between 108 and a variety of cyclic amines (Scheme 3). Through this method, sufficient quantities of analogues 85–87 were obtained for the current study. 2D NMR (NOESY) of the final compounds was employed to confirm the regiochemistry of amine substitution (see ESI‡).
For the synthesis of 6,7-dimethoxy-2,4-diaminoquinoline analogues (88–91) we modified our earlier adopted synthetic methodology,41 which contained a number of non-optimal synthetic steps.55,56 Thus, the key intermediate, dimethoxy-2,4-dichloroquinoline (110), was synthesized by heating 3,4-dimethoxyaniline (109) and malonic acid with phosphoryl chloride, following a reported procedure (Scheme 4).57 Compound 110 was subsequently heated with a variety of secondary amines to give the desired 2-substituted isomers 111a–c, together with minor amounts of the 4-substituted regioisomer and 2,4-disubstituted products. These minor side-products were readily removed by column chromatography. The conditions reported for this reaction in Scheme 4 were found to be optimum in order to obtain the maximum yield of the desired isomers, often together with recovery of the unreacted starting material 110. The final structures of the regioisomers were confirmed by either 2D NMR (NOESY) and/or single X-ray crystallography (for 111a, see Fig. SF1 ESI‡). Finally, as was developed for the isoquinolines, the 2-substituted quinolines (111a–c) were subjected to a Buchwald–Hartwig amination reaction, using either 1-benzyl-4-piperidylamine or 1-methyl-4-piperidylamine under palladium catalysis, in order to obtain the desired analogues 88–91. While the Buchwald–Hartwig coupling steps in both Schemes 3 and 4 proved suitable to provide sufficient material for SAR analysis, we note that the methods are not optimized to maximize product yield. Hence there is scope to screen other catalysts, bases and solvents to further improve this synthetic methodology towards diaminoquinolines and diaminoisoquinolines – scaffolds poorly represented in the literature.
Results and discussion
For the library of compounds synthesised, we measured and compared parasite killing effects with G9a inhibition. For select compounds, we also used a cell-viability counter screen to evaluate potential non-specific host toxicity using a HepG2 hepatoma cell line. We divide our discussion into key regions of SAR for this series.Quinazoline SAR: position 2
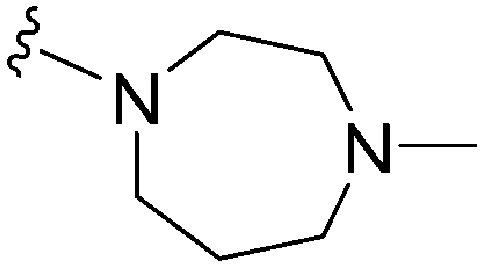
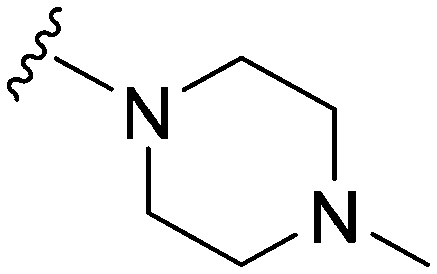
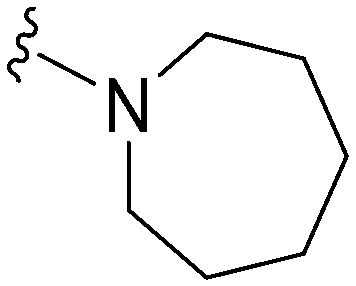
Initially, we explored the effect that the amine at position 2 had on parasite-killing and G9a inhibition, when 1-benzyl-4-piperidylamine was present at position 4 (Table 1). Previously, we have shown that decreasing the ring size at position 2, as in the case of compound 8, or removal of the basic nitrogen from N-methyl homopiperazine (9 and 10) does not affect the parasite-killing activity.33 However, analogues 9 and 10, lacking a basic nitrogen group in the ring, were found to be 4–5 fold less potent against G9a compared to their counterparts 1 and 2, which is in accordance with an earlier report.36 The protonated N-methylhomopiperazine is known to foster a hydrogen bond (H-bond) with the Asp1074 residue in the substrate binding pocket of the G9a (Fig. 2) thereby contributing to overall binding strength. However, this loss of binding contribution can be compensated by a ‘lysine mimic’ group at position 7, as evidenced by the high reported potencies of analogues such as 2, which contains a cyclohexyl ring at position 2.39 A variety of substituted rings that lacked such a basic nitrogen (e.g.9–16 and Table ST1 ESI,‡) exhibited poorer G9a inhibition than 1, whereas parasite-killing activity was less effected. In particular, bulky cyclic amines at position 2, such as in 14–16, were found to be suitable for maintaining high parasite-killing vs. G9a inhibition, especially in the absence of a ‘lysine mimic’.38 Interestingly, substitution with a primary acyclic amine instead of a cyclic amine at position 2 (17) resulted in poor potency against Plasmodium while maintaining good G9a inhibition. Together, these results suggest that the substituent at position 2 can have a significant effect on both parasite-killing and G9a inhibition. Importantly, removal of the basic centre from position 2, together with the use of bulky substituents, can be used to improve the parasite to G9a inhibition ratio.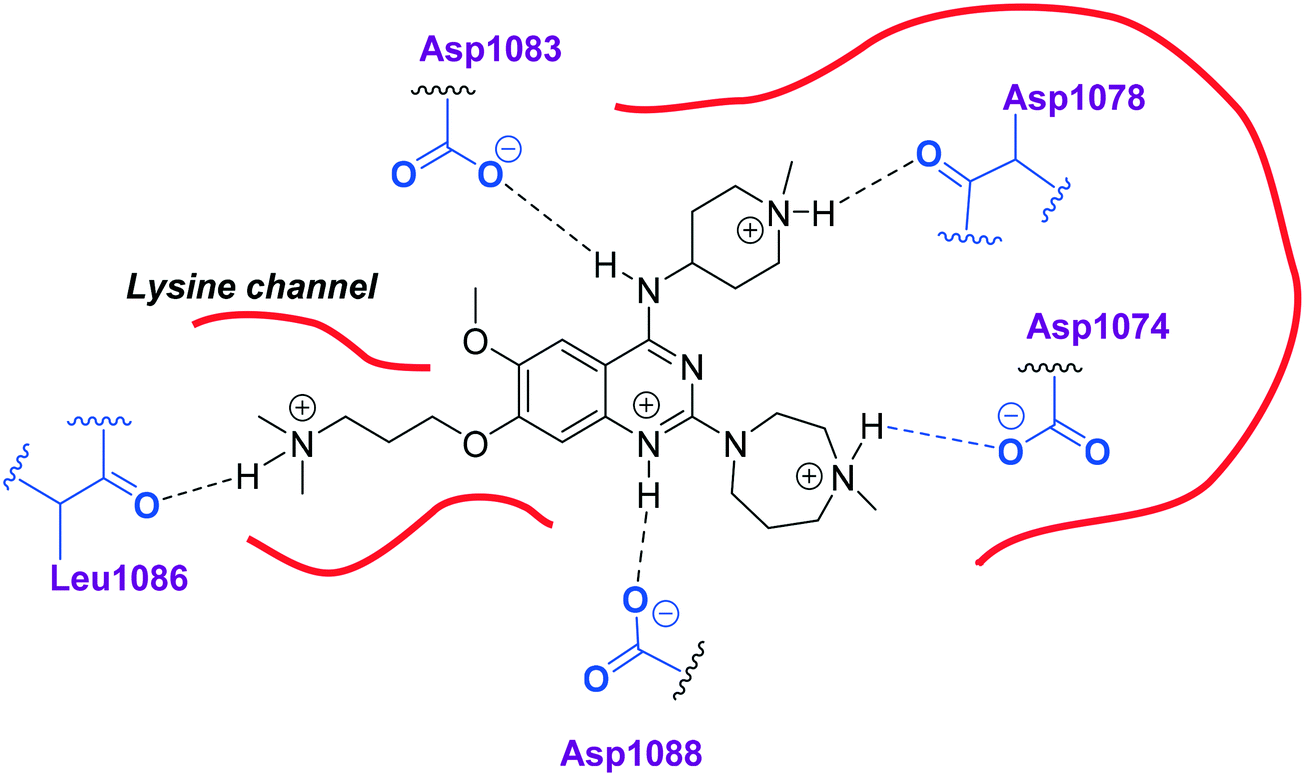 | ||
| Fig. 2 Two-dimensional diagram depicting key interactions of UNC0224 (66) in G9a substrate binding pocket (PDB ID 3K5K). | ||
Quinazoline SAR: position 4
Next we examined the effect of the substituent at position 4 of the diaminoquinazoline scaffold in conjunction with a variety of substituents at position 2. According to the crystal structure of 1 bound to GLP,58 a close homologue of G9a, the benzyl moiety of the 1-benzyl-4-piperidylamine is solvent exposed and thus, its removal or modification should not have considerable effect on G9a inhibitory activity. This fact is clearly apparent in previously described SAR studies of quinazoline G9a inhibitors.36,37 Indeed, analogues with methyl (18–20), fluoro (21–23) or methoxy (24) substitution on the phenyl ring of the 1-benzyl-4-piperidylamine displayed good G9a IC50 values, ranging from 78–169 nM (Table 2). However, once again the effect of a basic nitrogen within the substituent at position 2 was found to be key to potent G9a inhibition, where analogues 25–29 – possessing a piperidine in place of N-methylpiperazine – consistently exhibited lower potency against G9a (IC50 = 690 to 1214 nM). In contrast, all these analogues displayed good to moderate (IC50 < 150 nM) parasite-killing activity, (except 24 and 28) irrespective of the position 2 substituent. Together these results suggest that the benzyl group of 1-benzyl-4-piperidylamine can be modified without losing significant activity against either G9a or Plasmodium, thus providing a handle for tuning the physicochemical parameters of compounds for lead optimization.Interestingly, analogues with 1-benzyl-3-piperidylamine (30, 31) or 1-benzyl-3-pyrrolidinylamine (32–34) at position 4, were less potent against G9a compared to compounds bearing a 1-benzyl-4-piperidylamine in this position, for example 8vs.30 and 32, 10vs.31 and 33, and 15vs.34 (see also Table ST1, ESI‡). The protonated basic ring nitrogen of the piperidylamine moiety is expected to form an H-bond with the backbone carbonyl of Asp1078 in G9a (Fig. 2).37 However, this interaction is dependent on the conformation of the six-membered piperidylamine, which might present the protonated nitrogen N–H either towards (PDB 3K5K)37 or away (PDB 3RJW)39 from Asp1078. In analogues 30–34, the corresponding basic nitrogen is positioned differently within the rings and thus likely disrupts the H-bond with Asp1078. Such analogues have not been previously reported in the G9a inhibitor literature and therefore present a new aspect of SAR for this series. However, it should be noted that indolylamine based analogues such as 5 (A-366, Fig. 1) possess potent G9a activity59 despite the absence of a basic centre in the corresponding position; suggesting that maintenance of an interaction with Asp1078 is not critical for potent G9a inhibition per se, but can be compensated by other interactions; particularly those provided by a ‘lysine mimic’. In general, 30–34 retained good (93 nM) to moderate (330 nM) activity against Pf3D7, depending on the substituent at position 2. This suggests that an equivalent H-bond interaction is either maintained despite the different orientation in the PfHKMT target(s) or is not required for activity. The importance of this basic centre was further explored by testing analogues 35–37 with an acylated piperidylamine nitrogen. All such analogues were found to be completely inactive against G9a, while a few (35–37, see also, Table ST1 ESI‡) retained moderate parasite-killing activity, depending on the nature of the substituent at position 2.
Replacing 1-benzyl-4-piperidylamine with 1-methyl-4-piperidylamine at position 4 in 1 or 8, thus yielding 38 and 39 respectively, is known to retain G9a activity due to the solvent exposed nature of the terminal substituent group (vide supra).36,37 However, these compounds were both found to have low Pf3D7 activity. Both 38 and 39 have a calculated topological surface area (TPSA) identical to 1, but have a significantly lower clogP. Previously, diaminoquinazoline analogues possessing lower clogP values were found to have poor G9a activity in the cell based assays, suggesting poor permeability across cell membranes.38 In accordance with this, the poor parasite-killing activity exhibited by 38 and 39 may be due to their limited cellular uptake, rather than associated to a target-based effect. Indeed, analogues 40 and 41, with increased clogP were found to recover the parasite-killing activity and displayed Pf3D7 IC50 values comparable to 1. Conversely however, analogues 40 and 41 were weak inhibitors of G9a, either due to the absence of a basic centre (as in 40) or the presence of a bulky substituent (as in 41) at position 2 (vide supra). Remarkably, 42 and 43, previously reported to be weak inhibitors of G9a,36,37 displayed excellent parasite-killing activity. To further investigate the effect of increased clogP on parasite-killing activity we synthesized analogues having cyclohexylmethyl (44–47) or cyclohexyl (48–50) N-capping groups on the 4-substituent. These lipophilic rings have previously been reported to improve the cellular permeability of G9a inhibitors bearing a ‘lysine mimic’ at position 7.38 However, the parasite-killing activity of most of these analogues was less potent than 1, suggesting no direct correlation between clogP and the anti-Plasmodium activity. Broadly, analogues lacking a basic nitrogen in the 2-substituent ring exhibited better potency against Plasmodium compared to the analogues containing N-methylpiperazine (45 and 49) and N-methylhomopiperazine (44 and 48) at this position. A similar trend in parasite-killing activity was observed for molecules containing a 1-isopropyl-4-piperidylamine, cyclohexylamine or aniline at position 4 (51–59): analogues bearing a piperidine derivative at position 2 (such as 53, 54, 58 and 59) exhibited better anti-Plasmodium activity than comparable compounds with a piperazine or homopiperazine substituent in this position (e.g.51, 52, 55 and 57). As observed earlier, the trend for the G9a inhibition was opposite to the anti-Plasmodium activity in this regard: analogues with a piperazine or homopiperazine in the position 2, such as 44, 45, 48, 49, 51 and 52, were found to be more potent against G9a compared to those without such substituents (such as 46, 47, 54). Analogues having either a cyclohexylamine (55 and 56) or aniline (57–59) at position 4 were found to be devoid of G9a inhibition, while maintaining good to moderate (77–570 nM) parasite-killing activity, the precise potency of which depended on the substituent at position 2 (see also Table ST1 in ESI‡). This SAR can once again be justified through the necessity of a hydrogen bond with Asp 1078 for G9a inhibition, but not for anti-parasitic activity. Substitution of other amines such as isopropylamine, 1-methyl-3-pyrrolidinylamine or tetrahydro-2H-pyran-4-ylamine at position 4 (Table ST1, ESI‡) resulted in poor anti-Plasmodium and G9a inhibition.
In summary, modification of the benzyl moiety of 1-benzyl-4-piperidylamine at position 4 or replacing it with a range of other groups can be tolerated to retain both G9a and Pf3D7 activity, while other amine at position 4 are detrimental to G9a activity. This is presumably due to the loss of the H-bond with Asp1078. Pf3D7 activity does not follow this trend however and many a large variety of amines at position 4 are tolerated. In particular, an aromatic amine at this position can be used to achieve high selectivity in favour of anti-Plasmodium activity.
We note that methylation of the 4-amino group, or its substitution with an oxygen or sulphur, was found to dramatically decrease the parasite-killing activity of this series (Table ST2, ESI‡).33 For G9a inhibition, such a structural alteration eliminates a hydrogen donor interaction with Asp1083 (Fig. 2), and is thereby known to abolish G9a inhibition;37 which was further confirmed by the G9a inhibition data obtained by us (Table ST2, ESI‡). This highlights the importance of a hydrogen bond donor at position 4 for both anti-Plasmodium and G9a inhibition, thus making it an indispensable SAR feature for both targets. We note however that once again, a suitable ‘lysine mimic’ can potentially compensate for this effect, in terms of G9a potency (cf.5).59
Quinazoline SAR: position 7
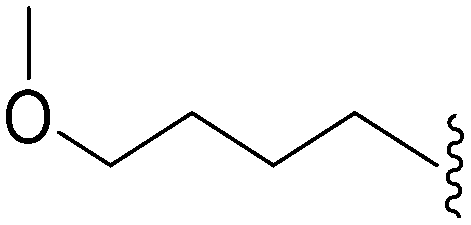
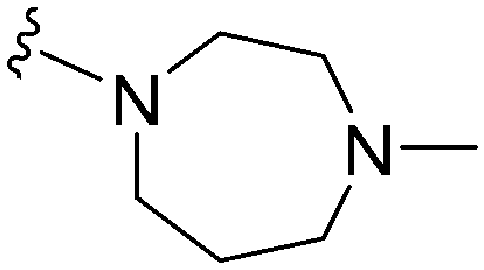
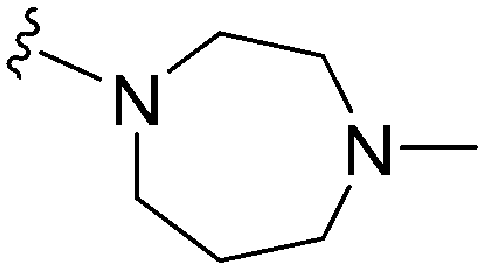
As already stated, installation of a ‘lysine mimic’ at position 7 has been found to be central to the development of potent G9a inhibitors.36–40,59 Interestingly, analogues devoid of the methoxy groups at position 6 and 7 were found to maintain moderate parasite-killing activity, but exhibited no G9a inhibition (Table ST3, ESI‡) as also reported earlier by us and others.41,60 This difference may indicate the inherent differences in the respective lysine binding channels of the G9a and putative PfHKMT target(s), since the methoxy groups are known to occupy regions close to the lysine binding channel in G9a (vide infra).36,37 Focusing specifically on position 7, our previous study reported 60 (Table 3), a positional isomer of 1, to be equally efficacious against Plasmodium.31,32 Interestingly, such ‘swapping’ of the terminal benzyl and methyl groups between positions 4 and 7 (1vs.60) resulted in a complete loss of G9a activity. Analogues 61 and 62 also maintained this selectivity for the parasite, exhibiting virtually no G9a inhibition. However, analogue 63 with a primary acyclic amine at position 2 lost activity against Plasmodium. This is in accordance with the similar effect observed above for analogue 17 (Table 1), more broadly suggesting a primary amine at position 2 to be deleterious to parasite activity.Analogues with linear 7-aminoalkoxy substituents (or ‘lysine mimics’) such as 64–67 (Table 3) exhibit potent activity against G9a due to the additional interactions in the lysine binding channel of this enzyme (Fig. 2).36–40,59 However, all these analogues were found to lack anti-Plasmodium activity as reported by us previously (see also Table ST4, ESI‡).33 We previously rationalized the poor parasite-killing activities of these analogues as attributed to their low clogP and/or high TPSA and hence poor cellular permeability. However, when considering such factors more broadly, it is apparent that the calculated physicochemical parameters for 64 and 65 are comparable to 1. Indeed, both these analogues have been reported to possess moderate G9a activity in cells as measured by the reduction of H3K9me2 levels in an In-Cell Western (ICW) assay, suggesting sufficient cellular permeability of these compounds, at least under the assay concentrations employed.38 Hence, we synthesized analogues 68–70 with a higher clogP while retaining the basic ‘lysine mimic’ group. All three analogues exhibited potent G9a activity, as expected, but showed only slight improvement in parasite-killing activity. In contrast, replacement of the basic ‘lysine mimic’ with less polar ether (71) or neutral hydrocarbon (72) chain further improved the parasite-killing activity while it significantly reduced the G9a potency of these analogues. Comparison of 72 and 69 is particularly interesting as both analogues have very similar clogP and TPSA values, but the former is ∼4 fold more potent against Plasmodium while reported to be considerably less potent against G9a.37 Together, this data suggests that unlike G9a, the lysine binding channel in the PfHKMT target(s) is better able to accommodate the hydrophobic benzyl or hydrocarbon chains. Indeed, computational analysis of lysine binding channels of various HKMTs suggests that they have diverse hotspot profiles that can be exploited for designing selective inhibitors.61
A number of recent reports suggest that for cases (in cancer) where the substrate histone lysine residues are mutated to either methionine (Met) or a hydrocarbon based residue such as norleucine (Nle) (e.g. H3K9M or H3K27M), these mutant histones act as potent inhibitors of the respective HKMTs: such as G9a inhibition by H3K9M or polycomb repressive complex 2 (PRC2) inhibition by H3K27M.62–68 Very recently Judge et al. have reported peptide inhibitors of SETD8 (a H4K20 methylase) by replacing the lysine (K20) residue of the substrate peptide to Nle/Met (H4K20Nle/Met) and other hydrophobic residues.69 Indeed, co-crystallized structures of such mutated peptide substrates with both human PRC2/G9a/SETD8 show Met and Nle to occupy the lysine binding channel in a manner similar to lysine in the non-mutated substrates.67–69 This data clearly suggests that ‘lysine mimic’ groups which interact with the lysine binding channels of HKMTs need not be limited to polar and basic sidechains. Given this, and given the fact that hydrocarbon chains were tolerated at position 7 for anti-parasite activity, analogues containing ‘Met or Nle mimics’ at position 7 were investigated. However, poor inhibition of G9a by 72 suggested that the length and nature of the ‘Nle mimic’ in context of diaminoquinazoline inhibitory scaffolds was not directly analogous to that in H3 peptides. Analogues 73–77 with a 6–8 carbon linear ‘Nle mimic’ all displayed very similar moderate IC50 values (∼231–268 nM) against the parasite. On the other hand, analogues with the linear or branched ‘Nle mimic’ containing 4–5 carbons (78–82) showed good parasite-killing activities, with 82 exhibiting an IC50 value (37 nM) comparable to 1. The G9a inhibitory SAR was found to be different however: a linear chain of 6 carbons was found to be optimum for G9a inhibition as evidenced by the low G9a IC50 values of 74, 75, and 77. Indeed, G9a inhibition seemed to be intolerant of chain length variations, as increasing (73, 76) or decreasing (78–82) the chain length led to a negative effect on inhibitor potency. To the best of our knowledge, analogues 74, 75 and 77 represent the first examples of non-peptidic small molecule G9a inhibitors possessing a hydrophobic ‘Nle mimic’ at position 7. Analogues 83 and 84 representing a ‘Met mimic’ showed only moderate parasite-killing activities and poor G9a inhibition. This is in accordance with the data reported for peptide based inhibitors where Met mutation was found to be less effective than Nle mutation.64 However, assessment of analogues possessing varying length ‘Met mimics’ and different position 2 substituents would need to be tested in order to draw firm conclusions over the SAR surrounding ‘Met mimics’.
SAR for the central heterocyclic ring
Finally, we examined the role of the central fused aromatic scaffold. We have previously synthesized and surveyed other fused heteroaromatic scaffolds, related to diaminoquinazolines, against G9a.41 Specifically, we examined cases where the fused benzenoid ring was replaced by a thiophene, furan, imidazole or a cyclopentane ring (Table ST5, ESI‡). All these analogues were found to be inactive against both G9a and Plasmodium suggesting both biological activities to be restricted to the fused six membered ring scaffold. Next, we tested the importance of pyrimidine ring nitrogens of the quinazoline scaffold, by comparing analogues based on diaminoquinolines and diaminoisoquinolines (Table 4). The protonated N-1 nitrogen of quinazoline interacts strongly with Asp1088 (Fig. 2) in the G9a pocket, while the N-3 nitrogen does not appear to form any specific interaction. Accordingly, isoquinoline analogues (85–87) lacking a basic centre at position 1 displayed poor activities against both G9a and the parasite while the quinoline compounds 88–91 were found to be potent against both targets. In particular, 88 displayed ∼5 fold increase in potency against G9a than the parental analogue 1; a fact that we have rationalised previously.41 This data highlights this SAR feature to clearly be conserved between G9a and the potential PfHKMT target(s) of this compound series.HepG2 cytotoxicity
Earlier, we screened several diaminoquinazoline analogues in a cell-viability assay to evaluate potential host toxicity using a HepG2 hepatoma cell line.33 The HepG2 cell line is routinely used to measure toxicity70–73 and displays slightly more sensitivity compared to the other commonly used cell lines.72 Of the select analogues examined in this assay, most display high anti-HepG2 IC50 values. No correlation is observed between the anti-Plasmodium and anti-HepG2 activities. For example, analogues 12, 40, 42 and 43 exhibit potent parasite-killing activity but are amongst the least toxic compounds studied, with anti-HepG2 IC50 > 10 μM. While lipophility can give rise to non-specific cellular toxicity,74 we did not observe any significant correlation between the cLogP and anti-HepG2 activity. Finally, no clear correlation is apparent between G9a inhibition and anti-HepG2 activity. For example, BIX01294 (1) has a G9a IC50 of 67 nM and a HepG2 IC50 of 4.8 μM, whereas compound 14 is inactive against G9a but has a HepG2 IC50 of 3.6 μM. The compounds that were most active (IC50s between 1.8–2.9 μM) against the HepG2 cell line had a (methylbenzyl)piperidin-4-yl group at position – 4 (such as 25–27) and benzyl/alkyl group at position 7 (such as 61, 62, 79, 81 and 82). Together, this data suggest that the reported analogues generally have a very promising differential activity for parasites over HepG2 human cells.
Further activity against Plasmodium
We have previously shown the diaminoquinazoline chemotype, as exemplified by analogues 1, 12, 40 and 60, to exhibit a fast parasite-killing phenotype, effective throughout the intra-erythrocytic parasite life cycle. Killing phenotypes are a function of the parasite molecular target(s) of a given compound75 and thus, in order to study whether the diaminoquinoline chemotype has a comparable target profile to the diaminoquinazolines, compounds 89 and 90 were employed in analogous phenotypic assays. Hence, highly synchronised parasites were treated with 89 and 90 for three distinct periods of the 12 hour intra-erythrocytic stage and re-invasion into the next cycle and out-growth two cycles later was quantified (Fig. 3). The data revealed 1, 89 and 90, to possess a similar erythrocytic stage-independent killing phenotype, suggesting a common target profile for the anti-Plasmodium activity of both the diaminoquinolines and the diaminoquinazolines.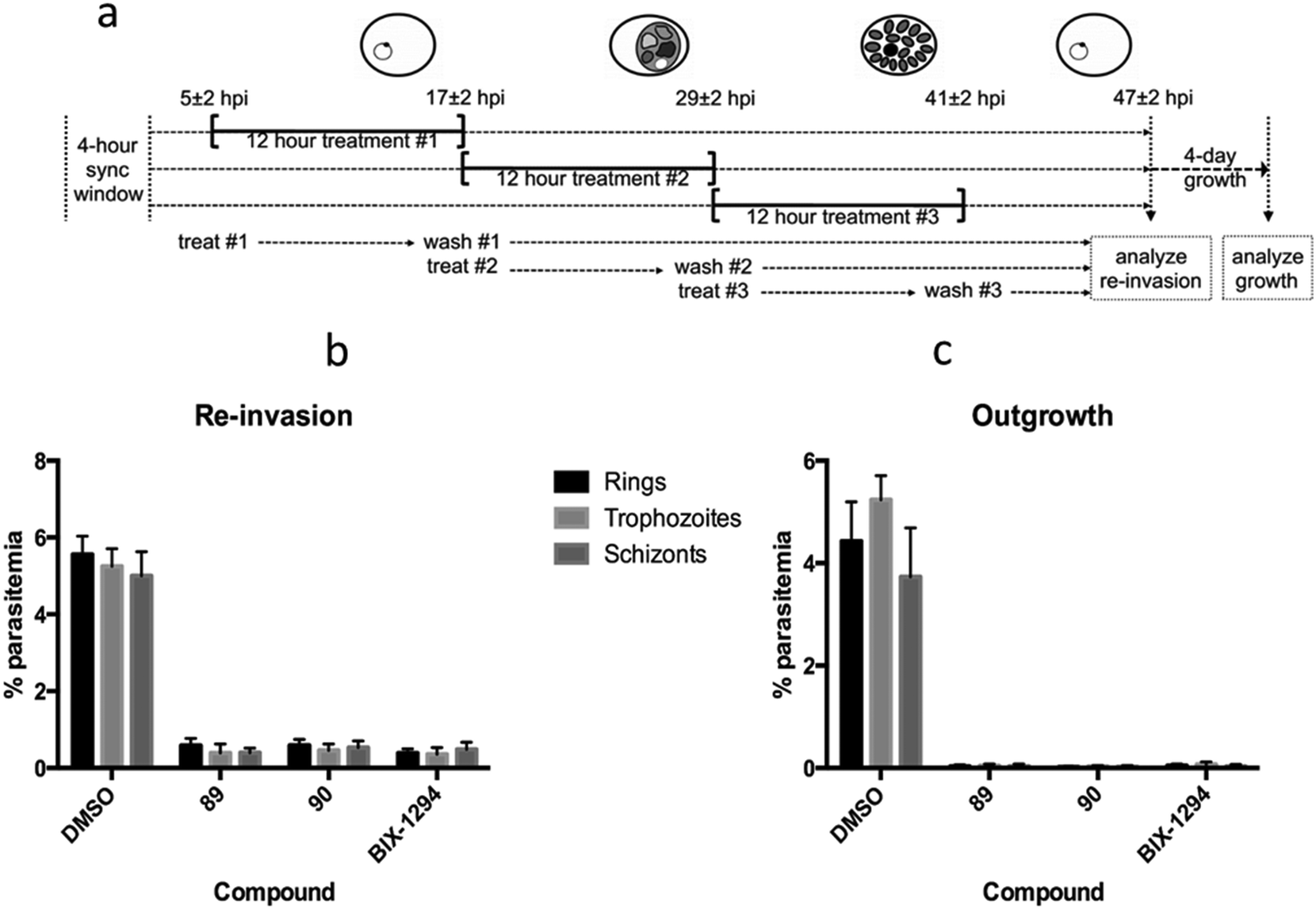 | ||
| Fig. 3 Stage-dependent antimalarial activity a) synchronised P. falciparum parasites were treated with DMSO or 10 × IC50 values of compounds 89, 90 or BIX01294 for three distinct 12 hour periods of the intra-erythrocytic life cycle b) re-invasion of treated parasites at 47 h post-invasion was quantified by flow cytometry. c) After 12 hour treatment, parasites were washed, diluted, and allowed to grow for four days, after which parasitemia was measured by flow cytometry. Data are the mean ± SD of 30000 RBCs from duplicate samples. | ||
Reduction in histone methylation levels in Plasmodium
Earlier, we demonstrated that BIX01294 and other diaminoquinazoline analogues decrease H3K4me3 and H3K9me3 levels in parasites in comparison to DMSO vehicle.31,33 Since, the diaminoquinolines such as 88–91 retain the parasite-killing activity, potentially by acting on the same PfHKMTs targets, we evaluated 89 and 90 in the same assay. Thus, malaria parasites (Pf3D7) were treated with 1, 89 and 90 and the histone H3K4me3 and H3K9me3 levels were analysed by western blot (Fig. 4) in the treated parasites, relative to control parasites treated with DMSO. As expected, the data demonstrates a decrease in H3K4me3 and H3K9me3 levels upon treatment of parasites with 1 and diaminoquinoline analogues 89 and 90 suggesting diaminoquinolines share the same target/mechanism as the diaminoquinazoline analogue BIX01294. Together, this supports our SAR studies demonstrating that both diaminoquinazoline and diaminoquinoline analogues retain the essential features for binding G9a and the related PfHKMT target in parasites.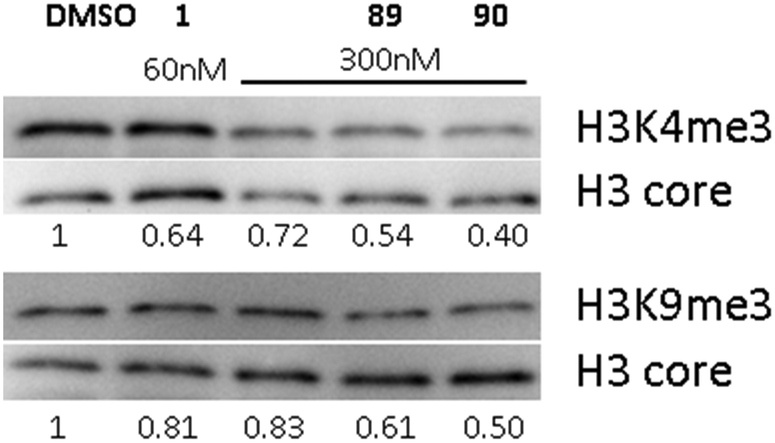 | ||
| Fig. 4 Effect of 1, 89 and 90 on the histone methylation levels in treated parasites. P. falciparum 3D7 parasites were treated with the indicated compounds or DMSO control for 12 hours. Specific histone H3K4me3 and H3K9me3 levels were quantified by densitometry, normalised to histone H3 core signal, and the resulting methylation levels relative to DMSO control-treated parasites are indicated below each pair of methylation-specific and corresponding core histone H3 bands. | ||
Conclusion
In summary, we have synthesized and tested a large number of diaminoquinazoline and related analogues against G9a and P. falciparum, in order to elucidate more complete SAR features for both targets (as summarized in Fig. 5). In addition to reconfirming some of the previously reported independent SAR features for these targets, our work provides new insight. For example, previous G9a SAR for this series36–40 has mostly focused on analogues possessing a ‘lysine mimic’ at position 7 and hence the effect of substituents at position 2 and 4 was not known in the absence of a position 7 lysine mimic. Our results suggest that diaminoquinazoline analogues display different SAR trends against G9a depending on whether a ‘lysine mimic’ is present or not. For instance, we found that in the absence of a ‘lysine mimic’ at position 7, basic centres at both position 2 and 4 and a hydrogen bond donor at position 4 are required for potent G9a inhibition. Additionally, the analogues lacking a ‘lysine mimic’ group do not tolerate bulky cyclic amine at position 2 and loose significant potency against G9a. Perhaps more importantly, the polar and basic ‘lysine mimic’ at position 7 – now well established for the widely employed G9a inhibitors UNC0638 and UNC064237,38 – can be replaced by hydrophobic ‘Nle mimics’, while retaining good G9a inhibition, as has been reported for the peptide-based inhibitors.62–69 Consequently, our SAR studies resulted in the discovery of first diaminoquinazoline G9a inhibitors bearing a 6-carbon ‘Nle mimic’ at position 7.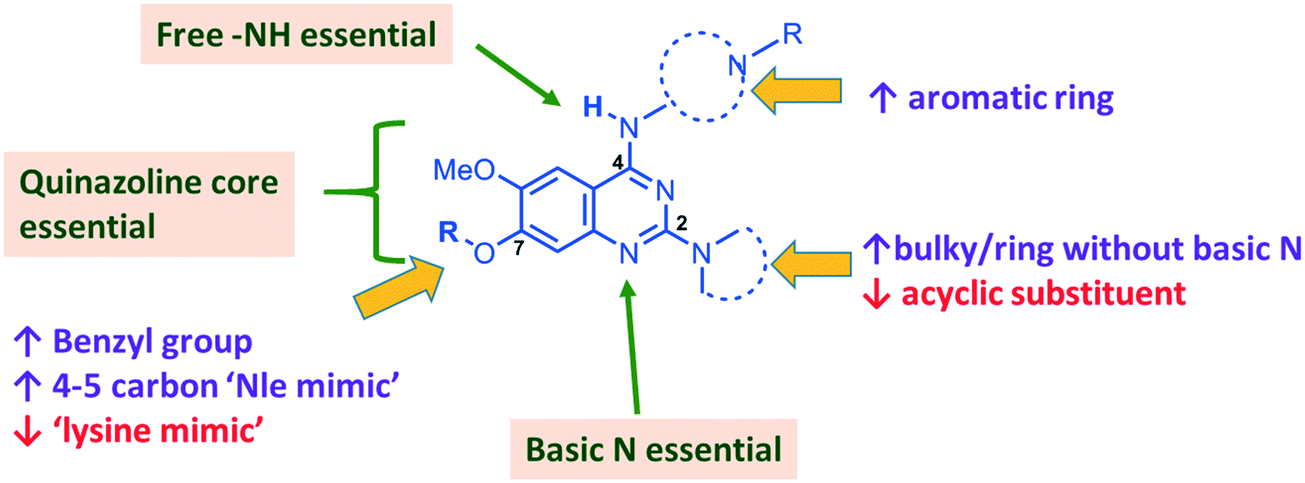 | ||
| Fig. 5 Summary of key SAR features of diaminoquinazoline analogues against G9a and Plasmodium and suggestions for designing parasite-selective analogues. Features highlighted in boxes are essential for both G9a and anti-Plasmodium activity. Acyclic substituent at position 2 and a ‘lysine mimic’ at position 7 were found to be unfavourable (↓) for parasite-killing activity. Conversely, bulky or a ring without basic centre at position 2, aromatic ring at position 4 and a benzyl/4-5 carbon ‘Nle-mimic’ at position 7 were found to impart selectivity in favour (↑) of the anti-Plasmodium activity. | ||
While conserved features of G9a and anti-parasitic SAR could be used to rationalise the likely PfHKMT target(s) of these inhibitors, clearly future optimization of this scaffold will need to diverge from activity on human (host) targets, such as G9a. We have identified key SAR features which demonstrate that high parasite vs. G9a selectivity can be achieved by selecting appropriate substituents at position 2, 4 and 7 of the quinazoline ring. For instance, a bulky substituent or a ring lacking a basic nitrogen at position 2 or an aromatic ring at position 7 can be used to design analogues with a high parasite-killing to G9a inhibition ratio. Similarly, we have shown that while a ‘lysine mimic’ substituent position 7 imparts high potency against G9a, it is deleterious to the anti-Plasmodium activity. Together, this data suggests that while broadly similar, the G9a and potential PfHKMT target(s) binding pockets and/or binding modes of the diaminoquinazoline analogues exhibit clear and exploitable differences. Thus, there remains significant potential in this series to further develop parasite selective analogues. Based on this, we believe this scaffold to have clear potential for development into a novel and much needed, new medicine for malaria.
Of course, a key question remains for this series: specifically which of the essential PfHKMTs are targeted by these compounds? To date, our study on PfSET7 represents the only successful report30 of recombinant expression of an active PfHKMT, suitable for biochemical characterization and study. Initial activity assays using PfSET7 have suggested PfSET7 not to be the target of 1 (data not shown), and therefore the other candidate targets are the remaining essential PfHKMTs: PfSET1, PfSET3, PfSET6, PfSET9, or PfSET10. Future studies aimed at robust biochemical characterisation of our lead inhibitors against all the essential PfHKMTs will be reported in due course, once protocols for successful protein generation have been optimized.
Experimental
Chemistry general procedures
All reactions were performed under an atmosphere of dry nitrogen unless otherwise stated. Flash column chromatography was carried out using Merck Kiesegel 60 silica gel (230–400 mesh, 0.040 0.063 mm). Thin layer chromatography (TLC) was performed on aluminium plates using Merck Kiesegel 60 F254 (230–400 mesh) fluorescent treated silica which were visualised under ultraviolet light (254 nm), or by staining with potassium permanganate or ninhydrin solution as appropriate. All 1H and 13C NMR spectra were recorded using Bruker 400 MHz spectrometers or Bruker AV 500. Chemical shifts (δ) are quoted in units of parts per million (ppm) downfield from tetramethylsilane and are referenced to a residual solvent peak. Coupling constants (J) are given in Hertz (Hz). The 1H NMR spectra are reported as follows: ppm (multiplicity, coupling constants J/Hz, number of protons). High and low resolution mass spectrometry (EI, ES, and CI) were recorded on Micromass Platform II and Micromass AutoSpec-Q spectrometers. All compounds tested in biological assays were >95% pure by LCMS. LCMS gradient: from 95:5, A:B to 5:95, A:B over 18 minutes, where A is water (0.1% formic acid), and B is methanol. Column: XBridge C18 columns with dimensions 4.6 mm × 100 mm.
:MeOH; 100:0 → 98:2) to afford titled compound as a white solid (85 mg, 80%). 1H NMR (400 MHz, CDCl3) 7.57 (s, 1H), 7.01 (s, 1H), 4.21 (s, 2H), 3.98 (s, 3H), 3.93 (s, 3H), 3.91 (s, 3H).
:MeOH; 100:0 → 99:1) to afford 107 (65 mg, 95%) as a white powder. 1H NMR (400 MHz, CDCl3) 7.48 (s, 1H), 7.43 (s, 1H), 6.98 (s, 1H), 4.05 (s, 3H), 4.03 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 154.41, 151.32, 148.10, 141.68, 136.28, 121.52, 118.37, 104.19, 104.15, 56.37, 56.28; HRMS (+ESI) m/z calcd for C11H10NO2Cl2 258.0089, found, 258.0095. MS (+ESI) m/z 258 [M + H]+, 260 (M + 2H)+.
:MeOH; 100:0 → 97:3) to obtain 108 as a brown solid (36 mg, 32%). 1H NMR (400 MHz, CDCl3) 7.38–7.27 (m, 5H), 6.87 (s, 1H), 6.84 (s, 1H), 6.83 (s, 1H), 4.83 (s, 1H, br), 4.27–4.20 (m, 1H), 4.00 (s, 3H), 3.98 (s, 3H), 3.61 (s, 2H), 2.96–2.93 (m, 2H), 2.30 (t, J = 11.9 Hz, 2H), 2.23–2.14 (m, 2H), 1.67 (ddd, J = 22.7, 11.4, 3.5 Hz, 2H); HRMS (+ESI) m/z calcd for C23H27N3O2Cl 412.1792, found, 412.1813.
:MeOH (7N NH3); 97:3 → 95:5) to obtain 85 as a dark green solid (16 mg, 32%). 1H NMR (400 MHz, CD2Cl2) 7.35–7.22 (m, 5H), 6.80 (s, 1H), 6.79 (s, 1H), 6.03 (s, 1H), 4.72 (s, 1H, br), 4.13–4.06 (m, 1H), 3.89 (s, 6H), 3.52 (s, 2H), 3.45–3.43 (m, 4H), 2.90–2.87 (m, 2H), 2.51–2.49 (m, 4H), 2.30 (s, 3H), 2.23–2.13 (m, 4H), 1.62–1.53 (m, 2H). 13C NMR (100 MHz, CD2Cl2) δ 155.09, 152.92, 152.67, 146.89, 136.96, 129.55, 128.53, 127.40, 110.36, 107.01, 105.29, 101.91, 88.81, 63.25, 56.42, 55.91, 55.30, 48.72, 46.26, 46.21, 32.79. HRMS (+ESI) m/z calcd for C28H38N5O2 476.3026, found, 476.3028.
:MeOH (7N NH3); 100:0 → 98:2) to obtain the pure product as a yellow solid (44 mg, 92%). 1H NMR (400 MHz, CD2Cl2) 7.36–7.22 (m, 5H), 6.78 (s, 1H), 6.77 (s, 1H), 6.05 (s, 1H), 4.70 (s, 1H, br), 4.13–4.06 (m, 1H), 3.95 (dt, J = 12.7, 4.4 Hz, 2H), 3.93 (s, 3H), 3.92 (s, 3H), 3.54 (s, 2H), 3.39–3.34 (m, 4H), 3.03 (ddd, J = 13.0, 9.9, 3.2 Hz, 2H), 2.91–2.89 (m, 2H), 2.25–2.11 (m, 4H), 1.98–1.94 (m, 2H), 1.60–1.52 (m, 4H); 13C NMR (100 MHz, CDCl3) δ 154.62, 152.34, 152.27, 146.31, 136.65, 129.28, 128.26, 127.19, 106.64, 104.96, 101.30, 89.04, 63.03, 56.15, 55.73, 55.50, 52.66, 48.12, 44.17, 32.36, 30.37; HRMS (+ESI) m/z calcd for C29H38N4O3 491.3022, found, 491.3008.
:MeOH (7N NH3); 98:2 → 96:4) to afford pure product as a yellow solid (15 mg, 42%). 1H NMR (400 MHz, CD2Cl2) 7.36–7.23 (m, 5H), 6.78 (s, 1H), 6.77 (s, 1H), 6.03 (s, 1H), 4.69 (s, 1H, br), 4.13–4.06 (m, 1H), 3.89 (s, 6H), 3.54 (s, 2H), 3.45–3.42 (m, 4H), 2.92–2.89 (m, 2H), 2.24–2.12 (m, 4H), 1.65–1.57 (m, 8H); 13C NMR (100 MHz, CD2Cl2) δ 155.52, 152.84, 152.64, 146.59, 137.20, 129.49, 128.51, 127.32, 106.58, 105.18, 101.96, 88.58, 63.31, 56.42, 55.87, 53.80, 48.77, 47.47, 32.90, 25.97, 25.32; HRMS (+ESI) m/z calcd for C28H37N4O2 461.2917, found, 461.2899.
:MeOH (7N NH3); 98:2 → 96:4) to yield desired isomer 111a as a white crystalline solid (111 mg, 17%) together with the minute quantity of the regioisomer (see ESI‡) and unreacted 110 (0.364 g, 47%). 1H NMR (400 MHz, CDCl3) δ 7.26 (s, 1H), 7.10 (s, 1H), 6.94 (s, 1H), 4.00 (s, 3H), 3.99 (s, 3H), 3.72–3.69 (m, 4H), 2.64–2.45 (m, 4H), 2.36 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 156.68, 153.01, 147.38, 145.13, 141.64, 115.70, 107.19, 106.64, 102.44, 56.08, 56.02, 54.94, 46.20, 45.26. HRMS (+ESI) m/z calcd for C16H21ClN2O3, 322.1322, found, 322.1333.
:MeOH (7N NH3); 98:2 → 90:10) to obtain 88 as a yellow solid (13.5 mg, 19%).1H NMR (400 MHz, CDCl3) δ 7.34–7.27 (m, 5H), 7.05 (s, 1H), 6.72 (s, 1H), 5.76 (s, 1H), 4.28 (s, 1H, br), 3.97 (s, 3H), 3.95–3.92 (m, 5 H), 3.75 (t, J = 6.3 Hz, 2H), 3.57 (s, 2H), 3.53–3.47 (m, 1H), 2.91 (d, J = 11.7 Hz, 2H), 2.75–2.73 (m, 2H), 2.60–2.57 (m, 2H), 2.38 (s, 3H), 2.24 (td, J = 11.5, 11.0, 2.7 Hz, 2H), 2.17–2.14 (m, 2H), 2.04 (p, J = 5.8 Hz, 2H), 1.70–1.62 (m, 2H). HRMS (+ESI) m/z calcd for C29H40N5O2, 490.3182, found, 490.3197.
Biology procedures
:1 and 2:1 dilutions of test compounds.
000 cells per well grown in DMEM. Cells were incubated with 1:1 dilutions of test compounds for three days and resulting cell viability was quantified using Promega CellTiterBlue.
:16 and allowed to grow without compound for an additional two cell cycles (4 days). Parasitemia after re-invasion or after 4 day growth was quantified on infected cells fixed in 0.025% glutaraldehyde and stained with 2× SYBR Green I (Lonza) in PBS.
:1000), H3 core (Abcam ab1791, 1:3000) and H3K9me3 (Abcam ab8898, 1:1000) diluted in TBS-T (50 mM Tris pH 7.5, 150 mM NaCl, 0.05% Tween-20, 5% BSA), followed by anti-mouse HRP (GE NA931V) or anti-rabbit HRP (GE NA934V) secondary antibodies. Blots were revealed using SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific) and quantified using Bio-Rad Image Lab software.
Abbreviations
| PfHKMT | P. falciparum histone lysine methyltransferase |
| ACTs | Artemisinin-based combination therapies |
| PfHDAC | P. falciparum histone deacetylases |
| TPSA | Total polar surface area |
| ICW | In-Cell Western assay |
| PRC2 | Polycomb repressive complex 2 |
| ECSD | Enzyme-coupled SAH detection assay |
| CLOT | Chemiluminescence-based oxygen tunnelling assay |
Conflict of interest declaration
We wish to confirm that there are no known conflicts of interest associated with this publication and there has been no significant financial support for this work that could have influenced its outcome.Author contributions
MF and AS conceptualized and designed the project. SS, ASL and GAL performed chemical synthesis, purification and characterization of compounds. PBC and NAM performed in vitro parasite growth/proliferation assay, HepG2 cytotoxicity assay and Western blot analysis. FL performed human G9a inhibition assays. MV and AS contributed to the assay designs and discussions throughout. AJW solved, refined and analyzed X-ray crystal structure of 111a. SS and MF compiled/analysed all data and drafted the manuscript. All authors participated in revision of the manuscript before submission.Acknowledgements
This work was funded by the French Parasitology Consortium ParaFrap (ANR-11-LABX0024), the Agence Nationale de Recherche (HypEpiC ANR-14 CE 160013 02), and by Cancer Research UK (C33325/A19435). N. M. was supported by a Institut Carnot Pasteur MI fellowship. S. S. acknowledges the European Commission for awarding a Marie Curie International Incoming Fellowship (Agreement No. 299857). A. L. was supported by an Imperial College Schrodinger Scholarship. The SGC is a registered charity (number 1097737) that receives funds from AbbVie, Bayer Pharma AG, Boehringer Ingelheim, Canada Foundation for Innovation, Eshelman Institute for Innovation, Genome Canada, Innovative Medicines Initiative (EU/EFPIA), Janssen, Merck & Co., Novartis Pharma AG, Ontario Ministry of Economic Development and Innovation, Pfizer, São Paulo Research Foundation-FAPESP, Takeda, and Wellcome Trust.References
- http://www.who.int/malaria/publications/world-malaria-report-2015/en/ (accessed on May 2016).
- I. Petersen, R. Eastman and M. Lanzer, FEBS Lett., 2011, 585, 1551–1562 CrossRef CAS PubMed.
- U. D'Alessandro and H. Buttiens, Trop. Med. Int. Health, 2001, 6, 845–848 CrossRef.
- N. Gogtay, S. Kannan, U. M. Thatte, P. L. Olliaro and D. Sinclair, Cochrane Database Syst. Rev., 2013, 10, Cd008492 Search PubMed.
- A. M. Dondorp, F. Nosten, P. Yi, D. Das, A. P. Phyo, J. Tarning, K. M. Lwin, F. Ariey, W. Hanpithakpong, S. J. Lee, P. Ringwald, K. Silamut, M. Imwong, K. Chotivanich, P. Lim, T. Herdman, S. S. An, S. Yeung, P. Singhasivanon, N. P. Day, N. Lindegardh, D. Socheat and N. J. White, N. Engl. J. Med., 2009, 361, 455–467 CrossRef CAS PubMed.
- H. Noedl, D. Socheat and W. Satimai, N. Engl. J. Med., 2009, 361, 540–541 CrossRef CAS PubMed.
- E. L. Flannery, A. K. Chatterjee and E. A. Winzeler, Nat. Rev. Microbiol., 2013, 11, 849–862 CrossRef CAS PubMed.
- J. Guizetti and A. Scherf, Cell. Microbiol., 2013, 15, 718–726 CrossRef CAS PubMed.
- A. P. Gupta, W. H. Chin, L. Zhu, S. Mok, Y.-H. Luah, E.-H. Lim and Z. Bozdech, PLoS Pathog., 2013, 9, e1003170 Search PubMed.
- Q. Wang, B. A. Rosa, B. Nare, K. Powell, S. Valente, D. Rotili, A. Mai, G. R. Marshall and M. Mitreva, PLoS Neglected Trop. Dis., 2015, 9, e0004026 Search PubMed.
- K. Trenholme, L. Marek, S. Duffy, G. Pradel, G. Fisher, F. K. Hansen, T. S. Skinner-Adams, A. Butterworth, C. J. Ngwa, J. Moecking, C. D. Goodman, G. I. McFadden, S. D. M. Sumanadasa, D. P. Fairlie, V. M. Avery, T. Kurz and K. T. Andrews, Antimicrob. Agents Chemother., 2014, 58, 3666–3678 CrossRef PubMed.
- S. D. M. Sumanadasa, C. D. Goodman, A. J. Lucke, T. Skinner-Adams, I. Sahama, A. Haque, T. A. Do, G. I. McFadden, D. P. Fairlie and K. T. Andrews, Antimicrob. Agents Chemother., 2012, 56, 3849–3856 CrossRef CAS PubMed.
- J. Marfurt, F. Chalfein, P. Prayoga, F. Wabiser, E. Kenangalem, K. A. Piera, D. P. Fairlie, E. Tjitra, N. M. Anstey, K. T. Andrews and R. N. Price, Antimicrob. Agents Chemother., 2011, 55, 961–966 CrossRef CAS PubMed.
- V. Patel, R. Mazitschek, B. Coleman, C. Nguyen, S. Urgaonkar, J. Cortese, R. H. Barker, E. Greenberg, W. Tang, J. E. Bradner, S. L. Schreiber, M. T. Duraisingh, D. F. Wirth and J. Clardy, J. Med. Chem., 2009, 52, 2185–2187 CrossRef CAS PubMed.
- S. Shen and A. P. Kozikowski, ChemMedChem, 2016, 11, 15–21 CrossRef CAS PubMed.
- M. Tachibana, K. Sugimoto, M. Nozaki, J. Ueda, T. Ohta, M. Ohki, M. Fukuda, N. Takeda, H. Niida, H. Kato and Y. Shinkai, Genes Dev., 2002, 16, 1779–1791 CrossRef CAS PubMed.
- A. Schaefer, S. C. Sampath, A. Intrator, A. Min, T. S. Gertler, D. J. Surmeier, A. Tarakhovsky and P. Greengard, Neuron, 2009, 64, 678–691 CrossRef CAS PubMed.
- I. Maze, H. E. Covington, D. M. Dietz, Q. LaPlant, W. Renthal, S. J. Russo, M. Mechanic, E. Mouzon, R. L. Neve, S. J. Haggarty, Y. Ren, S. C. Sampath, Y. L. Hurd, P. Greengard, A. Tarakhovsky, A. Schaefer and E. J. Nestler, Science, 2010, 327, 213–216 CrossRef CAS PubMed.
- H. E. Covington III, I. Maze, H. Sun, H. M. Bomze, K. D. DeMaio, E. Y. Wu, D. M. Dietz, M. K. Lobo, S. Ghose, E. Mouzon, R. L. Neve, C. A. Tamminga and E. J. Nestler, Neuron, 2011, 71, 656–670 CrossRef PubMed.
- J. Huang, J. Dorsey, S. Chuikov, L. Perez-Burgos, X. Zhang, T. Jenuwein, D. Reinberg and S. L. Berger, J. Biol. Chem., 2010, 285, 9636–9641 CrossRef CAS PubMed.
- Y. Kondo, L. Shen, S. Ahmed, Y. Boumber, Y. Sekido, B. R. Haddad and J.-P. J. Issa, PLoS One, 2008, 3, e2037 Search PubMed.
- Y. Kondo, L. Shen, S. Suzuki, T. Kurokawa, K. Masuko, Y. Tanaka, H. Kato, Y. Mizuno, M. Yokoe, F. Sugauchi, N. Hirashima, E. Orito, H. Osada, R. Ueda, Y. Guo, X. Chen, J. P. Issa and Y. Sekido, Hepatol. Res., 2007, 37, 974–983 CrossRef CAS PubMed.
- H. Ü. Kaniskan, K. D. Konze and J. Jin, J. Med. Chem., 2015, 58, 1596–1629 CrossRef CAS PubMed.
- X. Yi, X. J. Jiang, X. Y. Li and D. S. Jiang, Am. J. Transl. Res., 2015, 7, 2159–2175 Search PubMed.
- R. A. Copeland, M. E. Solomon and V. M. Richon, Nat. Rev. Drug Discovery, 2009, 8, 724–732 CrossRef CAS PubMed.
- E. Curry, I. Green, N. Chapman-Rothe, E. Shamsaei, S. Kandil, F. L. Cherblanc, L. Payne, E. Bell, T. Ganesh, N. Srimongkolpithak, J. Caron, F. Li, A. G. Uren, J. P. Snyder, M. Vedadi, M. J. Fuchter and R. Brown, Clin. Epigenet., 2015, 7, 1–12 CrossRef PubMed.
- L. Cui, Q. Fan, L. Cui and J. Miao, Int. J. Parasitol., 2008, 38, 1083–1097 CrossRef CAS PubMed.
- L. Jiang, J. Mu, Q. Zhang, T. Ni, P. Srinivasan, K. Rayavara, W. Yang, L. Turner, T. Lavstsen, T. G. Theander, W. Peng, G. Wei, Q. Jing, Y. Wakabayashi, A. Bansal, Y. Luo, J. M. Ribeiro, A. Scherf, L. Aravind, J. Zhu, K. Zhao and L. H. Miller, Nature, 2013, 499, 223–227 CrossRef CAS PubMed.
- J. C. Volz, R. Bartfai, M. Petter, C. Langer, G. A. Josling, T. Tsuboi, F. Schwach, J. Baum, J. C. Rayner, H. G. Stunnenberg, M. F. Duffy and A. F. Cowman, Cell Host Microbe, 2012, 11, 7–18 CAS.
- P. B. Chen, S. Ding, G. Zanghi, V. Soulard, P. A. DiMaggio, M. J. Fuchter, S. Mecheri, D. Mazier, A. Scherf and N. A. Malmquist, Sci. Rep., 2016, 6, 21802 CrossRef CAS PubMed.
- N. A. Malmquist, T. A. Moss, S. Mecheri, A. Scherf and M. J. Fuchter, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 16708–16713 CrossRef CAS PubMed.
- N. A. Malmquist, S. Sundriyal, J. Caron, P. Chen, B. Witkowski, D. Menard, R. Suwanarusk, L. Renia, F. Nosten, M. B. Jimenez-Diaz, I. Angulo-Barturen, M. Santos Martinez, S. Ferrer, L. M. Sanz, F. J. Gamo, S. Wittlin, S. Duffy, V. M. Avery, A. Ruecker, M. J. Delves, R. E. Sinden, M. J. Fuchter and A. Scherf, Antimicrob. Agents Chemother., 2015, 59, 950–959 CrossRef PubMed.
- S. Sundriyal, N. A. Malmquist, J. Caron, S. Blundell, F. Liu, X. Chen, N. Srimongkolpithak, J. Jin, S. A. Charman, A. Scherf and M. J. Fuchter, ChemMedChem, 2014, 9, 2360–2373 CrossRef CAS PubMed.
- L. Dembele, J. F. Franetich, A. Lorthiois, A. Gego, A. M. Zeeman, C. H. Kocken, R. Le Grand, N. Dereuddre-Bosquet, G. J. van Gemert, R. Sauerwein, J. C. Vaillant, L. Hannoun, M. J. Fuchter, T. T. Diagana, N. A. Malmquist, A. Scherf, G. Snounou and D. Mazier, Nat. Med., 2014, 20, 307–312 CrossRef CAS PubMed.
- S. Kubicek, R. J. O'Sullivan, E. M. August, E. R. Hickey, Q. Zhang, M. L. Teodoro, S. Rea, K. Mechtler, J. A. Kowalski, C. A. Homon, T. A. Kelly and T. Jenuwein, Mol. Cell, 2007, 25, 473–481 CrossRef CAS PubMed.
- F. Liu, X. Chen, A. Allali–Hassani, A. M. Quinn, G. A. Wasney, A. Dong, D. Barsyte, I. Kozieradzki, G. Senisterra, I. Chau, A. Siarheyeva, D. B. Kireev, A. Jadhav, J. M. Herold, S. V. Frye, C. H. Arrowsmith, P. J. Brown, A. Simeonov, M. Vedadi and J. Jin, J. Med. Chem., 2009, 52, 7950–7953 CrossRef CAS PubMed.
- F. Liu, X. Chen, A. Allali-Hassani, A. M. Quinn, T. J. Wigle, G. A. Wasney, A. Dong, G. Senisterra, I. Chau, A. Siarheyeva, J. L. Norris, D. B. Kireev, A. Jadhav, J. M. Herold, W. P. Janzen, C. H. Arrowsmith, S. V. Frye, P. J. Brown, A. Simeonov, M. Vedadi and J. Jin, J. Med. Chem., 2010, 53, 5844–5857 CrossRef CAS PubMed.
- F. Liu, D. Barsyte-Lovejoy, A. Allali-Hassani, Y. He, J. M. Herold, X. Chen, C. M. Yates, S. V. Frye, P. J. Brown, J. Huang, M. Vedadi, C. H. Arrowsmith and J. Jin, J. Med. Chem., 2011, 54, 6139–6150 CrossRef CAS PubMed.
- M. Vedadi, D. Barsyte-Lovejoy, F. Liu, S. Rival-Gervier, A. Allali-Hassani, V. Labrie, T. J. Wigle, P. A. Dimaggio, G. A. Wasney, A. Siarheyeva, A. Dong, W. Tempel, S. C. Wang, X. Chen, I. Chau, T. J. Mangano, X. P. Huang, C. D. Simpson, S. G. Pattenden, J. L. Norris, D. B. Kireev, A. Tripathy, A. Edwards, B. L. Roth, W. P. Janzen, B. A. Garcia, A. Petronis, J. Ellis, P. J. Brown, S. V. Frye, C. H. Arrowsmith and J. Jin, Nat. Chem. Biol., 2011, 7, 566–574 CrossRef CAS PubMed.
- F. Liu, D. Barsyte-Lovejoy, F. Li, Y. Xiong, V. Korboukh, X.-P. Huang, A. Allali-Hassani, W. P. Janzen, B. L. Roth, S. V. Frye, C. H. Arrowsmith, P. J. Brown, M. Vedadi and J. Jin, J. Med. Chem., 2013, 56, 8931–8942 CrossRef CAS PubMed.
- N. Srimongkolpithak, S. Sundriyal, F. Li, M. Vedadi and M. J. Fuchter, MedChemComm, 2014, 5, 1821–1828 RSC.
- A. Ma, W. Yu, Y. Xiong, K. V. Butler, P. J. Brown and J. Jin, MedChemComm, 2014, 5, 1892–1898 RSC.
- A. Ma, W. Yu, F. Li, R. M. Bleich, J. M. Herold, K. V. Butler, J. L. Norris, V. Korboukh, A. Tripathy, W. P. Janzen, C. H. Arrowsmith, S. V. Frye, M. Vedadi, P. J. Brown and J. Jin, J. Med. Chem., 2014, 57, 6822–6833 CrossRef CAS PubMed.
- F. Baur, D. Beattie, D. Beer, D. Bentley, M. Bradley, I. Bruce, S. J. Charlton, B. Cuenoud, R. Ernst, R. A. Fairhurst, B. Faller, D. Farr, T. Keller, J. R. Fozard, J. Fullerton, S. Garman, J. Hatto, C. Hayden, H. He, C. Howes, D. Janus, Z. Jiang, C. Lewis, F. Loeuillet-Ritzler, H. Moser, J. Reilly, A. Steward, D. Sykes, L. Tedaldi, A. Trifilieff, M. Tweed, S. Watson, E. Wissler and D. Wyss, J. Med. Chem., 2010, 53, 3675–3684 CrossRef CAS PubMed.
- S. R. Haadsma-Svensson, K. A. Cleek, D. M. Dinh, J. N. Duncan, C. L. Haber, R. M. Huff, M. E. Lajiness, N. F. Nichols, M. W. Smith, K. A. Svensson, M. J. Zaya, A. Carlsson and C.-H. Lin, J. Med. Chem., 2001, 44, 4716–4732 CrossRef CAS PubMed.
- L. Chen, G. Georges, A. Mertens and X. Wu, Isoquinoline aminopyrazole derivatives, their manufacture and use as pharmaceutical agents for the treatment of cancer, WO/2007/071348, 2007.
- E. Laborde, R. W. Macsata, F. Meng, B. T. Peterson, L. Robinson, S. R. Schow, R. J. Simon, H. Xu, K. Baba, H. Inagaki, Y. Ishiwata, T. Jomori, Y. Matsumoto, A. Miyachi, T. Nakamura, M. Okamoto, T. M. Handel and C. C. A. Bernard, J. Med. Chem., 2011, 54, 1667–1681 CrossRef CAS PubMed.
- M. H. P. Verheij, A. J. Thompson, J. E. van Muijlwijk-Koezen, S. C. R. Lummis, R. Leurs and I. J. P. de Esch, J. Med. Chem., 2012, 55, 8603–8614 CrossRef CAS PubMed.
- C. J. O'Brien, E. A. B. Kantchev, C. Valente, N. Hadei, G. A. Chass, A. Lough, A. C. Hopkinson and M. G. Organ, Chem. – Eur. J., 2006, 12, 4743–4748 CrossRef PubMed.
- E. A. Kantchev, C. J. O'Brien and M. G. Organ, Angew. Chem., Int. Ed., 2007, 46, 2768–2813 CrossRef CAS PubMed.
- M. G. Organ, M. Abdel-Hadi, S. Avola, I. Dubovyk, N. Hadei, E. A. Kantchev, C. J. O'Brien, M. Sayah and C. Valente, Chem. – Eur. J., 2008, 14, 2443–2452 CrossRef CAS PubMed.
- T. E. Barder and S. L. Buchwald, J. Am. Chem. Soc., 2007, 129, 5096–5101 CrossRef CAS PubMed.
- D. S. Surry and S. L. Buchwald, Angew. Chem., Int. Ed., 2008, 47, 6338–6361 CrossRef CAS PubMed.
- D. S. Surry and S. L. Buchwald, Chem. Sci., 2011, 2, 27–50 RSC.
- S. F. Campbell, J. D. Hardstone and M. J. Palmer, J. Med. Chem., 1988, 31, 1031–1035 CrossRef CAS PubMed.
- J. Bordner, S. F. Campbell, M. J. Palmer and M. S. Tute, J. Med. Chem., 1988, 31, 1036–1039 CrossRef CAS PubMed.
- A. B. Ahvale, H. Prokopcová, J. Šefčovičová, W. Steinschifter, A. E. Täubl, G. Uray and W. Stadlbauer, Eur. J. Org. Chem., 2008, 2008, 563–571 CrossRef.
- Y. Chang, X. Zhang, J. R. Horton, A. K. Upadhyay, A. Spannhoff, J. Liu, J. P. Snyder, M. T. Bedford and X. Cheng, Nat. Struct. Mol. Biol., 2009, 16, 312–317 CAS.
- R. F. Sweis, M. Pliushchev, P. J. Brown, J. Guo, F. L. Li, D. Maag, A. M. Petros, N. B. Soni, C. Tse, M. Vedadi, M. R. Michaelides, G. G. Chiang and W. N. Pappano, ACS Med. Chem. Lett., 2014, 5, 205–209 CrossRef CAS PubMed.
- D. Rotili, D. Tarantino, B. Marrocco, C. Gros, V. Masson, V. Poughon, F. Ausseil, Y. Chang, D. Labella, S. Cosconati, S. Di Maro, E. Novellino, M. Schnekenburger, C. Grandjenette, C. Bouvy, M. Diederich, X. Cheng, P. B. Arimondo and A. Mai, PLoS One, 2014, 9, e96941 Search PubMed.
- K. T. Nguyen, F. Li, G. Poda, D. Smil, M. Vedadi and M. Schapira, J. Chem. Inf. Model., 2013, 53, 681–691 CrossRef CAS PubMed.
- S. Bender, Y. Tang, A. M. Lindroth, V. Hovestadt, D. T. W. Jones, M. Kool, M. Zapatka, P. A. Northcott, D. Sturm, W. Wang, B. Radlwimmer, J. W. Hojfeldt, N. Truffaux, D. Castel, S. Schubert, M. Ryzhova, H. Seker-Cin, J. Gronych, P. D. Johann, S. Stark, J. Meyer, T. Milde, M. Schuhmann, M. Ebinger, C. M. Monoranu, A. Ponnuswami, S. Chen, C. Jones, O. Witt, V. P. Collins, A. von Deimling, N. Jabado, S. Puget, J. Grill, K. Helin, A. Korshunov, P. Lichter, M. Monje, C. Plass, Y. J. Cho and S. M. Pfister, Cancer Cell, 2013, 24, 660–672 CrossRef CAS PubMed.
- P. W. Lewis, M. M. Müller, M. S. Koletsky, F. Cordero, S. Lin, L. A. Banaszynski, B. A. Garcia, T. W. Muir, O. J. Becher and C. D. Allis, Science, 2013, 340, 857–861 CrossRef CAS PubMed.
- Z. Z. Brown, M. M. Muller, S. U. Jain, C. D. Allis, P. W. Lewis and T. W. Muir, J. Am. Chem. Soc., 2014, 136, 13498–13501 CrossRef CAS PubMed.
- Z. Z. Brown, M. M. Müller, H. E. Kong, P. W. Lewis and T. W. Muir, Angew. Chem., 2015, 127, 6557–6561 CrossRef.
- L. Jiao and X. Liu, Science, 2015, 350, aac4383 CrossRef PubMed.
- H. Jayaram, D. Hoelper, S. U. Jain, N. Cantone, S. M. Lundgren, F. Poy, C. D. Allis, R. Cummings, S. Bellon and P. W. Lewis, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 6182–6187 CrossRef CAS PubMed.
- N. Justin, Y. Zhang, C. Tarricone, S. R. Martin, S. Chen, E. Underwood, V. De Marco, L. F. Haire, P. A. Walker, D. Reinberg, J. R. Wilson and S. J. Gamblin, Nat. Commun., 2016, 7, 11316 CrossRef CAS PubMed.
- R. A. Judge, H. Zhu, A. K. Upadhyay, P. M. Bodelle, C. W. Hutchins, M. Torrent, V. L. Marin, W. Yu, M. Vedadi, F. Li, P. J. Brown, W. N. Pappano, C. Sun and A. M. Petros, ACS Med. Chem. Lett., 2016, 7, 1102–1106 CrossRef CAS PubMed.
- K. Zeilinger, N. Freyer, G. Damm, D. Seehofer and F. Knospel, Exp. Biol. Med., 2016, 241, 1684–1698 CrossRef CAS PubMed.
- S. S. Palabiyik, E. Karakus, Z. Halici, E. Cadirci, Y. Bayir, G. Ayaz and I. Cinar, Hum. Exp. Toxicol., 2016, 35, 1252–1263 CAS.
- W. G. Schoonen, J. A. de Roos, W. M. Westerink and E. Debiton, Toxicol. In Vitro, 2005, 19, 491–503 CrossRef CAS PubMed.
- S. Knasmuller, V. Mersch-Sundermann, S. Kevekordes, F. Darroudi, W. W. Huber, C. Hoelzl, J. Bichler and B. J. Majer, Toxicology, 2004, 198, 315–328 CrossRef CAS PubMed.
- J. Sikkema, F. J. Weber, H. J. Heipieper and J. A. M. D. Bont, Biocatalysis, 1994, 10, 113–122 CrossRef CAS.
- L. M. Sanz, B. Crespo, C. De-Cózar, X. C. Ding, J. L. Llergo, J. N. Burrows, J. F. García-Bustos and F.-J. Gamo, PLoS One, 2012, 7, e30949 CAS.
Footnotes |
| † The authors declare no competing interests. |
| ‡ Electronic supplementary information (ESI) available: Supplementary Tables ST1–ST5, experimental data for the representative diaminoquinazoline analogues, 2D NMRs of 85 and 111a and X-ray structure of 111a (Fig. SF1). The coordinates for 111a have been deposited with CCDC 1503377. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7md00052a |
| This journal is © The Royal Society of Chemistry 2017 |