
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe nature of the conserved basic amino acid sequences found among 437 heparin binding proteins determined by network analysis†
Timothy R.
Rudd
 *ab,
Mark D.
Preston
a and
Edwin A.
Yates
b
*ab,
Mark D.
Preston
a and
Edwin A.
Yates
b
aThe National Institute for Biological Standards and Control (NIBSC), Blanche Lane, South Mimms, Potters Bar, Hertfordshire EN6 3QG, UK. E-mail: tim.rudd@nibsc.org
bDepartment of Biochemistry, Institute of Integrative Biology, University of Liverpool, Liverpool, L69 7ZB, UK
First published on 14th March 2017
Abstract
In multicellular organisms, a large number of proteins interact with the polyanionic polysaccharides heparan sulphate (HS) and heparin. These interactions are usually assumed to be dominated by charge–charge interactions between the anionic carboxylate and/or sulfate groups of the polysaccharide and cationic amino acids of the protein. A major question is whether there exist conserved amino acid sequences for HS/heparin binding among these diverse proteins. Potentially conserved HS/heparin binding sequences were sought amongst 437 HS/heparin binding proteins. Amino acid sequences were extracted and compared using a Levenshtein distance metric. The resultant similarity matrices were visualised as graphs, enabling extraction of strongly conserved sequences from highly variable primary sequences while excluding short, core regions. This approach did not reveal extensive, conserved HS/heparin binding sequences, rather a number of shorter, more widely spaced sequences that may work in unison to form heparin-binding sites on protein surfaces, arguing for convergent evolution. Thus, it is the three-dimensional arrangement of these conserved motifs on the protein surface, rather than the primary sequence per se, which are the evolutionary elements.
1. Introduction
Heparan sulfate (HS) and heparin are closely related linear polyanionic carbohydrates (Scheme 1), which are members of a class of polysaccharide known as glycosaminoglycans (GAGs).1 Heparan sulfate is a ubiquitous element of mammalian cells and plays an important physiological role, including receptor–ligand clustering and signalling, cell-to-cell cross talk and adhesion, chemokine presentation, storage, cell adhesion and extracellular matrix (ECM) formation. Heparan sulfate is found on the surface of cells as a part of proteoglycans (HSPG), for example, syndecan and glypican,2 as well as being an integral component of the ECM, where HS is attached to proteoglycans such as agrin and perlecan.3 Heparan sulfate has also been found in the cell nucleus,4,5 although the functional significance of this remains unclear.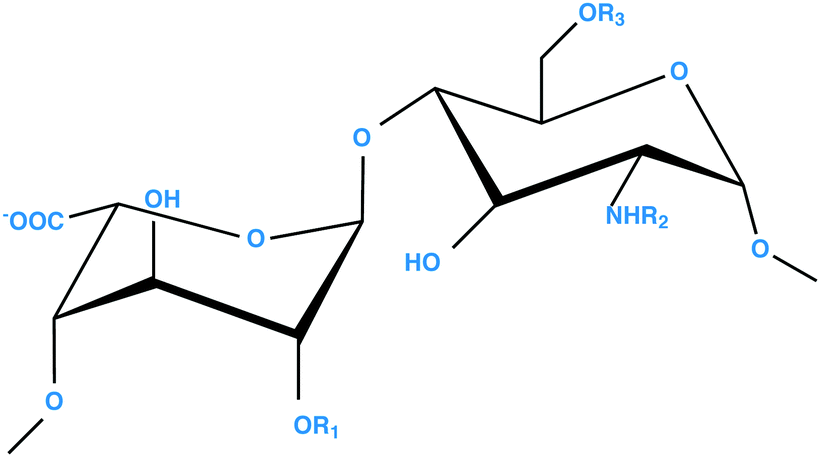 | ||
| Scheme 1 General repeating disaccharide structure of HS and heparin polysaccharides; [(−4) L-IdoA α(1→4) D-GlcN α(1−)], where R1 = H or SO3−, R2 = H/COCH3 or SO3− and R3 = H or SO3−. The α-L-IdoA residue can be replaced by its C-5 epimer, β-D-GlcA. HS has lower overall sulfation than heparin, possesses a more distinct domain structure and a higher proportion of GlcA residues. | ||
Owing to its abundance, relatively low cost and overall structural similarity, heparin is often used as an experimental proxy for HS. Heparin is readily available as a widely used pharmaceutical anticoagulant which originates in mast cells, where the polysaccharides are stored in intracellular granules as serglycin proteoglycans. Mast cells can be stimulated to eject their granules, a process termed degranulation, through physical/chemical damage or through interaction with IgE, cytokines and others agents.
Heparin is composed of the same disaccharide units, although in different proportions, and both HS and heparin share a common biosynthetic pathway.6 The polysaccharides comprise alternating disaccharides of an uronic acid linked 1→4 to α-D-glucosamine. The uronic acid can be present as β-D-glucuronic acid, or its C-5 epimer, α-L-iduronic acid, both of which can be O-sulfated at position 2. The glucosamine can be O-sulfated at positions 3 and 6, as well as N-acetylated, N-sulfated, or unsubstituted at position 2. The major repeating disaccharide unit of heparin is shown in Scheme 1. The large number of possible enzymatic modifications involved in the biosynthesis together with the non-template-driven nature of their biosynthesis, results in highly heterogeneous polysaccharides.7,8
The principal difference between HS and heparin resides in the organisation and content of their domain structures. The majority of the HS chain is composed of [(−4) D-GlcA α(1→4) D-GlcNAc α(1−)], disaccharide repeats containing glucuronic acid and N-acetyl glucosamine, exhibiting little or no sulfation. Interspersed between low sulfation domains are sequences with higher degrees of sulfation. It is in these high sulfation regions, where the majority of protein interactions are thought to occur,9,10 that have a structure more closely resembling that of heparin. Heparin consists of around 80% of the trisulfated disaccharide, [(−4) L-IdoA2-O-sulfate α(1→4) D-GlcN-sulfate,6-O-sulfate α(1−)]. Heparin is composed of around eighty percent of this trisulfated disaccharide, making it more homogenous than HS.7
It is often stated that heparan sulfate and heparin interact with numerous, key proteins primarily via the high sulfation regions in HS/heparin. This statement is perhaps tautological, since almost all experimental investigations have involved the selection of proteins bound to HS/heparin via elution from a heparin column using salt that inherently selects for high charge interactions.11 Ori et al.12 compiled a list of 435, non-redundant, human HS/heparin binding proteins (HEPbps) in the HS/heparin interactome, which include members of important protein families, such as growth factors, cytokines and morphogens. Heparan sulfate is a molecule that, in some manner, choreographs signalling pathways thereby allowing information to cross the cell membrane.11,13 Heparin binding proteins play a key role in controlling development, for example, via the Wnt, Hedgehog, transforming growth factor-beta and fibroblast growth factor (FGF) pathways.14 Furthermore, HS has been implicated in diseases such as Alzheimer's,15 cancer16 and sexually transmitted infections.17 Recently, Nunes et al.18 performed a study to examine the role of HEPbps in pancreatic diseases, concluding that a concerted network of highly connected HEPbps was important for distinguishing between normal and diseased pancreatic tissue. Chen et al.19 showed that the interaction between the cell surface HSPGs of two-breast cancer cell lines and their innate complement of HEPbps is a key component of tumourigenicity. Inhibition of the innate HEPbps of breast cancer cell lines by the addition of extraneous heparin perturbed the PI3K/Akt and Raf/MEK/ERK signalling pathways.
In evolutionary terms, HEPbps are thought to originate at the dawn of multicellular life, via colonies of communicating unicellular organisms. Monosiga brevicollis is one such organism and it is known to contain the biosynthetic machinery necessary to produce heparin/HS.12M. brevicollis also possesses receptor tyrosine kinases (RTK),20 and the HEPbps FGF family are ligands for RTKs in metazoans. Recently, Bertrand et al. found orthologous genes to the FGFs in M. Brevicollis and proposed that FGFs and their receptors originated in a eumetazoan ancestor.21 Finally, three GAG lyases have also been predicted in the proteome of the organism.22 These observations indicate that what is often considered a relatively simple organism possesses the full apparatus of a HSPG-mediated cell-signalling system. Furthermore, M. Brevicollis possesses lyases capable of causing GAGs to be shed into the environment and is, in principle, therefore, able to interact with its neighbours via protein and glycan communication. Such findings support the idea that HEPbps are crucial for, and may be a defining characteristic of, multicellular animal life.
Basic amino acids in HEPbps are postulated as being key to interactions with HS/heparin. Linhardt et al. published a number of studies investigating the heparin binding properties of the three basic amino acids,23–25 arginine, lysine and histidine. They concluded that the affinity between heparin and arginine is higher than that between heparin and lysine. Histidine exhibits low affinity and only at pH values at which it is protonated (below its pKa of ca. 6.5). The frequency, location and structure of basic amino acids in HEPbps are consequently likely to be important determinants of their binding properties.
Heparin binding sequences (HBSs) are amino acid sequences found in HEPbps that have been shown, or are predicted to be, the domains that bind to HS/heparin. Cardin and Weintraub26 reported two sequences, XBBBXXBX and XBBXBX in the heparin binding proteins: apo B; apo E; vitronectin; and platelet factor 4 (where B and X signify basic and hydropathic amino acids, respectively). These sequences were then used to predict HBSs in other proteins and a similar approach was used to propose the von Willebrand factor HBS – XBBBXXBBBXXBBX.27 Subsequently, Hileman et al. proposed the heparin-binding consensus sequence TXXBXXTBXXXTBB (T denotes a turn), combining secondary structure information and conserved sequence information. This sequence was proposed using the crystallographic/NMR structural data for FGF-1 and -2 and transforming growth factor (TGF). A recent theory proposed by Torrents et al., defines a minimal sequence, termed the “CPC clip motif” (C – cationic and P – polar residues), with this sequence working analogously to a staple; small points of contact pinning the polysaccharide to the protein.28 Even in combination, however, these studies have only surveyed a very small fraction of HEPbps, which may be too small for global features to become apparent. By examining all HEPbp sequences, it was thought that more general, underlying similarities may emerge.
The aim of this present study was to identify HBSs within all currently collated HEPbps. To do this, a sequence similarity metric paired with graph analysis29 was employed to investigate conserved sequences within HEPbps that contain basic amino acids. The similarity between amino acid sequences was determined here using the Levenshtein distance (DL).30Levenshtein distance is also called the edit distance and is defined as the minimum number of single letter elementary operations (insertions, deletions and replacements) required to convert one character string into another. This measure is used widely to compare strings of information, including in applications to protein interactions with small ligands31 within the field of protein interactions, which is reviewed in ref. 32. In the present work, a similarity matrix was created from the DL's to compare the basic sequences. The similarity matrices were transformed into a graph to visualise and analyse these data and this analysis allowed strongly conserved sequences to be extracted from among the highly variable 437 HEPbps, while excluding short, core regions. It is possible that a number of these sequences work in unison to form heparin-binding domains on protein surfaces. The results are consistent with convergent evolution, in which the three-dimensional arrangement of amino acids on the protein surface is the evolutionary element, rather than the primary sequence. Furthermore, when the human proteome was searched for the sequences found in the relatively small population of verified HEPbps, it became clear that many proteins may be able to interact with heparin/HS. Indeed, this may be an innate property of extracellular proteins. This calls into question the possible control mechanism behind protein:heparin/HS interactions; instead of considering a protein binding to a defined carbohydrate sequence, a more holistic concept should be considered.
2. Material and methods
2.1 Determination of heparin binding sequence (HBS) similarity matrices and the subsequent formation of networks
Four hundred and thirty seven HEPbp amino acid sequences were retrieved through UNIPROT.33 The original HEPbps list12 contained 435 proteins, from which FBS1 (Fibrosin-1, considered obsolete and removed from UNIPROT, accession number (a/no.) P62706) and IAPP (Islet Amyloid Polypeptide, a/no. P10997) were excluded. FGF11, 13, 19 and 21 (a/no. Q92914, Q92913, O95750 and Q9NSA1, respectively) were added, providing a final list containing 437 proteins.A search was made for seven amino acid sets within the HEPbps. The sets searched for were {B,X}, {B,X,A}, {B,X,P}, {B,X,S}, {B,X,P,A}, {B,X,P,S} and {B,X,A,S}, composed of the five different types of amino acid: basic (B); hydrophobic (X); polar (P); special (S); and acidic (A) (see ESI,† Table S1 for more details). In the text, these set names are abbreviated to BX, BXA, BXP, BXS, BXPA, BXPS and BXAS. These sets are neither exclusive nor are they exhaustive. Each HBS was read serially from the N- to C-terminus to identify amino acid sequences. Sequences had a minimum length of 3 amino acids.
For the group of amino acid sequences identified from each amino acid set, a similarity matrix (Scheme 2A) was calculated using a normalised Levenshtein distance. The Levenshtein distance was defined as the minimum number of elementary character operations (insert, delete or replace a single letter) required to transform one sequence into another:
| DL(a,b) := min(i(a,b) + d(a,b) + r(a,b)) | (1) |
| DLm(a,b) := 1 − (DL(a,b)/max length (a,b)) | (2) |
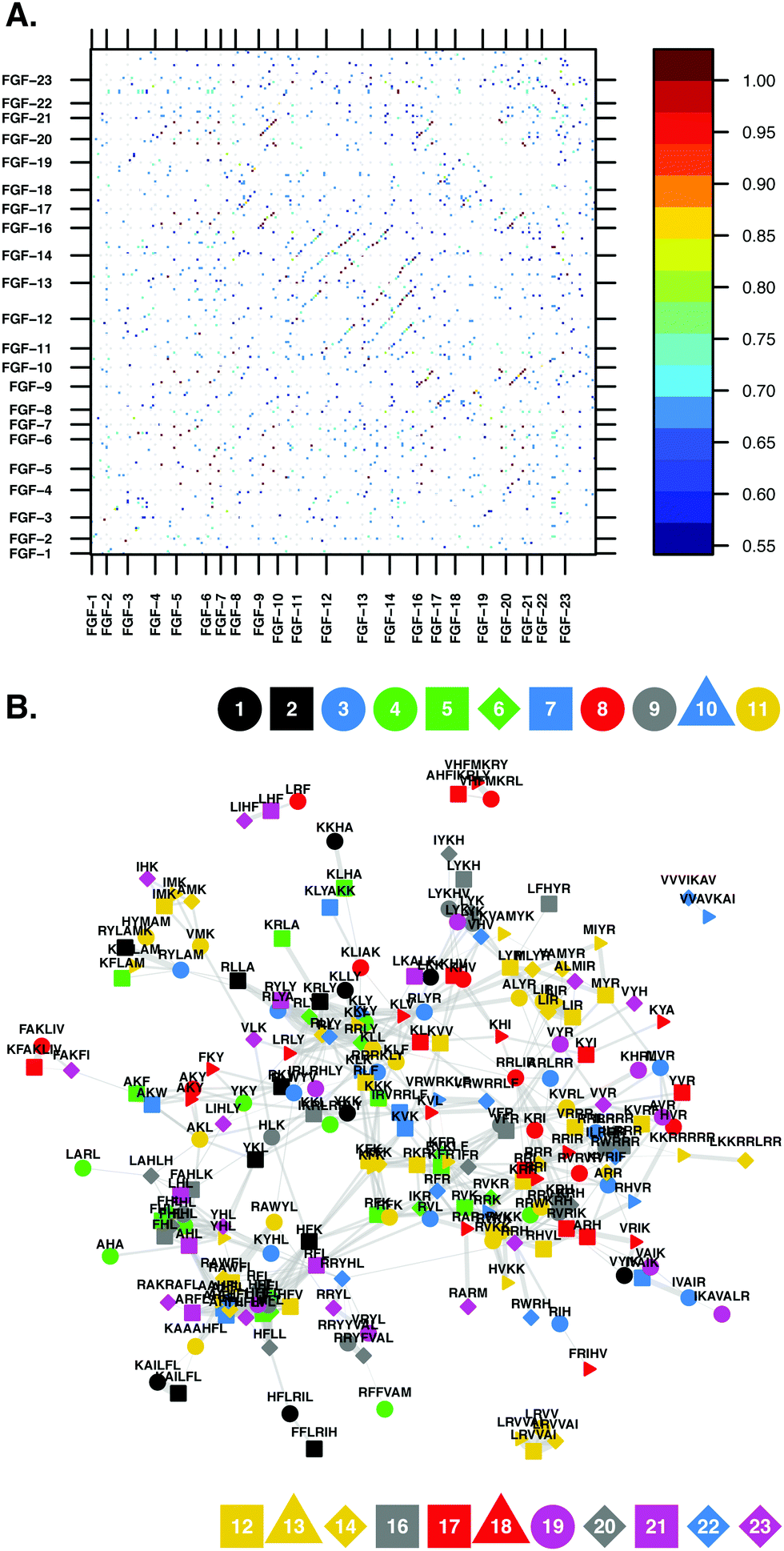 | ||
| Scheme 2 (A) Similarity matrix of the BX (basic and hydrophobic) amino acid-containing amino acid sequences, found in the FGF family. It is difficult to extract information from the data contained within the similarity matrix. The relationships between the proteins and basic amino acid containing sequences become clearer when the matrix is converted into a network, in the form of a graph. (B) Graph representations of the basic amino acid containing sequences found in the FGF family. The similarity cut-off (95% confidence interval of the Levenshtein matrix values) for the network was 0.57. The colour and shape of the vertices indicate which member of the FGF subfamily the sequence originated from, provided as a legend on the figure. The thickness of each edge is proportional to the similarity of the HBSs linked; the thicker the edge, the more similar are the vertices. | ||
The similarity matrices were visualised and further analysed in graph form (Schemes 2B and 3). Each node/vertex in the graph represents two properties: a sequence and the protein in which it is found. The edges of the graph are weighted by the similarity matrix (above the cut-off) or non-existent (below the cut-off). In the subsequent analysis a sequence within a protein is only compared with sequences from other proteins and not to sequences within the same protein. The graph layouts were determined using Fruchterman-Reingold and force-directed algorithms.34
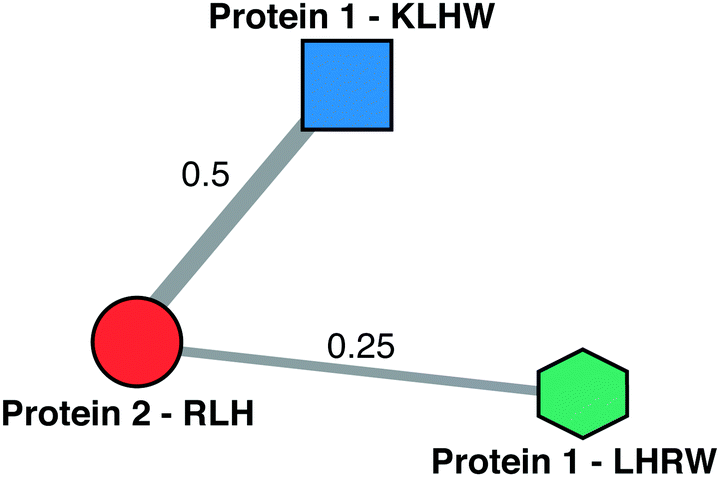 | ||
| Scheme 3 Schematic of a HEPbp HBS graph. The vertices represent a basic amino acid sequence from a heparin binding protein. The vertex contains both pieces of information. The connections between vertices is the similarity of the amino acid sequences. The width of the edges is proportional to the weight of the edge. In this analysis amino acid sequences arising from the same protein are not considered, i.e., there is no edge between the vertices belonging to protein 1 in the above schematic. | ||
If we consider only one component (sequence or protein) of vertex identity this reduces the number of vertices and therefore the complexity of the graphs. When there exist multiple edges between two vertices these are collapsed into one single edge with new weight defined as the sum of the weights of the original edges. A number of parameters are used to describe the resultant vertices and graphs, and are defined as:
Further information regarding graphs and their analysis can be found in ref. 29.
The resultant graphs were further refined by only considering the core of the graph; the highly connected heart of the network, which was defined using the closeness centrality.
The graphs were further collapsed by translating the devolving amino acids found in the basic amino acid containing sequences into their groups: B, X, P, S or A (see ESI,† Table S1). For example, the sequence LLR is converted to XXB. The multiple parallel edges were again collapsed into one single edge with weight equal to the sum of the constituent original edges.
2.2 Computation
The analyses were performed using R 3.1.2 “Pumpkin Helmet”35 running on a MacBook Pro (2.66 GHz Intel Core i7, 8 Gb RAM). Levenshtein distances were determined using the LevenshteinDist function from the RecordLinkage package.36 Networks were created using the igraph package34 and similarity matrices were plotted using the lattice package.37 Parallel processing in R was implemented using the foreach38 and doParallel39 packages.3. Results
3.1 Network description
Heparin binding protein amino acid sequences were decomposed into sequences comprising only amino acids contained within given amino acids subsets. Seven different amino acid subsets were considered, the simplest being amino acid sequences containing basic and hydropathic amino acids ({B,X} sequences). The other basic amino acid containing sequences considered were {B,X,A}, {B,X,P}, {B,X,S}, {B,X,P,A}, {B,X,P,S} and {B,X,A,S} sequences. These basic amino acid containing sequences were then compared using graphs derived from Levenshtein distance metric similarity matrices. A graph is composed of edges and vertices, a vertex represents a basic amino acid sequence and the protein from which that sequence originates. Vertices are connected by edges and, if two vertices are connected by an edge, this signifies that the similarity criteria was met for those two vertices and then the weight of the edge connecting them is the similarity value. The purpose of this analysis was to identify conserved basic amino acid sequences within the HEPbps; the hypothesis being that these sequences may be characteristic for HEPbps and form the heparin binding regions of the proteins. Two parameters were used to guarantee that only conserved sequences were considered. The first was an imposed similarity cut-off, i.e., two vertices were not considered to be connected if the similarity between the vertices was below the similarity cut-off. Second, the core of the graph, the highly connected heart of the network, was selected by using the closeness centrality.3.2 Similarity cut-off
For this study the similarity cut-off for the conserved basic amino acid sequence graphs was set at 0.7. The effect of varying the similarity cut-off can be observed in ESI,† Table S2. As expected, by increasing the similarity cut-off for the networks the number of vertices, unique sequences and the number of edges decreased. This is also true for the graphs network density and the average degree of the vertices within the graphs, while the transitivity of the graphs increased with the raising of the similarity cut-off, i.e., there is an increased probability that adjacent vertices of a vertex are connected.3.3 Network core selection
Unlike the analysis of a family of highly related proteins, such as the FGF family shown earlier (Scheme 2), the networks produced from the 437 HEPbps contain many isolated vertices, which are detached from the core of the graph. These vertices belong to sequences that are not highly conserved. When community analysis, using a walktrap algorithm, was performed on these networks many communities were found, for example, the BX graph contained 566 communities, the BXP graph 1311 communities and the BXPS graph 821 communities (Table 1), with most of these communities having a low number of members and a low average degree. The walktrap algorithm used to detect communities in a network is based on a random walk; short random walks tend to stay in the same community. The number of steps used by the algorithm can be defined. In this case seven steps were used, minimising the number of communities found while maximising the modularity of the network.| BX | BXP | BXS | BXA | BXPS | BXPA | BXAS | |
|---|---|---|---|---|---|---|---|
| a Even though a vertex is identified by the parent protein and sequence when determining the number of unique sequences, only the peptide sequences were considered. | |||||||
| Whole network | |||||||
| No. of starting sequences | 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 447 447 |
15426 |
15740 |
13536 |
14124 |
14479 |
16663 |
| No. of vertices | 8987 | 8439 | 10905 |
9801 | 3281 | 4724 | 7500 |
| No. of unique sequencesa | 4652 | 6099 | 7281 | 6154 | 2743 | 3837 | 5798 |
| No. of edges | 121007 |
39002 |
82277 |
81265 |
4774 | 9771 | 24852 |
| Network density | 3 × 10−3 | 1.10 × 10−3 | 1.38 × 10−3 | 1.69 × 10−3 | 8.87 × 10−4 | 8.76 × 10−4 | 8.84 × 10−4 |
| Average degree | 26.93 | 9.24 | 15.09 | 16.58 | 2.91 | 4.14 | 6.63 |
| Clustering coefficient – transitivity | 0.491 | 0.443 | 0.453 | 0.464 | 0.498 | 0.451 | 0.436 |
| No. of communities | 566 | 1311 | 1288 | 897 | 821 | 931 | 1160 |
| Closeness selected network core | |||||||
| No. of starting sequences | 10447 |
15426 |
15740 |
13536 |
14124 |
14479 |
16663 |
| No. of vertices | 8624 | 6740 | 9718 | 8875 | 1566 | 3100 | 5631 |
| No. of unique sequencesa | 4348 | 4606 | 6265 | 5347 | 1315 | 2473 | 4191 |
| No. of edges | 120742 |
37800 |
81409 |
80598 |
3513 | 8655 | 23563 |
| Network density | 3.25 × 10−3 | 1.66 × 10−3 | 1.72 × 10−3 | 2.05 × 10−3 | 2.86 × 10−3 | 1.80 × 10−3 | 1.49 × 10−3 |
| Average degree | 28 | 11.22 | 16.75 | 18.16 | 4.49 | 5.58 | 8.37 |
| Clustering coefficient – transitivity | 0.491 | 0.443 | 0.452 | 0.464 | 0.483 | 0.446 | 0.435 |
| No. of communities | 412 | 585 | 780 | 503 | 174 | 270 | 363 |
To isolate the highly conserved cores of the networks, the closeness measure of vertex centrality was used. This measure finds vertices that can ‘communicate’ quickly with the other vertices in the graph.29 The closeness of a vertex is defined as the reciprocal of the sum of the distance from the vertex of interest to all of the others in the graph. The closeness values for the HEPbps conserved basic amino acid sequence networks were bivariate; vertices with a higher closeness value residing in the core of the graph (ESI,† Fig. S1). After the isolated vertices were removed from the graphs, the number of communities found decreased. The majority of the communities had a large population and high average degree; for example, HEPbp BX HBS graph had 412 communities, HEPbp BXP HBS network 585 communities and HEPbp BXPS HBS graph 174 communities. Further information can be found in ESI,† Table S2 and Table 1.
3.4 Amino acid types
Historically, investigations looking for heparin-binding sequences within proteins have concentrated on amino acid types, i.e., basic, hydrophobic, etc. For the initial survey of the graphs we adopted the same approach. The sequence that each vertex represents was converted into its amino acid type. For example, the sequence LLR was converted to XXB.To find important sequences, the ratio of the degree centrality, the number of vertices incident on a node – how many sequences overcome the similarity cut-off to the number of sequences, was considered (ESI,† Fig. S2). The majority of sequences within the graphs have a low degree to number ratio. This can be seen in ESI,† Fig. S2, in which the density plot of the degree to the number ratio illustrates that there are two populations. The population with the high degree to number ratio comprise shorter sequences, containing 3 or 4 amino acids. Sequences that contain special amino acids; C, G or P, contain significant sequences which are much longer, i.e., in the BXPS graph vertices belonging to the sequence PSSSSPSSSPSSSSSBS have a high degree to number ratio. All of these sequences can be found in ESI,† Table S3. The total number of amino acid sequences found the various HEPbp HBS networks expressed as their amino acid type can be found in ESI,† Tables S7, S9, S11, S13, S15, S17 and S19.
3.5 Network centralities
The four centrality measures (eigenvector, degree, closeness and betweenness) give different, but related, insights into important network properties. The most informative and granular measure is the eigenvector centrality, as this identifies individual vertices that are connected to other important – highly connected – vertices. In the case of the networks being studied here, these are conserved sequences that are linked to other important conserved sequences. Table 2 contains the vertices that are in the top 1% by eigenvector centrality. These important vertices comprise a small fraction of the total number of vertices that compose the graphs; {B,X} 86 of 8987, {B,X,A} 89 of 6154, {B,X,P} 67 of 6099, {B,X,S} 97 of 7281, {B,X,P,A} 30 of 9771, {B,X,P,S} 16 of 4774 and {B,X,A,S} 56 of 5798. The sequences highlighted in the analysis of the {B,X} and {B,X,A} networks contained the amino acids L and R, with the sequence LLR (XXB) appear 33 and 22 times in the {B,X} and {B,X,A} graphs, respectively (Table 2). The important vertices found in the {B,X,P} and {B,X,P,A} networks were associated with the conserved amino acid sequence SYR (SXB), while the {B,X,S}/{B,X,P,S} networks had significant vertices containing the conserved amino acid sequence G?KG (S?BS), where ? was present as: A (X, prevalent in the {B,X,P,S} network); T, K, L, F, P (P, B, X, X, S, prevalent in the {B,X,S} network), and M (X). Finally, the sequence YCR (XSB) was important in the {B,X,S,A} network. Important sequences and the proteins that contain them as determined by the degree, closeness and betweenness centralities can be found in ESI,† Tables S4, S5 and S6, respectively.| BX | BXA | BXP | BXS | BXPA | BXPS | BXAS | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Seq. | n | Prot. | Seq. | n | Prot. | Seq. | n | Prot. | Seq. | n | Prot. | Seq. | n | Prot. | Seq. | n | Prot. | Seq. | n | Prot. |
| LLR | 33 | 5NTD | LLR | 22 | AACT | SYR | 7 | A1BG | GPKG | 9 | A1BG | SYR | 5 | FA12 | GAKG | 5 | C1QA | YCR | 4 | FGF9 |
| LLRL | 5 | A1BG | LRL | 15 | ABCBB | SYRT | 1 | FA12 | GKG | 8 | A2MG | LSYR | 1 | FBLN7 | GTKG | 4 | CO1A1 | LYCR | 3 | FGF16 |
| LMLR | 3 | AACT | LRLV | 3 | ABP1 | LSYR | 1 | FBLN7 | GLKG | 7 | APLP1 | SSYR | 1 | FBN1 | AGAKG | 2 | CO1A2 | FGF20 | ||
| LRL | 3 | ABP1 | LMLR | 3 | APLP1 | SSYR | 1 | FBN1 | GAKG | 7 | APOB | ESYR | 1 | FBN2 | KGAKG | 2 | CO5A1 | HGF | ||
| HLLR | 2 | APOB | LLRL | 3 | APOB | KSYR | 1 | FBN2 | GPRG | 7 | ATS3 | SEYR | 1 | HGF | GKG | 2 | CO5A3 | PLMN | ||
| ALLR | 2 | ATRN | LRAL | 3 | ATS8 | SYNR | 1 | HGF | PGPKG | 7 | C1QA | SYNR | 1 | HMGB1 | GKKG | 1 | CO8A | TPA | ||
| KLLR | 2 | CBPD | HLLR | 2 | BACE1 | ASYR | 1 | IBP5 | PKG | 5 | CAC1S | ASYR | 1 | ITA1 | TGAKG | 1 | COBA1 | UROK | ||
| RLLR | 2 | CHRD | LLDR | 2 | CAC1S | QSYR | 1 | ITA1 | RGPKG | 3 | CCL28 | QSYR | 1 | LAMA5 | GNKG | 1 | COBA2 | |||
| LLRI | 2 | CO3 | LREL | 2 | CBPD | SWYR | 1 | LAMA5 | GRG | 3 | CO1A1 | SWYR | 1 | LTBP1 | GAKA | 1 | CODA1 | |||
| LALR | 2 | CO4A | ALLR | 2 | CHRD | ISYR | 1 | LTBP1 | GPK | 2 | CO1A2 | ISYR | 1 | PLMN | PGAKG | 1 | CO1 | |||
| LLLR | 2 | CO9 | LDLR | 2 | CO3 | SAYR | 1 | NRP1 | AGPKG | 2 | CO3 | SAYR | 1 | TIMP3 | GLKG | 1 | HMGB1 | |||
| LLRH | 1 | COBA2 | LLRE | 2 | CO4A | PLMN | GMKG | 2 | CO3A1 | TPA | GAKS | 1 | LAMA4 | |||||||
| FLLR | 1 | COCA1 | LRVL | 2 | CO5 | TIMP3 | VGPKG | 2 | CO5A1 | TSP4 | GFKG | 1 | MBL2 | |||||||
| LRLR | 1 | COIA1 | LRLL | 2 | CO6A3 | TPA | GPKA | 2 | CO5A3 | PGBM | ||||||||||
| LKLR | 1 | COJA1 | FLLR | 1 | COBA2 | TSP4 | LPKG | 1 | CO6A3 | Q9HCS8 | ||||||||||
| LLRK | 1 | COMP | LERL | 1 | COMT | GKKG | 1 | CO9A1 | TSP1 | |||||||||||
| LLRV | 1 | COMT | LELR | 1 | CXCL6 | KPKG | 1 | COBA1 | ||||||||||||
| LLRF | 1 | CO1 | LRLR | 1 | CYR61 | PGAKG | 1 | COBA2 | ||||||||||||
| LLRR | 1 | ENOA | LLRK | 1 | DCC | GGKG | 1 | COCA1 | ||||||||||||
| LLRY | 1 | ENPP3 | LRV | 1 | ECM2 | GARG | 1 | CODA1 | ||||||||||||
| LHLR | 1 | FGFP3 | LKRL | 1 | ERBB2 | GVKG | 1 | COEA1 | ||||||||||||
| LLYR | 1 | FGFR4 | LRFL | 1 | FA11 | KGAKG | 1 | COIA1 | ||||||||||||
| VLLR | 1 | FSTL1 | LRIL | 1 | FBN1 | GHKG | 1 | COJA1 | ||||||||||||
| LLFR | 1 | HBEGF | LARL | 1 | FGF4 | GPKGR | 1 | COLQ | ||||||||||||
| LLHR | 1 | HFE | LRLY | 1 | FGF18 | GIKG | 1 | CO1 | ||||||||||||
| YLLR | 1 | INSR | KLLR | 1 | FGFR4 | LGPKG | 1 | COPA1 | ||||||||||||
| ITIH3 | RLLR | 1 | FSTL1 | MGPKG | 1 | CRLD2 | ||||||||||||||
| LAMA1 | LRLA | 1 | HFE | KGPKG | 1 | ERBB2 | ||||||||||||||
| LAMA2 | DLLR | 1 | ITIH3 | GPKGL | 1 | FINC | ||||||||||||||
| LAMA3 | LLRF | 1 | KALM | GPKH | 1 | HMGB1 | ||||||||||||||
| LAMA5 | LLRR | 1 | LAMA1 | GPKC | 1 | IBP4 | ||||||||||||||
| LGR4 | LRKL | 1 | LAMA2 | GPPKG | 1 | LAMA2 | ||||||||||||||
| LIFR | LHLR | 1 | LAMA3 | GFKG | 1 | LAMA5 | ||||||||||||||
| LIPC | LRRL | 1 | LAMA5 | VPKG | 1 | MBL2 | ||||||||||||||
| LPHN2 | FLRL | 1 | LGR4 | MMP9 | ||||||||||||||||
| MET | LLYR | 1 | LIFR | PAIRB | ||||||||||||||||
| MOT8 | LALR | 1 | LPHN2 | PCSK5 | ||||||||||||||||
| MRP6 | LYRL | 1 | MET | PEBP1 | ||||||||||||||||
| V2 | LLHR | 1 | MOT8 | PGBM | ||||||||||||||||
| NOGG | YLLR | 1 | MRP6 | POSTN | ||||||||||||||||
| PCOC2 | LLRD | 1 | V2 | S12A9 | ||||||||||||||||
| PCSK5 | NOGG | TSP1 | ||||||||||||||||||
| PERM | PCOC2 | TSP2 | ||||||||||||||||||
| PGBM | PCSK5 | XDH | ||||||||||||||||||
| PGS1 | PGBM | |||||||||||||||||||
| PLGF | PGS1 | |||||||||||||||||||
| PRG2 | PIGR | |||||||||||||||||||
| S12A9 | PLGF | |||||||||||||||||||
| S22AI | PRDX4 | |||||||||||||||||||
| SCN5A | PRELP | |||||||||||||||||||
| SEM5B | PRG2 | |||||||||||||||||||
| SLIT1 | PSN1 | |||||||||||||||||||
| SLIT2 | RL29 | |||||||||||||||||||
| TEN1 | S12A9 | |||||||||||||||||||
| TE | S20A2 | |||||||||||||||||||
| TENX | S22AI | |||||||||||||||||||
| THYG | SCN5A | |||||||||||||||||||
| TRFE | SLIT1 | |||||||||||||||||||
| TRFL | SLIT2 | |||||||||||||||||||
| TSP3 | TE | |||||||||||||||||||
| TSP4 | TENX | |||||||||||||||||||
| VGFR1 | TGM2 | |||||||||||||||||||
| WNT1 | THYG | |||||||||||||||||||
| XDH | TRFL | |||||||||||||||||||
| ZPI | TSP2 | |||||||||||||||||||
| TSP3 | ||||||||||||||||||||
| VGFR1 | ||||||||||||||||||||
| XDH | ||||||||||||||||||||
3.6 Communities
Another means of describing a graph is by determining the number of communities/clusters that the graph contains. The number of communities found in the networks is a measure of the diversity of the sequences the graphs represent. The method used here to determine the number of communities was based on a random walk, the number of steps taken was chosen by analysing the {B,X} network and determining the modularity of the clustered networks. The number of steps that produced the lowest modularity, before the modularity of the analyses converged, was 7.The networks formed of sequences that contain 4 different types of amino acid ({B,X,P,S}, {B,X,P,A}, and {B,X,A,S}) contain the fewest communities, BXPS, 174; BXPA, 270 and BXAS, 363. The most diverse network is formed by sequences that comprise basic, hydrophobic and special amino acids (BXS). This has 780 communities. The conserved sequences for the most significant communities can be found in Table 3. The gauge of significance used was size. The vertices that form the communities represent amino acid sequences that are very similar to each other, therefore, the greater the number of vertices that comprise a community, the more important is the conserved sequence.
| BX | BXP | BXA | BXS | BXPS | BXPA | BXAS | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Com1 | ----AAK--- | Com1 | --RRR-- | Com1 | --FRY | Com1 | --VVK--- | Com5 | -G-PGPKG--- | Com2 | -RDS- | Com1 | ---GRR-- |
| ----XXB--- | --BBB-- | --XBX | --XXB--- | -S-SSSBS--- | -BAP- | ---SBB-- | |||||||
| Com3 | ---LLR---- | Com3 | --FRI | Com3 | ----KKV--- | Com2 | ----KLL----- | Com13 | --LGR- | Com7 | ---RS--- | Com2 | ---KPC- |
| ---XXB---- | --XBX | ----BBX--- | ----BXX----- | --XSB- | ---BP--- | ---BSS- | |||||||
| Com4 | -----VKK | Com4 | ---KKL--- | Com7 | ----LLR--- | Com3 | ---LR---- | Com16 | ---GKKG | Com8 | KNEE- | Com3 | ----LLR-- |
| -----XBB | ---BBX--- | ----XXB--- | ---XB---- | ---SBBS | BPAA- | ----XXB-- | |||||||
| Com5 | ----RLL--- | Com6 | ---AKK-- | Com8 | --ARR-- | Com4 | ------KVV-- | Com17 | KVL-- | Com9 | --SLR-- | Com4 | ----LKK- |
| ----BXX--- | ---XBB-- | --XBB-- | ------BXX-- | BXX-- | --PXB-- | ----XBB- | |||||||
| Com6 | ----RA----- | Com11 | --RAA-- | Com9 | ---LKK--- | Com13 | --KII-- | Com18 | ---KLK- | Com10 | --RVS-- | Com5 | --KKI-- |
| ----BX----- | --BXX-- | ---XBB--- | --BXX-- | ---BXB- | --BXP-- | --BBX-- | |||||||
| Com8 | -FFH-- | Com12 | --VLK | Com12 | ----KKK--- | Com14 | -HPP-- | Com27 | -LLRL- | Com11 | --SKK | Com7 | ----VLK- |
| -XXB-- | --XXB | ----BBB--- | -BSS-- | -XXBX- | --PBB | ----XXB- | |||||||
| Com10 | --LHL-- | Com17 | --LVK-- | Com13 | ---RRR--- | Com15 | -PPR---- | Com30 | CIFK | Com13 | --AVK- | Com8 | --IR--- |
| --XBX-- | --XXB-- | ---BBB--- | -SSB---- | SXXB | --XXB- | --XB--- | |||||||
| Com11 | ------K--LL-- | Com21 | ---LR--- | Com16 | ---RLL---- | Com16 | --RVR-- | Com49 | -GRS- | Com21 | KKKS- | Com19 | --GGH---- |
| ------B--XX-- | ---XB--- | ---BXX---- | --BXB-- | -SBP- | BBBP- | --SSB---- | |||||||
| Com13 | --KKKK--- | Com25 | --RLL---- | Com18 | ----HFL- | Com20 | ---RRIP-- | Com60 | GRCC- | Com26 | SKL-- | Com21 | ------KKK-- |
| --BBBB--- | --BXX---- | ----BXX- | ---BBXS-- | SBSS- | PBX-- | ------BBB-- | |||||||
| Com18 | ---LLK-- | Com32 | --QQR- | Com20 | --HAA-- | Com21 | -CVR-- | Com30 | KAL-- | Com22 | -RAA-- | ||
| ---XXB-- | --PPB- | --BXX-- | -SXB-- | BXX-- | -BXX-- | ||||||||
| Com25 | ----HHL-- | Com33 | -LHV-- | Com25 | --HLA-- | Com22 | --RAA--- | Com37 | SRR-- | Com27 | --GGK- | ||
| ----BBX-- | -XBX-- | --BXX-- | --BXX--- | PBB-- | --SSB- | ||||||||
| Com26 | -KKR-- | Com34 | -QRVV-- | Com29 | --EIH- | Com24 | --LAH- | Com43 | --KKL | Com29 | --RLP-- | ||
| -BBB-- | -PBXX-- | --AXB- | --XXB- | --BBX | --BXS-- | ||||||||
| Com29 | --KVV-- | Com37 | ---KKK-- | Com32 | --LH--F | Com25 | ----AAR- | Com47 | ---RL-- | Com45 | --LLH- | ||
| --BXX-- | ---BBB-- | --XB--X | ----XXB- | ---BX-- | --XXB- | ||||||||
| Com32 | ---VVR--- | Com39 | --KKF | Com42 | --KEI | Com26 | ------L--KK----- | Com49 | -NKK- | Com46 | --DGK- | ||
| ---XXB--- | --BBX | --BAX | ------X--BB----- | -PBB- | --ASB- | ||||||||
| Com35 | ----LLH-- | Com43 | --RSS-- | Com48 | -LHD-- | Com31 | --RRRR- | Com48 | --VVR-- | ||||
| ----XXB-- | --BPP-- | -XBA-- | --BBBB- | --XXB-- | |||||||||
| Com37 | -IIR--- | Com46 | --KKK-- | Com49 | --RAA-- | Com32 | -CKGC | Com49 | ---RLL-- | ||||
| -XXB--- | --BBB-- | --BXX-- | -SBSS | ---BXX-- | |||||||||
| Com40 | --HAA--- | Com48 | --ASK | Com57 | -EER-- | Com34 | ---RGG-- | Com70 | --PRA-- | ||||
| --BXX--- | --XPB | -AAB-- | ---BSS-- | --SBX-- | |||||||||
| Com42 | IHH-- | Com58 | -AAR- | Com58 | -FFK-- | Com35 | --KKKA--- | Com80 | LHLL- | ||||
| XBB-- | -XXB- | -XXB-- | --BBBX--- | XBXX- | |||||||||
| Com74 | --KII-- | Com64 | --HHL | Com59 | --HVL-- | Com38 | --GHH-- | ||||||
| --BXX-- | --BBX | --BXX-- | --SBB-- | ||||||||||
| Com98 | -HLA-- | Com66 | -KSQ- | Com64 | --RRV-- | Com43 | --HLG-- | ||||||
| -BXX-- | -BPP- | --BBX-- | --BXS-- | ||||||||||
| Com69 | -KSS-- | Com75 | --LLH- | Com46 | ----GPPGPKG--- | ||||||||
| -BPP-- | --XXB- | ----SSSSSBS--- | |||||||||||
| Com80 | --SLH-- | Com83 | -FHI- | Com47 | --LLR-- | ||||||||
| --PXB-- | -XBX- | --XXB-- | |||||||||||
| Com83 | -VVKS | Com101 | -IHL-- | Com52 | --GRC--- | ||||||||
| -XXBP | -XBX-- | --SBS--- | |||||||||||
| Com98 | --KIT-- | Com120 | -H--VV | Com54 | --LLH--- | ||||||||
| --BXP-- | -B--XX | --XXB--- | |||||||||||
| Com99 | --RNT- | Com326 | -AAH- | Com56 | --GLH- | ||||||||
| --BPP- | -XXB- | --SXB- | |||||||||||
| Com100 | -KVT-- | Com60 | ---RLL---- | ||||||||||
| -BXP-- | ---BXX---- | ||||||||||||
| Com113 | -IYKT- | Com62 | --LHL- | ||||||||||
| -XXBP- | --XBX- | ||||||||||||
| Com120 | --VRT- | Com63 | --CRK-- | ||||||||||
| --XBP- | --SBB-- | ||||||||||||
| Com125 | -TARK | Com69 | --VVR- | ||||||||||
| -PXBB | --XXB- | ||||||||||||
| Com161 | --KVN- | Com76 | -----GPK---G---- | ||||||||||
| --BXP- | -----SSB---S---- | ||||||||||||
| Com96 | --KIG- | ||||||||||||
| --BXS- | |||||||||||||
| Com97 | --RHGY | ||||||||||||
| --BBSX | |||||||||||||
| Com101 | -IKK- | ||||||||||||
| -XBB- | |||||||||||||
| Com110 | -RGLPG--- | ||||||||||||
| -BSXSS--- | |||||||||||||
| Com114 | --GKK---- | ||||||||||||
| --SBB---- | |||||||||||||
| Com122 | --KGP-- | ||||||||||||
| --BSS-- | |||||||||||||
| Com132 | --FHL-- | ||||||||||||
| --XBX-- | |||||||||||||
| Com153 | -RLA-- | ||||||||||||
| -BXX-- | |||||||||||||
| Com179 | --PC-K | ||||||||||||
| --SS-B |
The significance cut-off was the 95th percentile. The distribution of community sizes had a positively skewed distribution, the number of significant communities found for the different graphs were, {B,X}, 21 of 421; {B,X,A}, 25 of 203; {B,X,P}, 30 of 585; {B,X,S}, 39 of 280; {B,X,P,S}, 9 of 174; {B,X,P,A}, 14 of 270 and {B,X,A,S}, 18 of 363. It is interesting to note that the core of the conserved sequences from the most significant communities are relatively short, three or four amino acids long, as seen in the eigenvector analyses, corresponding to small discrete areas on a protein surface. Tables can be found in ESI† that contain the conserved sequences, amino acid entropy and amino acid frequency for the significant communities found in networks formed from the {B,X}, {B,X,A}, {B,X,P}, {B,X,S}, {B,X,P,A}, {B,X,P,S} and {B,X,A,S} amino acid sets, ESI,† Tables S8, S10, S12, S14, S16, S18 and S20, respectively.
3.7 Conserved sequences in proteins
In order to validate this approach for identifying HBSs within HEPbps, the sequences extracted for a small set of proteins were compared against their experimentally determined HBSs. Molecular schematics and tables of the predicted HBSs can be found in ESI† (Fig. S3–S5 and Tables S21–S26). This approach has previously been used to identify HS/heparin binding sequences in H5N1 haemagglutinin (influenza A virus A/Cygnus olor/Italy/742/2006).40Fibroblast growth factors (FGFs) are a well-studied family of HEPbps. They are a group of 21 proteins that bind to HSPGs and FGF receptors (FGFRs) containing membrane-bound receptor tyrosine kinase. The HS binding of the family has been investigated using a mass spectroscopy “Protect and Label” strategy.41 The approach has been used to determine the HBS for FGF-1, -2, -3, -4, -6, -7, -9, -10, -17, -18 and -20.41–43 The principal example shown in the text is for FGF-1, colloquially termed acidic FGF. The network analysis method described here identifies sequences within this protein that are highly similar to sequences found in other proteins known to bind heparin/HS, see Fig. 1. The molecular representation of FGF-1 (Fig. 1), shows these conserved basic amino acid sequences creating an extended region around the protein (Fig. 1, lower network). Highlighted in this network are the amino acids (grey vertices) that are within 0.8 nm of the conserved amino acids and that arise in at least two of the seven sets: {B,X}; {B,X,A}; {B,X,P}; {B,X,S}; {B,X,P,A}; {B,X,P,S}; and {B,X,A,S}.
 | ||
| Fig. 1 Conserved basic amino acid containing sequences found in FGF-1. The table shows the sequences found for the different amino acid combinations. As a further selection criterion, an amino acid was only considered to be significant if it arose at least twice in the different amino acid groups, e.g., R50 appears in the BXA, BXPS and BXAS selections. The conserved amino acids are illustrated on the molecular structure of FGF-1 (1RG8).47 This structure was also represented as a network, the vertices of the network are the αC positions. In the bottom network, conserved basic amino acid containing residues are shown, along with any amino acid that is less than 0.8 nm away – the approximate length of a HS/heparin disaccharide. This reductionist view illustrates how the small basic amino acid containing sequences in unison can form an extended heparin-binding domain. The previously identified HBSs of FGF-1 can be found in Table 4. | ||
The approximate length of the heparin/HS disaccharide is 0.8 nm,7 and therefore a longer chain may lie across multiple connected vertices. These connected vertices would then form an extended heparin/HS binding domain. The ‘Protect and Label’ mass spectrometry performed on FGF-1 identified four heparin binding regions: KKPKLLY (amino acids (aa) 24–30); IKSTETGQYL (aa71–80); ISKKHAEKNWF (aa113–123); and VGLKKNGSCKRGPRTHYGQAILFLPL (aa124–150).42 The analysis described above identified amino acid sequences within each of the previously identified regions in FGF-1 that interact with HS/heparin (Fig. 1).
The network analysis was also validated against the FGF-2, FGF-7, FGF-9 and FGF-18 proteins. The conserved basic amino acid sequences of these proteins are shown in Fig. 2 with the ‘protect and label’ mass spectroscopy hits (Table 4).41,42 Furthermore, validation against FGF-3, -4, -6, -10, -17 and -20 are in ESI.†
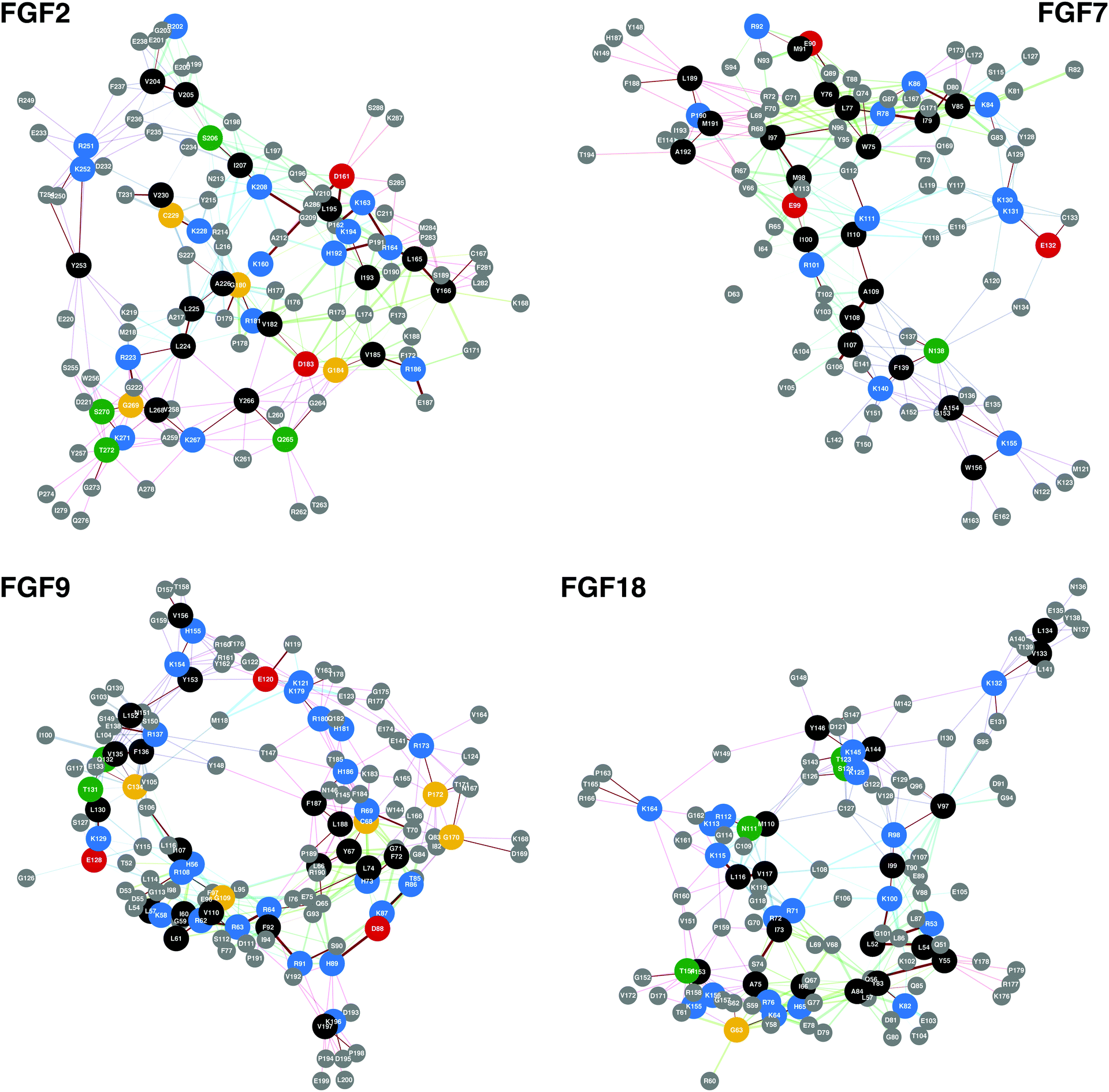 | ||
| Fig. 2 Conserved basic amino acid containing sequences found in members of the FGF family. In these networks conserved basic amino acid containing residues are shown along with any amino acid that is less than 0.8 nm away, the approximate length of a HS/heparin disaccharide. The previously identified HBSs, FGF-2, -7, -9 and -18 can be found in Table 4. The PDB files used to produce the networks were, FGF-2, 1BFF;53 FGF-7, 1QQK;54 FGF-9, 1IHK55 and FGF-18, 4CJM.56 | ||
| Amino acid sequence | Start | End | Ref. | |
|---|---|---|---|---|
| aa | aa | |||
| FGF-1 | KKPKLLY | 24 | 30 | Xu et al. protect and label ms42 |
| IKSTETGQYL | 71 | 80 | ||
| ISKKHAEKNWF | 113 | 123 | ||
| VGLKKNGSCKRGPRTHYGQAILFLPL | 124 | 150 | ||
| FGF-2 | KDPKRLYCKNGGFF | 160 | 173 | Ori et al. protect and label ms41 |
| LAMKEDGRLL | 216 | 225 | ||
| VALKRTGQY | 258 | 266 | ||
| KLGSKTGPGQKAIL | 267 | 280 | ||
| FGF-7 | YLRIDKRGKVKGTQEMKNNY | 76 | 95 | Xu et al. protect and label ms42 |
| LAMNKEGKLY | 119 | 128 | ||
| ASAKWTHNGGEMF | 152 | 164 | ||
| VALNQKGIPVRGKKTKKEQKTAHF | 165 | 188 | ||
| FGF-9 | HLEIFPNGTIQGTRKDHSRF | 73 | 92 | Xu et al. protect and label ms42 |
| KHVDTGRRY | 154 | 175 | ||
| VALNKDGTPREGTRTKRHQKF | 164 | 184 | ||
| THFLPRPVDPDKVPELY | 185 | 201 | ||
| FGF-18 | RIHVENQTRARDDVSRKQL | 34 | 52 | Xu et al. protect and label ms42 |
| GRRISARGEDGDKY | 70 | 83 | ||
| GSQVRIKGKETEFYL | 94 | 108 | ||
| CMNRKGKLVGKPDGTSKECVF | 109 | 129 | ||
| TKKGRPRKGPKTRENQQDVHFM | 154 | 175 | ||
| MKRYPKGQPELQKPF | 175 | 189 | ||
The above analysis indicates that the conserved amino acid containing sequences that are found in HEPbps form a significant part of the heparin binding regions of a protein. Further illustrations of this fact include, hepatoma-derived growth factor (HDGF), lymphotactin (chemokine (C motif) ligand (XCL1)) and interleukin-10 (IL10). Solution NMR analysis of hepatoma-derived growth factor indicated that it had a primary heparin binding site and then possibly a minor binding site at the N-terminal of the protein. The primary HBS consists of K 19, 61, 72, 78 and 80, as well as R 79. The secondary site, which resides in the flexible N-terminus of the molecule is formed of R2 and R6, and K8 and K11.44 The similarity analysis found all the members of the proposed principal binding site apart from K19. In fact, this amino acid was found by the analysis, but it only appeared once, in the BX group of amino acids. Of the minor binding site, only K11 was found to be significantly conserved, while K8 appeared once in the BXA amino acid group analysis. The network representation of HDGFs HBS highlights how the conserved basic amino acid containing sequences could come together to form the principal HBS, with the conserved sequence 28ARI30 linking the primary and secondary HBSs together (ESI,† Fig. S3).
Another example is lymphotactin, a small cytokine. Petersen et al.45 used backbone 1H and 15N chemical shift perturbations to identify the following amino acids as interacting with heparin, R39, R44, K46, K63, R64, K67, R78, R86, K87, and R91. Further use of site-directed mutagenesis identified R44 and R64 as the high affinity residues. All but three of these amino acids were identified by the similarity method employed in this manuscript, and these were K63, R64 and R70. The method was able to identify one of the high affinity binding residues and 70 percent of the total interacting residues (ESI,† Fig. S4).
The final example shown is interleukin-10 (IL-10), which is a cytokine involved in inflammation. It inhibits the production of inflammatory cytokines.46 It has been determined by NMR that IL-10 interacts with heparin via a binding site that comprises resides in helix D and the adjacent DE loop.46 The residues involved in the interaction are R120, R121, R124, R125, K135 and K137. The analyses shown in this manuscript identify all of these residues except K135. In particular, the analyses identify a domain comprising 8 basic amino acids, R42, R120, R122, R124, R125, R128, R127, H32 and H127 (ESI,† Fig. S5).
It should be noted that in the examples shown here, the FGFs, HDGF, XCL1 and IL-10, that the proteins contain conserved basic amino acid containing sequences that correspond to the experimentally determined HBSs, but there are also other conserved basic amino acid containing sequences that are found in these bona fide HEPbps. When heparin/HS binding studies are performed on these proteins the system may be in solution, for the case of NMR and MS studies, but this is still not the natural state of the system. Most of the proteins considered in this study are extracellular, either membrane bound or secreted in to the ECM. This environment is extremely crowded, being composed of many proteins and carbohydrates, of which proteoglycans are an important part. These additional conserved basic amino acid containing sequences found in the HEPbps may be related to the interaction of the HEPbp and its surrounds, for e.g., storage of the HEPbps in the ECM or control of HEPbps diffusion through the ECM, suggesting that there are primary and secondary HBS within HEPbps. In addition to other functions such stabilising the structure of the protein. The primary sites are related to a specific biological activity, i.e., the HBS related to a protein cell signal activity, while the secondary sites assist in the control and movement of the proteins though its environment. It is conceivable that a very large number of proteins interact with heparin/HS but, obviously, not all of them require heparin/HS for their biological activity.
3.8 Human proteome
The result of searching the human proteome for the conserved basic amino acid containing sequences found in the 437 HEPbp is interesting. From this analysis, two main pieces of information can be ascertained. The first, is the number of times a conserved basic amino acid containing sequence arose in the members of the human proteome. Unlike the earlier analysis performed, where discrete sequences were found in a set of HEPbp, this analysis searched for the sequences found in the earlier analysis in the entire human proteome. As a consequence of this, overlapping sequences will be found in proteins. For example, the sequence ARLLAR could have the sequences ARL, RLL, LLAR and LAR as hits. The second piece of information is the frequency with which a specific basic amino acid containing sequence appears in the human proteome.The median values for the number of different basic amino acid containing sequence types found per protein in the human proteome were: 42 BX, 61 BXA, 69 BXP, 64 BXS, 67 BXPA, 32 BXPS and 70 BXAS. With the 99th percentile values being: 213 BX, 333 BXA, 398 BXP, 316 BXS, 311 BXPA, 156 BXPS and 347 BXAS. The unique list, a combination of all proteins with more than or equal to the 99th percentile for the different amino acid combinations, contained 329 proteins, which can be found in ESI,† Table S27. Of these 329 proteins, 17 are found in the 437 HEPbps that were originally analysed, they are: APOB, apolipoprotein B-100; ATS9, a disintegrin and metalloproteinase with thrombospondin motifs 9; CAC1S, voltage-dependent L-type calcium channel subunit alpha-1S; CO6A3, collagen alpha-3 (VI) chain; COCA1, collagen alpha-1 (XII) chain; FBN1, fibrillin-1; LAMA1, laminin subunit alpha-1; LAMA2, laminin subunit alpha-2; LAMA3, laminin subunit alpha-3; LAMA5, laminin subunit alpha-3; NAV2, neuron navigator 2; PGBM, basement membrane-specific heparan sulfate proteoglycan core protein; STAB2, stabilin-2; TEN1, teneurin-1; TENX, tenascin-X; THYG, thyroglobulin and VWF, von Willebrand factor. Many of these are integral components of the extracellular matrix. For example, STAB2 is a large transmembrane receptor that acts as a scavenger for heparin and other GAGs, which may assist in maintaining tissue integrity by supporting extracellular matrix turnover. If the 437 bona fide HEPbp are considered as a whole, they have a higher median number of basic amino acid containing sequences than the whole human proteome, apart from sequences comprised of BXPA amino acids and the 99th percentiles values are all higher: median – 45 BX, 66 BXA, 75 BXP, 74 BXS, 58 BXPA, 34 BXPS and 77 BXAS and 99th percentile – 240 BX, 352 BXA, 429 BXP, 445 BXS, 327 BXPA, 191 BXPS and 462 BXAS. If one makes the selection criteria a little milder, the 95th percentile, then that pushes the number of proteins up to 1518, which is approaching ∼14% of the human proteome. This suggests that many proteins found in humans possibly interact with HS/heparin. This is not an absolute measure of heparin binding, as the analysis finds overlapping sequences. It does though provide a measure of the propensity of a protein to interact with heparin/HS.
These data support the conjecture that if many proteins can bind to these polyanions, then the mechanism of control may not lie at the level of the protein, but in the sequences found in the polysaccharide chains. This would go some way to explaining why so much energy has been committed to produce the many HS/heparin biosynthetic enzymes (4 enzymes for chain initiation, 2 enzymes for chain extension and 16 enzymes for chain modification – a total of 22 enzymes for a single polysaccharide chain).
The significant conserved basic amino acid containing sequences all appear more than 2000 times (99th percentile, 2857 BX, 3271 BXP, 2826 BXS, 3083 BXA, 3816 BXPA, 3818 BXPS and 3361 BXAS) in the human proteome. The median value was considerably lower than that (median – 42 BX, 69 BXP, 64 BXS, 61 BXA, 67 BXPA, 32 BXPS and 70 BXAS). All the significant sequences were tri-peptides. The sequences on the whole contained either arginine or lysine, with only two histidine-containing sequences found in the significant populations, HLL and LLH. The unique list, a combination of all sequences with more than or equal to the 99th percentile for the different amino acid combinations, contained 98 sequences, as follows; ARR, KLA, LAK, LLR, LKL, KAA, RLA, KLK, RRA, ARL, KKL, RAA, RVL, KRK, LRR, ALK, LRA, RAL, RKL, KVL, ALR, LRK, LKA, RLL, LKK, KAL, KLL, RLR, AAR, RKK, ARA, VLK, LLK, KKK, RRL, LRL, LAR, LKR, KRL, VLR, HLL, LRV, RRR, EKK, EER, ELK, KEE, KLE, KEL, EKE, LEK, EEK, ELR, LKE, ERL, REE, LER, LRE, EKL, RLE, KEK, LKD, REL, RSS, LQR, LRS, SSR, RLS, LSR, SRL, KSL, SLR, LKS, RSL, LQK, LSK, SLK, SRS, RLG, GRR, LRG, GLR, GRG, RLP, RLK, PRP, LRP, RGL, PRL, LPR, GRL, PPR, KLR, GKL, LVK, LLH, LGK and LGR.
4. Conclusions
These analyses indicate that basic regions, and therefore heparin binding sites within HEPbps, are highly variable, containing only small conserved motifs at the heart of the HBS. It is likely that many of these small basic sequences work in unison via multiple heparin binding sites on a protein surface. This implies that there is agility and leeway in the composition of the complementary protein binding surface, comparable to the latitude observed in binding sequences of HS. These data preclude the notion of there being a single, universal HBS in the family of HEPbps, since many amino acid sequence combinations are able to fulfil the same role.Considering basic amino acid sequences found within HEPbps is a first step to understanding the biochemistry of these interactions. There are other facets to the interaction between heparin/HS and their binding proteins that are likely to have influence, including post-translational modifications, GAG heterogeneity, cationic association and the possibility that, in some cases, HEPbps may be active independent of the presence of heparin/HS.
Differing post-translation modification has been shown to regulate the interaction between the protein and carbohydrate. For example, glycosylation of the protein ligand in FGFR-1 alters the affinity of the interaction.48 Not all GAGs are equivalent however. Both heparin and HS are heterogeneous polysaccharides with their disaccharide sequences dependent on the organ from which they originate.49 As polyanionic polysaccharides, both heparin and HS are associated with different cations that modify their conformation.50 For example, it has been shown that a biologically inactive carbohydrate is activated by the addition of the appropriate cation.51 In some cases (e.g., FGF-1 and -2) heparin/HS dependant signalling pathways have been stimulated by non-GAG materials, including sulfated plant polysaccharides. For these FGFs, such proxy-GAG carbohydrates only need to either thermally stabilise or induce the correct conformational change in the HEPbp for signalling to be maintained.52
It is difficult to rationalise an explicit control mechanism for systems regulated by protein and HS/heparin interactions. The innate elasticity of the HBSs within HEPbps, coupled to the heterogeneity found in heparin and HS precludes this. Instead of focusing on the interaction between a single protein and HS/heparin to understand biological processes, these analyses may indicate that a holistic view, taken over all the molecular interactions may be more appropriate. Specifically, they indicate that HEPbps interact with HS/heparin in a multitude of ways, and in complex networks, which enables them both to perform many tasks and for these capabilities to be both interdependent in complex ways but, also backed-up by robust systems. The network analyses above utilise a multi-dimensional technique to interrogate this multi-faceted interactome.
These multi-dimensional network analyses of HEPbp sequences have identified HBSs on a family-wide scale. They have indicated that HBSs may be composed of multiple, small, independent basic amino acid stretches that work in unison to form the HBS regions. A single universal HBS is therefore unlikely; rather many arrangements of amino acids may fulfil the same task. These observations lead to two logical inferences: that HEPbps possess an agility in their heparin/HS interactions; and that there may be a higher degree of convergent evolution in HBPs than previously thought. These analyses provide both an insight and springboard into the HEPbp, heparin and HS interactomes, as well as a validated technique for investigating protein sequences at a phenotypic level.
Acknowledgements
This article is dedicated to Prof. Benito Casu (1926-2016); a charming and intelligent person.Notes and references
- D. L. Rabenstein, Nat. Prod. Rep., 2002, 19, 312–331 RSC.
- S. Sarrazin, W. C. Lamanna and J. D. Esko, Cold Spring Harbor Perspect. Biol., 2011, 3, 1–33 Search PubMed.
- L. Schaefer and R. M. Schaefer, Cell Tissue Res., 2010, 339, 237–246 CrossRef CAS PubMed.
- I. Kovalszky, A. Hjerpe and K. Dobra, Biochim. Biophys. Acta, 2014, 1840, 2491–2497 CrossRef CAS PubMed.
- M. D. Stewart and R. D. Sanderson, Matrix Biol., 2014, 35, 56–59 CrossRef CAS PubMed.
- P. Carlsson and L. Kjellén, Handb. Exp. Pharmacol., 2012, 207, 23–41 CAS.
- G. Venkataraman and R. Sasisekharan, Curr. Opin. Chem. Biol., 2000, 4, 626–631 CrossRef.
- T. R. Rudd and E. a Yates, Mol. Biosyst., 2012, 8, 1499–1506 RSC.
- J. T. Gallagher, J. E. Turnbull and M. Lyon, Int. J. Biochem., 1992, 24, 553–560 CrossRef CAS PubMed.
- J. E. Turnbull and J. T. Gallagher, Biochem. J., 1991, 553–559 CrossRef CAS.
- M. C. Z. Meneghetti, A. J. Hughes, T. R. Rudd, H. B. Nader, A. K. Powell, E. a. Yates and M. a. Lima, J. R. Soc., Interface, 2015, 12, 20150589 CrossRef PubMed.
- A. Ori, M. C. Wilkinson and D. G. Fernig, J. Biol. Chem., 2011, 286, 19892–19904 CrossRef CAS PubMed.
- J. Gallagher, Int. J. Exp. Pathol., 2015, 96, 203–231 CrossRef CAS PubMed.
- X. Lin, Development, 2004, 131, 6009–6021 CrossRef CAS PubMed.
- H. Cui, C. Freeman, G. A. Jacobson and D. H. Small, IUBMB Life, 2013, 65, 108–120 CrossRef CAS PubMed.
- E. H. Knelson, J. C. Nee and G. C. Blobe, Trends Biochem. Sci., 2014, 39, 277–288 CrossRef CAS PubMed.
- V. Tiwari, E. Maus, I. M. Sigar, K. H. Ramsey and D. Shukla, Glycobiology, 2012, 22, 1402–1412 CrossRef CAS PubMed.
- Q. M. Nunes, V. Mournetas, B. Lane, R. Sutton, D. G. Fernig and O. Vasieva, Pancreatology, 2013, 13, 598–604 CrossRef CAS PubMed.
- Y. Chen, M. Scully, G. Dawson, C. Goodwin, M. Xia, X. Lu and A. Kakkar, Thromb. Haemostasis, 2013, 109, 1148–1157 CrossRef CAS PubMed.
- G. Manning, S. L. Young, W. T. Miller and Y. Zhai, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 9674–9679 CrossRef CAS PubMed.
- S. Bertrand, T. Iwema and H. Escriva, Mol. Biol. Evol., 2014, 31, 310–318 CrossRef CAS PubMed.
- F. Williams, H. A. Tew, C. E. Paul and J. C. Adams, Matrix Biol., 2014, 37, 60–68 CrossRef CAS PubMed.
- R. E. Hileman, J. R. Fromm, J. M. Weiler and R. J. Linhardt, BioEssays, 1998, 20, 156–167 CrossRef CAS PubMed.
- J. R. Fromm, R. E. Hileman, E. E. O. Caldwell, J. M. Weiler and R. J. Linhardt, Arch. Biochem. Biophys., 1995, 323, 279–287 CrossRef CAS PubMed.
- J. R. Fromm, R. E. Hileman, E. E. Caldwell, J. M. Weiler and R. J. Linhardt, Arch. Biochem. Biophys., 1997, 343, 92–100 CrossRef CAS PubMed.
- A. D. Cardin and H. J. Weintraub, Arterioscler., Thromb., Vasc. Biol., 1989, 9, 21–32 CrossRef CAS.
- M. Sobel, D. F. Soler, J. C. Kermode and R. B. Harris, J. Biol. Chem., 1992, 267, 8857–8862 CAS.
- M. Torrent, M. V. Nogués, D. Andreu and E. Boix, PLoS One, 2012, 7, e42692 CAS.
- G. A. Pavlopoulos, M. Secrier, C. N. Moschopoulos, T. G. Soldatos, S. Kossida, J. Aerts, R. Schneider and P. G. Bagos, BioData Min., 2011, 4, 10 CrossRef PubMed.
- V. I. Levenshtein, Soviet Physics - Doklady, 1966, 10, 707–710 Search PubMed.
- M. Veeramalai and D. Gilbert, Bioinformatics, 2008, 24, 2698–2705 CrossRef CAS PubMed.
- L. Tan, J. Batista and J. Bajorath, Chem. Biol. Drug Des., 2010, 76, 191–200 CAS.
- A. Bateman, M. J. Martin, C. O’Donovan, M. Magrane, R. Apweiler, E. Alpi, R. Antunes, J. Arganiska, B. Bely, M. Bingley, C. Bonilla, R. Britto, B. Bursteinas, G. Chavali, E. Cibrian-Uhalte, A. Da Silva, M. De Giorgi, T. Dogan, F. Fazzini, P. Gane, L. G. Castro, P. Garmiri, E. Hatton-Ellis, R. Hieta, R. Huntley, D. Legge, W. Liu, J. Luo, A. Macdougall, P. Mutowo, A. Nightingale, S. Orchard, K. Pichler, D. Poggioli, S. Pundir, L. Pureza, G. Qi, S. Rosanoff, R. Saidi, T. Sawford, A. Shypitsyna, E. Turner, V. Volynkin, T. Wardell, X. Watkins, H. Zellner, A. Cowley, L. Figueira, W. Li, H. McWilliam, R. Lopez, I. Xenarios, L. Bougueleret, A. Bridge, S. Poux, N. Redaschi, L. Aimo, G. Argoud-Puy, A. Auchincloss, K. Axelsen, P. Bansal, D. Baratin, M. C. Blatter, B. Boeckmann, J. Bolleman, E. Boutet, L. Breuza, C. Casal-Casas, E. De Castro, E. Coudert, B. Cuche, M. Doche, D. Dornevil, S. Duvaud, A. Estreicher, L. Famiglietti, M. Feuermann, E. Gasteiger, S. Gehant, V. Gerritsen, A. Gos, N. Gruaz-Gumowski, U. Hinz, C. Hulo, F. Jungo, G. Keller, V. Lara, P. Lemercier, D. Lieberherr, T. Lombardot, X. Martin, P. Masson, A. Morgat, T. Neto, N. Nouspikel, S. Paesano, I. Pedruzzi, S. Pilbout, M. Pozzato, M. Pruess, C. Rivoire, B. Roechert, M. Schneider, C. Sigrist, K. Sonesson, S. Staehli, A. Stutz, S. Sundaram, M. Tognolli, L. Verbregue, A. L. Veuthey, C. H. Wu, C. N. Arighi, L. Arminski, C. Chen, Y. Chen, J. S. Garavelli, H. Huang, K. Laiho, P. McGarvey, D. A. Natale, B. E. Suzek, C. R. Vinayaka, Q. Wang, Y. Wang, L. S. Yeh, M. S. Yerramalla and J. Zhang, Nucleic Acids Res., 2015, 43, D204–D212 CrossRef PubMed.
- G. Csárdi and T. Nepusz, InterJournal Complex Syst., 2006, 1695, 1695 Search PubMed.
- R Development Core Team, 2016.
- M. Sariyar and A. Borg, R J., 2010, 2, 61–67 Search PubMed.
- D. Sarkar, Lattice, Springer Science + Business Media, 2008 Search PubMed.
- Revolution Analytic and S. Weston, 2015.
- R. Calaway, Revolution Analytics and S. Weston, 2015, 1–4.
- M. A. Skidmore, A. Kajaste-Rudnitski, N. M. Wells, S. E. Guimond, T. R. Rudd, E. A. Yates and E. Vicenzi, Med. Chem. Commun., 2015, 6, 640–646 RSC.
- A. Ori, P. Free, J. Courty, M. C. Wilkinson and D. G. Fernig, Mol. Cell. Proteomics, 2009, 8, 2256–2265 CAS.
- R. Xu, A. Ori, T. R. Rudd, K. A. Uniewicz, Y. A. Ahmed, S. E. Guimond, M. A. Skidmore, G. Siligardi, E. A. Yates and D. G. Fernig, J. Biol. Chem., 2012, 287, 40061–40073 CrossRef CAS PubMed.
- Y. Li, C. Sun, E. A. Yates, C. Jiang, M. C. Wilkinson and D. G. Fernig, Open Biol., 2016, 6, 150275 CrossRef PubMed.
- S. C. Sue, J. Y. Chen, S. C. Lee, W. G. Wu and T. H. Huang, J. Mol. Biol., 2004, 343, 1365–1377 CrossRef CAS PubMed.
- F. C. Peterson, E. S. Elgin, T. J. Nelson, F. Zhang, T. J. Hoeger, R. J. Linhardt and B. F. Volkman, J. Biol. Chem., 2004, 279, 12598–12604 CrossRef CAS PubMed.
- G. Kunze, S. Köhling, A. Vogel, J. Rademann, D. Huster, S. Ko, A. Vogel and D. Huster, J. Biol. Chem., 2016, 291, 3100–3113 CrossRef PubMed.
- M. J. Bernett, T. Somasundaram and M. Blaber, Proteins, 2004, 57, 626–634 CrossRef CAS PubMed.
- L. Duchesne, B. Tissot, T. R. Rudd, A. Dell and D. G. Fernig, J. Biol. Chem., 2006, 281, 27178–27189 CrossRef CAS PubMed.
- T. Toida, H. Yoshida, H. Toyoda, I. Koshiishi, T. Imanari, R. E. Hileman, J. R. Fromm and R. J. Linhardt, Biochem. J., 1997, 322(Pt 2), 499–506 CrossRef CAS PubMed.
- T. R. Rudd, S. E. Guimond, M. A. Skidmore, L. Duchesne, M. Guerrini, G. Torri, C. Cosentino, A. Brown, D. T. Clarke, J. E. Turnbull, D. G. Fernig and E. A. Yates, Glycobiology, 2007, 17, 983–993 CrossRef CAS PubMed.
- S. E. Guimond, T. R. Rudd, M. A. Skidmore, A. Ori, D. Gaudesi, C. Cosentino, M. Guerrini, R. Edge, D. Collison, E. McInnes, G. Torri, J. E. Turnbull, D. G. Fernig and E. A. Yates, Biochemistry, 2009, 48, 4772–4779 CrossRef CAS PubMed.
- T. R. Rudd, K. A. Uniewicz, A. Ori, S. E. Guimond, M. a Skidmore, D. Gaudesi, R. Xu, J. E. Turnbull, M. Guerrini, G. Torri, G. Siligardi, M. C. Wilkinson, D. G. Fernig and E. A. Yates, Org. Biomol. Chem., 2010, 8, 5390–5397 CAS.
- J. S. Kastrup, E. S. Eriksson, H. Dalbøge and H. Flodgaard, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1997, 53, 160–168 CrossRef CAS PubMed.
- S. Ye, Y. Luo, W. Lu, R. B. Jones, R. J. Linhardt, I. Capila, T. Toida, M. Kan, H. Pelletier and W. L. McKeehan, Biochemistry, 2001, 40, 14429–14439 CrossRef CAS PubMed.
- A. N. Plotnikov, A. V. Eliseenkova, O. A. Ibrahimi, Z. Shriver, R. Sasisekharan, M. A. Lemmon and M. Mohammadi, J. Biol. Chem., 2001, 276, 4322–4329 CrossRef CAS PubMed.
- A. Brown, L. E. Adam and T. L. Blundell, Protein Cell, 2014, 5, 343–347 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6mb00857g |
| This journal is © The Royal Society of Chemistry 2017 |