
Assessment of exposure to airborne carbon nanotubes by laser-induced breakdown spectroscopy analysis of filter samples

Abstract
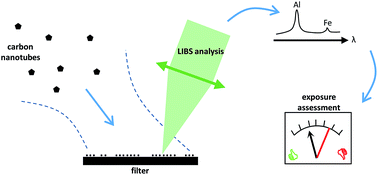
Exposure assessment is a key step in the evaluation of the risk induced by the handling of engineered nanomaterials. It is a very complex task, because several properties of nanoparticles are assumed to have an effect on their hazards. For exposure monitoring at the workplace, real-time onsite measurements are commonly implemented to measure the particles size and number density, whereas the sampled material is subsequently analysed by electron microscopy. A complementary approach would consist in doing onsite chemical analysis of the filter samples, in order to routinely monitor a potential chronic exposure. Laser-induced breakdown spectroscopy (LIBS) has distinctive advantages for this purpose. Therefore, this work aims at evaluating the performances of LIBS to assess the exposure to airborne carbon nanotubes (CNTs) at the workplace. As carbon is a ubiquitous element in the environment, our strategy was to target metal impurities in CNTs, aluminum and iron in our case. Then, we proceeded in three steps. First, we optimized the choice of the filter type to get the lowest detection limit for both elements. Secondly, this filter was used to quantitatively measure deposited CNTs. Eventually, we conducted an onsite measurement campaign in an industrial CNT production plant to evaluate the exposure in a real situation. We demonstrated that we could reach a detection limit for CNTs compliant with the current NIOSH recommendation of 1 μg m−3, and that the detected CNTs during the onsite campaign in areas accessible to workers were at an extremely low concentration, several orders of magnitude lower than this recommendation.
- This article is part of the themed collection: Young Analytical Scientists
 Please wait while we load your content...
Please wait while we load your content...

