
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLignin depolymerization to monophenolic compounds in a flow-through system†
Ivan
Kumaniaev‡
a,
Elena
Subbotina‡
a,
Jonas
Sävmarker
*b,
Mats
Larhed
*b,
Maxim V.
Galkin
 *a and
Joseph S. M.
Samec
*a
*a and
Joseph S. M.
Samec
*a
aDepartment of Organic Chemistry, Stockholm University 106 91, Stockholm, Sweden. E-mail: maxim.galkin@su.se; joseph.samec@su.se
bScience for Life Laboratory, Department of Medicinal Chemistry, Biomedical Center (BMC), Uppsala University, Box 574, 75 1 23 Uppsala, Sweden. E-mail: jonas.savmarker@orgfarm.uu.se; mats.larhed@orgfarm.uu.se
First published on 15th November 2017
Abstract
A reductive lignocellulose fractionation in a flow-through system in which pulping and transfer hydrogenolysis steps were separated in time and space has been developed. Without the hydrogenolysis step or addition of trapping agents to the pulping, it is possible to obtain partially depolymerized lignin (21 wt% monophenolic compounds) that is prone to further processing. By applying a transfer hydrogenolysis step 37 wt% yield of lignin derived monophenolic compounds was obtained. Pulp generated in the process was enzymatically hydrolyzed to glucose in 87 wt% yield without prior purification.
Economical utilization of lignin as a value-added product at a commercial scale is a key to make a lignocellulosic biorefinery cost effective. The ability to deconstruct lignin first from the intact biomass revolutionizes the conventional concept of the biorefinery by capturing high-value products from lignin as the first step and enhancing downstream of the cellulosic residue (improving ethanol production, etc.).1–3
The most prevalent lignin depolymerization target is the ether bond of the β-O-4′ motifs. There are a number of different strategies available where some are focused on the in situ stabilization1 of the monophenolic compounds released during pulping2–14 and the rest are focused on obtaining lignin that is structurally close to a native one or, at least, have an equivalent of the β-O-4′ motif preserved in its structure.15–19 These strategies allow one to obtain monophenolic products in near theoretical yields to produce 45 to 55 mol% of monophenolic compounds, along with enzyme-digestible pulps. The dominant lignin depolymerization strategy is the hydrogenolysis of lignin during the biomass fractionation process (reductive catalytic fractionation or catalytic upstream biorefining). Such processes usually involve mixing a heterogeneous metal catalyst with a solid biomass in a batch reactor. These reductive catalytic fractionation methods are complicated by catalyst recovery, requirement of hydrogen gas or other additives, mass transfer problems, and pulp contamination by the catalyst; these issues limit and restrict implementation of the method.20
A process that would not require an additional trapping agent for the stabilization of generated monophenolic compounds, no addition of hydrogen gas or hydrogen source,21 where biomass intercalation and hydrogenolysis steps are separated time- and space-wise but still in the same flow through system would avoid most of the mentioned drawbacks.22
We report here an application of a flow-through system23 for biomass fractionation where the lignin released during the solvolysis step is mainly fragmented to monophenolic compounds, dimers, and trimers and to a lesser extent higher oligomers. The hemicellulose fraction is used as an internal reducing agent and hydrogen donor. The application of a transfer hydrogenolysis step increased the yield of monophenolic compounds to 83% of the theoretical maximum.
The system used consists of a percolation reactor filled with woody biomass and a fixed catalytic bed reactor filled with Pd/C (Scheme 1).24 For biomass delignification, organosolv pulping conditions were chosen. When continuous removal of the pulping media takes place an addition of Brønsted or Lewis acids is advantageous.25 Therefore, a methanol–water solution of phosphoric acid (2.8 g L−1) was used. The influence of water to methanol ratio, and the influence of the concentration and the nature of an acid on the biomass components during the pulping process have been studied elsewhere.26–28 The results from the optimization of the pulping conditions are in good agreement with those reported previously (ESI, Tables S1–S6,† see ref. 26–28).
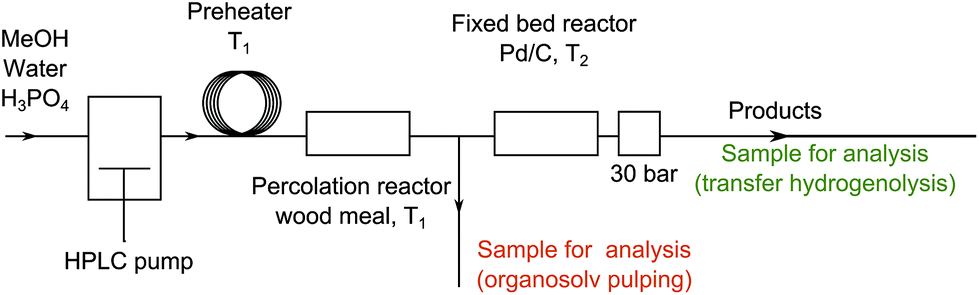 | ||
| Scheme 1 Schematic representation of the flow-through system used in the study; back pressure regulator (BPR) pressure for optimized reaction conditions. | ||
To obtain monophenolic compounds in high yields, the effect of temperature, flow rate, and process time on the fractionation of birch wood and the following reduction step was examined. When the temperature at which birch wood was subjected to percolation with MeOH–H2O (7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3, v/v) was increased from 160 to 220 °C and the temperature of the reactor with Pd/C was kept constant (180 °C), the yield of monophenolic compounds changed from 17 to 24 wt% with a maximum of 27 wt% yield at 200 °C (Table 1, entries 2–4). The increase in temperature improves delignification where higher temperature signifies repolymerization reactions and therefore decreases the yield.29 A similar effect was observed for the temperature of the reactor with Pd/C, where both too high and too low temperatures impaired productivity (Table 1, entries 3, 5, and 6). The optimal flow rate was found to be 0.3 mL min−1 (31 wt% yield), where lowering the rate (0.1 mL min−1) leads to over hydrogenation of the monophenolic compounds (Table 1, entries 7–9).
:3, v/v) was increased from 160 to 220 °C and the temperature of the reactor with Pd/C was kept constant (180 °C), the yield of monophenolic compounds changed from 17 to 24 wt% with a maximum of 27 wt% yield at 200 °C (Table 1, entries 2–4). The increase in temperature improves delignification where higher temperature signifies repolymerization reactions and therefore decreases the yield.29 A similar effect was observed for the temperature of the reactor with Pd/C, where both too high and too low temperatures impaired productivity (Table 1, entries 3, 5, and 6). The optimal flow rate was found to be 0.3 mL min−1 (31 wt% yield), where lowering the rate (0.1 mL min−1) leads to over hydrogenation of the monophenolic compounds (Table 1, entries 7–9).
| Entry | Time (h) | T 1 (°C) | T 2 (°C) | Flow rate (mL min−1) | Yielda (wt%) |
|---|---|---|---|---|---|
| General conditions: Non-dewaxed oven-dry birch wood meal (0.150 g, 2 wt% residual moisture), Pd/C 5 wt% (0.150 g), 2.8 g L−1 H3PO4 in MeOH–H2O 7:3 v/v.a The yield of monophenolic compounds estimated from the 1H NMR data (ESI 4.2.1), represented as wt% of total lignin (ESI 3.2.2).b Impregnated wood meal.c Average of 4 (entry 1) and 3 (entry 11) repetitions. The yield of monophenolic compounds estimated from the GC-MS data (ESI 4.3). For entry 1: 4-(3-hydroxyprop-1-en-1-yl)-2,6-dimethoxyphenol (1a), 2,6-dimethoxy-4-(3-methoxyprop-1-en-1-yl)phenol (2a), and 2-methoxy-4-(3-methoxyprop-1-en-1-yl)phenol (2b); for entry 11: 4-(3-hydroxypropyl)-2,6-dimethoxyphenol (3a), 4-(3-hydroxypropyl)-2-methoxyphenol (3b), 2,6-dimethoxy-4-(3-methoxypropyl)phenol (4a), 2-methoxy-4-(3-methoxypropyl)phenol (4b), 2,6-dimethoxy-4-propylphenol (5a), and 2-methoxy-4-propylphenol (5b).d Dewaxed wood meal.e Non-dewaxed wood meal. |
|||||
| Organosolv pulping (no Pd/C) | |||||
| 1b | 1–3 | 200 | — | 0.3 | 21 ± 2.8c |
| Organosolv pulping followed by transfer hydrogenolysis (Pd/C) | |||||
| 2 | 3 | 180 | 180 | 0.2 | 17 |
| 3 | 3 | 200 | 180 | 0.2 | 27 |
| 4 | 3 | 220 | 180 | 0.2 | 24 |
| 5 | 3 | 200 | 200 | 0.2 | 22 |
| 6 | 3 | 200 | 160 | 0.2 | 22 |
| 7 | 3 | 200 | 180 | 0.3 | 31 |
| 8 | 3 | 200 | 180 | 0.5 | 21 |
| 9 | 3–6 | 200 | 180 | 0.1 | 0 |
| 10b,d | 3 | 200 | 180 | 0.3 | 29 |
| 11b,e | 3 | 200 | 180 | 0.3 | 39/37 ± 0.6/1.5c |
To increase the permeability of the biomass pulping solution, dewaxed biomass is commonly used.30 Consequently, one set of samples was dewaxed prior to the experiment to investigate the effects of waxes on the delignification and catalyst activity. Interestingly, the lower yield of monophenolic compounds was obtained when dewaxed biomass was used (Table 1, entry 10, and ESI 3.1.1 and Table S6†). The effect of wood meal pre-soaking was also tested. When the biomass was left in the pulping solution overnight at RT, the yield of monophenolic compounds increased to 37 wt% (Table 1, entry 11). The solvent mixture in the current continuous flow system may operate in different phases due to temperature, flow rate, and pressure that may vary within the system. This would be of great interest to study in the future.
Additional studies were performed (NMR, SEC, GC-MS, etc.) to gain further insight into the process of lignin depolymerization using the optimized process conditions. Besides the possibility to control the pulping and catalytic parameters separately, the system allowed both rapid heating and cooling of the reaction media. Thereby, we could study the nature of lignin directly after pulping.
By monitoring the reaction, we found that the delignification of the biomass takes place within the first 90 min (Fig. 1, ESI, Fig. S12 and S13†). However, in the lignin reaction with Pd/C, it is apparent that the product desorption from the catalyst bed leads to a delay in the release of lignin products from the second reactor. In fact, it takes approximately 180 min for the lignin products to be discharged from the reactor.
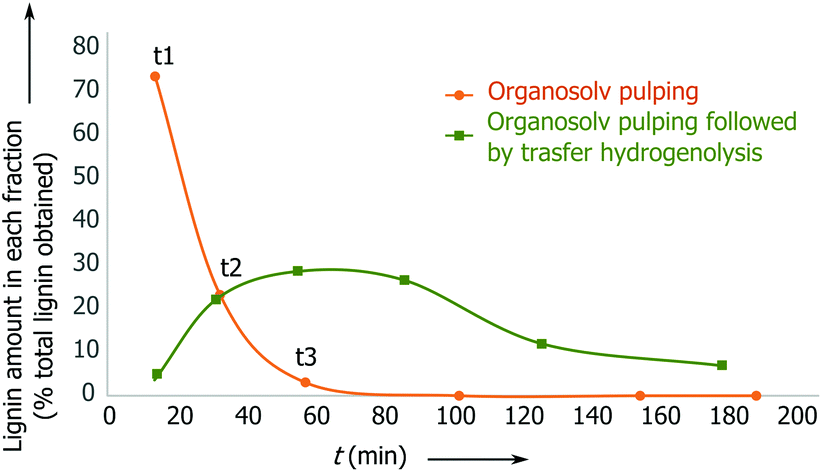 | ||
| Fig. 1 Reaction concentration profile for birch wood pulping with and without the catalyst present: Pd/C 5 wt% (0.150 g), 2.8 g L−1 H3PO4 in MeOH–H2O 7:3 v/v. | ||
Previously Rinaldi's group has demonstrated that lignin fragments released at the beginning of the pulping process display a bimodal distribution of monophenolic compounds and oligomeric fragments up to trimers, whereas the lignin fragments obtained after the prolonged reaction time represent higher oligomers with a considerable fraction greater than 3 kDa.6 Notably, the apparent Mw distribution of lignin fragments released in the present process changes insignificantly with the time passed from the beginning of the pulping (Fig. 2, t1 and t2), where the main part of the lignin belongs to monophenolic compounds and dimer fractions and the rest belongs to higher oligomers. At the end of pulping there is an increase in the apparent molecular weight of the released lignin fragments (from 823 − t1 to 1340 Da − t3). However, this part accounts for only 5 wt% of the total lignin. These results clearly show that the lignin depolymerization to a large extent takes place during the solvolysis and partially during the transfer hydrogenolysis step (vide infra, Fig. 3).
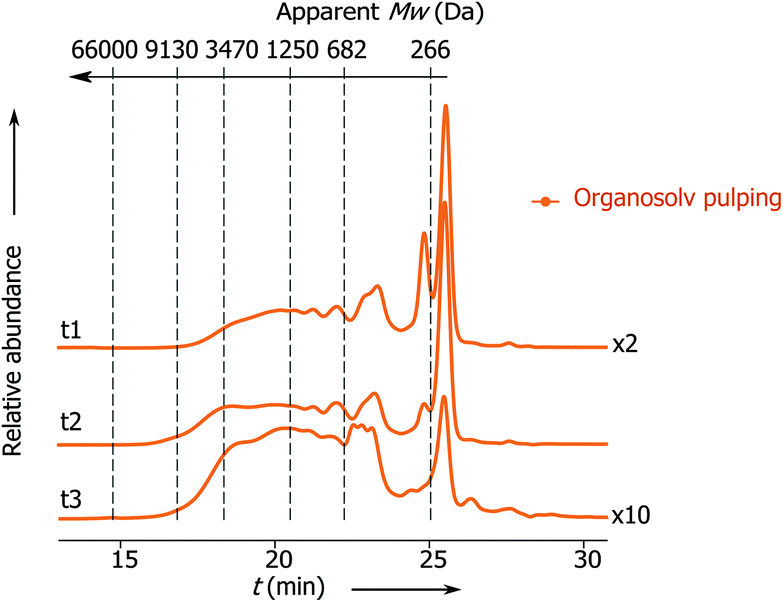 | ||
| Fig. 2 SEC chromatograms: apparent Mw distribution of lignin obtained at different times from the solvolysis step: t1 from 0 to 15 min; t2 from 15 to 30 min and t3 from 30 to 60 min. For conditions see Table 1, entry 1. | ||
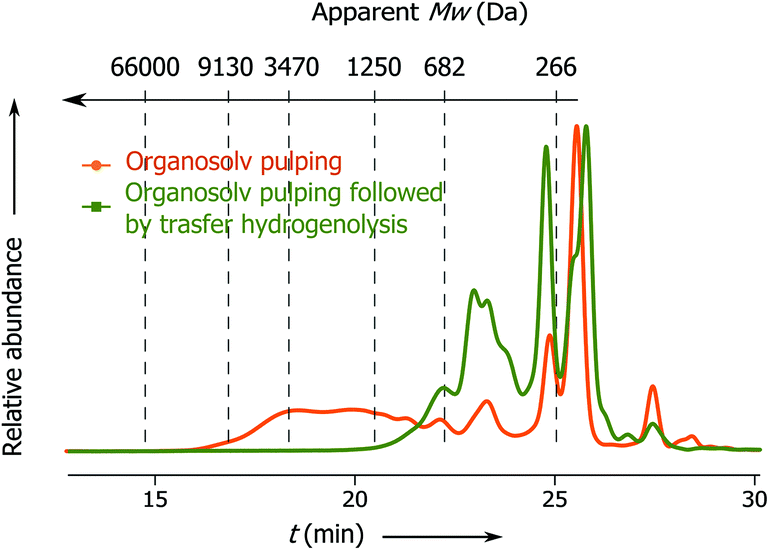 | ||
| Fig. 3 SEC chromatograms of lignin obtained after 3 h reaction with and without Pd/C. | ||
Analysis of the obtained hydrolyzed lignin by 2-D NMR spectroscopy (HSQC and HMBC) revealed a decrease in the occurrence of native ether bonds (Fig. 4) and the appearance of alkene functionalities. The signals of the new alkene functionalities were ascribed to three major lignin monophenolic compounds (1a, 2a, and 2b; Table 1, entry 1; Fig. 4) where the most abundant 2,6-dimethoxy-4-(3-methoxyprop-1-en-1-yl)phenol (2a) was obtained in up to 15 wt% yield. The identification and quantification of the monophenolic compounds are described in ESI 4.2 and 4.3†.
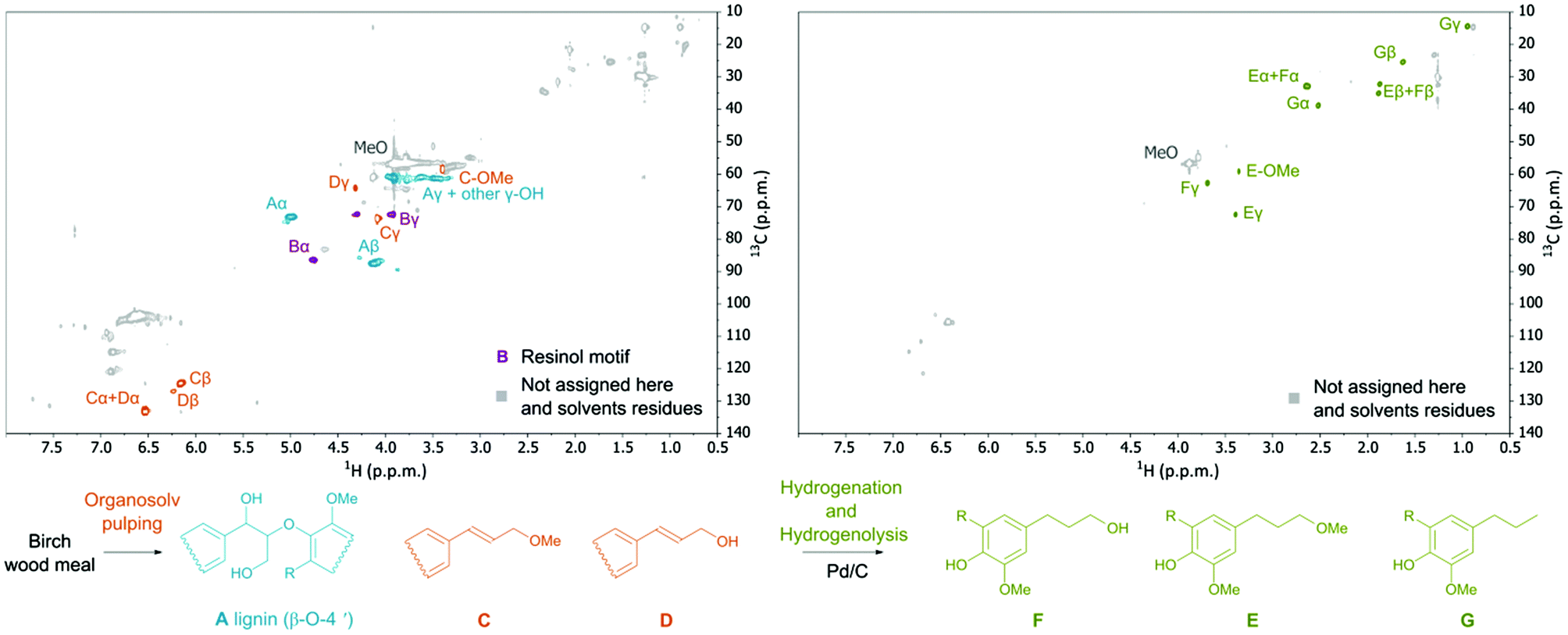 | ||
| Fig. 4 Complete HSQC spectra of: organosolv lignin (left) and organosolv lignin after Pd/C treatment (right). Structures found in the dissolved lignin: (A) β-O-4′, (C) and (D) methoxy and hydroxy propenyl phenol fragments including compounds 1a, 2a, and 2b; F: compounds 3a and 3b, E: compounds 4a and 4b, and G: compounds 5a and 5b. Reaction conditions: Non-dewaxed, flushed with the solvent system (RT, 0.3 mL min−1, 10 min) and left overnight before the reaction of wood meal, 2.8 g L−1 H3PO4 in MeOH–H2O 7:3 v/v; preheater on; T1 = 200 °C; T2 = 180 °C; flow rate 0.3 mL min−1; reaction time 3 h. | ||
It is gratifying that lignin undergoes solvolysis under the applied fractionation conditions and then the homolysis of the β-O-4′ bonds starts from the free phenol group-peeling (Fig. 5).31–34 Reducing sugars released from biomass can act as an internal reducing agent to react with the radicals of sinapyl and coniferyl alcohols to yield the corresponding alcohols (1a and 1b) as products.35–37 This homolytic pathway has been extensively studied (predominantly utilizing model compounds).31–38 Thereby, the reaction allows lignin depolymerization to monomers starting from the free phenolic OH groups (of the β-O-4′ motif) and the reaction will maintain only in a continuous sequence of monomers joined with β-O-4′ bonds. Therefore, we attribute the presence of higher oligomers in the lignin obtained after pulping partially to the inability of the homolysis reaction to take place at β-O-4′ motifs with other types of surroundings and to a lower extent to repolymerization reactions of reactive radicals.38,39 It is evident that the catalyst has an important role in transforming these less available β-O-4′ bonds (Fig. 3 and 4).
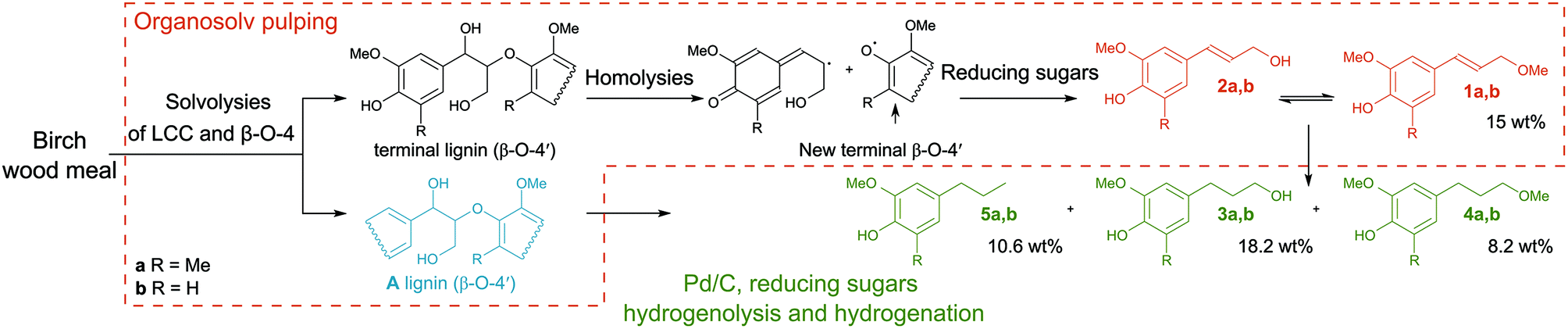 | ||
| Fig. 5 Proposed mechanism of the β-O-4′ bond cleavage. Organosolv pulping step: the formation of coniferyl and sinapyl alcohols and consequently their ethers in the absence of catalyst is attributed to the homolysis reaction with the following reduction of the radicals with reducing sugars (ESI Fig. S32†). Catalytic hydrogenation and hydrogenolysis steps: cleavage of the non-terminal β-O-4′ bonds and saturation of unsaturated intermediates. The yield of monophenolic compounds estimated from GC-MS data (ESI 4.3†). | ||
In contrast to direct biomass hydrogenolysis, our approach is compatible with most polysaccharide depolymerization techniques and does not need tedious pulp and catalyst separation. The analysis of all the fractions obtained after the process revealed that 92 wt% of cellulose was intact and present in the solid residue. As a result, the treatment of birch wood under mild enzymatic hydrolysis conditions gave glucose in 95% yield, which corresponds to 87 wt% of the original glucose content in the woody biomass (Table 2). In the present system the hemicellulose fraction is used as an internal reducing agent and hydrogen donor (ESI 5†). The yield of lignin derived monophenolic compounds is comparable to the best methodologies previously reported (38–55%) as well as a cellulose yield (74–94%).18,24,40
| Before the fractionation | After the fractionation | ||
|---|---|---|---|
| Birch wood meala | Solid fractionc | Liquid fraction | |
| For reaction conditions see Table 1 entry 11.a For analysis details, see ESI, section 3.b Total lignin: ASL and AIL.c ESI, section 4.6.d Methyl-α-D-xylopyranoside, methyl-β-D-xylopyranoside and xylose (ESI, section 4.5).e The yield of monophenolic compounds estimated from GC-MS data (ESI 4.3).f For details of C-balance calculation, see ESI, section 4.7 and ref. 41.g Extractive-free basis C-balance. | |||
| Extractives | 3 | ND | ND |
| Xylans | 25 (27) | 0 (0) | 2d (2) |
| Glucans | 38 (41) | 35 (37) | 0 (0) |
| Lignin | 23b (32) | 1.5 (2) | 9 (12)e |
| Sum | 89 (100)g | 36.5 (39) | 11 (14) |
| 47.5 (53) | |||
The present approach results in a very good yield of monophenolic compounds 37 wt% (83% theoretical yield, ESI 3.5 and 4.3.2†) and has several advantages where the cellulose fraction has not been contaminated by the catalyst and thereby is easily enzymatically hydrolyzed to glucose without prior purifications in 87 wt% yield (ESI 3.2.1 and 6†). In addition, the use of the flow-through system enables optimizing and studying both processes separately where we found that organosolv pulping and transfer hydrogenolysis should be performed under different conditions; and depolymerized lignin (21 wt% monophenolic compound yield) can be obtained without the palladium catalyzed step. We anticipate that the demonstrated approach fulfills an important gap in second generation biorefineries towards the production of liquid biofuels and chemicals from lignocellulose.
Conflicts of interest
We have no conflict of interest to declare.Acknowledgements
This work was supported by the Swedish Energy Agency. We would like to acknowledge Vanhälls Såg AB for providing biomass samples and Magnus Fagrell for providing the RedHeater continuous flow system.Notes and references
- C. Xu, R. A. D. Arancon, J. Labidid and R. Luque, Chem. Soc. Rev., 2014, 43, 7485–7500 RSC.
- M. V. Galkin and J. S. M. Samec, ChemSusChem, 2016, 9, 1544–1558 CrossRef CAS PubMed.
- R. Rinaldi, R. Jastrzebski, M. T. Clough, J. Ralph, M. Kennema, P. C. A. Bruijnincx and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2016, 55, 8164–8215 CrossRef CAS PubMed.
- C. Li, X. Zhao, A. Wang, G. W. Huber and T. Zhang, Chem. Rev., 2015, 115, 11559–11624 CrossRef CAS PubMed.
- P. J. Deuss and K. Barta, Coord. Chem. Rev., 2016, 306, 510–532 CrossRef CAS.
- P. Ferrini and R. Rinaldi, Angew. Chem., Int. Ed., 2014, 53, 8634–8639 ( Angew. Chem. , 2014 , 126 , 8778–8783 ) CrossRef CAS PubMed.
- Q. Song, F. Wang, J. Cai, Y. Wang, J. Zhang, W. Yua and J. Xu, Energy Environ. Sci., 2013, 6, 994–1007 CAS.
- S. Vanden Bosch, W. Schutyser, R. Vanholme, T. Driessen, S.-F. Koelewijn, T. Renders, B. De Meester, W. J. J. Huijgen, W. Dehaen, C. M. Courtin, B. Lagrain, W. Boerjanbc and B. F. Sels, Energy Environ. Sci., 2015, 8, 1748–1763 CAS.
- P. J. Deuss, M. Scott, F. Tran, N. J. Westwood, J. G. de Vries and K. Barta, J. Am. Chem. Soc., 2015, 137, 7456–7467 CrossRef CAS PubMed.
- D. M. Miles-Barrett, A. R. Neal, C. Hand, J. R. D. Montgomery, I. Panovic, O. S. Ojo, C. S. Lancefield, D. B. Cordes, A. M. Z. Slawin, T. Lebl and N. J. Westwood, Org. Biomol. Chem., 2016, 14, 10023–10030 CAS.
- P. J. Deuss, C. W. Lahive, C. S. Lancefield, N. J. Westwood, P. C. J. Kamer, K. Barta and J. G. de Vries, ChemSusChem, 2016, 9, 2974–2981 CrossRef CAS PubMed.
- P. J. Deuss, C. S. Lancefield, A. Narani, J. G. de Vries, N. J. Westwood and K. Barta, Green Chem., 2017, 19, 2774–2782 RSC.
- V. M. Roberts, V. Stein, T. Reiner, A. Lemonidou, X. Li and J. A. Lercher, Chem. – Eur. J., 2011, 17, 5939–5948 CrossRef CAS PubMed.
- M. V. Galkin, A. T. Smit, E. Subbotina, K. A. Artemenko, J. Bergquist, W. J. J. Huijgen and J. S. M. Samec, ChemSusChem, 2016, 9, 3280–3287 CrossRef CAS PubMed.
- A. Rahimi, A. Ulbrich, J. J. Coon and S. S. Stahl, Nature, 2014, 515, 249–252 CrossRef CAS PubMed.
- J. S. Luterbacher, J. M. Rand, D. M. Alonso, J. Han, J. T. Youngquist, C. T. Maravelias, B. F. Pfleger and J. A. Dumesic, Science, 2014, 343, 277–280 CrossRef CAS PubMed.
- J. S. Luterbacher, A. Azarpira, A. H. Motagamwala, F. Lu, J. Ralph and J. A. Dumesic, Energy Environ. Sci., 2015, 8, 2657–2663 CAS.
- L. Shuai, M. T. Amiri, Y. M. Questell-Santiago, F. Héroguel, Y. Li, H. Kim, R. Meilan, C. Chapple, J. Ralph and J. S. Luterbacher, Science, 2016, 354, 329–333 CrossRef CAS PubMed.
- K. H. Kim, T. Dutta, E. D. Walter, N. G. Isern, J. R. Cort, B. A. Simmons and S. Singh, ACS Sustainable Chem. Eng., 2017, 5, 3913–3919 CrossRef CAS.
- C. Xu, R. Arneil, D. Arancon, J. Labidi and R. Luque, Chem. Soc. Rev., 2014, 43, 7485–7500 RSC.
- C. Chesi, I. B. D. de Castro, M. T. Clough, P. Ferrini and R. Rinaldi, ChemCatChem, 2016, 8, 2079–2088 CrossRef CAS.
- B. Yang and C. E. Wyman, Flow-through operation removes lignin before it can condense onto the biomass surface, enhancing the accessibility of cellulose and its digestibility, Biotechnol. Bioeng., 2004, 86, 88–95 CrossRef CAS PubMed.
- General reference to flow chemistry: M. B. Plutschack, B. Pieber, K. Gilmore and P. H. Seeberger, Chem. Rev., 2017, 117, 11796–11893 CrossRef CAS PubMed.
- Selected articles on the reductive catalytic fractionation (RCF) of lignocellulosic biomass catalyzed by palladium: (a) T. Renders, W. Schutyser, S. Van den Bosch, S.-F. Koelewijn, T. Vangeel, C. M. Courtin and B. F. Sels, ACS Catal., 2016, 6, 2055–2066 CrossRef CAS; (b) F. Gao, J. D. Webb, H. Sorek, D. E. Wemmer and J. F. Hartwig, ACS Catal., 2016, 6, 7385–7392 CrossRef CAS; (c) X. Huang, O. M. Morales Gonzalez, J. Zhu, T. I. Korányi, M. D. Bootb and E. J. M. Hensen, Green Chem., 2017, 19, 175–187 RSC; (d) S. Van den Bosch, W. Schutyser, S.-F. Koelewijn, T. Renders, C. M. Courtin and B. F. Sels, Chem. Commun., 2015, 51, 13158–13161 RSC; (e) I. Klein, C. Marcum, H. Kenttäma and M. M. Abu-Omar, Green Chem., 2016, 18, 2399–2405 RSC.
- Organosolv pulping can be performed under autocatalytic conditions. The deacetylation of hemicellulose decreases the pH value of the liquor providing the acid catalyst for solvolytic bond cleavage (see ref. 6 and 24c). Under the flow conditions the formed acids need to be recycled, which is theoretically possible, but was not tested.
- (a) K. Atsushi, K. Makiko, S. Ryo, S. Kaori and W. Takashi, Green Chem., 2015, 17, 2780–2783 RSC; (b) W. Schutyser, S. Van den Bosch, T. Renders, T. De Boe, S.-F. Koelewijn, A. Dewaele, T. Ennaert, O. Verkinderen, B. Goderis, C. M. Courtin and B. F. Sels, Green Chem., 2015, 17, 5035–5045 RSC; (c) K. Barta, T. D. Matson, M. L. Fettig, S. L. Scott, A. V. Iretskii and P. C. Ford, Green Chem., 2010, 12, 1640–1647 RSC.
- X. Huang, X. Ouyang, B. Hendriks, O. Morales, J. Zhu, T. I. Korányi, M. Boot and E. J. M. Hensen, Faraday Discuss., 2017, 202, 141–156 RSC.
- T. Renders, S. Van den Bosch, T. Vangeel, T. Ennaert, S.-F. Koelewijn, G. Van den Bossche, C. M. Courtin, W. Schutyser and B. F. Sels, ACS Sustainable Chem. Eng., 2016, 4, 6894–6904 CrossRef CAS.
- Kraft Pulping: A Compilation of Notes, ed. A. Mimms, TAPPI Press, 1993, pp. 50–70 Search PubMed.
- A. T. Smit, W. J. J. Huijgen and R. J. H. Grisel, WO2014/126471, 2014 Search PubMed.
- H. Nimz, Angew. Chem., Int. Ed. Engl., 1966, 5, 843 CrossRef.
- S. Li, K. Lundquist and U. Westermark, Nord. Pulp Pap. Res. J., 2000, 15, 292–299 CAS.
- T. Kishimoto and Y. Sano, Holzforschung, 2001, 55, 611–616 CrossRef CAS.
- T. Kishimoto and Y. Sano, Holzforschung, 2002, 56, 623–631 CrossRef CAS.
- Y. Sano and T. Kishimoto, J. Wood Chem. Technol., 2003, 23, 279–292 CrossRef.
- T. Kishimoto and Y. Sano, J. Wood Chem. Technol., 2003, 23, 233–248 CrossRef CAS.
- J. Kajimoto and Y. Sano, Japan TAPPI J., 2001, 55, 1470–1479 CrossRef CAS.
- S. Omori, M. Aoyama and A. Sakakibara, Holzforschung, 1998, 52, 391–397 CrossRef CAS.
- T. Kishimoto and Y. Sano, Holzforschung, 2002, 56, 623–631 CrossRef CAS.
- Alternative approach: S. Van den Bosch, T. Renders, S. Kennis, S.-F. Koelewijn, G. Van den Bossch, T. Vangeel, A. Deneyer, D. Depuydt, C. M. Courtin, J. M. Thevelein, W. Schutyser and B. F. Sels, Green Chem., 2017, 19, 3313–3326 RSC.
- (a) P. Ferrini, C. A. Rezende and R. Rinaldi, ChemSusChem, 2016, 9, 3171–3180 CrossRef CAS PubMed; (b) C. Chesi, I. B. D. de Castro, M. T. Clough, P. Ferrini and R. Rinaldi, ChemCatChem, 2016, 8, 2079–2088 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7gc02731a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |