
A quest for polycarbonates provided via sustainable epoxide/CO2 copolymerization processes†
Stephanie J.
Poland  *a
and
Donald J.
Darensbourg
b
*a
and
Donald J.
Darensbourg
b
*a
and
Donald J.
Darensbourg
b
Abstract
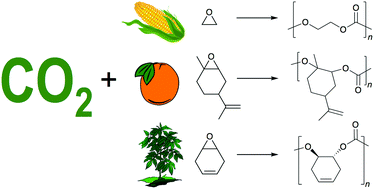
The present review highlights advances in the copolymerization of carbon dioxide (CO2) and epoxides (oxiranes) to produce polycarbonates. Specifically, focus has been given to epoxide starting materials that have been generated from renewable feedstocks including straight-chain alkylene oxides, cyclohexadiene oxide, and limonene oxide, among several others. These renewable feedstocks are attractive green alternatives to traditional petroleum-based materials, however, some limitations do exist regarding the availability of monomer feedstocks as well as economic factors. Recent advances for each of these polymeric products will be discussed in detail. The use of industrial post-combustion CO2 waste streams will also be commented upon.
- This article is part of the themed collection: 2017 Green Chemistry Hot Articles
 Please wait while we load your content...
Please wait while we load your content...

