
Crystal lattice distortion in ultrathin Co(OH)2 nanosheets inducing elongated Co–OOH bonds for highly efficient oxygen evolution reaction†
 *a
*a
Abstract
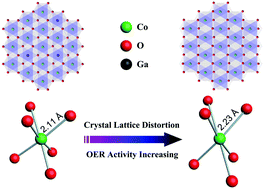
The exploration of highly efficient nonprecious metal-based electrocatalysts for the oxygen evolution reaction (OER) is of great importance for potential applications in sustainable energy conversion. Recently, the layered metal hydroxide (LMH) family is receiving extensive research attention owing to its unique structural properties. However, the gained electrocatalytic performance of LMH-based catalysts for the OER is still far from the state-of-the-art requirements because of its finite number and poor reactivity of exposed active sites. In response, we synthesized crystal lattice distorted ultrathin cobalt hydroxide (denoted as CLD-u-Co(OH)2) nanosheets with a great number of efficient catalytic active sites through the introduction of Ga into ultrathin Co(OH)2, followed by a selective removal of Ga, denoted as the “introduction @ removal” process. As a result, the crystal lattice distortion confined inside CLD-u-Co(OH)2 generates abundant elongated Co–OOH bonds on exposed (1![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) 0) facets serving as efficient catalytic active sites for the OER. Besides, the optimized amount of “introduction @ removal” of Ga (4 at%) allows for an exquisite balance between distortion engineering and electrical conductivity, synergistically. The as-prepared CLD-u-Co(OH)2 achieves an overpotential of 265 mV at a current density of 10 mA cm−−2, an unexpectedly small Tafel slope of 47 mV dec−−1, and a long-term stability (beyond 20 h) in basic media. It is mainly attributed to abundant catalytic active sites, robust reactivity per site, and good electrical conductivity. Furthermore, the green and sustainable engineering of crystal lattice distortion to improve the intrinsic electrocatalytic activity of CLD-u-Co(OH)2 nanosheets presented in this work may provide a promising strategy to design and synthesize newly highly efficient LMH-based electrocatalysts for the OER.
0) facets serving as efficient catalytic active sites for the OER. Besides, the optimized amount of “introduction @ removal” of Ga (4 at%) allows for an exquisite balance between distortion engineering and electrical conductivity, synergistically. The as-prepared CLD-u-Co(OH)2 achieves an overpotential of 265 mV at a current density of 10 mA cm−−2, an unexpectedly small Tafel slope of 47 mV dec−−1, and a long-term stability (beyond 20 h) in basic media. It is mainly attributed to abundant catalytic active sites, robust reactivity per site, and good electrical conductivity. Furthermore, the green and sustainable engineering of crystal lattice distortion to improve the intrinsic electrocatalytic activity of CLD-u-Co(OH)2 nanosheets presented in this work may provide a promising strategy to design and synthesize newly highly efficient LMH-based electrocatalysts for the OER.
- This article is part of the themed collection: 2017 Green Chemistry Hot Articles
 Please wait while we load your content...
Please wait while we load your content...

