
Tuneable 3D printed bioreactors for transaminations under continuous-flow†

Abstract
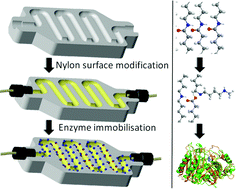
A method to efficiently immobilize enzymes on 3D printed continuous-flow devices is presented. Application of these chemically modified devices enables rapid screening of immobilization mechanisms and reaction conditions, simple transfer of optimised conditions into tailored printed microfluidic reactors and development of continuous-flow biocatalytic processes. The bioreactors showed good activity (8–20.5 μmol h−1 mgenz−1) in the kinetic resolution of 1-methylbenzylamine, and very good stability (ca. 100 h under flow).
- This article is part of the themed collection: 2017 Green Chemistry Hot Articles
 Please wait while we load your content...
Please wait while we load your content...

