
A highly stable Ru/LaCO3OH catalyst consisting of support-coated Ru nanoparticles in aqueous-phase hydrogenolysis reactions†
 *a
*a
Abstract
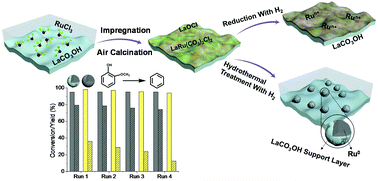
Hydrothermal reduction under aqueous conditions is widely used to convert biomass into more valuable products. However, the harsh conditions inherent in the process can irreversibly alter the intrinsic structure of the support, as well as dissolve the metal ions into the aqueous solution. In this work, for the first time we have synthesized a new highly hydrothermally stable Ru/LaCO3OH catalyst mostly consisting of Ru nanoparticles partially encapsulated by the LaCO3OH support with a strong metal–support interaction (SMSI), which confers high stability and activity to the catalyst under hydrothermal reduction conditions in the hydrogenolysis of the biomass model molecules guaiacol and glycerol. During impregnation, the RuCl3·3H2O precursor initially reacts with LaCO3OH to form a LaRu(CO3)2Cl2 complex and LaOCl. XPS demonstrated that Ru was present in the oxidized state, TEM and XRD showed the absence of Ru0, and the XRD pattern showed the presence of the characteristic lattice fringe of LaOCl. While the LaRu(CO3)2Cl2 complex was resistant to H2 reduction at 350 °C, the complex underwent facile reduction to Ru0 under hydrothermal conditions at 240 °C. In the subsequent process, LaRu(CO3)2Cl2 and LaOCl underwent hydrolysis, forming crystalline LaCO3OH (confirmed by Ag+ titration and XRD patterns), Ru(OH)3, and HCl. The Ru(OH)3 was reduced in situ to Ru0 nanoparticles, as revealed by XPS and TEM analysis. The simultaneous hydrothermal reduction of Run+ species and the formation of crystalline LaCO3OH result in the formation of Ru nanoparticles encapsulated by a protective LaCO3OH layer, as evidenced by HRTEM and DRIFTS CO adsorption measurements. The preparation of catalysts with this unique structure comprising metal nanoparticles protected by the support itself, which confers additional stability, is a novel strategy to prepare hydrothermally stable catalysts.
- This article is part of the themed collection: 2017 Green Chemistry Hot Articles
 Please wait while we load your content...
Please wait while we load your content...

