
An ionic liquid catalyzed probase method for one-pot synthesis of α,β-unsaturated esters from esters and aldehydes under mild conditions†
 b
Chunshan
Li
*ab
b
Chunshan
Li
*ab
Abstract
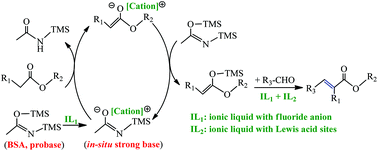
A one-pot synthesis of α,β-unsaturated esters from unactivated esters and aldehydes using strong bases, such as sodium alkoxide and potassium tert-butoxide, was reported. However, the ionic liquid (IL) catalyzed probase method for producing α,β-unsaturated esters was not reported until now. In this work, a series of ILs with fluoride anions were firstly prepared and used as catalysts in combination with the probase N,O-bis(trimethylsilyl) acetamide (BSA) for the α,β-unsaturated esters synthesis. This process could also be promoted through the introduction of another IL with Lewis acid sites. The yield and selectivity of the product could reach up to 84.2% and 95.0%, respectively, when [Bmim]F was used in combination with [Bmim]Cl/AlCl3 (the molar fraction of AlCl3 is 0.67). The mechanism investigation through GC-MS indicates that BSA would convert into onium amide, which acted as a strong base for α-H abstraction, with the catalysis of [Bmim]F. Meanwhile, [Bmim]Cl/AlCl3 played an important role in the condensation step between enolates and aldehydes. On the basis of mechanism insights, kinetic and thermodynamic studies were also carried out for a better understanding of this new route.
- This article is part of the themed collection: 2017 Green Chemistry Hot Articles
 Please wait while we load your content...
Please wait while we load your content...

