
Direct use of humic acid mixtures to construct efficient Zr-containing catalysts for Meerwein–Ponndorf–Verley reactions†
 *a
*a
Abstract
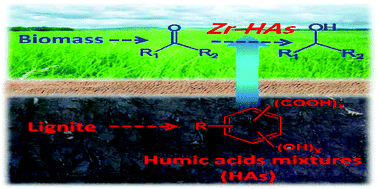
With the increasing demands for energy and carbon resources, exploration of novel utilization approaches of fossil resources and promotion of the conversion of sustainable resources become critical issues facing human society. In this study, we used humic acids (HAs), which are important derivatives from the low-rank coal, and the transition metal zirconium (Zr) to construct novel Zr-containing catalysts (Zr-HAs) for Meerwein–Ponndorf–Verley (MPV) reactions of biomass-derived platforms. Both commercial HAs and HAs extracted directly from lignite without any purification were used as raw materials to prepare the catalysts. The results showed that Zr-HAs catalysts were highly efficient for the conversion of furfural, with a high furfuryl alcohol yield of up to 97%, and also effective for the conversion of other carbonyl compounds with different structures under mild conditions. This novel strategy to construct catalysts using HAs as raw materials may be beneficial for both value-added utilization of low-rank coal and the conversion of biomass resources.
- This article is part of the themed collection: 2017 Green Chemistry Hot Articles
 Please wait while we load your content...
Please wait while we load your content...

