
Chemisorption of CO2 by chitosan oligosaccharide/DMSO: organic carbamato–carbonato bond formation†
 *a
Khaleel I.
Assaf,
b
Sanaa K.
Bardaweel,
c
Mahmoud
Masri,
e
Thomas
Brück,
e
Bernhard
Rieger
f
and
Ala'a F.
Eftaiha
*d
*a
Khaleel I.
Assaf,
b
Sanaa K.
Bardaweel,
c
Mahmoud
Masri,
e
Thomas
Brück,
e
Bernhard
Rieger
f
and
Ala'a F.
Eftaiha
*d
Abstract
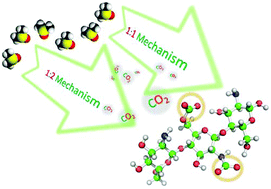
A newly formed bond of organic carbamato–carbonato emerged upon bubbling CO2 in a low molecular weight chitosan hydrochloride oligosaccharide CS·HCl/DMSO binary mixture. The aforementioned bond was detected and confirmed using attenuated total reflectance-Fourier transform Infrared (ATR-FTIR) spectroscopy, with two prominent peaks at 1551 cm−1 and 1709 cm−1 corresponding to ionic organic alkylcarbonate (RCO3−) and carbamate (RNH–CO2− NH3+–R), respectively. 1H–, 13C–, and 1H–15N heteronuclear single quantum coherence (HSQC) NMR experiments were also employed. According to 13C NMR, two newly emerged peaks at 157.4 ppm and 161.5 ppm attributed for the carbonyl carbon within the sequestered species RCO3− and RNH–CO2− NH3+–R, respectively. Upon CO2 bubbling, cross peaks obtained from 1H–15N HSQC at 84.7 and 6.8 ppm correlated to the ammonium counterpart chemical shift bound to the proton resonances. Volumetric uptake of CO2 was measured using an ATR-FTIR autoclave equipped with a silicon probe. The equilibrium sorption capacity was 0.6 and 0.2 bars through the formation of RCO3− and RNH–CO2− NH3+–R, respectively. Moreover, physisorption by the dried DMSO contributed to additional 0.4 bars. Density functional theory (DFT) calculations supported the occurrence of the suggested dual mechanisms and confirmed the formation of carbonate at C-6 of the glucosamine co-monomer. Moreover, CS·HCl/DMSO showed a slight impact on cell proliferation after 48 hours; this was a clear evidence of its non-toxicity. The biodegradation test revealed that a degradation of about 80% of CS·HCl/DMSO was achieved after 33 days; these results indicated that this method is suitable for green industry. CS·HCl/DMSO showed modest activities against Staphylococcus aureus and Escherichia coli. In addition, CS·HCl/DMSO demonstrated a significant antifungal activity against Aspergillus flavus in comparison with Fluconazole.
 Please wait while we load your content...
Please wait while we load your content...

