
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Organic electrolyte solutions as versatile media for the dissolution and regeneration of cellulose
Matthew T.
Clough

Max-Planck-Institut für Kohlenforschung, Kaiser-Wilhelm-Platz 1, 45470 Mülheim-an-der-Ruhr, Germany. E-mail: matthew.clough@syngenta.com
First published on 31st August 2017
Abstract
Organic electrolyte solutions – mixtures of a (room-temperature) ionic liquid with a neutral, organic, polar co-solvent – are attracting increasing attention as solvents for the regeneration and derivatisation of cellulose. Despite advantages (in comparison to simple ionic liquid analogues) associated with rapid or instantaneous dissolution, reduced viscosity, enhanced thermal stability and fine-tunable physicochemical properties, a firm understanding of the precise solvent–solute interactions and the relative kinetic versus thermodynamic contributions to dissolution remains elusive. The incorporation of a co-solvent introduces an additional layer of complexity, therefore an informed choice of both ionic liquid and co-solvent is necessary in order to achieve the desired properties. This article first provides an overview of the structure and bonding properties of native and non-native cellulose allomorphs, and a brief history of strategies for cellulose dissolution. Subsequently, organic electrolyte solutions are introduced as versatile solvents for cellulose, and the underpinning thermodynamic, kinetic and mechanistic phenomena behind cellulose solubility are critically discussed. The final sections summarize recent advances in the development of organic electrolyte technologies for derivatisation and regeneration of cellulose and critically discuss whether organic electrolytes can, or could be, rightly regarded as ‘green solvents’.
 Matthew T. Clough | Matthew T. Clough (United Kingdom, 1989) obtained his PhD from Imperial College London, UK (2015), supervised by Prof. Tom Welton and Dr. Patricia Hunt, on a combined experimental and computational project investigating the thermal and chemical stabilities of ionic liquids and their mixtures with carbohydrates. Subsequently, he was a Postdoctoral Research Fellow at the Max-Planck-Institut für Kohlenforschung, Mülheim-an-der-Ruhr, Germany (2015–2017), in the groups of Dr. Roberto Rinaldi and Prof. Ferdi Schüth, where his research centred on the development and characterisation of solvent systems for cellulose, and catalytic valorisation of lignocellulosic biomass. In April 2017 he joined Syngenta Ltd., UK, as a Senior Analytical Chemist. |
1. Introduction
Motivated by the pressing need for sustainable alternatives to fossil fuel resources in order to meet mankind's burgeoning material demands, lignocellulosic biomass has received widespread and growing interest in recent years. Biomass is primarily composed of three tightly-interwoven biopolymers: (i) cellulose, a linear and regular carbohydrate polymer that offers important structural integrity to the cell walls of plant species;1,2 (ii) lignin, a complex and irregular aromatic polymer constructed from para-(propenylated) phenol units that contributes toward cell wall rigidity;3,4 and (iii) hemicellulose, a comparatively low-molecular-weight branched polymer of (functionalised) pentose and hexose sugars, serving as a cross-linking motif between cellulose microfibrils and lignin.5 To achieve the maximum economic potential of an integrated biorefinery, challenges associated with the separation, dissolution and upgrading of these three biopolymers must be overcome, exploiting differences in solubility and reactivity under process conditions.6,7An estimated 56 × 109 tonnes of biomass is generated globally each year (as of 2005),1 of which cellulose accounts for the largest mass fraction (typically ≥40 wt% of dry biomass).2 In addition to its extraordinary abundance, the high carbon content and homopolymeric structure of cellulose render it an attractive candidate for valorisation to paper products, textile fibres, biofuels and composite materials (Fig. 1). However, despite the high concentration of hydrophilic hydroxyl substituents along the polymer backbone, cellulose exhibits vanishingly low solubility in water and the majority of conventional solvents because of the extensive network of hydrogen bonds (and van der Waals interactions).
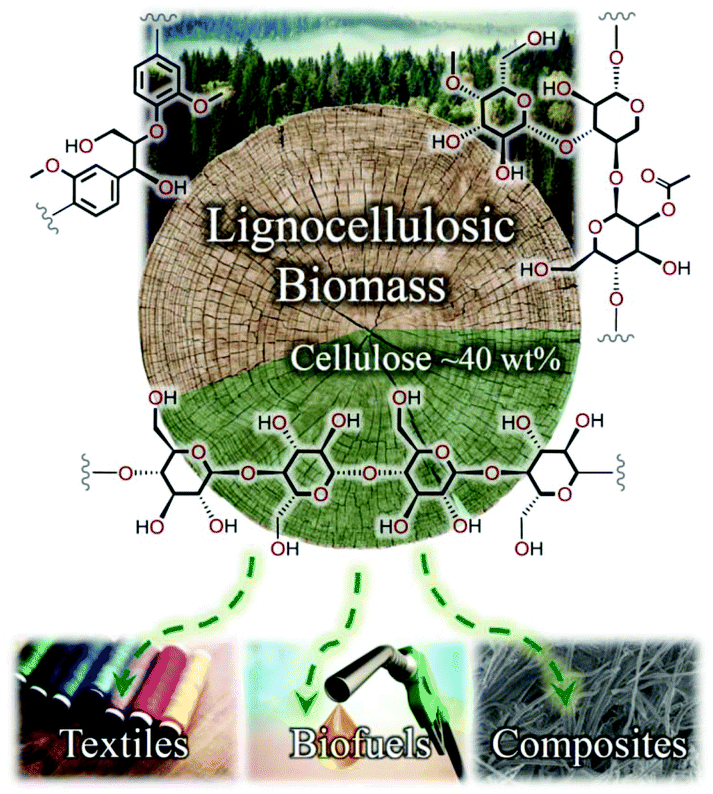 | ||
| Fig. 1 Selected target materials from cellulose: regeneration (e.g., electrospinning) into textile fibres; depolymerisation and fermentation to biofuels; or derivatisation, mixing and fabrication into composite materials (or hydrogels). | ||
Despite significant historical advances in the development of cellulose dissolution technologies, leading to established and successful large-scale industrial procedures (e.g., the Lenzing process, Austria), frequently-encountered problems associated with solvent toxicity, instability and limited recyclability point out the need for fundamental and applied research toward more sustainable alternatives. A solvent for cellulose should ideally meet several important criteria: (i) high solubility; (ii) rapid dissolution; (iii) facile solvent recyclability; (iv) minimal toxicity and flammability; and (v) insusceptibility to side reactions between solvent and solute. In this context, so-called ‘organic electrolyte solutions’ (OESs), mixtures of a (room-temperature) ionic liquid with an organic co-solvent, are promising candidates and have received growing attention since their first reports in the literature.8
This article is divided into six sections. Initially, a succinct overview of the key structural and bonding properties of cellulose is provided. Secondly, the evolution of cellulose dissolution strategies through the 19th and 20th centuries is described. Thirdly, the properties and behaviour of OES–cellulose systems are reviewed in detail. Next, examples of the successful application of OESs to cellulose derivatisation and valorisation are evaluated. The fifth section addresses the important question as to whether OESs can be considered ‘green solvents’ and, finally, key conclusions and future prospects of OESs are discussed.
The primary purpose of this review is to clarify the thermodynamic, kinetic and mechanistic properties that underpin cellulose dissolution in OES solvents, and to describe OES-based methodologies for the dissolution and valorisation of isolated cellulose.
OES solvents have been successfully applied to the fractionation of lignocellulose, for example purification of cellulose pulp by selective extraction of hemicelluloses in [C2C1im][OAc]–EtOH/acetone (IONOCELL-P9). Moreover, inclusion of an organic co-solvent (e.g., ethanolamine10) during pretreatment has been found to influence the efficiency of enzymatic hydrolysis of cellulose. Nevertheless for brevity, these topics are excluded from this review, instead focusing on procedures for the dissolution and valorisation of pre-isolated cellulosic materials. The interested reader is directed to a recent review article on the fractionation of biomass in ionic liquids and OESs.11
2. Cellulose structure and bonding
As the largest single component of hardwoods (43–47 wt%), softwoods (40–44 wt%) and bagasse (ca. 40 wt%),2 cellulose is the world's most abundant bio-renewable resource. It is a strong candidate for targeted upgrading towards textile fibres,12–18 biofuels,19–23 (nano)composites24–28 or hydrogels,29,30 not only because of its high natural abundance but also because of its well-defined structure of repeating glucopyranose residues, linked via β-1,4-glycosidic bonds. The β-configuration at the anomeric carbon gives rise to an extended linear geometry (differentiating cellulose from starch, with α-1,4 configuration and helical structure). The number of glucopyranose residues in a single cellulose chain (denoted the ‘degree of polymerisation’, DP) may reach 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 (e.g., in cotton or bacterial cellulose). Agglomeration of approximately 40–70 individual cellulose strands gives rise to a single ‘microfibril’, whereby strands are held together through an ordered arrangement of intramolecular and intermolecular hydrogen bonds and van der Waals interactions.31 Effective dissolution of cellulose hinges on the solvent's ability to dismantle this network of interactions and peel away individual strands from the microfibril.
000 (e.g., in cotton or bacterial cellulose). Agglomeration of approximately 40–70 individual cellulose strands gives rise to a single ‘microfibril’, whereby strands are held together through an ordered arrangement of intramolecular and intermolecular hydrogen bonds and van der Waals interactions.31 Effective dissolution of cellulose hinges on the solvent's ability to dismantle this network of interactions and peel away individual strands from the microfibril.
Various crystalline allomorphs of cellulose exist either in native organisms (cellulose Iα and Iβ, collectively referred to as cellulose I), or are attainable through annealing or treatment (cellulose II, IIII, IIIII, IVI and IVII).2 In native cellulose I, each glucopyranose residue participates in two intramolecular hydrogen bonds with neighbouring monomers (specifically O3H⋯O5 and O2H⋯O6), and one intermolecular hydrogen bond (linking O3 and O6 groups) with an adjacent strand in the same sheet, via the all-equatorial hydroxyl substituents. The arrangement of the heavy atom skeleton is identical in cellulose Iα and Iβ although the hydrogen-bonding patterns are subtly different. In contrast to cellulose II, neither cellulose Iα nor Iβ contain inter-sheet hydrogen bonds (sheets instead interact via van der Waals interactions).32 In nature, cellulose Iα is always accompanied to some degree by cellulose Iβ. The proportion of cellulose Iα may be as high as ca. 70% in primitive organisms (e.g., bacteria) and as low as 20% in higher plants. Diagrams showing representations of structures, hydrogen-bonding patterns and interconversion pathways of different cellulose allomorphs are displayed in Fig. 2.
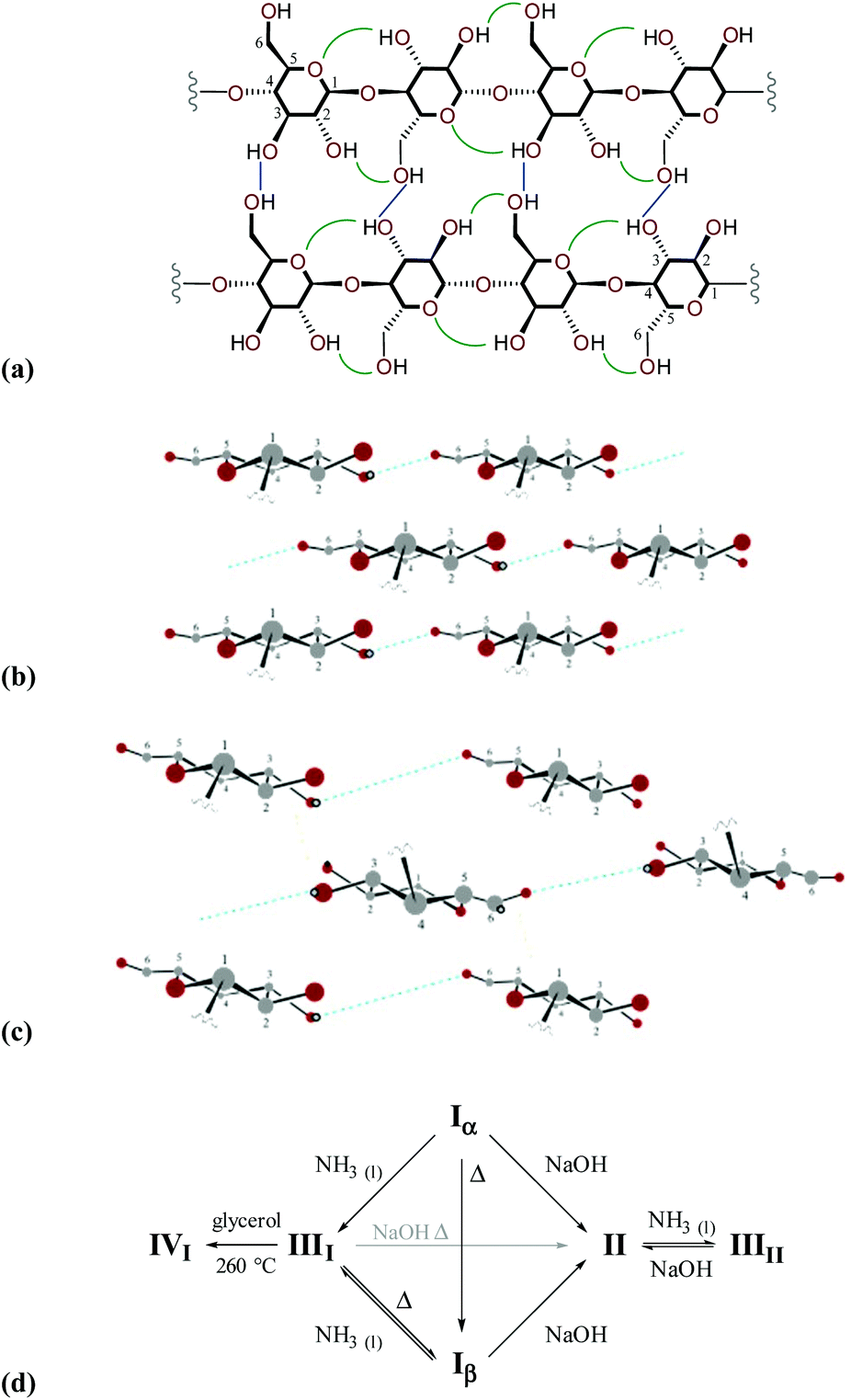 | ||
| Fig. 2 (a) Diagram highlighting the network of intramolecular (green) and intermolecular (blue) hydrogen-bonding interactions within a sheet of cellulose I; comparison of the arrangement of sheets within a microfibril of cellulose I (b) and cellulose II (c) (only hydrogen atoms that participate in hydrogen-bonding are shown, for clarity); and (d) routes for interconversion of cellulose allomorphs (adapted from D. Klemm et al., in Biopolymers Online, Wiley-VCH Verlag GmbH & Co. KGaA, 2005).2 | ||
Although naturally-occurring cellulose I is metastable, it typically undergoes a transformation to cellulose II during regeneration or mercarisation, suggesting that biosynthesis of cellulose fibrils does not proceed via the crystallisation of pre-formed chains. Instead, it has been proposed that chain growth occurs by sequential addition of glucopyranose units concurrent with crystallisation into fibrils. In higher plants, biosynthesis occurs with the participation of cellulose synthase complexes tethered to the cell plasma membrane (the interested reader is directed to recent review literature33,34).
When characterizing the mechanism of cellulose dissolution or derivatisation in a given solvent system, an understanding of the changes in (hydrogen-)bonding properties and specific solvent–solute interactions is essential. A brief overview of the history of cellulose dissolution strategies is provided in the next section, highlighting differences between early derivatizing solvent systems and more recent (formally non-derivatizing) technologies.
3. Evolution of cellulose dissolution strategies
Dissolution is likely to be a crucial stage of any cellulose valorisation technology. However, as discussed in the previous section, dissolving cellulose is challenging on account of the need to break the ordered and extensive network of strong intramolecular and intermolecular hydrogen-bonding35 and van der Waals32 interactions. Consequently, cellulose does not dissolve in the great majority of conventional solvents.Fig. 3 shows the evolution of cellulose dissolution technologies through the 19th and 20th centuries. Early technologies typically involved derivatisation to enhance its solubility (usually in water). In this manner, cellulose was shown to be soluble as cellulose nitrate (Schönbein, 1846), in the form of organo-soluble cellulose acetate (Schützenberger, 1865), or as cellulose xanthogenate, attainable by treatment with NaOH and CS2 (Cross, 1892).36 The latter procedure is the so-called ‘Viscose’ process and is still conducted today at industrial scale. Cellulose was shown to be solubilised by a cuprammonium hydroxide solution (Schweizer, 1857),37 whereby in the presence of ‘Schweizer's reagent’ ([Cu(NH3)4](OH)2), cellulose reacts to form a complex with copper involving bidentate interaction of the O2 and O3 hydroxyl residues (shown in Fig. 3). In each of the above processes, the added functional group enhances the solubility of the resultant polymer in the solvent media.
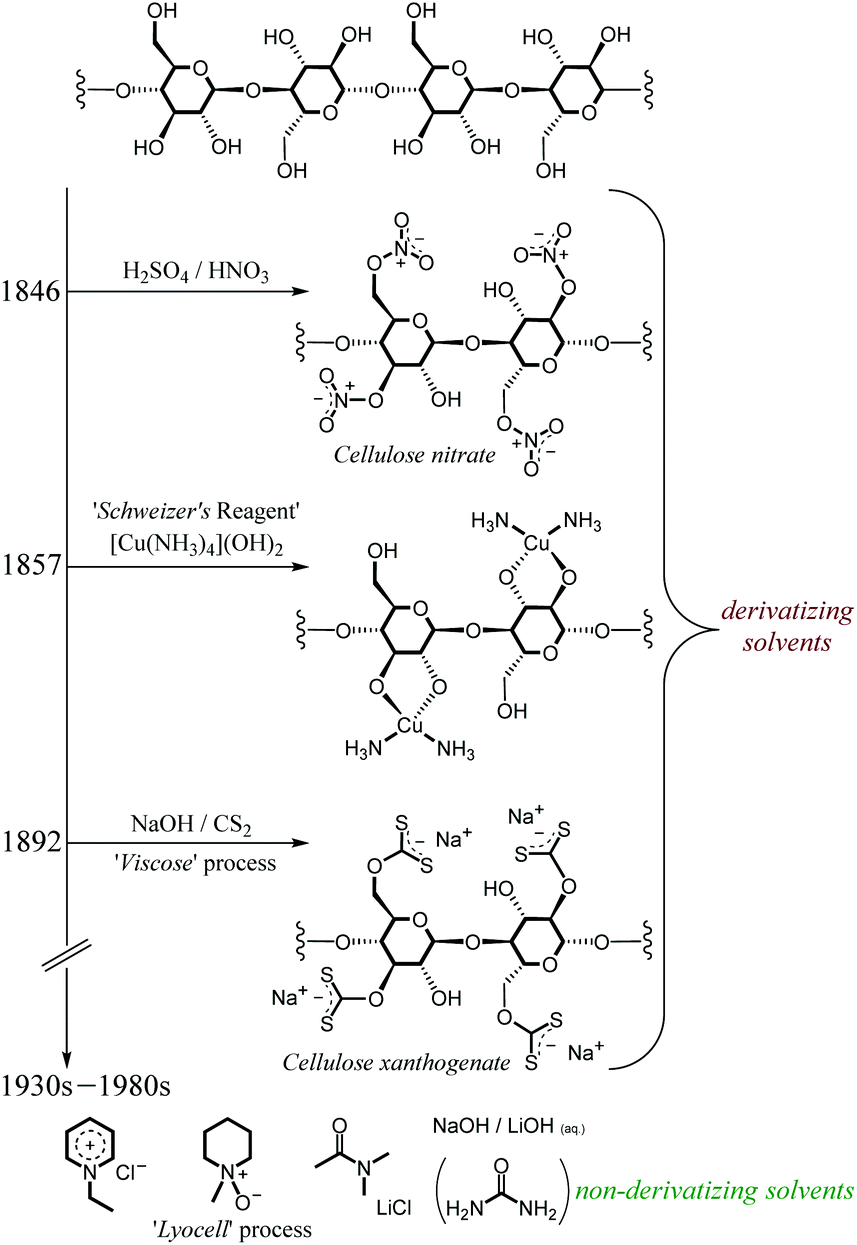 | ||
| Fig. 3 A simplified diagram representing the evolution of cellulose dissolution strategies through the 19th and 20th centuries. | ||
In the intervening years, cellulose has been shown to be soluble in a range of (formally non-derivatising) solvents, among them 1-ethylpyridinium chloride (Gränacher, 1934),38N-methylmorpholine-N-oxide (‘Lyocell’ process, American Enka, 1972),36 LiCl in dimethylacetamide (DMA) (McCormick, 1978),39–44 and simple aqueous solutions of group I metal hydroxides (with45,46 or without47–49 urea) (Kamide, 1987)49. However, such technologies encounter problems associated with solvent toxicity/flammability,36 severe thermal instability50–53 or poor recyclability.
Contemporary ionic liquids (ILs), salts that are molten at (or close to) ambient temperatures, were first successfully applied to the dissolution of cellulose by Swatloski et al. in 2002.54 The authors noted that up to 25 wt% of high-molecular-weight cellulose (DP ≈ 1000) dissolved in the archetypal IL 1-butyl-3-methylimidazolium chloride, [C4C1im]Cl, assisted by 3–5-second microwave heating pulses, leading to a clear viscous solution. Moreover, this article first demonstrated the strong sensitivity of IL–cellulose mixtures to the anti-solvent effect of water, a recurrent theme in subsequent literature.55–59 It is now established that pairing strongly hydrogen bond-basic anions (e.g. Cl−, [OAc]−, [HCO2]− or [(CH3O)2PO2]−), contributing to ‘Kamlet–Taft’ (KT) β values ≥0.8 (ref. 60–62) with moderately hydrogen bond-acidic cations (e.g., 1-alkyl-3-methylimidazolium, [CnC1im]+; 1,8-diazabicyclo[5.4.0]undecenium, [HDBU]+; tetraalkyl-phosphonium) generates ILs that are effective cellulose-dissolving agents (Fig. 4).27,54,61–72 Hauru et al. proposed that the difference between hydrogen bond basicity and acidity is a useful parameter for predicting the ability of a solvent to dissolve cellulose, whereby effective solvents exhibit β − α values in the range ca. 0.35–0.9.73 Although the associated cost74,75 and susceptibility to undergoing side reactions with cellulose76–82 have to be considered carefully, ILs could nevertheless offer profound benefits over traditional cellulose dissolution systems on account of their typically low toxicities (low vapour pressures) and their potential for continuous recycling.83–85
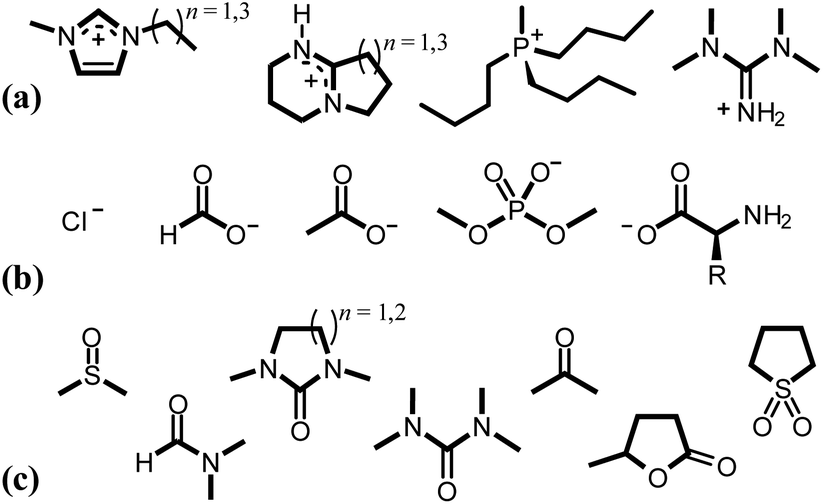 | ||
| Fig. 4 Typical cations (a), anions (b) and co-solvents (c) incorporated into (IL and) OES solvents for the dissolution of cellulose. | ||
Questions on the mechanism of cellulose dissolution in ILs, and the specific roles of the cation and anion in dismantling the network of hydrogen-bonded strands, have formed the basis of various experimental72,86–89 and theoretical55,59,90–97 studies. There is a broad concensus that bonding interactions between the IL anion and cellulose are the predominant driving force for dissolution of the biopolymer.59,87,90,92 It has been suggested that in [C4C1im]Cl–cellulose mixtures, the anion interacts with the equatorial hydroxyl substituents in a specific and directional manner (with 1:1 stoichiometry between Cl− anions and hydroxyl residues),87 although a Molecular Dynamics (MD) investigation of cellulose dissolved in the same IL demonstrated interactions of Cl− from both axial and equatorial directions.91 In addition, MD simulations have provided evidence that the cation occupies positions at the upper and lower faces of cellulose, interacting in a relatively non-specific/non-directional manner via hydrophobic or dispersion interactions.59,90,94,96,97 The cation may intercalate between individual cellulose strands,96 aiding in separation of the chains. Dissolution of cellulose in ILs is likely to be accompanied by increased polymer conformational flexibility55,86,95 and disruption of the O3H⋯O5 intramolecular hydrogen bonds between neighbouring glucopyranose monomers.55 Therefore, the chemical and electronic properties of both the anion and cation are of critical importance for effective cellulose dissolution in an IL.
4. Organic electrolyte solutions and cellulose: thermodynamic and mechanistic considerations
Although electrolytes of inorganic salts have been known as solvents for cellulose for many years (e.g., LiCl in DMA),39–44 analogous ‘organic electrolyte solutions’ (OESs) – mixtures incorporating a room-temperature ionic liquid (IL) and a polar organic co-solvent – were characterized in detail only recently (Rinaldi, 2011).8 Remarkably, it was shown by Rinaldi that complete dissolution of 10 wt% Avicel cellulose was possible even at very low mole fractions of the IL, e.g., χIL = 0.08 ([C4C1im]Cl–DMSO), 0.10 ([C4C1im]Cl–DMF) or 0.18 ([C4C1im]Cl with 1,3-dimethyl-2-imidazolidinone, DMI).8 Moreover, it was observed that complete dissolution was achieved far more rapidly in the OES than in the corresponding neat IL (e.g., <3 min for 10 wt% cellulose in 50:50 wt/wt [C4C1im]Cl–DMI, cf. >10 h for 5 wt% cellulose in [C4C1im]Cl65). The inclusion of a co-solvent has, in one instance, been shown to enhance the solubility of cellulose98 and may even enable dissolution where the neat IL is incapable of doing so.99 Example ions and co-solvents that contribute to effective cellulose dissolution are shown in Fig. 4. A flow diagram to aid in the appropriate choice of cation, anion and co-solvent combinations for cellulose dissolution is shown in Fig. 5.
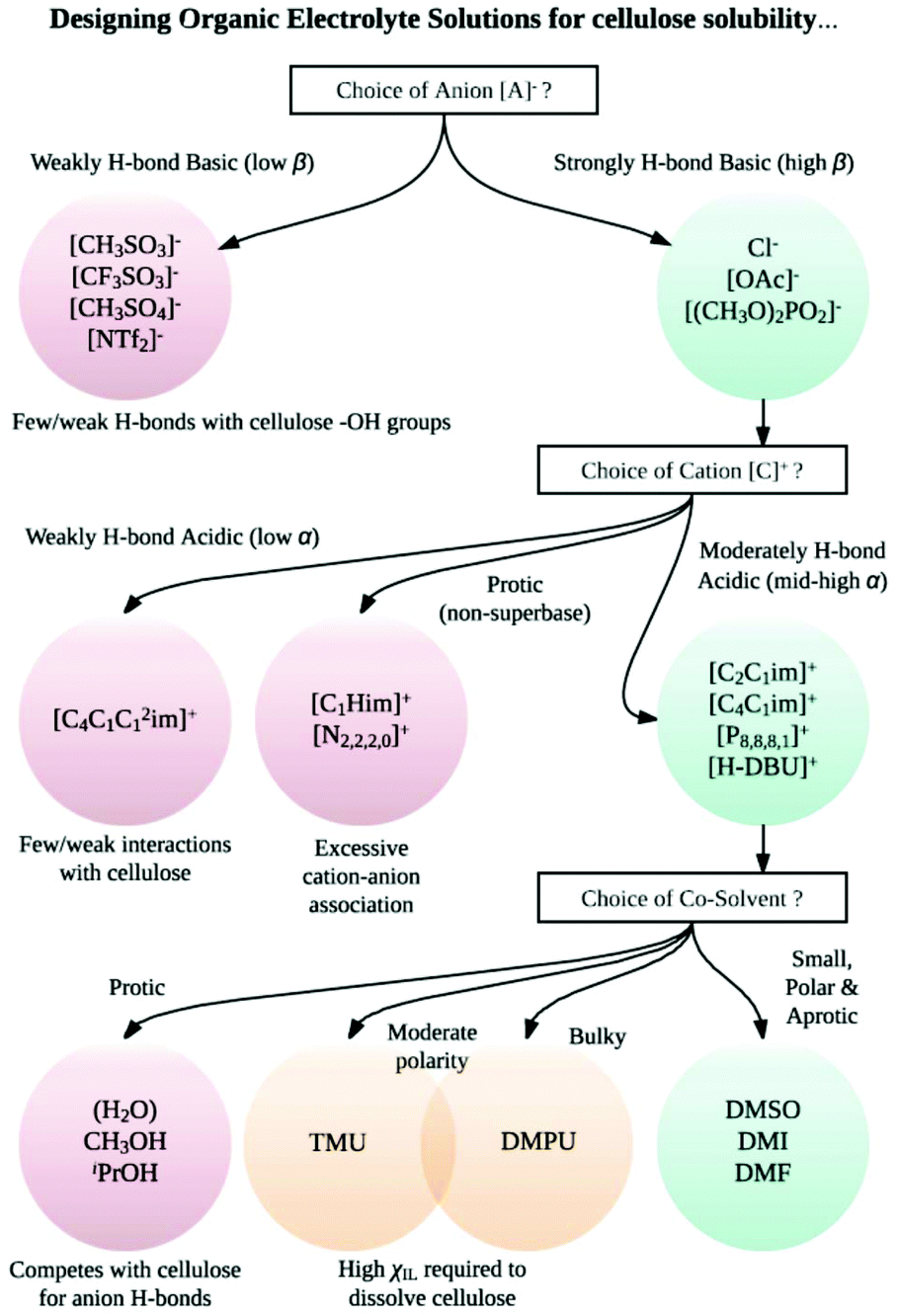 | ||
| Fig. 5 A flow diagram to aid in the selection of sensible cation–anion–co-solvent combinations for effective cellulose dissolution. The diagram is intended to highlight key structural and electronic factors that influence cellulose solubility, and is not exhaustive. [NTf2]− = bis(trifluoromethanesulfonyl)imide; [OAc]− = acetate; [C4C1C12im]+ = 1-butyl-2,3-dimethylimidazolium; [C1Him]+ = 1-methylimidazolium; [N2,2,2,0]+ = triethylammonium; [C2C1im]+ = 1-ethyl-3-methylimidazolium; [C4C1im]+ = 1-butyl-3-methylimidazolium; [P8,8,8,1]+ = tri(n-octyl)methylphosphonium; [H-DBU]+ = 1,8-diazabicyclo[5.4.0]undecenium; TMU = 1,1,3,3-tetramethylurea; DMPU = 1,3-dimethyl-3,4,5,6-tetrahydro-2-pyrimidinone; DMI = 1,3-dimethyl-2-imidazolidinone. | ||
A variety of different experimental methods for dissolving cellulose in OESs have been described. In many instances cellulose was added – either in small aliquots or as a single addition – to a pre-mixed solution containing the desired stoichiometry of IL and co-solvent.99–102 For instance, Huang et al. described addition of various cellulose materials to mixtures of tetra(n-butyl)ammonium acetate and DMSO at 25 °C.101 The authors observed that the time required to dissolve 4 wt% of microcrystalline cellulose (MCC) was influenced by the mass fraction of IL in the solvent, such that complete dissolution was most rapid (T < 2 min) at WIL = 0.15. Understandably, dissolution time increased incrementally with the DP of the cellulosic material. Comparative studies of this type are of great value in elucidating the little-known effects of e.g. IL mole fraction and DP on the practicalities associated with preparing OES-cellulose mixtures.
Another method for preparation of OES–cellulose mixtures is the initial dispersion of cellulose in the co-solvent, followed by addition of the IL. In one early example, cellulose was suspended in DMSO prior to the addition of tetra(n-butyl)ammonium fluoride (TBAF), after which complete dissolution was attained within 15 min.103 Similarly, by first preparing a suspension of Avicel in DMI at 100 °C, agglomeration of biopolymer fibres was prevented; upon subsequent addition of the IL, complete dissolution was attained in a remarkably short time frame (<3 min with [C4C1im]Cl or instantaneous with [C2C1im][OAc]).8 In preparing dope solutions for electrospinning of cellulose fibers (refer to section 5.1, below), Härdelin et al. describe treating small pre-dried square pieces of pulp material first with co-solvent, followed after 1 minute by addition of the IL.104 The authors observed that complete dissolution was less time-consuming following this procedure, and they rationalised that absorption of the co-solvent enhanced the rate of ion diffusion into the pulp. Future investigations into the exact influence of co-solvent pretreatment on the uptake of IL into the microfibrils would be of significant interest.
An important, recurring observation in studies of OESs and cellulose is that the specific electronic (and steric) properties of the co-solvent are of critical importance in determining cellulose solubility. For example, from the outset it was noted that OESs derived from DMI and 1,1,3,3-tetramethylurea (TMU), close structural analogues, required vastly different quantities of [C2C1im][OAc] for complete dissolution of 10 wt% cellulose (χIL = 0.18 and 0.59, respectively).8 Similarly Holding and co-workers demonstrated that, although OES solvent systems composed of tri(n-octyl)methylphosphonium acetate and DMSO were capable of dissolving up to 19 wt% cellulose, the analogous mixtures with DMF exhibited negligibly low cellulose solubility.99 Pinkert put forward a qualitative argument for the difference in co-solvent capability of DMI and TMU, suggesting that the increased structural rigidity of DMI and static nature of the N π-orbitals (which can readily hybridize to sp2) enables DMI to better assist with correct positioning of the ions at the hydrophilic and hydrophobic sites of cellulose.105 However, gas-phase Natural Bonding Orbital (NBO) calculations suggested that the energies of DMI and TMU frontier molecular orbitals are approximately equal.106 More likely, differences in dipole moment and subtle steric properties explain the contrasting abilities of co-solvent species to aid in cellulose dissolution (Fig. 6).106
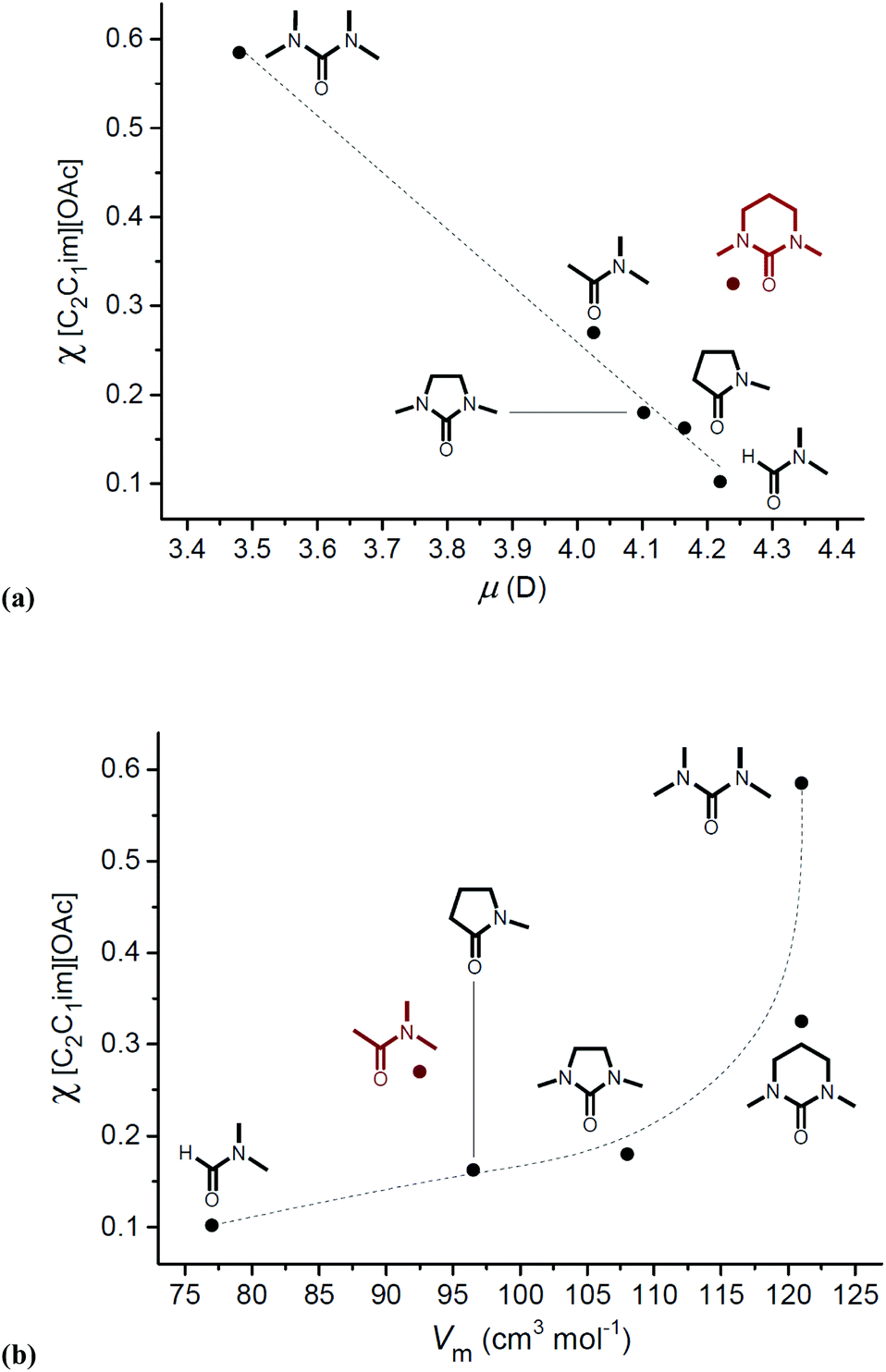 | ||
| Fig. 6 Understanding the factors contributing to an effective OES co-solvent: relationship between minimum [C2C1im][OAc] mole fraction required for 10 wt% cellulose dissolution, χIL, and the co-solvent dipole moment (μ), (a), and molar volume (Vm), (b). Adapted with permission from J. Chem. Eng. Data, 2012, 57, 1341–1343. Copyright 2012 American Chemical Society. | ||
The capabilities of IL solvents to dissolve cellulose have been shown to correlate62 with the KT empirical solvation parameters α (hydrogen bond acidity),107β (hydrogen bond basicity)60 and π* (polarity/polarisability).108 Interestingly, for OES solvents derived from [C2C1im][OAc] and DMI, the β and π* values do not change significantly over the range χIL = 0.1–1.0. The hydrogen bond basicity (β, largely influenced by the nature of the anion) and polarisability properties are therefore not significantly perturbed up to χDMI ≈ 0.90. By contrast, the hydrogen bond acidity (α, largely influenced by the nature of the cation) appears to partially decrease in the direction χIL = 1.0 → 0.1.8 Below χIL ≈ 0.1 the β, α (and to a lesser extent π*) values fall sharply, associated with the observed loss in cellulose solubility (Fig. 7).8 Gericke et al. studied the influence of adding aliquots of a co-solvent (or co-solvent binary mixture) to 10 wt% cellulose solutions in the ILs 1-allyl-3-methylimidazolium chloride ([C1![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C2C1im]Cl), [C4C1im]Cl and [C2C1im][OAc].109 It was concluded that, in order to be miscible with the initial mixtures of IL and cellulose, a co-solvent must typically have solvent parameter values within certain thresholds (β > 0.4, π* > 0.8, normalized empirical polarity (ETN) > 0.3). Strongly dipolar and hydrogen-bond-basic co-solvents (e.g. DMSO, DMF) could be added in ≥1:1 wtco-solvent/wtIL–cellulose quantities before cellulose precipitation occurred, for all three investigated ILs. By contrast, protic co-solvents, characterized by high α values, caused immediate and permanent cellulose precipitation at 0.2:1 wtco-solvent/wtIL–cellulose, attributed to the tendency of the co-solvent to compete with cellulose for interactions with the IL anions.
C2C1im]Cl), [C4C1im]Cl and [C2C1im][OAc].109 It was concluded that, in order to be miscible with the initial mixtures of IL and cellulose, a co-solvent must typically have solvent parameter values within certain thresholds (β > 0.4, π* > 0.8, normalized empirical polarity (ETN) > 0.3). Strongly dipolar and hydrogen-bond-basic co-solvents (e.g. DMSO, DMF) could be added in ≥1:1 wtco-solvent/wtIL–cellulose quantities before cellulose precipitation occurred, for all three investigated ILs. By contrast, protic co-solvents, characterized by high α values, caused immediate and permanent cellulose precipitation at 0.2:1 wtco-solvent/wtIL–cellulose, attributed to the tendency of the co-solvent to compete with cellulose for interactions with the IL anions.
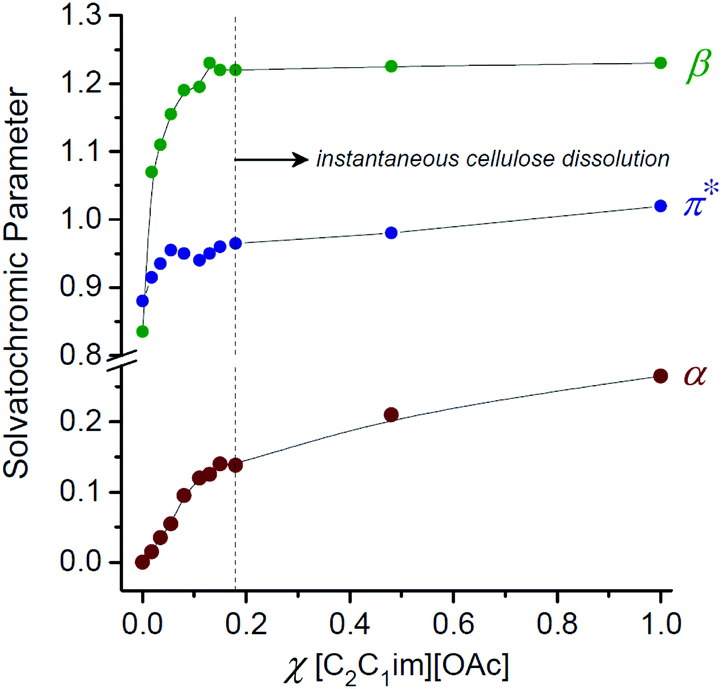 | ||
| Fig. 7 The influence of solvent composition on the Kamlet–Taft empirical solvation parameters α, β and π*, for [C2C1im][OAc]–1,3-dimethyl-2-imidazolidinone (DMI) solvents. Adapted from ref. 8 with permission from the Royal Society of Chemistry. | ||
Although OESs have emerged as an independent class of solvent for cellulose, the mechanisms that underpin dissolution remain only partially understood. As for pure ILs,59,87,90,92 there is almost unanimous agreement that strong, directional hydrogen-bonding interactions that form between the anions and the hydroxyl residues of cellulose are the driving force behind dissolution in an OES.101,110–113 Nevertheless, the precise role of the co-solvent in enabling rapid or instantaneous cellulose dissolution is the subject of some debate.
Xu et al. demonstrated that the addition of aliquots of DMSO to [C4C1im][OAc] brought about a considerable increase in ion conductivity (maximum conductivity occurred at χDMSO/χIL ≈ 5).110 They hypothesized that preferential solvation of cations by DMSO helped to increase the availability of dissociated or ‘free’ [OAc]− anions, which were capable of accepting hydrogen bonds with the hydroxyl units of cellulose more readily. Similarly, from results of MD simulations on [C4C1im][OAc]–DMSO and [C4C1im][OAc]–DMF systems, Zhao and colleagues suggested a partial separation of the cation–anion association by the co-solvent. Once again, a partial breakdown in associative interactions between cations and anions was thought to indirectly account for rapid dissolution of cellulose, though in this instance no preferential solvation of cations or anions by DMSO or DMF was evidenced by the simulations.111
Subsequent studies have challenged the concept of ion pair loosening. Andanson et al. undertook a comparison of [C4C1im][OAc] vs. [C4C1im][OAc]–DMSO (50:50 mol/mol), with the use of both experimental (viscometry, conductivity) and computational (MD) techniques.114 Conductivity was certainly enhanced with the inclusion of DMSO, in agreement with Xu et al.110 However, this was attributed to improved mass transport of ions (lower viscosity) and not due to a significant change to either ion–ion or ion–cellulose interactions, on the basis that the Walden plot indicated no increase in the number of charge carriers with increased DMSO concentration (Fig. 8).114 This was further supported by an analysis of the radial distribution functions from MD simulations, which indicated that inclusion of DMSO did not perturb the pre-existing network of hydrogen bonds between cations and anions.114 An MD study of the dissolution of a 36-strand cellulose microfibril in equimolar [C4C1im][OAc]–DMSO indicated that DMSO does not establish specific associative interactions with either ions or cellulose.113 On this basis, Velioglu et al. referred to DMSO as an ‘innocent’ co-solvent.
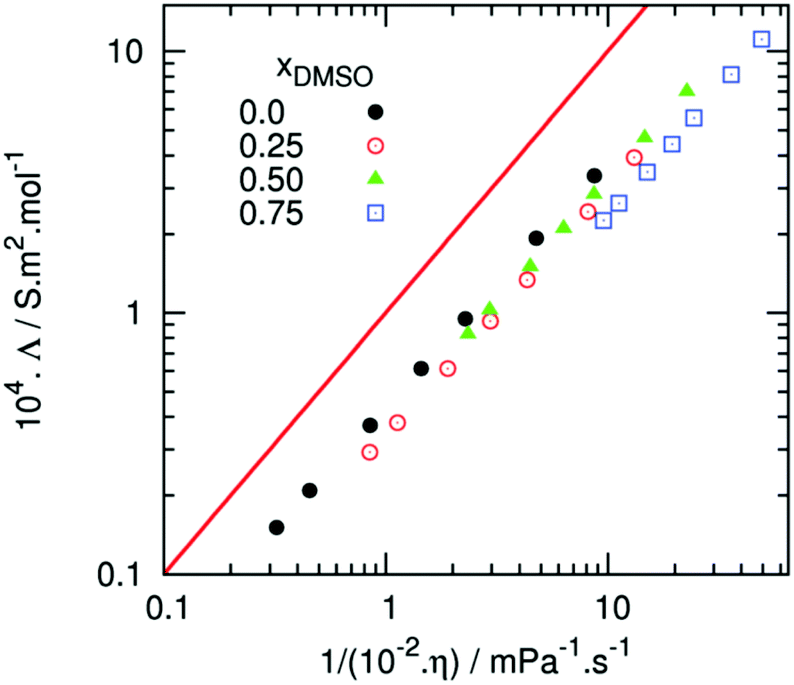 | ||
| Fig. 8 Walden plot comparing the viscosity and conductivity values for binary [C4C1im][OAc]–DMSO mixtures (χDMSO = 0, 0.25, 0.5, 0.75), reproduced from ref. 107 with permission from the Royal Society of Chemistry. The red line denotes the reference ionicity of 0.01 M aqueous KCl, at 25 °C. | ||
Overall, considering this collective data on [C4C1im][OAc]–DMSO OESs, despite apparent separation of [C4C1im][OAc] ion pairs, closer inspection indicates that strong cation–anion association is not significantly perturbed by addition of DMSO to the parent IL.
The extent of ion pair loosening may vary considerably between different IL–co-solvent combinations; trends that have been observed for [C4C1im][OAc]–DMSO OESs therefore may not be replicated in all instances. As an example, Huang et al. recently proposed that mixtures of tetra(n-butyl)ammonium acetate and DMSO exhibit distinct ‘ion-split’ and ‘ion-paired’ stages (in the absence of cellulose), at different mass fractions of the IL (0.05 ≤ WIL ≤ 0.15 and 0.15 ≤ WIL ≤ 0.25, respectively).101 The authors recognised that balancing the ion concentration and ion ‘mobility’ was the key to ensuring maximum solubility of cellulose, occuring at WIL = ca. 0.15 for this specific OES. A recent NMR relaxometry investigation highlighted a similar duality for 1-allyl-3-methylimidazolium chloride–DMSO mixtures: at low χDMSO, evidence suggested that the co-solvent molecules insert into the IL phase and loosen the cation–anion interactions, whereas high χDMSO values (>0.5) promote clustering of ions, restricting the ability of ions to form favourable hydrogen-bonding interactions with cellulose.115 These data confirm a phenomenon noted in the earlier literature – that subtle differences in electronic and steric properties of the IL ions and co-solvent may strongly influence the cellulose-solvating behaviour of the resultant OES.8,105,106
The above-described improvement in mass transport of ions in an OES, relative to a simple IL, probably explains the high rate at which cellulose dissolves,8 whereby cations and anions can rapidly orientate themselves along the cellulose backbone and participate in favourable hydrogen-bonding interactions. Although this kinetic effect has been noted in multiple literature reports, thermodynamic data relating to dissolution of cellulose in OESs is relatively scarce. Polymer dissolution in a molecular solvent is usually an endothermic event (favoured by operating at higher temperatures).116 However, cellulose dissolution in ILs is typically exothermic (ΔH < 0 kJ mol−1, whereby formation of ion–cellulose interactions more than compensates for breaking of cellulose–cellulose and ion–ion interactions). Dissolution of any polymer is usually accompanied by a small entropic gain (ΔS > 0 J mol−1).117 Therefore, to maintain spontaneous cellulose solubility in an OES (ΔG < 0 kJ mol−1), the enthalpy term must remain approximately neutral or exothermic after dilution by the co-solvent (ΔG = ΔH − TΔS).
Andanson et al. used solution calorimetry to compare the heat of cellulose dissolution in [C2C1im][OAc] against dissolution in equimolar [C2C1im][OAc]–DMSO.89 The inclusion of 50 mol% DMSO brought about a surprisingly minimal reduction in exothermicity (at 80 °C), from −132 ± 8 J g−1 to −106 J g−1, despite a significant dilution of ions. In a subsequent study, solution calorimetry was also used to measure and compare the enthalpies of mixing for cellobiose (as a model for cellulose) in IL and OES solvents, ΔmixH(CB).118 The mole fractions of IL ([C4C1im]Cl or [C4C1im][OAc]) and co-solvent (DMSO, DMI or DMF) were systematically varied, and the ΔmixH(CB) values were determined (at 25 °C). The ΔmixH(CB) profiles as a function of χIL for mixtures with DMSO, DMI and DMF were strikingly different (Fig. 9). For the [C4C1im]Cl–DMI and [C4C1im][OAc]–DMSO series, ΔmixH(CB) rose sharply towards endothermic values from χIL = 0.1 → 0. However, in agreement with the observation of Andanson et al.,89 changes in ΔmixH(CB) in the broad range χIL = 0.1–1 were comparatively small for all mixture series. Therefore despite a small loss in exothermicity by inclusion of a co-solvent, the dissolution of cellulose in an OES is likely to be rapid on account of both thermodynamic and kinetic favourability. When χIL falls below a particular threshold (dependent on the specific choice of IL and co-solvent), cellulose solubilisation is no longer thermodynamically preferable, and spontaneous dissolution of cellulose does not occur.
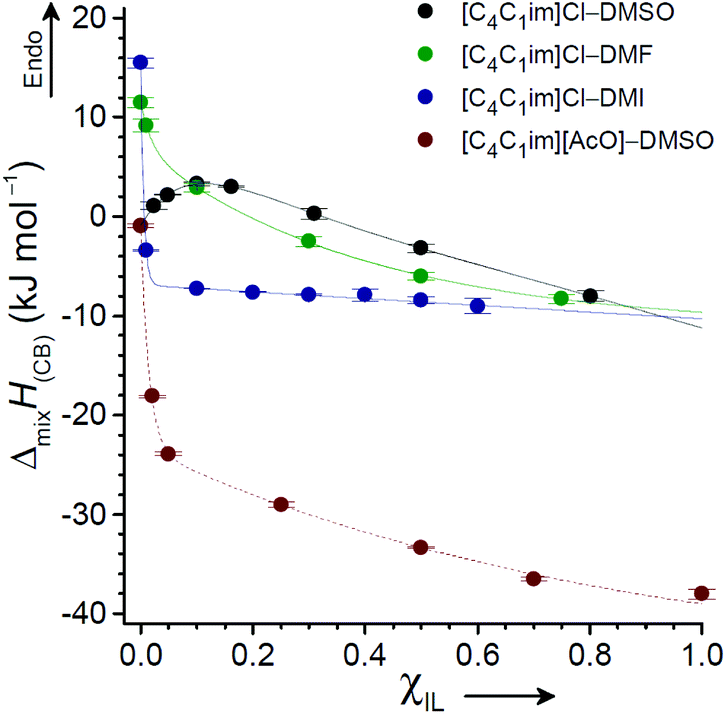 | ||
| Fig. 9 Relationship between χIL and the heat of cellobiose mixing, ΔmixH(CB), for OES solvents derived from [C4C1im]Cl or [C4C1im][OAc] with DMSO, DMI or DMF. Measurements were obtained at 25 °C. Adapted with permission from ChemSusChem, 2015, 8, 1577–1584. Wiley-VCH Verlag GmbH & Co. KGaA, 2015. | ||
5. Cellulose derivatisation and valorisation
Building on the success of ILs for the dissolution, derivatisation and regeneration of cellulosic materials,27,62,71 OESs have received growing attention on account of the additional ‘handle’ for controlling the physicochemical properties of the solvent. Foremost, the ability to modify viscoelastic and rheological properties by inclusion of a co-solvent, whilst still retaining high cellulose solubility, is an attractive aspect. Key examples of the implementation of OES systems toward cellulose derivatisation and valorisation are summarized in the following subsections.5.1 Fibre electrospinning
Continuous and regulated production of biopolymer fibres can be achieved with the use of an ‘electrospinning’ apparatus. Electrospinning functions by applying a high voltage difference between a solvent–polymer solution, contained within a syringe, and a collection vessel. A syringe pump regulates the rate at which the solvent–polymer solution is dispensed from the syringe, through a narrow ‘spinneret’ (often a needle). A pendant droplet forms at the spinneret and, once the voltage difference becomes great enough, the droplet deforms into a conical tip denoted a ‘Taylor cone’.119 A thin and heavily-charged stream of solvent–polymer solution is then drawn towards the collection vessel. When a volatile organic solvent is used, solvent evaporation occurs at this stage. Conversely with (non-volatile) IL or OES solvents,13,14,18,120,121 the solution is discharged into a collection bath (e.g. water14 or EtOH122), causing precipitation of the polymer fibres. Therefore, use of an IL (or OES incorporating a relatively non-volatile co-solvent) mitigates environmental concerns associated with the release of volatile and hazardous organic components, which are typically not recycled.123 A simplified diagram representing an IL or OES electrospinning apparatus is shown in Fig. 10.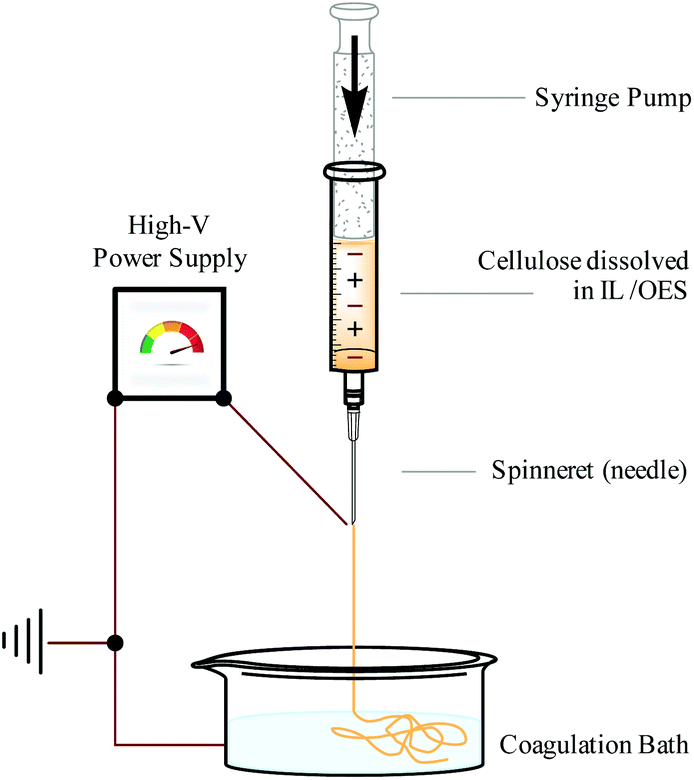 | ||
| Fig. 10 Apparatus (simplified) for continuous and regulated electrospinning of cellulose fibers from an IL or OES solvent system, adapted from ref. 10 with permission from the Royal Society of Chemistry. | ||
Electrospun fibers typically exhibit the beneficial properties of high porosity,124 large specific surface area125 and well-regulated fibre diameter. Such properties are readily controllable by modification of process parameters (e.g. the field strength, feed rate, distance between spinneret and collection vessel), solution parameters (e.g. concentration, polymer molecular weight, surface tension, viscosity),126–129 or ambient conditions.130,131 Considering solution parameters, it is established that low viscosity is a key property that broadly differentiates OESs from their corresponding ILs. For example, Lv et al. studied solutions of a cotton-derived cellulose (DP = 650) in [C4C1im]Cl–DMSO and [C1C2C1im]Cl–DMSO solvents, at 25 °C.132 For mixtures incorporating 1.5 wt% cellulose, exponential reduction in viscosity was observed in the range χDMSO 0 → 0.7, mirroring the behaviour of OES solvents without cellulose. In a later investigation of [C4C1im]Cl–DMSO–cellulose mixtures by Saba et al. (5 wt% cellulose, DP = 500), non-linear viscosity behaviour was observed in the range 5–8 wt% DMSO, at temperatures of 50–80 °C (Fig. 11).133 The non-linearity was attributed to microscopic aggregation or phase separation in this region. Although in one instance a higher viscosity was observed for an OES–cellulose mixture ([C2C1im][OAc]/DMSO 90:10 wt/wt) than the equivalent neat IL–cellulose solution,104 the general trend of decreasing viscosity upon addition of the molecular co-solvent still holds.
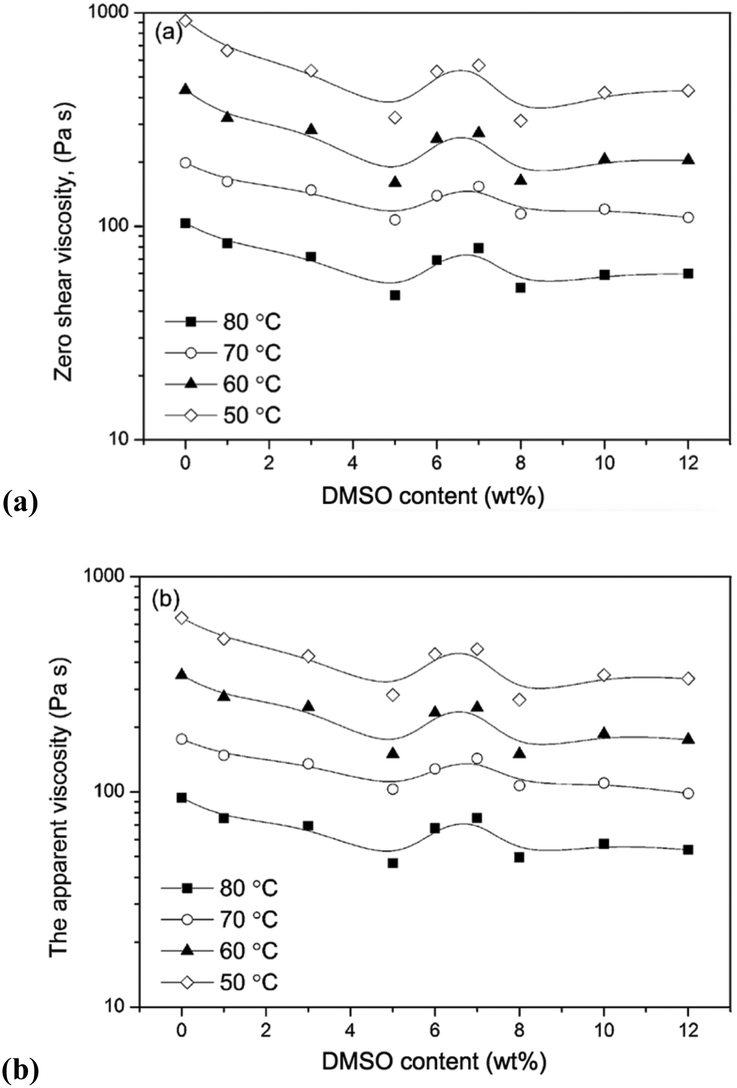 | ||
| Fig. 11 Influence of DMSO content (wt%) on zero-shear-rate viscosity, (a), and apparent viscosity at shear rate 1 s−1, (b), for [C4C1im]Cl–DMSO–cellulose ternary mixtures (5 wt% cellulose), at 50–80 °C (solid lines are provided to guide eyes). Adapted from ref. 126 with permission from the Royal Society of Chemistry. | ||
Prior to OESs emerging as an independent class of solvents for cellulose,8 the inclusion of a co-solvent had been used as a strategy for controlling the rheological properties of IL–cellulose mixtures for electrospinning processes. For example, Xu and co-workers explored the influence of DMSO concentration on the electrospinning properties of cellulose (from cotton linters, DP = 1600) dissolved in [C1C2C1im]Cl.12 DMSO reduced the viscosity,114,132–134 surface tension and entanglement density, thereby contributing toward a continuous electrospinning jet. The authors identified that a sufficient cellulose loading was necessary to ensure effective entanglement. At a fixed cellulose loading of 5 wt%, excessive quantities of DMSO (e.g., 8:1 DMSO/[C1C2C1im]Cl wt/wt) led to electrospraying of the mixture. Insufficient DMSO (e.g. 1:1 DMSO/[C1C2C1im]Cl wt/wt) afforded a mixture with poor electrospinning capability, on account of the high viscosity. Therefore, it is worth noting that the solution parameters cannot be considered in isolation when developing OES–cellulose electrospinning processes.12
Härdelin et al. conducted a detailed investigation of the influence of DMA, DMF and DMSO co-solvents on the ability of [C2C1im][OAc] to electrospin cellulose fibres (from dissolving pulp, DP = 750).104 Interestingly, although each of the three co-solvents brought about a reduction in the surface tension of the mixture, DMA and DMF had a significantly more pronounced effect than DMSO. The OES–cellulose mixtures incorporating DMSO exhibited the greatest degree of ‘shear thinning’, leading to optimal fibre formation (Fig. 12).
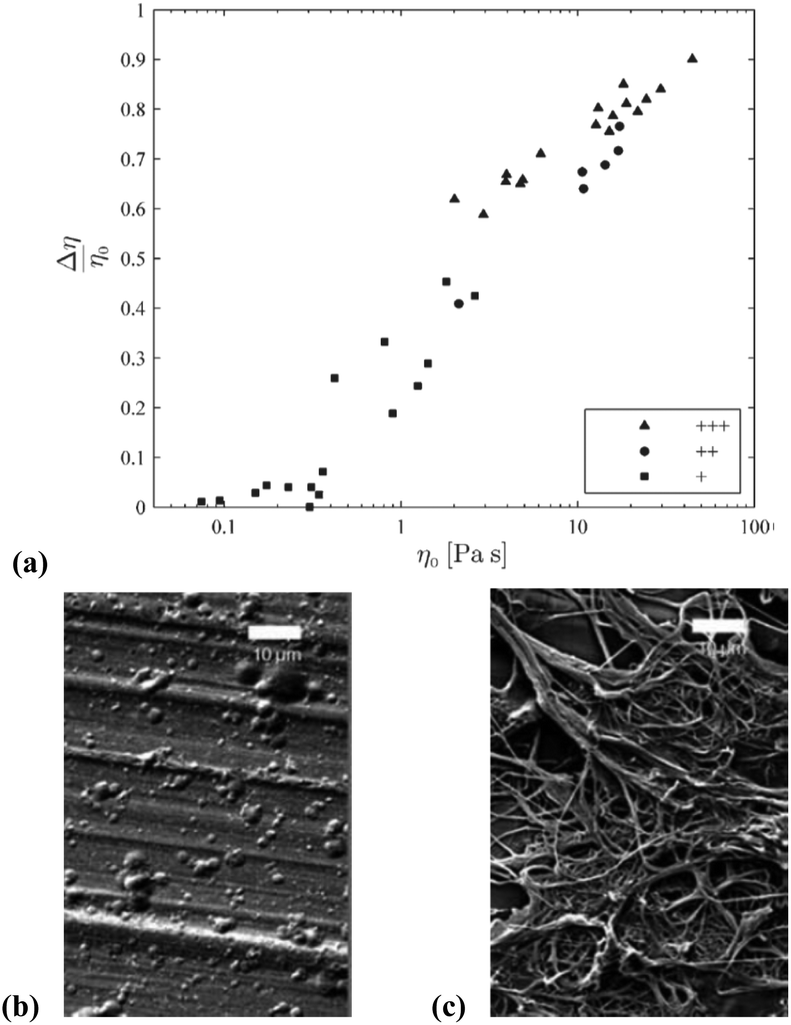 | ||
| Fig. 12 (a) Relationship between shear thinning of OES–cellulose mixtures, and the propensity for fibre formation: + denotes no fibre formation; ++ denotes partial fibre formation; +++ denotes good fibre formation. Scanning Electron Microscopy (SEM) images for example ‘+’ and ‘+++’ cellulose electrospinning experiments are shown in (b) and (c), respectively (scale bars denote 10 μm). Reproduced and adapted with permission from J. Appl. Polym. Sci., 2012, 125, 1901–1909. Wiley-VCH Verlag GmbH & Co. KGaA, 2012. | ||
Therefore, in analogy to the differing abilities of co-solvent species to aid and promote cellulose dissolution (see section 4, above), the specific choice of co-solvent plays an important role in determining the precise solution parameters for cellulose electrospinning procedures. Whilst the inclusion of a co-solvent may offer an additional handle for controlling viscoelastic properties of the mixture, the solution parameters must be considered collectively in order for successful electrospinning of cellulose fibres to be achieved.
5.2 Solution-state NMR spectroscopy of cellulose
The low viscosities of OES–cellulose mixtures offer potential benefits beyond tailored electrospinning. Typically, cellulose solutions are of prohibitively high viscosity for high-resolution NMR spectroscopy measurements, with broad and poorly-resolved 1H signals. Because of this, whilst solid-state (e.g., 13C ‘Cross-Polarisation–Magic-Angle Spinning’) NMR spectroscopy has been successfully applied to cellulose,135,136 solution-state measurements are comparatively uncommon.Nevertheless, Holding et al. were able to collect high-resolution 1D and 2D NMR data of high-Mw cellulose, at ≥8 wt% concentration, dissolved in tri(n-octyl)methylphosphonium acetate–DMSO-d6 (50:50 wt/wt).137 The sharpening of cellulose 1H signals in this low-viscosity medium enabled glucopyranose residues at the reducing and non-reducing ends to be distinguished from those embedded within the chain. Cellulose DP values could therefore be semi-quantified via integration of the respective signals. Whilst the DP values of low-Mw celluloses (DP ≤ 100), calculated by NMR, aligned closely with the results of Gel Permeation Chromatography (GPC) analysis, the increased difficulty in spectral resolution for long-chain cellulose polymers (up to DP > 600) led to poor correlation between NMR and GPC assays. In spite of this, such methodologies employing OES solvents hold promise for future in situ NMR analyses of cellulose depolymerisation and derivatisation reactions.
5.3 Cellulose derivatisation
The addition of a co-solvent offers a dual benefit for homogeneous cellulose derivatisation reactions performed in IL-type solvents: reduced viscosity, and improved miscibility with hydrophobic reagents.109 The co-solvent can aid in the miscibility of common derivatisation agents for cellulose, such as acetic anhydride or acetyl chloride. As early as 1999, Heinze and co-workers demonstrated the use of an OES-type solvent, tetra(n-butyl)ammonium fluoride trihydrate with DMSO, to dissolve high-Mw cellulose (DP > 600).138 This solvent system was successfully used to carboxymethylate138 and acylate103 cellulose samples. Nevertheless, the use of such systems was hampered by hydrolysis of anhydrides and acid chlorides as a result of residual water in the solvent,103 and the high energy input required for solvent recycling.139More recently, homogeneous acetylation of microcrystalline cellulose (MCC, 5 wt%) was performed in solvent systems comprised of triethyl(n-octyl)ammonium chloride with acetone or DMA (9:10 wt/wt). Results were compared against reactions carried out in conventional LiCl/DMA and [C4C1im]Cl solvents.140 Strikingly, solvents containing tetraalkylammonium salts were advantageous because they gave rapid dissolution of cellulose, without the need for prior activation or prolonged heating. Using acetyl chloride as the derivatising agent, a similar degree of substitution was observed when comparing reactions performed in the tetraalkylammonium systems against those using LiCl/DMA and [C4C1im]Cl.
Although examples of procedures for the derivatisation of cellulose using OESs are currently scarce, the potential benefits associated with improved IL–reagent miscibility are significant, serving as another example of how inclusion of a co-solvent could tailor the properties of an IL solvent toward improved outcomes.
5.4 Phase separation
Although OES solvents are capable of dissolving high quantities of cellulose at remarkably low values of χIL, excessive dilution of the ions (e.g., χIL ≤ 0.08 for [C4C1im]Cl–DMSO, 10 wt% Avicel cellulose8) leads to systems where cellulose dissolution is no longer thermodynamically favourable. Recently, OES–cellulose mixtures that exhibit thermally triggered and reversible phase separation were reported.141 The mixtures, derived from [C2C1im][OAc], DMI and MCC, could be easily handled as a single, homogeneous and low-viscosity mixture at elevated temperatures. Upon cooling, the mixtures passed through a well-defined ‘temperature of phase separation’ (TPS), and separated into two distinct liquid phases. Cellulose was found to be wholly retained within the lower IL-enriched phase, whereas the upper phase was dominated by the co-solvent, DMI. Interestingly, the TPS parameter could be tailored over a wide range of temperatures (ΔTPS ≈ 75 °C) by small changes to the IL and DMI mole fractions (Δχ ± 0.05) (Fig. 13a). Although such phase-separating OES–cellulose mixtures have not yet been applied to cellulose dissolution or derivatisation, it can be envisaged that they may find use for derivatisation to high-value-added products (e.g., chiral stationary phases for liquid chromatography), exploiting trivial separation of the cellulosic product from residual lipophilic reagents.141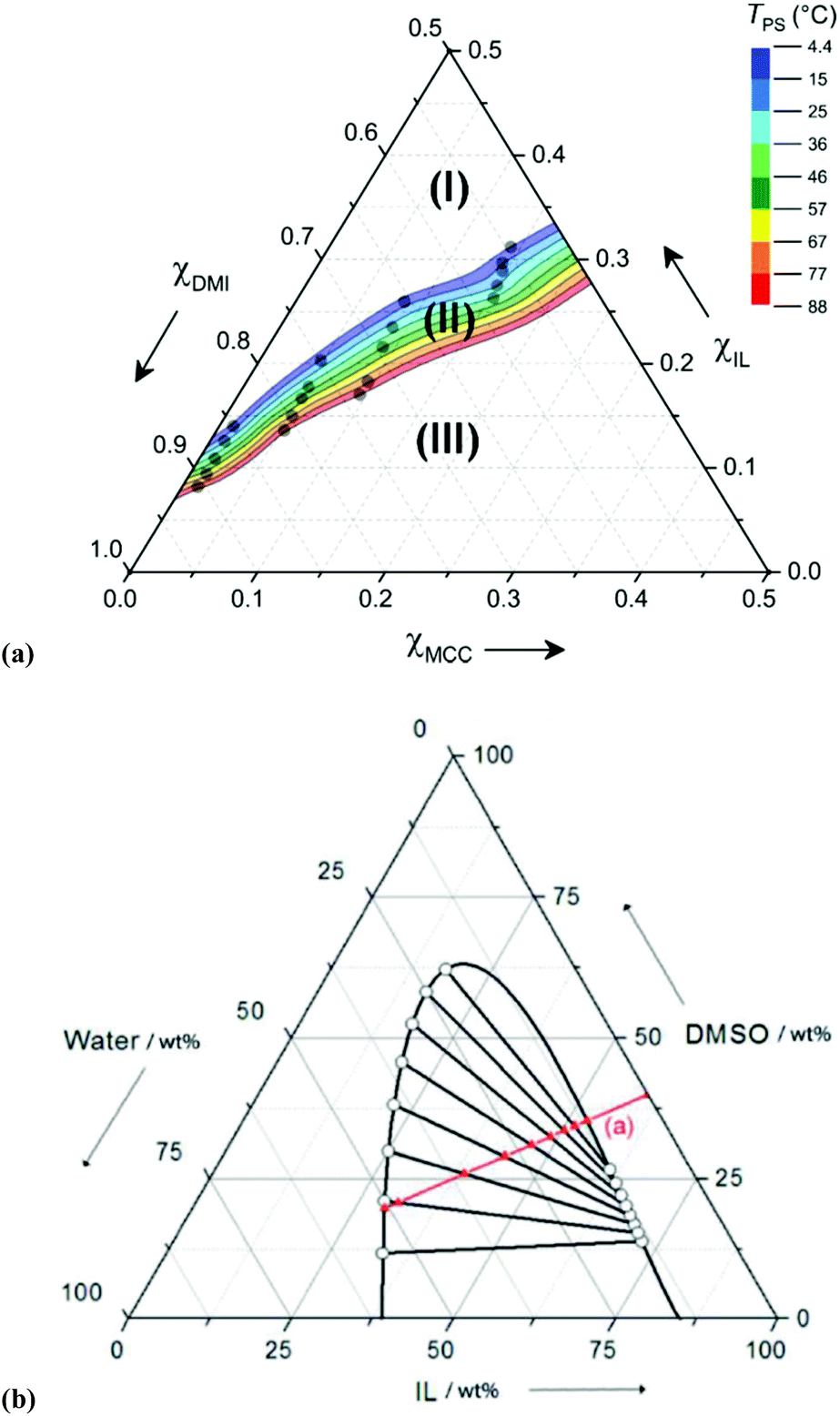 | ||
| Fig. 13 (a) Composition diagram for thermally triggered phase-separating [C2C1im][OAc]–DMI–microcrystalline cellulose blends. For T > 4 °C, no phase separation is observed in region I, region II denotes phase separation, and region III indicates compositions at which cellulose is partially insoluble;141 (b) composition diagram for phase-separation of [P8881][OAc]–DMSO mixtures upon addition of water, line ‘(a)’ (in the absence of cellulose). Ternary mixtures that lie within the biomodal curve are biphasic, consisting of an IL-enriched upper phase and a water-enriched lower phase.99 Reproduced with permission from ChemSusChem, 2016, 9, 3324–3329, and ChemSusChem, 2014, 7, 1422–1434, respectively. Wiley-VCH Verlag GmbH & Co. KGaA, 2016, 2014. | ||
Holding and co-workers reported a somewhat different phase separation phenomenon, enabling recovery of the expensive IL component tri(n-octyl)methylphosphonium acetate, [P8881][OAc], from OES mixtures with DMSO.99 The addition of water or aqueous sodium acetate to blank OES solvents brought about division into two phases, namely, an upper phase rich in the IL and a lower aqueous-rich phase (Fig. 13b). This strategy was then applied to 5 wt% MCC solutions in [P8881][OAc]–DMSO (55:45 wt/wt), whereby addition of 10 wt% aqueous sodium acetate caused simultaneous phase separation and precipitation of the MCC. The costly phosphonium IL could be recovered in close to quantitative yield and high-purity, and the cellulose could be obtained as a white powder having undergone significant conversion from cellulose I to cellulose II (cf.Fig. 2).
Therefore, a variety of phase separation protocols could be envisaged for versatile and sustainable recovery of high-purity IL and cellulosic materials from complex mixtures. Added to those above, various promising strategies have been put forward, incorporating (e.g.) kosmotropic salt solutions,142–145 hydrophobic or fluorinated anions,146–148 or distillable ILs.67 These separation methods are likely to be of increasing interest within the field of organic electrolyte solutions.
6. Organic electrolyte solutions – green solvents?
If OESs are to find suitability as solvents for cellulose beyond the laboratory setting, they must not only fulfil various economic criteria, but also satisfy the condition of being ‘green solvents’. To state this another way, the OESs themselves – or the processes in which they are involved – ought to meet requirements of (among other factors) waste reduction, energy efficiency and hazard minimization. Questions as to whether ILs are (or could be) green solvents, for example as solvents for catalysis,149 have been previously addressed.With reference to the 12 established ‘principles of green chemistry’,150,151 OESs could be considered as advantageous or disadvantageous relative to the parent IL, depending on the principle in question or the process/technology in which the solvent is to be used. In the paragraphs below, selected relevant principles of green chemistry are addressed individually, and the extent to which OESs could prove beneficial or problematic for the dissolution and regeneration of cellulose (compared to exisiting alternative solvents) is critically examined.
6.1 Principle 1 – Prevent waste
If a volatile co-solvent (e.g., acetone or DMI) is used, an OES solvent is likely to generate a greater quantity of non-recoverable waste (in the form of solvent vapours) relative to a pure IL with negligibly low vapour pressure under typical conditions of cellulose dissolution or electrospinning.152,153 However, the co-solvent could in principle be recovered and purified by distillation, whereas the IL is susceptible to gradual (irreversible) decomposition during repeat cellulose dissolution–regeneration cycles.76–82 Therefore, so long as vaporisation of co-solvent is minimized via carrying out a procedure in a closed system, diluting an IL with co-solvent could bring about a significant net reduction in waste by minimizing the fraction of the solvent that undergoes unintended degradation. IL and OES solvents could both offer a potential reduction in waste in comparison to existing industrial technologies (e.g., the Viscose process).6.2 Principle 3 – Design less hazardous chemical syntheses
Relatively few examples of cellulose derivatization procedures using OES solvents have been shown in the literature.103,138–140 The flammability, volatility and toxicity of any co-solvent species will present a significant hazard over and above the analogous procedure performed in a pure IL. Nevertheless, the use of an OES–cellulose mixture that exhibits phase separation could offer simplified separation of cellulosic products from unspent toxic organo-soluble reagents.6.3 Principle 4 – Use renewable feedstocks
Interesting preliminary studies have pointed towards the potential for renewable co-solvents to be used in OES–cellulose mixtures. For example, Gale et al. recently identified γ-valerolactone (GVL, Fig. 4c) as a promising co-solvent when paired with [C2C1im][OAc].102 GVL may itself be prepared from biomass,7 and its credentials as a green solvent have already been established.1546.4 Principles 8 and 12 – Use safer solvents and reaction conditions and minimize the potential for accidents
A severe disadvantage associated with OES solvents is the inclusion of often volatile, flammable and toxic organic species. This presents a significant hazard at large scale. OESs could be regarded as green solvents (according to principles 8 and 12) only where relatively benign co-solvents are used. Accordingly, future research must centre on co-solvents that share properties with DMSO, in contrast to solvents such as DMF or N-methylpyrrolidinone (which, e.g., present a risk to reproductive health). In most instances an OES solvent would pose less of a safety risk than N-methylmorpholine N-oxide or CS2, used for the established Lyocell and Viscose processes, respectively. However, great care would need to be taken to ensure that toxic co-solvent residues did not find their way into the cellulosic product.6.5 Principle 9 – Increase energy efficiency
The low viscosities of OES–cellulose blends – in comparison to those based on pure ILs – could bring about a substantial reduction in the energy costs associated with e.g. handling or stirring. Moreover, the high rate of cellulose dissolution (at close to ambient temperatures) in OES solvents may also improve energy efficiency, minimizing the extent of heating and stirring required in order to obtain homogenous mixtures. However, the need to prepare mixtures of IL and co-solvent in careful stoichiometric quantities prior to the dissolution of cellulose will likely add at least one extra process stage.6.6 Principle 11 – Analyze in real time to prevent pollution
As has been demonstrated in recent literature, the low viscosities of OES–cellulose mixtures make possible certain analyses (e.g. 2D NMR spectroscopy137,155) that are not feasible in pure IL–cellulose blends. Therefore, OESs could represent a more green alternative to pure ILs with respect to in-line analysis of cellulose dissolution and processing, thereby minimizing sampling and affiliated pollution.In summary, questions as to whether OES-based technologies could be sustainable or ‘green’ processes must be answered on a case-by-case basis, taking into consideration the specific ions and co-solvent that are incorporated into the mixture. Certainly, there is scope for OESs to improve upon existing technologies in the field of cellulose dissolution, derivatisation and regeneration. In the scientific literature to date, attempts to evaluate the green credentials of OES solvents are scarce,102 yet such studies will become increasingly relevant in future.
7. Conclusions
The development of effective, stable and recyclable solvent systems for cellulose has posed a challenge to chemists for many decades. In order to peel away and solvate individual cellulose strands from the solid state, without chemical modification of cellulose itself, the solvent must be capable of participating in strong hydrogen-bonding interactions with the polymer strands. Throughout the 20th and 21st centuries various non-derivatising cellulose solvents have emerged, among which ‘organic electrolyte solutions’ (OESs) can be considered as a class of solvent in their own right, though sharing certain properties with the ionic liquids (ILs) from which they are derived.The rapidity with which high quantities of cellulose dissolve in an OES is best explained as a delicate balance between thermodynamic and kinetic properties. So long as the condition of a negative Gibbs free energy is satisfied, enabling the spontaneous dissolution of cellulose, dilution with a co-solvent and the accompanying reduction in viscosity may enhance the mass transport of the ions. Consequently, diffusion of ions into the microfibril and formation of hydrogen bonds between ions and cellulose will occur more rapidly with the aid of the co-solvent. Though the co-solvent appears to ‘loosen’ the ion pairs, closer inspection of mixtures of [C4C1im][OAc] with DMSO114 suggests that ion–ion interactions are (at least in this instance) not significantly perturbed in the presence of the co-solvent. However, the microscopic and macroscopic properties of the OES hinge upon the specific electronic (and structural) properties of the chosen co-solvent. Therefore, an alternative choice of co-solvent could lead to an OES in which the associative interactions between cations and anions are more heavily disrupted.155 In this regard, OESs may be considered to share the reputation of ILs as ‘designer solvents’, whereby physicochemical properties of the solvent can be fine-tuned by choice of cation, anion and co-solvent.
OES solvents have been successfully applied to the electrospinning of high-quality cellulose fibres, to accurate solution-state NMR measurements of cellulose, and also to cellulose functionalisation. Most of these methods benefit from the low viscosity (compared to the parent IL) of the OES solvent. Furthermore, phase-separable OES-cellulose mixtures extend the range of possibilities for cellulose derivatisation, purification, and solvent recycling over and above conventional ILs. Future research is likely to be similarly directed towards improved process sustainability and the development of green OES solvents. As has been demonstrated for ILs,156ab initio computational calculations could be able to aid the identification of sensible IL–co-solvent combinations, mitigating the need for time-consuming experimental trial and error.
In summary, OESs represent attractive and versatile alternatives to simple ILs for the dissolution, derivatisation and regeneration of cellulose. In light of the additional ‘handle’ for modifying physicochemical properties by inclusion of a co-solvent, and the flexibility that this affords, they may be expected to attract increasingly widespread interest in the coming years.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was performed as part of the Cluster of Excellence ‘Tailor-Made Fuels from Biomass’ (DFG). MTC wishes to thank Dr Heitor F. N. de Oliveira and Dr Roberto Rinaldi for helpful discussions. Open Access funding provided by the Max Planck Society.References
- D. Klemm, B. Heublein, H. P. Fink and A. Bohn, Angew. Chem., Int. Ed., 2005, 44, 3358–3393 CrossRef CAS PubMed.
- D. Klemm, H.-P. Schmauder and T. Heinze, in Biopolymers Online, Wiley-VCH Verlag GmbH & Co. KGaA, 2005 Search PubMed.
- R. Vanholme, B. Demedts, K. Morreel, J. Ralph and W. Boerjan, Plant Physiol., 2010, 153, 895–905 CrossRef CAS PubMed.
- R. Rinaldi, R. Jastrzebski, M. T. Clough, J. Ralph, M. Kennema, P. C. A. Bruijnincx and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2016, 55, 8164–8215 CrossRef CAS PubMed.
- M. Pauly, S. Gille, L. Liu, N. Mansoori, A. de Souza, A. Schultink and G. Xiong, Planta, 2013, 238, 627–642 CrossRef CAS PubMed.
- R. Rinaldi, Angew. Chem., Int. Ed., 2014, 53, 8559–8560 CrossRef CAS PubMed.
- J. S. Luterbacher, J. M. Rand, D. M. Alonso, J. Han, J. T. Youngquist, C. T. Maravelias, B. F. Pfleger and J. A. Dumesic, Science, 2014, 343, 277–280 CrossRef CAS PubMed.
- R. Rinaldi, Chem. Commun., 2011, 47, 511–513 RSC.
- C. Froschauer, M. Hummel, M. Iakovlev, A. Roselli, H. Schottenberger and H. Sixta, Biomacromolecules, 2013, 14, 1741–1750 CrossRef CAS PubMed.
- P. Weerachanchai and J.-M. Lee, ACS Sustainable Chem. Eng., 2013, 1, 894–902 CrossRef CAS.
- Q. Hou, M. Ju, W. Li, L. Liu, Y. Chen and Q. Yang, Molecules, 2017, 22, 490 CrossRef PubMed.
- S. Xu, J. Zhang, A. He, J. Li, H. Zhang and C. C. Han, Polymer, 2008, 49, 2911–2917 CrossRef CAS.
- L. Meli, J. Miao, J. S. Dordick and R. J. Linhardt, Green Chem., 2010, 12, 1883–1892 RSC.
- S.-L. Quan, S.-G. Kang and I.-J. Chin, Cellulose, 2010, 17, 223–230 CrossRef CAS.
- L. J. Hauru, M. Hummel, A. Michud and H. Sixta, Cellulose, 2014, 21, 4471–4481 CrossRef CAS.
- M. Hummel, A. Michud, M. Tanttu, S. Asaadi, Y. Ma, L. J. Hauru, A. Parviainen, A. T. King, I. Kilpeläinen and H. Sixta, in Ionic Liquids for the Production of Man-Made Cellulosic Fibers: Opportunities and Challenges, Springer, Berlin, Heidelberg, 2015, pp. 1–36 Search PubMed.
- L. K. J. Hauru, M. Hummel, K. Nieminen, A. Michud and H. Sixta, Soft Matter, 2016, 12, 1487–1495 RSC.
- R. De Silva, K. Vongsanga, X. Wang and N. Byrne, Cellulose, 2016, 23, 2741–2751 CrossRef CAS.
- R. D. Perlack, L. L. Wright, A. F. Turhollow, R. L. Graham, B. J. Stokes and D. C. Erbach, Biomass as feedstock for a bioenergy and bioproducts industry: the technical feasibility of a billion-ton annual supply, DTIC Document, 2005 Search PubMed.
- J. Pickett, Sustainable biofuels: prospects and challenges, The Royal Society, 2008 Search PubMed.
- R. Rinaldi, R. Palkovits and F. Schüth, Angew. Chem., Int. Ed., 2008, 47, 8047–8050 CrossRef CAS PubMed.
- T. Searchinger, R. Heimlich, R. A. Houghton, F. Dong, A. Elobeid, J. Fabiosa, S. Tokgoz, D. Hayes and T.-H. Yu, Science, 2008, 319, 1238–1240 CrossRef CAS PubMed.
- T. R. Brown and R. C. Brown, Biofuels, Bioprod. Biorefin., 2013, 7, 235–245 CrossRef CAS.
- I. Siró and D. Plackett, Cellulose, 2010, 17, 459–494 CrossRef.
- R. J. Moon, A. Martini, J. Nairn, J. Simonsen and J. Youngblood, Chem. Soc. Rev., 2011, 40, 3941–3994 RSC.
- H. A. Khalil, A. Bhat and A. I. Yusra, Carbohydr. Polym., 2012, 87, 963–979 CrossRef.
- M. Gericke, P. Fardim and T. Heinze, Molecules, 2012, 17, 7458–7502 CrossRef PubMed.
- Y. Habibi, Chem. Soc. Rev., 2014, 43, 1519–1542 RSC.
- X. Shen, J. L. Shamshina, P. Berton, G. Gurau and R. D. Rogers, Green Chem., 2016, 18, 53–75 RSC.
- X. Shen, J. L. Shamshina, P. Berton, J. Bandomir, H. Wang, G. Gurau and R. D. Rogers, ACS Sustainable Chem. Eng., 2016, 4, 471–480 CrossRef CAS.
- A. C. O'Sullivan, Cellulose, 1997, 4, 173–207 CrossRef.
- Y. Nishiyama, P. Langan and H. Chanzy, J. Am. Chem. Soc., 2002, 124, 9074–9082 CrossRef CAS PubMed.
- S. Li, L. Bashline, L. Lei and Y. Gu, The Arabidopsis Book/American Society of Plant Biologists, 2014, vol. 12, p. e0169 Search PubMed.
- J. T. McNamara, J. L. Morgan and J. Zimmer, Annu. Rev. Biochem., 2015, 84, 895 CrossRef CAS PubMed.
- E. Dinand, M. Vignon, H. Chanzy and L. Heux, Cellulose, 2002, 9, 7–18 CrossRef CAS.
- T. Liebert, Cellulose Solvents: For Analysis, Shaping and Chemical Modification, American Chemical Society, 2010 Search PubMed.
- E. Schweizer, J. Prakt. Chem., 1857, 72, 109–111 CrossRef.
- C. Gränacher, U.S. Patent, 1943176, 1934 Search PubMed.
- C. McCormick, D. Lichatowich and M. Fooladi, in Proceedings of the 5th International Symposium on Controlled Release of Bioactive Materials, 1978, vol. 3 Search PubMed.
- C. McCormick and D. Lichatowich, J. Polym. Sci., Polym. Lett. Ed., 1979, 17, 479–484 CrossRef CAS.
- C. L. McCormick, P. A. Callais and B. H. Hutchinson Jr., Macromolecules, 1985, 18, 2394–2401 CrossRef CAS.
- Y. Nishio, S. K. Roy and R. S. J. Manley, Polymer, 1987, 28, 1385–1390 CrossRef CAS.
- T. Dawsey and C. L. McCormick, J. Macromol. Sci., Rev. Macromol. Chem., 1990, 30, 405–440 CrossRef.
- S. Spange, A. Reuter, E. Vilsmeier, T. Heinze, D. Keutel and W. Linert, J. Polym. Sci., Part A: Polym. Chem., 1998, 36, 1945–1955 CrossRef CAS.
- J. Cai and L. Zhang, Macromol. Biosci., 2005, 5, 539–548 CrossRef CAS PubMed.
- N. Isobe, K. Noguchi, Y. Nishiyama, S. Kimura, M. Wada and S. Kuga, Cellulose, 2013, 20, 97–103 CrossRef CAS.
- A. Isogai and R. Atalla, Cellulose, 1998, 5, 309–319 CrossRef CAS.
- K. Kamide, K. Okajima and K. Kowsaka, Polym. J., 1992, 24, 71–86 CrossRef CAS.
- K. Kamide and K. Okajima, Cellulose dope, process for preparation and method for application thereof, U.S. Pat, 4634470, 1987 Search PubMed.
- S. Dorn, F. Wendler, F. Meister and T. Heinze, Macromol. Mater. Eng., 2008, 293, 907–913 CrossRef CAS.
- R. Kotek, Regenerated cellulose fibers, 2007 Search PubMed.
- C. Woodings, in Kirk-Othmer Encyclopedia of Chemical Technology, John Wiley & Sons, Inc., 2000 Search PubMed.
- T. Rosenau, A. Potthast, H. Sixta and P. Kosma, Prog. Polym. Sci., 2001, 26, 1763–1837 CrossRef CAS.
- R. P. Swatloski, S. K. Spear, J. D. Holbrey and R. D. Rogers, J. Am. Chem. Soc., 2002, 124, 4974–4975 CrossRef CAS PubMed.
- B. Mostofian, X. Cheng and J. C. Smith, J. Phys. Chem. B, 2014, 118, 11037–11049 CrossRef CAS PubMed.
- H. Liu, K. L. Sale, B. A. Simmons and S. Singh, J. Phys. Chem. B, 2011, 115, 10251–10258 CrossRef CAS PubMed.
- Y. Zhao, J. Wang, H. Wang, Z. Li, X. Liu and S. Zhang, J. Phys. Chem. B, 2015, 119, 6686–6695 CrossRef CAS PubMed.
- J. Shi, K. Balamurugan, R. Parthasarathi, N. Sathitsuksanoh, S. Zhang, V. Stavila, V. Subramanian, B. A. Simmons and S. Singh, Green Chem., 2014, 16, 3830–3840 RSC.
- H. M. Cho, A. S. Gross and J.-W. Chu, J. Am. Chem. Soc., 2011, 133, 14033–14041 CrossRef CAS PubMed.
- M. J. Kamlet and R. W. Taft, J. Am. Chem. Soc., 1976, 98, 377–383 CrossRef CAS.
- M. A. Ab Rani, A. Brandt, L. Crowhurst, A. Dolan, M. Lui, N. H. Hassan, J. P. Hallett, P. A. Hunt, H. Niedermeyer, J. M. Perez-Arlandis, M. Schrems, T. Welton and R. Wilding, Phys. Chem. Chem. Phys., 2011, 13, 16831–16840 RSC.
- A. Brandt, J. Gräsvik, J. P. Hallett and T. Welton, Green Chem., 2013, 15, 550–583 RSC.
- Y. Fukaya, A. Sugimoto and H. Ohno, Biomacromolecules, 2006, 7, 3295–3297 CrossRef CAS PubMed.
- Y. Fukaya, K. Hayashi, M. Wada and H. Ohno, Green Chem., 2008, 10, 44–46 RSC.
- A. Pinkert, K. N. Marsh, S. Pang and M. P. Staiger, Chem. Rev., 2009, 109, 6712–6728 CrossRef CAS PubMed.
- A. Xu, J. Wang and H. Wang, Green Chem., 2010, 12, 268–275 RSC.
- A. W. T. King, J. Asikkala, I. Mutikainen, P. Järvi and I. Kilpeläinen, Angew. Chem., Int. Ed., 2011, 50, 6301–6305 CrossRef CAS PubMed.
- K. Ohira, Y. Abe, M. Kawatsura, K. Suzuki, M. Mizuno, Y. Amano and T. Itoh, ChemSusChem, 2012, 5, 388–391 CrossRef CAS PubMed.
- H. Wang, G. Gurau and R. D. Rogers, Chem. Soc. Rev., 2012, 41, 1519–1537 RSC.
- A. Parviainen, A. W. T. King, I. Mutikainen, M. Hummel, C. Selg, L. K. J. Hauru, H. Sixta and I. Kilpelainen, ChemSusChem, 2013, 6, 2161–2169 CrossRef CAS PubMed.
- X. Yuan and G. Cheng, Phys. Chem. Chem. Phys., 2015, 17, 31592–31607 RSC.
- M. Abe, K. Kuroda, D. Sato, H. Kunimura and H. Ohno, Phys. Chem. Chem. Phys., 2015, 17, 32276–32282 RSC.
- L. K. Hauru, M. Hummel, A. W. King, I. Kilpeläinen and H. Sixta, Biomacromolecules, 2012, 13, 2896–2905 CrossRef CAS PubMed.
- L. Chen, M. Sharifzadeh, N. Mac Dowell, T. Welton, N. Shah and J. P. Hallett, Green Chem., 2014, 16, 3098–3106 RSC.
- A. George, A. Brandt, K. Tran, S. M. S. N. S. Zahari, D. Klein-Marcuschamer, N. Sun, N. Sathitsuksanoh, J. Shi, V. Stavila, R. Parthasarathi, S. Singh, B. M. Holmes, T. Welton, B. A. Simmons and J. P. Hallett, Green Chem., 2015, 17, 1728–1734 RSC.
- G. Ebner, S. Schiehser, A. Potthast and T. Rosenau, Tetrahedron Lett., 2008, 49, 7322–7324 CrossRef CAS.
- T. Liebert and T. Heinze, BioResources, 2008, 3, 576–601 Search PubMed.
- M. Schrems, G. Ebner, F. Liebner, E. Becker, A. Potthast and T. Rosenau, in Cellulose Solvents: For Analysis, Shaping and Chemical Modification, American Chemical Society (ACS), 2010, pp. 149–164 Search PubMed.
- O. Hollóczki, D. S. Firaha, J. Friedrich, M. Brehm, R. Cybik, M. Wild, A. Stark and B. Kirchner, J. Phys. Chem. B, 2013, 117, 5898–5907 CrossRef PubMed.
- M. T. Clough, K. Geyer, P. A. Hunt, J. Mertes and T. Welton, Phys. Chem. Chem. Phys., 2013, 47, 20480–20495 RSC.
- M. T. Clough, K. Geyer, P. A. Hunt, S. Son, U. Vagt and T. Welton, Green Chem., 2015, 17, 231–243 RSC.
- M. T. Clough, J. A. Griffith, O. Kuzmina and T. Welton, Green Chem., 2016, 18, 3758–3766 RSC.
- N. V. Plechkova and K. R. Seddon, Chem. Soc. Rev., 2008, 37, 123–150 RSC.
- J. Sun, J. Wang, W. Cheng, J. Zhang, X. Li, S. Zhang and Y. She, Green Chem., 2012, 14, 654–660 RSC.
- C. Maton, N. De Vos and C. V. Stevens, Chem. Soc. Rev., 2013, 42, 5963–5977 RSC.
- J. S. Moulthrop, R. P. Swatloski, G. Moyna and R. D. Rogers, Chem. Commun., 2005, 1557–1559 RSC.
- R. C. Remsing, R. P. Swatloski, R. D. Rogers and G. Moyna, Chem. Commun., 2006, 1271–1273 RSC.
- T. G. A. Youngs, J. D. Holbrey, C. L. Mullan, S. E. Norman, M. C. Lagunas, C. D'Agostino, M. D. Mantle, L. F. Gladden, D. T. Bowron and C. Hardacre, Chem. Sci., 2011, 2, 1594–1605 RSC.
- J. M. Andanson, A. A. H. Padua and M. F. Costa Gomes, Chem. Commun., 2015, 51, 4485–4487 RSC.
- H. Liu, K. L. Sale, B. M. Holmes, B. A. Simmons and S. Singh, J. Phys. Chem. B, 2010, 114, 4293–4301 CrossRef CAS PubMed.
- A. S. Gross, A. T. Bell and J.-W. Chu, J. Phys. Chem. B, 2011, 115, 13433–13440 CrossRef CAS PubMed.
- H. Liu, G. Cheng, M. Kent, V. Stavila, B. A. Simmons, K. L. Sale and S. Singh, J. Phys. Chem. B, 2012, 116, 8131–8138 CrossRef CAS PubMed.
- Y. Zhao, X. Liu, J. Wang and S. Zhang, Carbohydr. Polym., 2013, 94, 723–730 CrossRef CAS PubMed.
- B. D. Rabideau, A. Agarwal and A. E. Ismail, J. Phys. Chem. B, 2013, 117, 3469–3479 CrossRef CAS PubMed.
- V. S. Bharadwaj, T. C. Schutt, T. C. Ashurst and C. M. Maupin, Phys. Chem. Chem. Phys., 2015, 17, 10668–10678 RSC.
- B. Mostofian, J. C. Smith and X. Cheng, Cellulose, 2014, 21, 983–997 CrossRef CAS.
- B. D. Rabideau, A. Agarwal and A. E. Ismail, J. Phys. Chem. B, 2014, 118, 1621–1629 CrossRef CAS PubMed.
- D. L. Minnick, R. A. Flores, M. R. DeStefano and A. M. Scurto, J. Phys. Chem. B, 2016, 120, 7906–7919 CrossRef CAS PubMed.
- A. J. Holding, M. Heikkilä, I. Kilpeläinen and A. W. T. King, ChemSusChem, 2014, 7, 1422–1434 CrossRef CAS PubMed.
- A. J. Holding, A. Parviainen, I. Kilpelainen, A. Soto, A. W. T. King and H. Rodriguez, RSC Adv., 2017, 7, 17451–17461 RSC.
- Y.-B. Huang, P.-P. Xin, J.-X. Li, Y.-Y. Shao, C.-B. Huang and H. Pan, ACS Sustainable Chem. Eng., 2016, 4, 2286–2294 CrossRef CAS.
- E. Gale, R. H. Wirawan, R. L. Silveira, C. S. Pereira, M. A. Johns, M. S. Skaf and J. L. Scott, ACS Sustainable Chem. Eng., 2016, 4, 6200–6207 CrossRef CAS.
- G. T. Ciacco, T. F. Liebert, E. Frollini and T. J. Heinze, Cellulose, 2003, 10, 125–132 CrossRef CAS.
- L. Härdelin, J. Thunberg, E. Perzon, G. Westman, P. Walkenström and P. Gatenholm, J. Appl. Polym. Sci., 2012, 125, 1901–1909 CrossRef.
- A. Pinkert, J. Chem. Eng. Data, 2012, 57, 1338–1340 CrossRef CAS.
- R. Rinaldi, J. Chem. Eng. Data, 2012, 57, 1341–1343 CrossRef CAS.
- R. W. Taft and M. J. Kamlet, J. Am. Chem. Soc., 1976, 98, 2886–2894 CrossRef CAS.
- M. J. Kamlet, J. L. Abboud and R. W. Taft, J. Am. Chem. Soc., 1977, 99, 6027–6038 CrossRef CAS.
- M. Gericke, T. Liebert, O. A. E. Seoud and T. Heinze, Macromol. Mater. Eng., 2011, 296, 483–493 CrossRef CAS.
- A. Xu, Y. Zhang, Y. Zhao and J. Wang, Carbohydr. Polym., 2013, 92, 540–544 CrossRef CAS PubMed.
- Y. Zhao, X. Liu, J. Wang and S. Zhang, J. Phys. Chem. B, 2013, 117, 9042–9049 CrossRef CAS PubMed.
- F. Huo, Z. Liu and W. Wang, J. Phys. Chem. B, 2013, 117, 11780–11792 CrossRef CAS PubMed.
- S. Velioglu, X. Yao, J. Devémy, M. G. Ahunbay, S. B. Tantekin-Ersolmaz, A. Dequidt, M. F. Costa Gomes and A. A. Pádua, J. Phys. Chem. B, 2014, 118, 14860–14869 CAS.
- J.-M. Andanson, E. Bordes, J. Devémy, F. Leroux, A. A. Pádua and M. F. C. Gomes, Green Chem., 2014, 16, 2528–2538 RSC.
- C. Zhang, H. Kang, P. Li, Z. Liu, Y. Zhang, R. Liu, J.-f. Xiang and Y. Huang, Cellulose, 2016, 23, 1165–1175 CrossRef CAS.
- P. Handbook, Wiley, New York, 1999 Search PubMed.
- B. A. Miller-Chou and J. L. Koenig, Prog. Polym. Sci., 2003, 28, 1223–1270 CrossRef CAS.
- H. F. N. de Oliveira and R. Rinaldi, ChemSusChem, 2015, 8, 1577–1584 CrossRef PubMed.
- G. Taylor, Proc. R. Soc. London, Ser. A, 1964, 280, 383–397 CrossRef.
- Y. Zheng, J. Miao, N. Maeda, D. Frey, R. J. Linhardt and T. J. Simmons, J. Mater. Chem. A, 2014, 2, 15029–15034 CAS.
- J. L. Shamshina, O. Zavgorodnya, J. R. Bonner, G. Gurau, T. Di Nardo and R. D. Rogers, ChemSusChem, 2017, 10, 106–111 CrossRef CAS PubMed.
- G. Viswanathan, S. Murugesan, V. Pushparaj, O. Nalamasu, P. M. Ajayan and R. J. Linhardt, Biomacromolecules, 2006, 7, 415–418 CrossRef CAS PubMed.
- H. S. Wang, G. D. Fu and X. S. Li, Recent Pat. Nanotechnol., 2009, 3, 21–31 CrossRef CAS PubMed.
- K. Yoon, B. S. Hsiao and B. Chu, J. Mater. Chem., 2008, 18, 5326–5334 RSC.
- V. Kalra, J. H. Lee, J. H. Park, M. Marquez and Y. L. Joo, Small, 2009, 5, 2323–2332 CrossRef CAS PubMed.
- A. Frenot and I. S. Chronakis, Curr. Opin. Colloid Interface Sci., 2003, 8, 64–75 CrossRef CAS.
- J. M. Deitzel, J. Kleinmeyer, D. Harris and N. C. Beck Tan, Polymer, 2001, 42, 261–272 CrossRef CAS.
- D. Li and Y. Xia, Adv. Mater., 2004, 16, 1151–1170 CrossRef CAS.
- M. G. McKee, J. M. Layman, M. P. Cashion and T. E. Long, Science, 2006, 311, 353–355 CrossRef CAS PubMed.
- S. De Vrieze, T. Van Camp, A. Nelvig, B. Hagström, P. Westbroek and K. De Clerck, J. Mater. Sci., 2009, 44, 1357–1362 CrossRef CAS.
- F. Lu, L. Wang, C. Zhang, B. Cheng, R. Liu and Y. Huang, Cellulose, 2015, 22, 3077–3087 CrossRef CAS.
- Y. Lv, J. Wu, J. Zhang, Y. Niu, C.-Y. Liu, J. He and J. Zhang, Polymer, 2012, 53, 2524–2531 CrossRef CAS.
- H. Saba, Y. Yongbo, W. Jianning, X. Xiaolin, W. Kaijian, Z. Yumei and W. Huaping, RSC Adv., 2015, 5, 8318–8322 RSC.
- L. Wang, L. Gao, B. Cheng, X. Ji, J. Song and F. Lu, Carbohydr. Polym., 2014, 110, 292–297 CrossRef CAS PubMed.
- R. Atalla and D. L. VanderHart, Solid State Nucl. Magn. Reson., 1999, 15, 1–19 CrossRef CAS PubMed.
- M. Foston, Curr. Opin. Biotechnol., 2014, 27, 176–184 CrossRef CAS PubMed.
- A. J. Holding, V. Mäkelä, L. Tolonen, H. Sixta, I. Kilpeläinen and A. W. T. King, ChemSusChem, 2016, 9, 880–892 CrossRef CAS PubMed.
- T. Heinze, T. Liebert, P. Klüfers and F. Meister, Cellulose, 1999, 6, 153–165 CrossRef CAS.
- T. Heinze and T. Liebert, Prog. Polym. Sci., 2001, 26, 1689–1762 CrossRef CAS.
- C. Achtel and T. Heinze, Macromol. Chem. Phys., 2016, 217, 2041–2048 CrossRef CAS.
- H. F. N. de Oliveira, M. T. Clough and R. Rinaldi, ChemSusChem, 2016, 9, 3324–3329 CrossRef CAS PubMed.
- C. L. Louros, A. F. M. Cláudio, C. M. Neves, M. G. Freire, I. M. Marrucho, J. Pauly and J. A. Coutinho, Int. J. Mol. Sci., 2010, 11, 1777–1791 CrossRef CAS PubMed.
- N. J. Bridges, K. E. Gutowski and R. D. Rogers, Green Chem., 2007, 9, 177–183 RSC.
- K. E. Gutowski, G. A. Broker, H. D. Willauer, J. G. Huddleston, R. P. Swatloski, J. D. Holbrey and R. D. Rogers, J. Am. Chem. Soc., 2003, 125, 6632–6633 CrossRef CAS PubMed.
- V. Najdanovic-Visak, J. N. C. Lopes, Z. P. Visak, J. Trindade and L. P. Rebelo, Int. J. Mol. Sci., 2007, 8, 736–748 CrossRef CAS.
- K. Fukumoto and H. Ohno, Chem. Commun., 2006, 3081–3083 RSC.
- K. Fukumoto and H. Ohno, Angew. Chem., Int. Ed., 2007, 46, 1852–1855 CrossRef CAS PubMed.
- Y. Kohno, H. Arai, S. Saita and H. Ohno, Aust. J. Chem., 2012, 64, 1560–1567 CrossRef.
- T. Welton, Green Chem., 2008, 10, 483–483 RSC.
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
- P. T. Anastas and J. C. Warner, Green chemistry: Theory and practice, 1998, pp. 29–56 Search PubMed.
- A. Deyko, K. R. J. Lovelock, P. Licence and R. G. Jones, Phys. Chem. Chem. Phys., 2011, 13, 16841–16850 RSC.
- J. P. Armstrong, C. Hurst, R. G. Jones, P. Licence, K. R. J. Lovelock, C. J. Satterley and I. J. Villar-Garcia, Phys. Chem. Chem. Phys., 2007, 9, 982–990 RSC.
- I. T. Horváth, H. Mehdi, V. Fábos, L. Boda and L. T. Mika, Green Chem., 2008, 10, 238–242 RSC.
- M. T. Clough, C. Fares and R. Rinaldi, ChemSusChem, 2017 DOI:10.1002/cssc.201701042.
- H. Niedermeyer, C. Ashworth, A. Brandt, T. Welton and P. A. Hunt, Phys. Chem. Chem. Phys., 2013, 15, 11566–11578 RSC.
| This journal is © The Royal Society of Chemistry 2017 |