
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A continuous flow process for the production of 2,5-dimethylfuran from fructose using (non-noble metal based) heterogeneous catalysis†
Max
Braun
 * and
Markus
Antonietti
* and
Markus
Antonietti
Max-Planck-Institute of Colloids and Interfaces, 14424 Potsdam, Germany. E-mail: max.braun@mpikg.mpg.de
First published on 11th July 2017
Abstract
The abundant carbohydrate fructose is converted into two biofuel molecules, namely 2,5-dimethylfuran (DMF) and ethyl levulinate (EL) in a simple cascade flow reactor. With an overall yield of 85% (38.5% of 2,5-dimethylfuran and 47% of ethyl levulinate), the main remainder is unconverted fructose. The two column flow reactor set-up enables the adjustment of temperatures and reaction times in such a way that the reactive intermediate hydroxymethylfurfural (5-HMF) is generated in optimal yields and converted into the stable DMF immediately. The process is so simple and fast (<20 min) that economic and sustainable production of these fuels and platform chemicals can be envisioned. A remaining minor char formation is regarded to be the major problem which has to be addressed by catalyst development.
Introduction
Biofuels of the first and the second generation are well placed in society, but the controversial discussion on their impact has already reached public media. Their ecobalances are simply not very favourable, and even basic energy balances throughout their generation are worse than those commonly known. Using for instance cassava as a non-food industrial plant and as an example for modern bioethanol fermentation,1 the overall efficiency is only around 40%, i.e. only a minority of the energy in sugar is ending up in bioethanol. The rather slow and dilute fermentation and the slow and inefficient separation of water and ethanol as cost drivers are not even considered here. The reasons for this inefficiency are essentially elemental rules of thermodynamics and reaction engineering. Bioethanol in addition is a “disappointing” fuel: its volumetric energy density is just 2/3 of the one of gasoline, i.e. both the driving range of a given car and fuel consumption are negatively perceived. All these problems are to be addressed by more meaningful biofuels of a new generation: first they should optimally be based on lignocellulosic biomass, if not even agro-industrial side products (such as corn stover2 or bagasse3), as well as on space time efficient wet-chemical continuous processes. Additionally, ideal candidates should rely, by design, only on simple purification processes, must be non-toxic and non-groundwater endangering, and should have high energy densities and good combustion behaviour. Indeed, in the meantime, some of these potential candidates are analysed in detail, and to our opinion, γ-valerolactone, ethyl levulinate and especially 2,5-dimethylfuran stand out. However, until today, no efficient and simple continuous flow system has been presented.The here discussed 2,5-dimethylfuran (DMF) is made in a two-step process. First, the production of 5-hydroxymethylfurfural (5-HMF) by the dehydration of fructose is highlighted, followed by its reduction with an active hydrogenation catalyst to DMF. It is the inherent advantage of reaction thermodynamics that the energy efficiency of these two steps in theory is close to 100%; both reactions are therefore energetically neutral. The product is in addition not water miscible, was already favourably tested in combustion engines, and has a volumetric energy density of 30.0 MJ L−1, very close to gasoline (32.2 MJ L−1), whereas ethanol only exhibits 21.3 MJ L−1.4 In addition, DMF is a potential entry scheme for fine chemical refining as it can be easily turned into p-xylene, a product significantly more valued than combustion fuels, with a demand of currently 40 Mt per annum.5 The single step from lignocellulosic biomass to DMF and potentially p-xylene, as well as γ-valerolactone is summarized in Scheme 1. It is important to remember that the saccharification of wood is a pre-WWII technology, with a known cost structure.6 These processes are currently challenged by more modern techniques, such as mechano-chemistry, where cellulose is molecularly depolymerized in a solid state reaction.7 In spite of the potential simplicity of the process, its application is currently just in the development stage.8 In the present contribution, it is intended to analyse how flow chemistry in green solvents (ethanol) with overall reaction times in the minute scale can contribute to a more economic and more side-product-poor production of DMF. A key step is the development of a heterogeneous catalyst system based on common, sustainable transition metal elements which withstand the conditions of hydrothermal or solvothermal processes on a longer run.9 This problem is usually underestimated by experts on gas-phase heterogeneous catalysis as superhot aqueous solvents dissolve most standard choices from most oxidic supports over polymer resins to many metals. It is underlined that the choice of most of the chemicals and catalysts is restricted by our decision that they can only be obtained from green or at least sustainable sources, e.g. cheap and abundant nickel is preferred over the more active but rare palladium. This study therefore aims to depict an easy but also sustainable method in terms of choosing the right catalysts and solvents as well as materials, combinations and conditions to result in a profitable process of production.
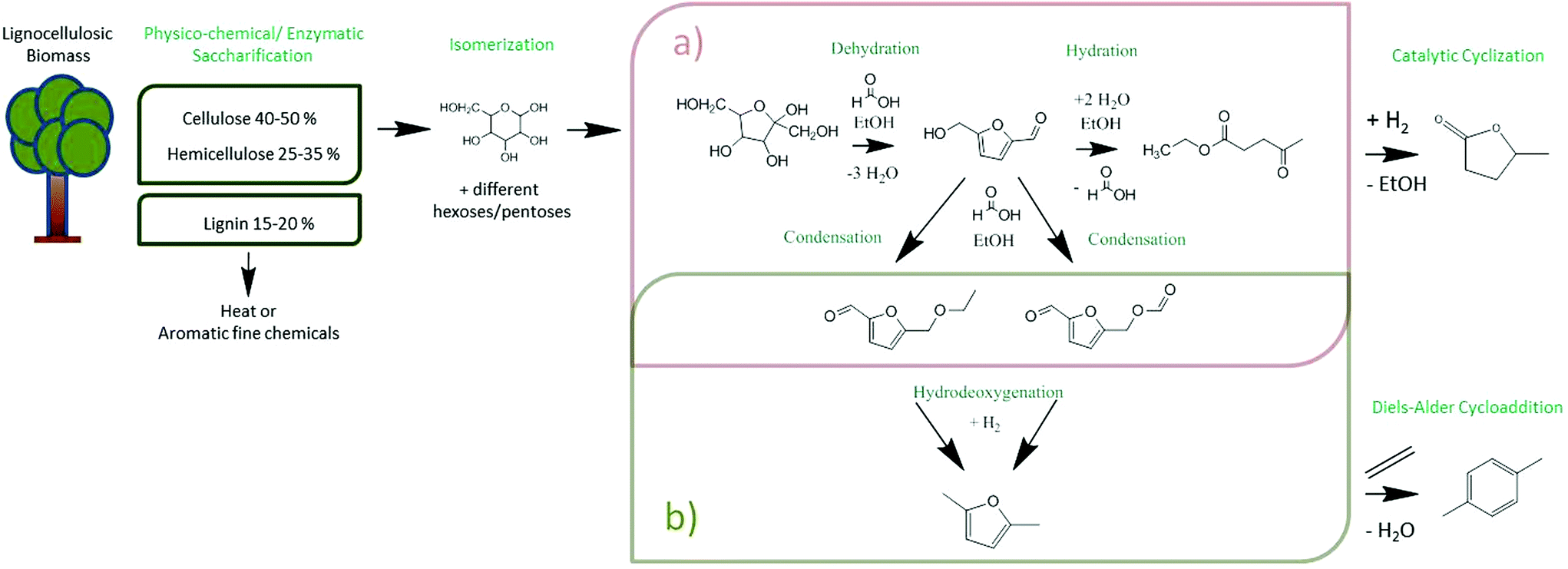 | ||
| Scheme 1 Reaction scheme showing possible reaction pathways during the dehydration and hydrodeoxygenation step. (a) Represents the reaction pathways present in the Amberlyst 15 column; (b) represents the hydrodeoxygenation reaction performed in a Ni@WC column. | ||
Results and discussion
Reaction mechanism
During the conversion of fructose, different intermediates and molecules on the way to DMF and EL were observed. The qualitative results and therefore the reaction mechanism will be discussed first and are illustrated in Scheme 1. This overview drafts the full cascade from the raw biomass to possible products, whereas the reactions in our two catalyst columns are highlighted. The release of three water molecules from fructose (or other hexoses) to form 5-HMF under acidic conditions is a rather simple process, slightly exothermic, but also entropically driven.10 Under acidic conditions, 5-HMF is an unstable intermediate and undergoes a variety of follow-up reactions, including charring and resin formation.11 This is why flow chemistry and the control of residence time are of absolute importance for a successful reaction design based on 5-HMF. At the short times under consideration, 5-HMF can be hydrated and it splits to levulinic acid and formic acid in good yields, depending on the reaction conditions. In ethanol, which is used as a renewable solvent, levulinic acid is esterified to ethyl levulinate. The ethyl levulinate will turn out to be the main side product next to 5-HMF. Further possible 5-HMF derivatives (like 5-ethoxymethylfurfural)12 or esters with the involved acid are also likely to form.13Since the mechanism of dehydration of fructose is well understood, Assary et al. (2012)10 published a helpful paper explaining the mechanism on a thermodynamic level. The whole process starts with the pyranose form of fructose, which is first protonated and releases a water molecule. The left positive charge can be shifted within the molecule. The acidic catalyst protonates the molecule further and another water molecule can be released, whereas the following deprotonation enables the formation of a double bond within the ring system. The third alcohol moiety is as well eliminated by dehydration and the aromatic character of the furan ring is formed by the last deprotonation. Furthermore, the hydrodeoxygenation and therefore the removal of the next two oxygen atoms within the molecule was described by Saha et al. (2015)14 in a review. Since HMF possesses an aldehyde as well as an alcohol group, the first step suggested is the hydration of the aldehyde to a further alcohol group resulting in 2,5 bis(hydroxymethyl)furan. Elemental hydrogen dissociates on nickel and hence offers this proton for the reaction. This intermediate can be hydrogenated and a water molecule is released into the solvent. After repeating this step with the other alcohol moiety left in the molecule, DMF is the result. The aromatic ring structure is not further hydrogenated and the biofuel is left in solution. There are also other reaction mechanisms to consider, for instance, dehydration of the alcohol before hydrating the aldehyde or forming an ether with the present formic acid. In our system, the formation of two condensates was observed. HMF underwent etherification with ethanol, as well as with formic acid. Nevertheless, the hydrodeoxygenation was successful. The exact mechanism needs to be investigated in future research studies.
The 5-HMF products are not isolated, but an in-flow, second step hydrogenation, using elemental hydrogen is pursued. The hydrogenolysis results in 2,5-dimethylfuran.15 The specific relative energy content of DMF is beyond the elimination of water and increased as compared to 5-HMF, with the energy coming from the added hydrogen. Also here, the reaction is partly driven by the entropic release of water. In spite of a potential biorefinery scheme, all possible side products also undergo hydrogenation and therefore do not have to be separated.
Ethyl levulinate for instance can be reduced to γ-valerolactone over RANEY® Nickel (already shown with a commercial catalyst by our group),16 another high value chemical and/or fuel molecule. Isolation or co-isolation in this case only depends on the hydrogenation conditions being selective or non-selective. As such, DMF can be easily isolated and stored, and is stable, useful as a fuel molecule, and also a valuable platform molecule. Future processes can expand the product spectrum by simply adding a further step, e.g. a Diels–Alder-reaction with ethylene in a flow reactor, to upgrade 2,5-DMF to p-xylene.5 This step would also remove the last oxygen present in the molecule to generate a pure hydrocarbon, usually synthesized from fossil resources and highly valued because it is the source of terephthalic acid, and for example useful in polymerization reactions.
Dehydration of fructose using Amberlyst 15
In a first set of experiments, the conditions of hexose dehydration were optimized (Scheme 1a). For that, we used 2.4 g of Amberlyst 15 at its maximum usage temperature (110 °C) under atmospheric pressure and varied the flow rates with an external piston pump accordingly to adjust different residence times (4 to 40 min) in a 250 mm × 4.6 mm packed column. The relationship of fructose used and the mass of Amberlyst 15 catalyst is 5.2 × 10−6 molfru per gcat per minute at 0.25 mL min−1 flow rate. Amberlyst 15 was chosen according to the already known excellent activity for dehydration reactions as well as to select a heterogeneous catalyst. All residence times were calculated using the reactor volume and flow rates. Since the first step of the fructose conversion takes place under acidic conditions, the dehydration results in 5-HMF, which can further hydrolyse to levulinic acid. The resulting HMF product molecules are in any case simultaneously esterified with ethanol or even formic acid. Under the given conditions, mainly the ethyl-derivative is formed, as proven by analysing the pure reference sample in comparison with the main peak in the GC-MS chromatogram. The product to reaction time curve is shown in Fig. 1. A trend of increasing product concentration with a lower flow rate and therefore higher residence time is clearly seen. Until a residence time of about 6 minutes, the increase of HMF derivative concentration is nearly linear, but then flattens with a further increase of time. 5-HMF is only an intermediate in a cascade of possible conversions, and the saturation of the 5-HMF level reflects its further conversion to levulinic acid. This hydrolysis occurs with water generated from the dehydration of fructose. With ethanol as a solvent, levulinic acid is in situ esterified under the given conditions to ethyl levulinate. Indeed, the yield of the ethyl levulinate rises linearly (7% to 52%) with decreasing flow rate and therefore a longer contact time with the catalyst. Under the given conditions, it is not further converted and represents the main follow-up product from 5-HMF. The selectivity and productivity at this level can only be controlled by the contact time: at shorter times, we have a high selectivity for 5-HMF, but lower conversions, while at higher reaction times, the overall conversion of fructose to these two products increases to about 89% (41.5 min residence time). Interestingly, no 5-HMF condensation products preceding char formation are found, underlining the big advantage of a flow system when compared to a batch reactor. All observations underline the importance of the reaction regime in a flow system and open up new possibilities for the adjustment of products. | ||
Fig. 1 Yield of HMF derivatives (mainly 5-ethoxymethylfurfural) ( ) as well as the yield of ethyl levulinate (■) during the conversion of the 0.05 M fructose/0.5 M formic acid/ethanol mixture using a 2.4 g Amberlyst 15 packed fixed bed column at 110 °C under atmospheric pressure with different residence times, adjusted by different flow rates. All data were obtained using a GC-MS system (ESI-S1†). ) as well as the yield of ethyl levulinate (■) during the conversion of the 0.05 M fructose/0.5 M formic acid/ethanol mixture using a 2.4 g Amberlyst 15 packed fixed bed column at 110 °C under atmospheric pressure with different residence times, adjusted by different flow rates. All data were obtained using a GC-MS system (ESI-S1†). | ||
Hydrodeoxygenation using 10 wt% Ni@WC
The second step in the flow reactor cascade is a hydrogenation reaction over a nickel–tungsten-carbide-based catalyst (Ni@WC) (Scheme 1b). As compounds derived from biomass always contain a high amount of oxygen, the heating value of these molecules is decreased. By a hydrodeoxygenation step, the combustion properties can be increased, using hydrogen as a reactant. Removing two further oxygen atoms from 5-HMF additionally lowers the boiling point of the substance and improves the research octane number to 119. Pure tungsten carbide shows restricted hydrogenation reactivity under the applied mild conditions, as recently shown by our group,17 but therefore shows an electron transfer to activate a deposited catalyst via metal–metal heterojunction effects. This allows the use of relatively cheap and abundant nickel for more effective hydrogenation, while the electron flow from the nickel to the WC makes the nanoparticles unusually stable against harsh conditions and solvents.18 Pure commercial RANEY® Nickel is not only water sensitive, but shows a too high hydrogenation activity and is known to be non-selective for DMF production. The combination of 10 wt% nickel nanoparticles and tungsten carbide however gives extraordinary selectivity to DMF with complete conversion and yield at 150 °C, 30 bar and 0.5 mL min−1 flow rate, using 1.68 g catalyst packed into a 70 mm column placed into the H-CUBE Pro hydrogenation reactor (ESI-S2†). The relationship between fructose and the mass of the Ni@WC catalyst is 1.4 × 10−5 molfru per gcat per minute. Additionally, it could have also been shown that the hydrodeoxygenation was not only possible with pure 5-HMF, but also with the ester formed. Under the same conditions, 5-ethoxymethylfurfural was converted to DMF. The contact time in the applied small column is about 3 minutes and hence unusually small compared to other chemical processes, which usually take hours to complete the reaction.The reactivity was tested as well with the main side product, ethyl levulinate. A 0.05 M solution in ethanol was run under the same conditions as optimized before (1.68 g Ni@WC, 150 °C, 30 bar, 0.5 mL min−1). By using pure tungsten carbide as well as Ni@WC as the catalyst, the solution was not hydrogenated and no products were obtained. By using RANEY® Nickel on the other hand, full conversion to γ-valerolactone and traces of 2,5-dimethyltetrahydrofuran were found (ESI-S3†). The combination of Ni@WC with the rather electron poor nickel did not convert the ethyl levulinate, and this molecule was therefore left as such in the final reaction mixture.
Combination of dehydration and hydrodeoxygenation in one continuous flow system
As both catalysts (Amberlyst 15 and Ni@WC) are working independent of each other but can set-up a reaction cascade, the two packed columns were combined to one continuous flow process (Scheme 1(a) and (b)). The conditions were kept similar to the ones in the single step experiments, using 2.4 g of Amberlyst 15 at 110 °C, 30 bar and 0.25 mL min−1 in a 250 mm column with a residence time of about 16.6 min. The reaction temperature using the packed cartridge (Ni@WC, 1.68 g) was 150 °C. The lowest flow rate possible for the hydrogenation reactor (H CUBE Pro) is about 0.25 mL min−1, since flow restrictions of the pump and operation recommendation of the system need to guarantee a good hydrogen saturation within the liquid feed and over the Ni@WC catalyst. Since both reactions are performed in separate columns, the temperature can be adjusted individually. The flow and the pressure are provided by using one piston pump and are therefore the same in both columns. The hydrogen is produced in situ by water electrolysis and fed in after the dehydration step. This set-up is drafted in Scheme 2. By applying the mentioned conditions, DMF and ethyl levulinate were obtained as the main products from a solution of fructose in ethanol (ESI-S4†). The fructose solution was overall converted to give products in 85% of yield compared to the starting material, with the single yield of 38.5% of DMF and 47% of ethyl levulinate within a combined reaction time of about 20 min. Such a high conversion with such a low residence time in a continuous flow system was, to our knowledge, not observed before. A very interesting and similar cascade system using zeolite and copper based catalysts was introduced by Xiang et al. (2016).19 The temperature used for the dehydration of fructose is 140 °C and therefore 30 °C higher than our approach using 110 °C. Also the second step using the copper based catalyst requires a temperature of 240 °C, whereas we were just using 150 °C, which basically saves the energy input. The water that is used as a co-solvent in the process also has two negative effects. Firstly, it helps HMF to undergo a consecutive hydration reaction forming EL. Secondly, water is always difficult to separate from the product mixture. In more simple words in regard to our research: turning a sugar solution within 20 minutes in a rather low-tech flow set-up into a mixture of fuel molecules is rather appealing and can be regarded as a process qualifying as a next generation thermochemical biofuel process. | ||
| Scheme 2 Reactor set-up of combined columns of Amberlyst 15 and Ni@WC. (a) Represents the external Amberlyst 15 column connected to the H CUBE Pro system. (2.4 g Amberlyst 15, 110 °C, 30 bar, 0.25 mL min−1, residence time: 16.6 min) (b) represents the integrated column in the H CUBE Pro system (1.68 g Ni@WC, 150 °C, 30 bar, 0.25 mL min−1, hydrogen, residence time: 3 min). | ||
Recently, the engineering of the process has been tackled more and more, which shows how important the topic is today. Jeong et al. (2015)20 introduced a very interesting idea by using a cascade reactor system on the basis of capillaries as well. These columns are functionalized and deposited with the catalysts and are needed to be held in place by magnets to not leach out. This can be a drawback, since with a scale up, this approach may be difficult to realize. To simply place the catalyst in between filters and flush the reactants through the bed is a well-known and easy technique to scale up, as long as the pressure drop over the bed stays small, which can be realized with bigger particles or pellets.
As the conversion of both the single run using Amberlyst 15 and Ni@WC as well as the combined run showed high conversion rates (up to 85%), the selectivity was nevertheless affected in our system. Assuming a full conversion of the HMF derivative to DMF or ethyl levulinate under the same conditions as applied in the single run, but with an increase to 30 bar pressure, especially in the Amberlyst 15 column, the yield of the ring product dropped from 53% to about 38%. This 15% loss of the furan ring product is speculated to be found pro rata in the ethyl levulinate as a consecutive reaction with a yield of 47% and hence an increase of about 30%. The additional 15% increase comes most likely from the increased pressure and therefore a more effective conversion in general, when pressure is applied. Since the diffusion rate towards the catalytic surface depends on the pressure difference in the system, a bigger discrepancy also results in a higher diffusion throughout the stagnant liquid layer around the catalyst and therefore the concentration of hydrogen on the surface.
Stability of the continuous and combined system
Since sufficient activity as well as selectivity are given, the stability is another important property for a possible application in industry. Over a continuous run of 7 hours (Fig. 2/ESI-S6†), the reactivity did not show a significant difference. The concentrations of DMF can be considered as constant over all this time and give a first hint towards the longevity and stable run, which is important for an industrial process. A slight decrease of the ethyl levulinate concentration without the increase of DMF possibly hints towards the formation of side products like humins and coke, which is judged by us as the major remaining problem for a potential scale up. Analysing the catalyst and checking for the differences in appearance, a strong color change from grey to black can indeed be observed for the Amberlyst 15. To check for the formation of humins, a blank test was run additionally using granular, non-catalytic silica in a 250 mm column under the standard conditions of the fructose dehydration. After several hours of the continuous run, the silica clearly showed the formation of a brown surface layer. Most likely this film consists of adsorbed and dehydrated fructose with the highly surface active reaction products clogging the particles together. This phenomenon is very likely to also happen using the Amberlyst 15 catalyst, whereas the surface is even more hydrophobic. Fig. 3C clearly shows a caking of the primary single μm particles. Interestingly, the baking process occurs at the entrance side of the column, indicating humin formation. The rest of the Amberlyst 15 particles are still free and accessible, which was also confirmed by titration before and after the reaction of 7 hours (ESI-S9†). The titration showed a slight decrease of 11% acidic sites. The elemental analysis showed as well a slight increase of the carbon content (3 wt%), as well as a decrease of the sulfur content (ESI-T1†), which underlines the formation of higher mass molecules and a little leaching of the sulfonic acid groups. The back pressure of the column (about 3 bars) still did not increase after all this time, which hints that the clogging is still far off. This agglomeration together with the “coating” is very likely to decrease the longevity of the solid acid catalyst when used beyond the applied 7 hours scale. To avoid these phenomena, further acid catalysts and especially supports with different wettabilities have to be explored.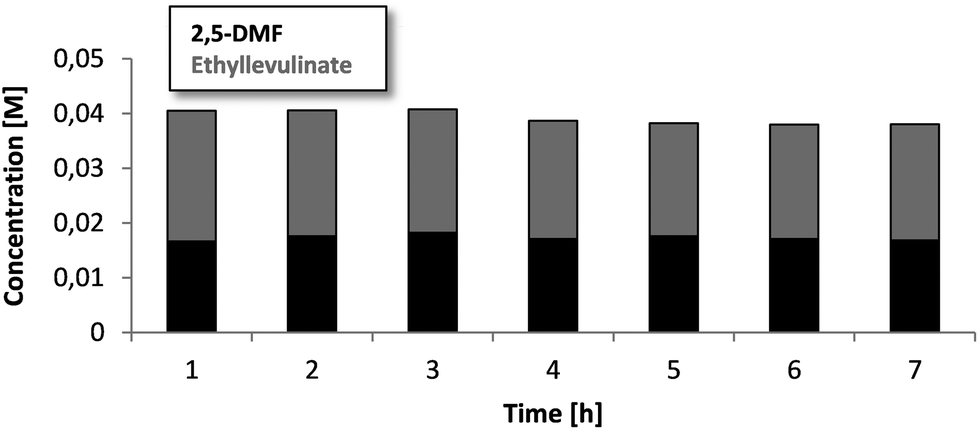 | ||
| Fig. 2 Concentration trend of DMF and ethyl levulinate over 7 h of a continuous run in a combined system with about 85% conversion. | ||
 | ||
| Fig. 3 A: Blank granular silica before and after the reaction with fructose solution at 110 °C and 0.25 mL min−1 in a 250 mm column. B: Amberlyst 15 catalyst before and after reactions with fructose solution at 110 °C and 0.25 mL min−1 in a 250 mm column. C: Enlarged picture of Amberlyst 15 spheres after usage in the reaction. | ||
Separation of products
Since DMF (bp: 95 °C) and ethyl levulinate (bp: 205 °C) are produced with a good conversion and yield and both are valuable products for further upgrade, a possible separation was explored. Hence both boiling points are separated by about 110 °C and it is fairly easy to isolate the molecules by distillation. Additionally, the evaporation of DMF requires three times less energy compared to ethanol, even if the boiling point is 14 °C higher than DMF.21 These facts encouraged us to use a simple rotary evaporator, whereas all DMF is found in the distillate, due to its higher volatility, and pure ethyl levulinate stays in the bottom of the flask (ESI-S5†). By using ethanol as a solvent, this separation step is easy to perform and outrivals water, which would be hard to remove. Both the products have a significant value higher than the raw material glucose.Experimental
Catalyst selection and characterization
Since the upgrade of cellulosic biomass is a multistep process and comprises a bunch of consecutive reactions with partially unwanted side reactions, the catalysts for the two step continuous process need to be carefully selected and optimized. The hydrolysis of wood into carbohydrates and the related processes were already invented at the beginning of the twentieth century. Glucose/fructose isomerization, e.g. in acidic solution or by glucose-isomerase, is also commonplace and commercial.22 For the first triple dehydration steps towards 5-HMF, the commercial Amberlyst 15 (ESI-S7A†) was selected, known as one of the strongest solid acid catalysts on the market. Here, sulfonic acid groups are bound to a macroreticular styrene-divinylbenzene support. We chose 300 μm diameter particles in the hydrogen form and packed them into a 250 mm empty HPLC column with a diameter of 4.6 mm (volume of 4.15 mL). As backpressure always plays a crucial role in continuous flow systems, powder-like catalysts often have this as the main disadvantage; however, due to the chosen particle size, backpressure effects were negligible in the present system. The packed column (packed with 2.1 g catalyst, dead volume about 2.5 mL) was conditioned below 100 °C with ethanol before used in the reaction. Because Amberlyst 15 has a maximum thermal stability of 120 °C, the reaction was done at 110 °C, while reaction time was varied with the flow rate.After the first flow step, the intermediate is not isolated, but the flow is further directed into a second smaller column. Here, the hydrodeoxygenation was performed using elemental hydrogen over a Ni@WC catalyst. This catalyst was synthesized using tungsten mono-carbide (WC) nanoparticles with particle sizes of 175 nm ± 25 nm as a hydrothermally stable support on which nickel nanoparticles were deposited by a wet impregnation method in the hydrogen oven. WC is a metallic ceramic in which the catalytic nanoparticles are stabilized by electron transfer-heterojunction effects and are stable under hydrothermal conditions as such.23 The tungsten carbide (2 g) was dispersed in ethanol (20 ml) together with nickel(II) chloride (0.43 g). After intense stirring and removal of ethanol in a rotary evaporator, the dry powder was placed into a hydrogen furnace for the reduction of nickel ions to Ni(0). The application of 400 °C for 3 h under a 5% H2/Ar atmosphere gave a 10 wt% Ni@WC powder (ESI-S7B†).
ICP measurements approved the relative Ni content as 8.3 wt%, which is in agreement with the measurement failure. Also after the reaction, this wt % did not decrease (ESI-T2†). 1.5 g of this catalyst was placed into a 70 mm cartridge with a diameter of 3.0 mm and therefore a volume of 0.424 mL. The crystalline size of the tungsten carbide was calculated to be 21.3 nm and the one of nickel to be 27 nm, according to XRD and the Scherrer equation. The particle size of the nickel was proven to be about 35 nm using the SEM (Fig. 4). The nitrogen sorption of the Ni@WC material was measured and gave a specific surface area of 9 m2 g−1.
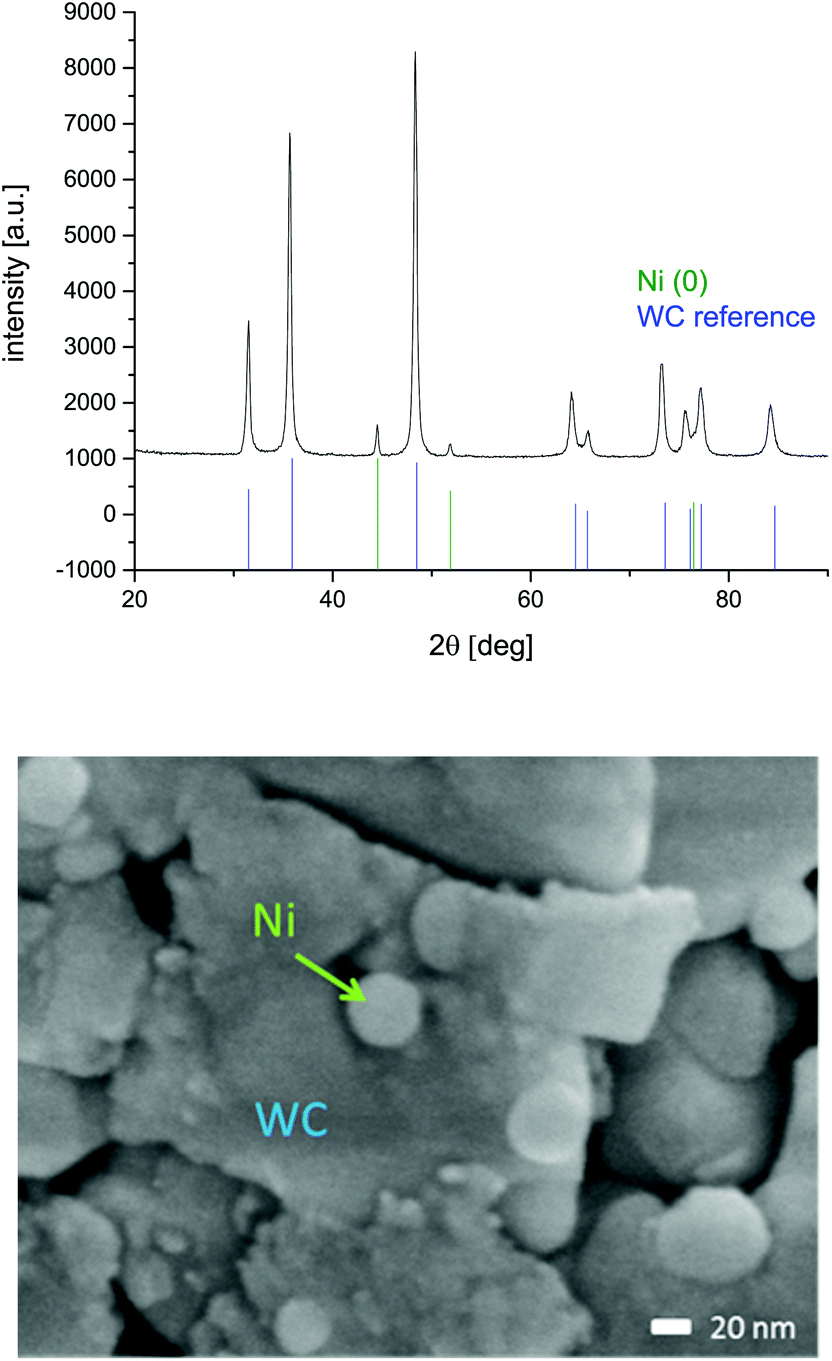 | ||
| Fig. 4 XRD-diffractogram of Ni@WC catalyst before application. Nickel and WC references are extracted from the PDF2 database. Additonally the scanning electron microscopy picture of 10 wt% Ni@WC is given (ESI-S8†). | ||
Substrate preparation
In compliance with the rules of green chemistry, ethanol was chosen as a solvent (200 ml) for fructose (1.8 g, 0.05 M). Alcohol as a solvent brings the advantage of a more simplified analysis in the GC-MS system as well as a more simple separation of the products by evaporation. To facilitate the solubility of sugars in alcohol at room temperature, formic acid (0.5 M) was added in ten times molar excess due to limited hydrogen bond formation. The solution was stirred until all the sugar crystals were dissolved. Formic acid also occurs as a side-product during the formation of levulinic acid and is therefore anyway present in the reaction and the circulating solvent. Since the sulfonic acid groups of Amberlyst 15 have very strong acidic properties, formic acid is believed to just play a minor role as a homogeneous catalyst,24 but certainly helps for a potential glucose-fructose equilibration.Conclusions
In conclusion, it was shown that under the given mild conditions, a combination of commercial Amberlyst 15 (110 °C, 30 bar, 0.25 mL min−1) and synthesized Ni@WC (150 °C, 30 bar, 0.25 mL min−1, hydrogen) in a simple cascade flow reactor provided a good conversion of fructose with about 85% yields of DMF (38%) and ethyl levulinate (47%) with a total reaction time of 20 min. This simple set-up enables the continuous synthesis of DMF as well as ethyl levulinate, two platform chemicals which are easy to separate by distillation. The stability of the catalysts was tested under continuous run conditions and did not show a significant decrease of the product concentration within 7 h. The currently used polymeric solid acid catalyst for dehydration is however considered to be the weak point to work on, as it showed indications for the formation of humins, pointing to a potential clogging at extended timescales. In general, we presented a highly worthwhile process, using non-noble metal catalysis in a wet flow chemistry set-up in the minute scale. All chemicals used are non-toxic and the whole process can be considered as green.Further work is currently expanded in three directions. On the one hand using saccharide/glucose is meaningful, and the isomerization of glucose to fructose could be integrated into the flow set-up. Expansion to lignocellulosic biomass or similar waste streams can be considered, but wood sugar is preferentially made in an upstream step. It, however, can be used without purification, as 2-methylfuran, the product of xylose, is again easily separable at the end. The development of alternative catalyst supports that avoid the tendency of humin deposition or the use of tungsten is another task for catalyst optimization. Furthermore, DMF cannot only be used as a fuel, but also upgraded further, e.g. in a Diels–Alder reaction to produce non-oxygen containing hydrocarbons as p-xylene. Also, this step could be easily integrated into a cascade flow process in the future.
Acknowledgements
We acknowledge the Max Planck Society for financial support, as well as UNICAT, the German Excellence Cluster for Catalysis. The technical staff at the MPI is thanked for standard analysis. Open Access funding provided by the Max Planck Society.Notes and references
- T. L. Nguyen, S. H. Gheewala and S. Garivait, Environ. Sci. Technol., 2007, 41, 4135–4142 CrossRef CAS PubMed.
- J. B. Binder and R. T. Raines, J. Am. Chem. Soc., 2009, 131, 1979–1985 CrossRef CAS PubMed.
- S. Dutta, S. De, M. I. Alam, M. M. Abu-Omar and B. Saha, J. Catal., 2012, 288, 8–15 CrossRef CAS.
- S. Zhong, R. Daniel, H. Xu, J. Zhang, D. Turner, M. L. Wyszynski and P. Richards, Energy Fuels, 2010, 24, 2891–2899 CrossRef CAS.
- C. L. Williams, C.-C. Chang, P. Do, N. Nikbin, S. Caratzoulas, D. G. Vlachos, R. F. Lobo, W. Fan and P. J. Dauenhauer, ACS Catal., 2012, 2, 935–939 CrossRef CAS.
- (a) J. F. Saeman, J. L. Bubl and E. E. Harris, Ind. Eng. Chem., Anal. Ed., 1945, 17, 35–37 CrossRef CAS; (b) S. T. Moe, K. K. Janga, T. Hertzberg, M.-B. Hägg, K. Øyaas and N. Dyrset, Energy Procedia, 2012, 20, 50–58 CrossRef CAS; (c) E. C. Sherrard and F. W. Kressman, Ind. Eng. Chem., 1945, 37, 5–8 CrossRef CAS.
- M. D. Kaufman Rechulski, M. Käldström, U. Richter, F. Schüth and R. Rinaldi, Ind. Eng. Chem. Res., 2015, 54, 4581–4592 CrossRef CAS.
- I. Micromidas, Vol. 2017, 2013.
- T. I. Kosolapova, Carbides; properties, production, and applications, Plenum Press, New York, 1971 Search PubMed.
- R. S. Assary, T. Kim, J. J. Low, J. Greeley and L. A. Curtiss, Phys. Chem. Chem. Phys., 2012, 14, 16603–16611 RSC.
- M. M. Titirici and M. Antonietti, Chem. Soc. Rev., 2010, 39, 103–116 RSC.
- (a) C. M. Lew, N. Rajabbeigi and M. Tsapatsis, Ind. Eng. Chem. Res., 2012, 51, 5364–5366 CrossRef CAS; (b) Z. Wang and Q. Chen, Green Chem., 2016, 18, 5884–5889 RSC.
- T. Thananatthanachon and T. B. Rauchfuss, Angew. Chem., Int. Ed., 2010, 49, 6616–6618 CrossRef CAS PubMed.
- B. Saha and M. M. Abu-Omar, ChemSusChem, 2015, 8, 1133–1142 CrossRef CAS PubMed.
- (a) J. Luo, L. Arroyo-Ramírez, J. Wei, H. Yun, C. B. Murray and R. J. Gorte, Appl. Catal., A, 2015, 508, 86–93 CrossRef CAS; (b) S. De, B. Saha and R. Luque, Bioresour. Technol., 2015, 178, 108–118 CrossRef CAS PubMed.
- V. Molinari, M. Antonietti and D. Esposito, Catal. Sci. Technol., 2014, 4, 3626–3630 CAS.
- M. Braun and D. Esposito, ChemCatChem, 2017, 9, 393–397 CrossRef CAS.
- Y.-B. Huang, M.-Y. Chen, L. Yan, Q.-X. Guo and Y. Fu, ChemSusChem, 2014, 7, 1068–1072 CrossRef CAS PubMed.
- X. Xiang, J. Cui, G. Ding, H. Zheng, Y. Zhu and Y. Li, ACS Sustainable Chem. Eng., 2016, 4, 4506–4510 CrossRef CAS.
- G.-Y. Jeong, A. K. Singh, S. Sharma, K. W. Gyak, R. A. Maurya and D.-P. Kim, NPG Asia Mater., 2015, 7, e173 CrossRef CAS.
- Y. Roman-Leshkov, C. J. Barrett, Z. Y. Liu and J. A. Dumesic, Nature, 2007, 447, 982–985 CrossRef CAS PubMed.
- L. Hobbs, in Starch, Academic Press, San Diego, 3rd edn, 2009, pp. 797–832 Search PubMed.
- X.-H. Li and M. Antonietti, Chem. Soc. Rev., 2013, 42, 6593–6604 RSC.
- N. Jiang, R. Huang, W. Qi, R. Su and Z. He, BioEnergy Res., 2012, 5, 380–386 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7gc01055a |
| This journal is © The Royal Society of Chemistry 2017 |