
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structural models of the biological oxygen-evolving complex: achievements, insights, and challenges for biomimicry
Satadal
Paul
,
Frank
Neese
and
Dimitrios A.
Pantazis
 *
*
Max Planck Institute for Chemical Energy Conversion, Stiftstr. 34-38, 45470 Mülheim an der Ruhr, Germany. E-mail: dimitrios.pantazis@cec.mpg.de
First published on 17th March 2017
Abstract
The oxygen-evolving complex (OEC) in Photosystem II (PS-II) of oxygenic photosynthesis catalyzes the oxidation of water into dioxygen, protons and electrons, a reaction that underpins solar to chemical energy conversion in the biosphere. The inorganic core of the OEC is an oxo-bridged cluster that comprises four Mn and one Ca ions, Mn4CaO5. Deciphering the structure of this cluster and its immediate environment has been the aim of intense experimental efforts that span decades of research. The constantly improving structural definition of the OEC in the last fifteen years has offered opportunities to better understand its properties and function; it has also provided ever clearer and more well-defined targets for biomimetic synthetic chemistry. Here we present a critical overview of the most recent advances in molecular structural models of the OEC, focusing mostly on successful research efforts reported after the availability of atomically resolved crystallographic models of PS-II. We delineate the properties that have been targeted in biomimetic studies and analyze which structural aspects have by now been reproduced in synthetic systems. In combination with in-depth theoretical studies, the availability of novel synthetic structural analogues has led to considerable insight into structure–property correlations despite the lack of catalytic activity. Nevertheless, there are important features of the OEC that remain inaccessible to synthetic chemistry. Principal among them are the unique type of restricted structural flexibility and the highly structured and stable ligand sphere which enable the tightly controlled interplay of geometry, spin state and reactivity that is the hallmark of the OEC.
Introduction
The oxygen-evolving complex (OEC) of Photosystem II (PS-II) splits water into dioxygen, protons and electrons, generating the reducing equivalents and proton gradient that drive the rest of the photosynthetic machinery in cyanobacteria, algae and plants.1–6 Tremendous effort has been directed towards synthesizing models of the inorganic core of the OEC.7–21 Replicating the structural and electronic properties of the natural system in synthetic analogues would allow us to gain insight into the properties and function of the biological catalyst itself, as well as to understand how certain structural and electronic aspects may relate to activity, as part of the greater drive towards the development of artificial photosynthesis.22–30 The OEC contains a Mn4CaO5 cluster,31,32 often discussed in terms of a Mn3CaO4 unit connected with a dangling fourth manganese via one of the unit's oxo bridges and an additional bis-μ-oxo bridge (Fig. 1a). The metal ions are coordinated to mostly carboxylate ligands (side chains of aspartate and glutamate residues), one histidine, and four water molecules or water-derived ligands. Dioxygen evolution is the final step of a catalytic cycle comprising distinct metastable oxidation states known as Si states, where the subscript indicates the number of stored oxidizing equivalents (Fig. 1b).33–38 Driven by photo-induced charge separation at the reaction center chlorophylls of PS-II, the OEC cycles from the S0 state with individual manganese oxidation states of Mn(III)3Mn(IV), via a series of metal-centered oxidation to the all-Mn(IV) S3 state; the fourth oxidation, which may or may not be metal-centered, leads to a transient S4 state that evolves O2 and resets the catalyst to S0.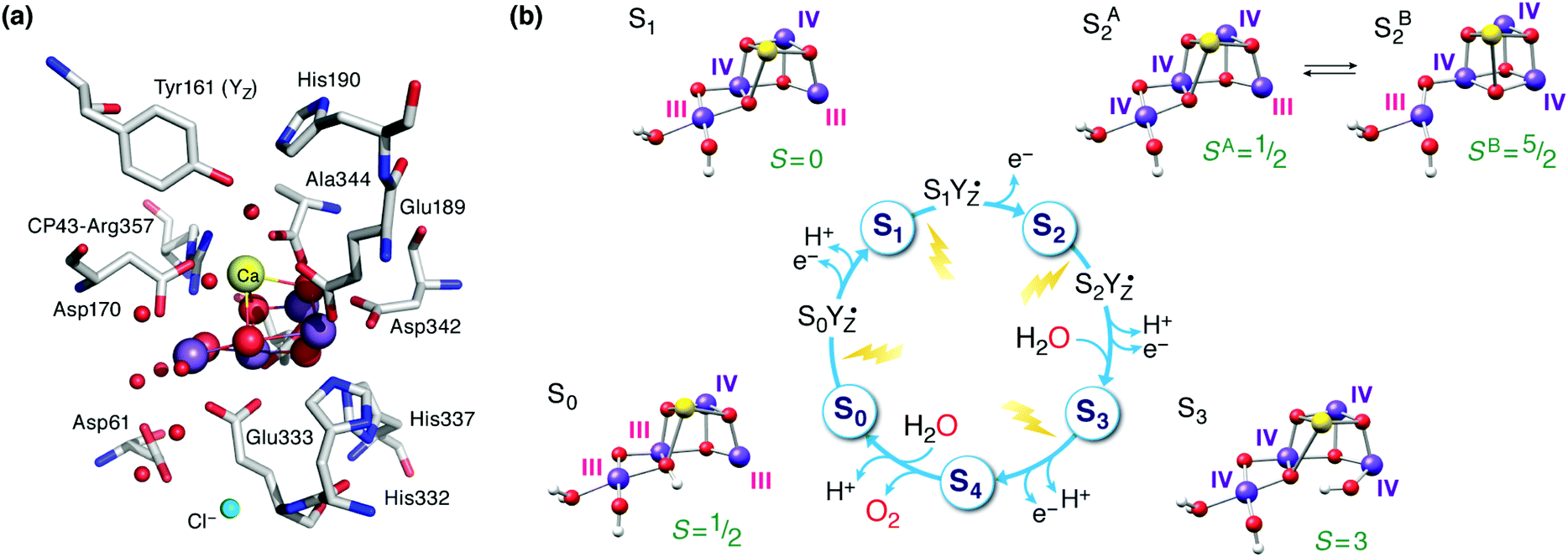 | ||
| Fig. 1 (a) The manganese–calcium cluster of the oxygen-evolving complex and its protein environment.41 See Fig. 2b for a schematic diagram and labelling of the inorganic core. (b) The cycle of Si states with structural, individual Mn oxidation states, and total spin state assignments.42 | ||
Even the above cursory structural description of the OEC is in fact the combined product of enormous research efforts pursued internationally over many years. For example, although various structural elements of the OEC were known for a long time and techniques ranging from EXAFS to EPR spectroscopy provided important information about the possible arrangements of the metal ions, the first crystallographic model of PS-II only appeared in 2001.39 During the 1980s and 1990s the elemental composition and the fact that the manganese ions should be close enough and magnetically interacting, and hence at least partially connected in an uncertain topology by an uncertain number of oxygen and/or carboxylate bridges pretty much summarized the available information about the structure of the OEC. During this time hypotheses regarding the structure of the OEC were stimulated by molecules that were synthesized and structurally characterized, i.e. the availability of any given connectivity pattern would inspire speculation about whether this pattern might mimic the OEC. This period of time for synthetic inorganic chemistry was characterized by the absence of specific targeted structural motifs: the topology of the OEC core was unknown so there was no directed effort towards a synthetic structural goal. By surveying the literature of that period one would be excused to form the impression that any molecule that contained more than one Mn could be considered a potential mimic of the OEC. Regarding the suggestions about the structure of the OEC itself, the absence of concrete structural data behind the ideas circulated at the time led Wieghardt to characterize the then available models as “an aesthetically pleasing combination of experimental facts, thoughtful deduction, and a lot of imagination”.40
Important and beautiful chemistry resulted this way nevertheless, vastly enriching the library of manganese complexes, especially of oligonuclear compounds. Early synthetic endeavors encompassed oxo-bridged mixed-valent dimeric manganese complexes, and gradually extended to achieve successful preparation and characterization of manganese complexes of varying nuclearity, composition and architecture. Manganese complexes of this time and the insights gained from them have been extensively reviewed.7,10,40,43,44 The main focus of the present review will instead be on more recent achievements and on synthetic efforts that were expressly guided by a structure-oriented biomimetic approach inspired by modern knowledge on the topology of the OEC.
The protein crystallography era was inaugurated in 2001 by the first X-ray diffraction model of PS-II.39 This low-resolution model (3.8 Å) could not offer an atomistic model of the inorganic core, but for the first time it looked like the structure of the OEC was within reach. An important milestone was reached in 2004 with the 3.5 Å resolution model of Ferreira et al.,45 known more colloquially as the “London model”. This offered the first, incomplete yet daringly specific suggestion about the topology of the inorganic core, proposing the presence of a Mn3CaO4 cubane subunit with a fourth Mn ion attached to one of the four O bridges of the cube (Fig. 2a). Thus, the London model provided a clear and well-defined target for structural biomimetic chemistry, but also formed the basis for a series of computational studies.46–50 The crystallographic view on the OEC was refined by the “Berlin model” of 3.0 Å resolution51 that offered an improved view of the immediate environment of the cluster and was incorporated with great success in mechanistic studies by Siegbahn.52–54 EXAFS studies have provided valuable information about intermetallic distances and gave rise to numerous structural interpretations, but without converging to consensual three-dimensional models.36,55–64 The first atomic-resolution (1.9 Å) model of the OEC was reported in 2011 by Umena et al.41 This model established the contemporary view of the inorganic core with the five oxo bridges and altered connectivity compared to the original Ferreira et al. model (Fig. 2b). Still, a 1.95 Å resolution model that is free from radiation-damage64–67 and can be safely attributed to predominantly42,68,69 the S1 state of the OEC was only reported in 2015.70
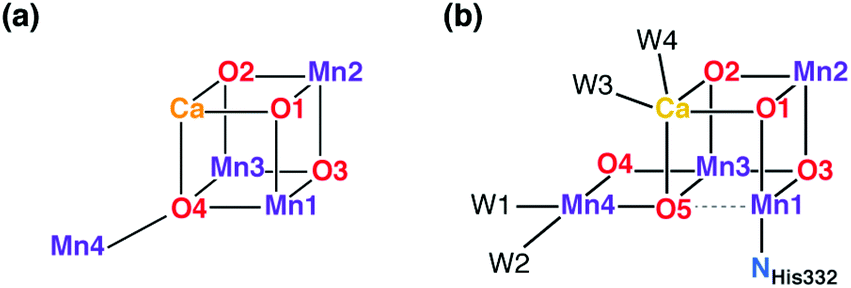 | ||
| Fig. 2 Schematic connectivity diagram of the London model for the OEC core (a)45 and of the current model for the Mn4CaO5 cluster (b).41,70 | ||
Inspired by the 2011 crystallographic model,41 a staggering volume of subsequent experimental and theoretical studies (too many to recount here) began to refine it and address additional structural and electronic aspects of the OEC in various Si states. A landmark achievement in structural insight was the structural interpretation in 2012 of the two well-known distinct EPR signals of the S2 state (a multiline signal at g ≈ 2 arising from a spin S = 1/2 state and a signal at g ≥ 4.1 arising from an S = 5/2 state) in terms of valence isomeric forms of the inorganic core that differ in their oxo-bridge connectivity.71 The two forms are interconvertible via a shift in the bonding of the central O5 bridge: the high-spin form (SB2 in Fig. 1b) contains a “closed” Mn3CaO4 cubane with a five-coordinate dangling Mn(III) ion, whereas in the low-spin form (SA2) the cubane is “opened” by the presence of the Jahn–Teller distorted five-coordinate Mn(III) ion at the Mn1 site (see Fig. 2b for labeling), resulting in rearrangement of the bonding and formation of a Mn4–O5 bond instead.71–73 The emergence of the high-spin closed-cubane topology in the S2 state has been proposed to be mechanistically important in subsequent catalytic progression.6,74–78 There is reasonably broad convergence of opinion on the topology of the S0, S1, and S2 states (depicted in Fig. 1b) arising from many recent studies, but the details of the S2–S3 transition and the precise nature of the S3 state remain under active investigation.74,77,79–98 There is essentially no structural information about states past the S3 and hence the identity of S4 and of the intermediates involved in dioxygen formation and release remain debated.99–103
A point that is important to clarify from the outset because it is highly relevant for the present review concerns the precise bonding topology in the inorganic core. The description of the cluster in terms of a “3 + 1” model for the arrangement of Mn ions (a topology already included in the multitude of possible interpretations of EXAFS data for a long time55) is intimately connected with the analysis of the S2 state multiline EPR signal by Britt and coworkers.104 The seemingly analogous description of the cluster in terms of a cuboidal oxo-bridged Mn3Ca unit plus a “dangler” Mn ion reflects the original London model of the inorganic core (Fig. 2a), but strictly speaking this terminology is not entirely appropriate for current models of the OEC—at least not for the connectivity in the lower Si states. In other words, the (still valid in most cases)105,106 “trimer–monomer” description of the magnetic topology of the OEC and the “cubane–dangler” description of the geometric structure are neither synonymous nor equally applicable. Besides, it is already obvious from the core models shown in Fig. 1b that the inorganic core in general does not contain a “proper”, fully bonded Mn3CaO4 cubane in most of the observable states. This point will prove important in the discussion of current synthetic models.
It is useful to keep the above evolution of structural ideas and models in mind when reviewing the efforts of synthetic chemists to create structural analogues of the OEC, because for a long time these efforts were inspired by models that eventually proved to be inaccurate or of only indirect relevance to the current detailed view of the OEC. Pecoraro and Hsieh appropriately remarked on this point: “at times, it seems as if nothing in photosynthetic research stands the test of time…”.9 In the present review we wish to provide a critical perspective on the recent evolution in the geometry, electronic structure and associated properties of synthetic systems from recent progress towards improved structural mimics of the OEC. At this point in time there is no convergence of structural analogy and water-oxidizing ability in synthetic models. This means that existing Mn complexes that have been implicated in oxygen evolution do not, in general, resemble the OEC in terms of geometric structure. In addition, it is sometimes unclear whether such complexes are genuine molecular catalysts107–109 or whether dioxygen evolution can always be attributed to true water oxidation.110–112 Given that such non-biomimetic manganese-based systems and their chemistry have been extensively reviewed in recent literature,18–21,113–116 we will not discuss them here. Similarly, we will not address heterogeneous systems,17,117–121 although it should be recognized that specific structural motifs related to the OEC can be present in Mn or Mn/Ca oxides.17,122
Our point of departure is a series of synthetic tetramanganese clusters. From the first generation of oxo-bridged tetramanganese clusters the presentation of systems proceeds to oxomanganese cubanes incorporating calcium and the description of structures where a fifth ion is attached to the Mn3Ca unit. Through experimental characterization and the significant input of theoretical chemistry many of these structural analogues of the OEC have already contributed significant insights into the properties of the biological system.
Targeted structural features
Before we proceed it is useful to highlight a few salient properties of the OEC that serve as individual or combined targets for structural biomimetic chemistry. These include:• The stoichiometry of four manganese and one calcium ions.
• Incorporation of Ca2+ within an oxo-bridged metal framework.
• A Mn3CaO4 substructure that may or may not be present as a fully bonded unit in some of the Si states depending on the Mn oxidation states.
• A fourth Mn ion (“dangler Mn”) external to the above cuboidal unit, connected with it via oxo bridges.
• Almost exclusively carboxylate and water-derived ligands.
• In addition to the above structural features, a model should contain the physiologically relevant oxidation states of Mn ions (exclusively III and IV)42 and the ligand framework would ideally support access to multiple oxidation states.
All of the above structural features have important implications for the electronic structure of the cluster, its magnetic and spectroscopic properties, and ultimately for its catalytic function, although the latter cannot be meaningfully considered in isolation from the protein matrix of the OEC and the rest of the PS-II machinery.
Overview of synthetic models
Tetramanganese complexes
First we would like to present a few tetranuclear Mn complexes that have been historically important in biomimetic synthetic chemistry, even though most of them would not be recognized today as direct mimics of the OEC. A variety of systems with metal oxidation states ranging from Mn(II)4 to Mn(IV)4 pervade the literature of tetranuclear manganese chemistry. Reviews on tetranuclear Mn-oxo clusters provide a classification in terms of commonly observed and structurally distinct patterns shown in Fig. 3.7,10,123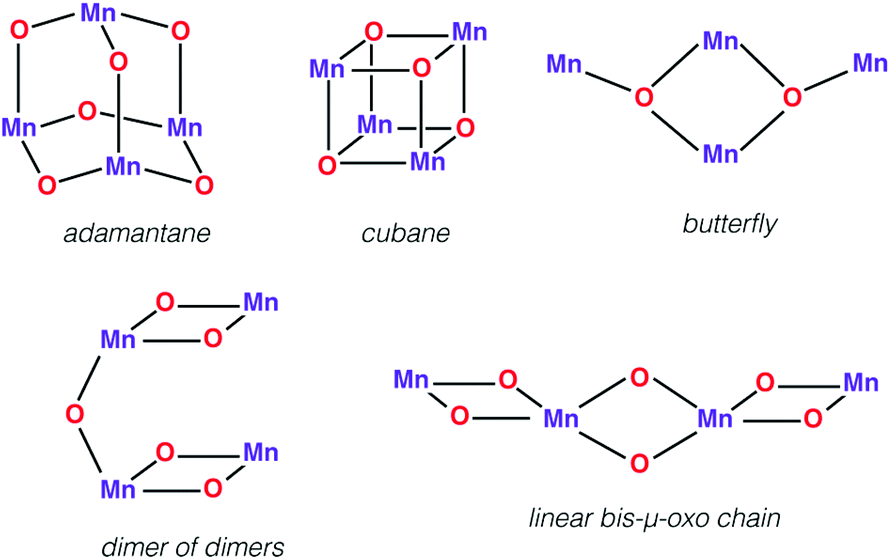 | ||
| Fig. 3 Representative topologies of oxo-bridged tetramanganese clusters. | ||
One of the earliest models with a tetramanganese motif was the Mn(IV)4O64+ adamantane stabilized by three chelating 1,4,7-triazacyclononane ligands (Fig. 4a).124 The complex by Wieghardt and co-workers shows weak ferromagnetic interaction between Mn ions that changes to weak antiferromagnetic upon protonation, as observed by Hagen et al.125 Similar adamantane-shaped tetranuclear complexes were reported by Armstrong and co-workers.126
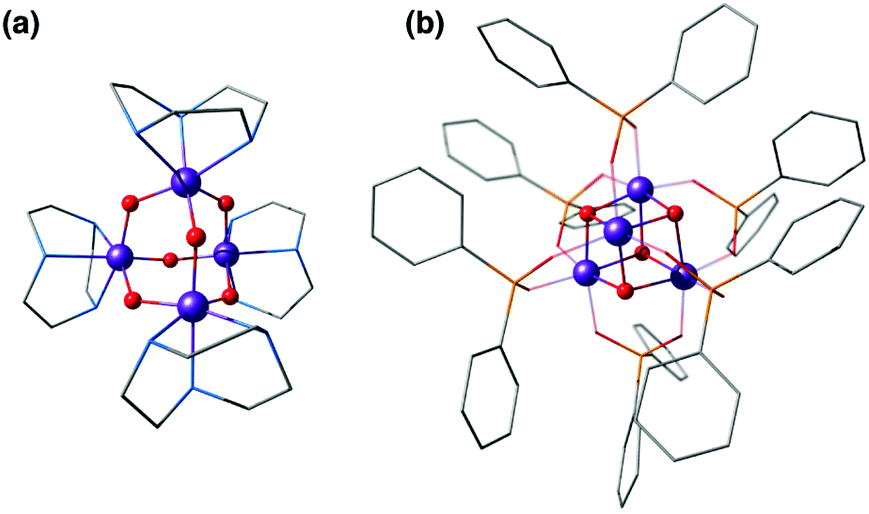 | ||
| Fig. 4 Mn4O6 adamantane complex by Wieghardt and co-workers124 (a) and Mn4O4 cubane complex by Dismukes and co-workers127 (b). | ||
A Mn4(μ3-O)4 cubane core has been one of the early suggestions for the OEC structure in different Si states of the catalytic cycle.128,129 A cuboidal arrangement of Mn ions was accessed by Christou and coworkers who synthesized the distorted cubane with stoichiometry [Mn(III)3Mn(IV)(μ3-O)3X]6+ (X = Cl or Br).43,130–132 Magnetochemistry revealed a high-spin (S = 9/2) ground state with well separated excited states resulting from antiferromagnetic interaction between the unique Mn(IV) and the three ferromagnetically coupled Mn(III) ions,132–134 while the near-parallel alignment of the three Jahn–Teller axes of the Mn(III) ions leads to high magnetic anisotropy.133,134 Low-temperature X-band EPR spectra showed two sets of signals, a broad peak at g ≈ 6 and another well resolved 16-line signal with 55Mn hyperfine structure centered at g ≈ 2, reminiscent of the two EPR signals in the S2 state of the OEC.131,135
Over the following years analogous compounds with varying terminal ligands (Cl−, pyridines, acetylacetonates, dibenzoylmethane, etc.) and the anionic μ3-X positions (X = I−, Cl−, Br−, F−, OAc−, OH−, OMe−, OPh−, NO3−, N3−, and NCO−) further extended this class of complexes.131,134,136–143 Some of them, for example a compound reported by Wang et al., were also shown to mimic the S1 to S2 oxidation step.142
An all-μ-oxo bridged Mn(III)2Mn(IV)2 cubane complex was reported by Dismukes and co-workers (Fig. 4b).127,144–146 Despite being a mixed valent species, the compound Mn4O4L6, (L = Ph2PO2−), showed almost equivalent Mn–O bond lengths, which was attributed to valence delocalization at elevated temperature (298 K). Lowering the temperature to 150 K led to a small differentiation of Mn–O bonds147 and the bond length inequivalence was also shown to depend on the ligand L.148,149 The crystal structure of the singly oxidized product of the complex [Mn4O4]7+ demonstrates a trigonal distortion compared to the tetragonal symmetry of the parent complex, i.e. [Mn4O4]6+.150 In this oxidized form the Mn(III)Mn(IV)3 valence distribution was assigned to the manganese ions, although no significant axial tetragonal distortion is observed for Mn(III). A similar Mn4O4 cubane with two Mn(III) and two Mn(IV) was prepared by Kanady et al.151 An exciting feature of the Dismukes cubane is that it can undergo photo-rearrangement to yield O2 and a diarylphosphinate ligand and ultimately decays to a butterfly-shaped product (Fig. 5). The idea of a reversible cubane/butterfly rearrangement was actually one of the early proposed mechanisms for the catalytic cycle of the OEC and the butterfly-shaped structure was one of the suggested structural models for the lower Si states.129,152 Theoretical studies have described the electronic structure and magnetic properties of these complexes, as well as the possible pathways for dioxygen evolution.153–155
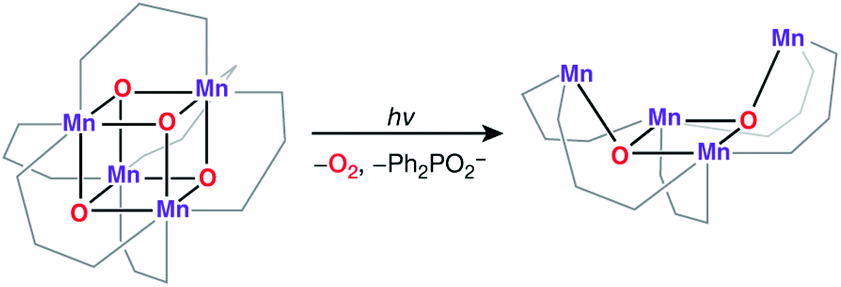 | ||
| Fig. 5 Photoinduced opening of the Dismukes cubane to a butterfly core. | ||
Another example of the butterfly type of complex is the series [Mn4(μ3-O)2(μ-O2CR)7(bpy)2]n+ (R = Me, Et, Ph) spanning oxidation states from Mn(II)2Mn(III)2 to Mn(III)4, which contains a non-planar Mn4 unit, with two Mn atoms forming a central Mn2O2 diamond core and the other two Mn ions being ligated to the oxo bridges (Fig. 6).156,157 The presence of two types of Mn–Mn distance in the OEC inferred from EXAFS, at ca. 2.7 Å and 3.3 Å,40 is fulfilled by the butterfly-shaped complex that contains two different Mn–Mn distances of 2.85 Å and 3.30 Å. Many similarly shaped tetranuclear clusters have been synthesized with bidentate chelating ligands.136,140,158–164 It should be noted that without the presence of at least one Mn(IV) such complexes do not correspond to any one of the physiological states of the OEC.
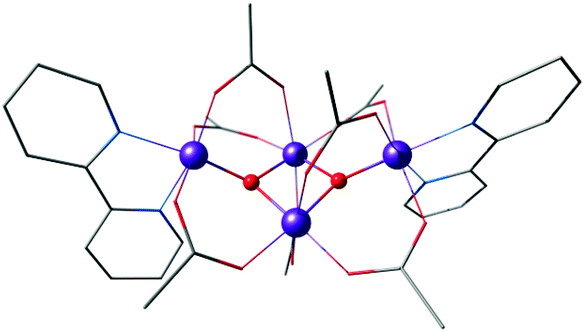 | ||
| Fig. 6 Butterfly type complex by Vincent et al.156 | ||
The “dimer of dimers” geometry has been another possible model of the OEC discussed on the basis of EXAFS data.56,57 This motif was influential and enjoyed popularity for a long time, until the 3 + 1 arrangement of the Mn ions began receiving stronger support.39,104 The archetypal form (Fig. 3) involves two Mn2O2 rings bridged by an oxo, which would justify the presence of both short and long Mn–Mn distances as deduced from EXAFS measurements of the OEC. Several synthetic realizations of this type of core have been reported, with various ligands bridging the two Mn2O2 rings symmetrically or unsymmetrically, and the manganese ions in various oxidation states.165–169 These are discussed in the comprehensive review by Mukhopadhyay and Armstrong.7Fig. 7a shows an example of this type of core, in this case a complex by Chen et al.169 that features two μ-O bridged Mn(IV)2O2 units with each Mn coordinating a terpyridine ligand.
 | ||
| Fig. 7 Dimer of dimers complex by Chen et al.169 (a) and a bis-μ-oxo chain by Philouze et al.170 (b). | ||
A closely related type of topology is that of three joined bis-μ-oxo units. The example of the Mn(IV)4 compound reported by Philouze et al.,170 shown in Fig. 7b, utilizes bipyridine ligands and comprises a chain of bis-μ-oxo units with Mn–Mn distances ca. 2.75 Å. Antiferromagnetic coupling between the Mn ions leads to a diamagnetic (S = 0) ground state. This complex gained high relevance to the OEC because under γ-ray irradiation at cryogenic temperatures it is reduced to the Mn(IV)3Mn(III) form that shows an 18-line X-band EPR signal similar to the multiline signal of the S2 state of the OEC.167 Hence the compound serves as a spectroscopic mimic for understanding the S2 state EPR multiline signal in the OEC135 and has been successfully employed in the evaluation of quantum chemical methods that were developed to target the spin states and EPR parameters of oligonuclear manganese systems.171
All of the complexes described above are characterized by a high degree of symmetry and the presence of predominantly N-donor ligands, whereas the core of the OEC is in fact asymmetric and ligated by carboxylates and only one N-donor. In the following we describe how the efforts to introduce calcium to the cluster led to more diverse structures and eventually to the synthesis of carboxylate-bridged Mn3CaO4 cubanes.
Manganese–calcium cubanes
The calcium ion is indispensable for water oxidation in the OEC and specifically for advancement to the S3 state of the catalytic cycle.172 Its close association with the Mn ions first became apparent from EXAFS, which suggested a Mn–Ca distance of 3.4 Å.173 Gerey et al. have very recently compiled a comprehensive review of manganese–calcium heterometallic compounds;14 here we focus on selected examples. A tetramanganese structure attached to Ca was reported by Jerzykiewicz et al.,174 but the first high-valent Ca-incorporating oxomanganese cubane was prepared by Christou and co-workers—albeit as a substructure of a higher nuclearity complex. The structure consists of a Mn13Ca2 core held together by bridging oxo, hydroxo and methoxy ions and showed a Mn–Ca distance of ca. 3.5 Å (Fig. 8a).175,176 The whole structure can be visualized as a combination of four subunits, two of which are Mn3CaO4 cubanes attached by Mn–O linkages to two other Mn3O4. Thus, a portion of the compound, the Mn3CaO4 cubane subunit linked to a fourth manganese center via an oxo bridge, resembles the OEC.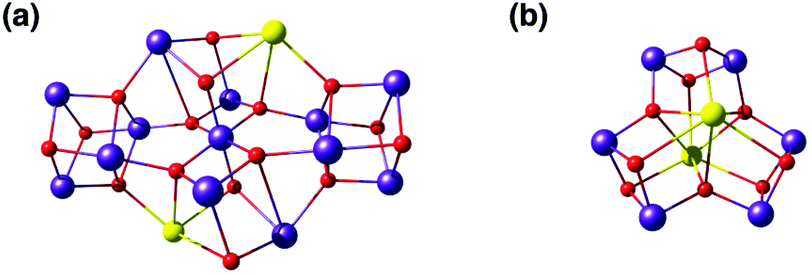 | ||
| Fig. 8 Cores of polynuclear complexes by Mishra et al.175 (a) and by Chen et al.177 (b) containing Mn3Ca cubanes as integrated substructures (ligands omitted for clarity). | ||
Chen et al. reported a family of heteronuclear Mn(IV)Ca-oxido complexes with carboxylate ligands which contain fused manganese/calcium cuboidal moieties and water molecules on Ca2+ similar to the OEC of PSII.177 The complex contains three [Mn2Ca2O4] distorted cubanes sharing a trigonal bipyramidal [Ca2O3] central motif (Fig. 8b).
Christou's group also reported the synthesis, structure and physical properties of a diamagnetic Mn(III)4Ca complex possessing the correct 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 Mn:Ca ratio, but apart from the stoichiometry the complex can hardly be regarded as a structural mimic of the OEC.178 Other Mn/Ca complexes with exact Mn4Ca stoichiometry that are not topological analogues of the OEC were reported by Powell and co-workers, but in lower Mn oxidation states.179
:1 Mn:Ca ratio, but apart from the stoichiometry the complex can hardly be regarded as a structural mimic of the OEC.178 Other Mn/Ca complexes with exact Mn4Ca stoichiometry that are not topological analogues of the OEC were reported by Powell and co-workers, but in lower Mn oxidation states.179
The first distinct cuboidal Mn(IV)3CaO4 unit was synthesized by Agapie and co-workers (Fig. 9).151 In subsequent work, the Ca2+ ion of this heterometallic oxomanganese cubane cluster was replaced by different metals such as Mn3+, Sr2+, Zn2+, Sc3+, Y3+, Gd3+, Ln3+ to investigate the role of redox-inactive metal in tuning the redox potentials of the clusters.180–182 In all compounds the approximately octahedral environment of Mn ions is stabilized by coordination of three alkoxide groups and three pyridine groups attached to an appropriately designed 1,3,5-triarylbenzene spacer. In the lanthanoid series, the crystal structure of Dy3+-substituted cubane also shows two water molecules coordinated to Dy3+, reminiscent of the two Ca-bound water molecules in the OEC (Fig. 1a and 2b).182
 | ||
| Fig. 9 Mn(IV)3CaO4 cubane complex by Kanady et al.151 | ||
Attachment of a “dangler” ion
Critical roles have been attributed to the fourth manganese of the OEC in the bioassembly of the cluster, solvent/substrate attachment, and catalytic function.49,52,84,91,183–193 Although an architecture containing a Mn3CaO4 cubane attached to another manganese ion was discussed already (Fig. 8), these complexes had overall much higher nuclearity than the biological cluster. Similarly, an example of incorporated cubane attached to another metal comes from Chen et al. (Fig. 10).194 Instead of calcium the complex incorporates strontium and the fifth ion, in this case another strontium, does not adopt the same connectivity mode as in the OEC, being rather a component of the second, similar cubane. The core of the complex is composed of two Mn(IV)3SrO4 units connected by one μ2-oxo and two μ4-oxo moieties (Fig. 10).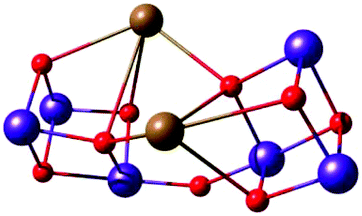 | ||
| Fig. 10 Core of the Mn6Sr2 complex by Chen et al.194 | ||
A complex with a unique dangling Ca2+ ion attached to a distinct Mn3Ca cubane was first reported by Mukherjee et al. (Fig. 11).195 The complex is composed of an asymmetric Mn(IV)3CaO4 core where the metal ions are bridged exclusively by carboxylate/carboxylic acid ligands. The external seven-coordinate Ca2+ is connected to one of the oxo bridges and to six carboxylates, three of which bridge it to Mn ions of the cubane. Dominant ferromagnetic coupling results in a high-spin, S = 9/2 ground state. The magnetic and spectroscopic (EPR) properties of this molecule have been carefully characterized,195 and thus the complex served as an invaluable reference in quantum chemical analysis of the electronic properties of manganese–calcium cubanes.196
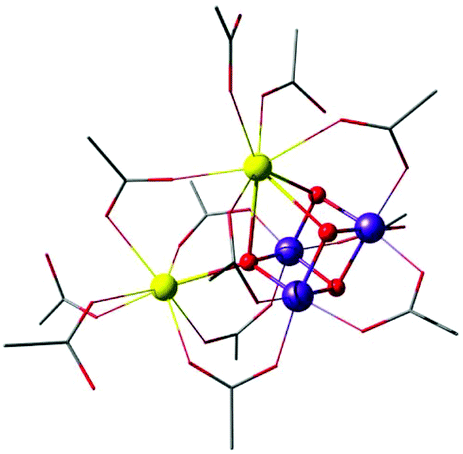 | ||
| Fig. 11 Mn3CaO4 cubane complex with attached Ca2+ ion, by Mukherjee et al.195 (tert-butyl groups on carboxylate ligands simplified for clarity). | ||
Agapie and co-workers described a rational way to increase the basicity of μ3-oxo ligands by modulating ligand scaffolds and desymmetrizing the Mn3CaO4 cluster.181 In this way they could obtain a Ag–Mn3CaO4 complex, where the dangling Ag+ is linked to the cubane via a μ4-oxo, a μ2-alkoxide, and a pyridine (Fig. 12).181 Thus, the synthesis of [Mn(IV)3CaAgO4] from [Mn(IV)3CaO4] provided an example of a systematic approach for the synthesis of the asymmetric pentanuclear core structure of the OEC.
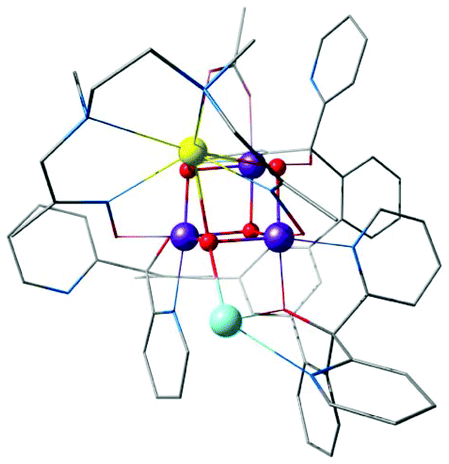 | ||
| Fig. 12 Mn3CaO4–Ag complex by Kanady et al.181 | ||
The latest addition in this series came from Zhang's laboratory; it contains a manganese ion attached to a Mn3CaO4 cubane complexes and represents currently the closest mimic of the OEC.197 This is the first complex to contain a manganese ion attached to the Mn3CaO4 cuboidal unit, thus correctly reproducing the metal stoichiometry of the biological cluster (Fig. 13). Importantly, it was synthesized with a Mn(III)2Mn(IV)2 oxidation state distribution that mirrors that of the S1 state of the OEC and exhibits similar redox properties as the OEC, being able to span several oxidation states. This is a key feature that differentiates this complex from the other cubane-incorporating complexes. Access to multiple oxidation states had previously been demonstrated with a pentanuclear manganese helicate complex, which however does not progress beyond Mn(III)5 within its original ligand framework.198,199 In the one electron oxidized state, the complex by Zhang et al. shows two simultaneous EPR signals, a major one at g ≈ 4.9 and a secondary multiline signal at g ≈ 2, which are reminiscent of the two signals in the S2 state of the OEC.197
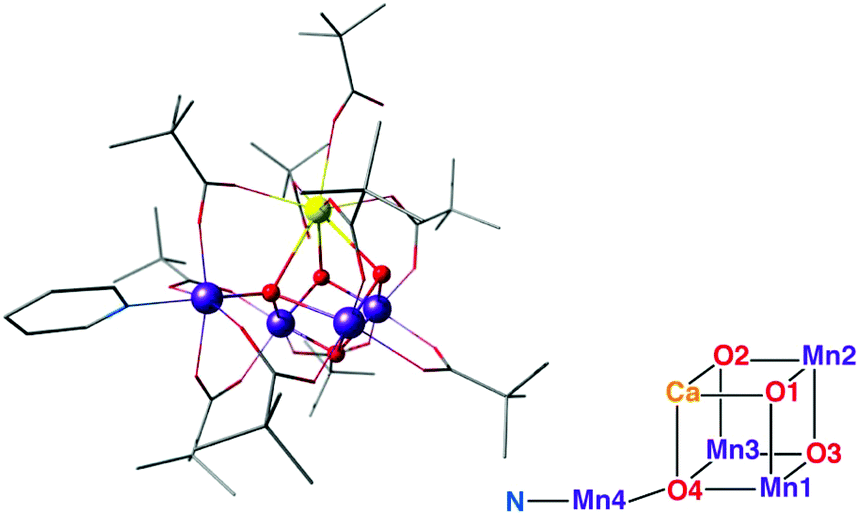 | ||
| Fig. 13 The Mn3CaO4–Mn complex of Zhang et al.,197 with schematic depiction of the inorganic core in analogy to Fig. 2. | ||
The limits of structural analogies should also be recognized so that future challenges can be defined: comparison of the core in Fig. 13 with that in Fig. 2 shows that this synthetic model still does not exactly reproduce the stoichiometry and the connectivity of the OEC. Thus, similarities in observable properties should not be automatically assumed to have the same structural origin. Nevertheless, this complex satisfies many of the target points listed in the previous section and represents the current pinnacle of structural biomimicry.
Discussion of specific examples
In the preceding section we traced, in an inevitably selective manner, the historical development of structural models for the OEC. Here we will discuss in greater depth selected models that incorporate Mn3CaO4 cubane units and discuss the insights obtained from them so far in terms of correlating structure and composition with observed properties.Mn3CaO4 and Mn3CaO4–Ca complexes
We discuss these complexes from the Agapie and Christou groups together because despite the fundamentally different ligand framework, the electronic structure and properties of the inorganic core are similar and are principally defined by the three Mn ions of the Mn3CaO4 cubane.The average Mn–Mn, Mn–Ca and Mn–μ3-O distances in the [Mn3CaO4]6+ core of the complex by Kanady et al. (Fig. 9) as obtained from X-ray diffraction data are 2.834 Å, 3.231 Å and 1.872 Å, consistent with the presence of three Mn(IV) ions.151 Comparison of these distances with experimental data (EXAFS-derived distances and crystallography) on the S1 state of the OEC reveals that the corresponding distances are longer in the natural system. The shorter Mn–Ca distances in the synthetic complex compared to the S1 state of the OEC presumably stem from the constraints imposed by the bridging acetate ligands. However, unlike Mn(IV) ions in the synthetic cubane, the presence of Jahn–Teller distorted Mn(III) in the S1 state of OEC can also be the source of this difference.14 In the S2 state of the OEC, the cluster exists in two interconvertible forms, the open cubane form with the Mn(III) located at the Mn1 site within the Mn3Ca unit, and the closed cubane form with a dangling Mn(III) and a “genuine” Mn3(IV)CaO4 cubane.71 These two valence isomers can be oxidised to the all-Mn(IV) S3 state,82 subject to appropriate ligand binding and deprotonation.74 Hence, the comparison of the synthetic Mn(IV)3CaO4 core with the OEC only finds relevance in the closed-cubane form of the OEC in the S2 and S3 states.
The asymmetric cubane bound to an external Ca ion, [Mn(IV)3Ca2O4(O2CBut)8(ButCO2H)4], reported by Mukherjee et al.195 (Fig. 11) mirrors the OEC with respect to the peripheral carboxylate ligands and obviously the Mn3CaO4 subunit, which as noted above is relevant for the closed cubane forms of the S2 and S3 states of the OEC. The attachment of the external Ca causes a lowering of the threefold symmetry of the [Mn3CaO4] cubane, leading to variance in the Mn–Mn separation and slight changes in the Mn–Ca distances. Whilst the complex by Kanady et al.151 features almost equivalent Mn–Mn distances of ca. 2.83 Å, here the Mn–Mn distances within the cube are 2.74 Å and 2.86 Å,195 highly reminiscent of the 2.73 Å and 2.82 Å Mn–Mn distances in 2:1 ratio suggested from EXAFS on the S2 state of the OEC.200 The Mn–Ca separation of 3.39–3.45 Å within the cubane is similar to the distances (3.3–3.4 Å) in the crystallographic model of the S1 state of the OEC and the EXAFS-deduced average distance (ca. 3.4 Å).63,70 Mn–μ3,4–oxo bond lengths are also comparable to those in the OEC.
A DFT study on the Mn3CaO4 complex of Kanady et al.151 revealed small splitting between high spin and low spin states of the complex, consistent with small values (≤8.0 cm−1) of the pairwise exchange coupling constants (Table 1).196 The weak ferromagnetic interaction of manganese spins indeed leads to closely spaced states in the spin ladder with a total high-spin (S = 9/2) ground state. As discussed earlier, the closed cubane form of the OEC in the S2 and S3 states adopts the same valence state distribution and geometry. However, despite little differences in the Mn–Mn distances in the synthetic system compared to the OEC, the synthetic model shows much weaker magnetic coupling between manganese ion pairs compared to the OEC in either the S2 or the S3 closed cubane states (intra-cubane J values for the latter are reported to be up to 36 cm−1).71,82 The presence of an external dangling Mn ion in the OEC obviously acts as a major source of this distinction, as can be understood from the study of magnetic interactions in oxo-bridge protonated species.196 This revealed that the attachment of Lewis acids to an oxo bridge decreases the extent of exchange coupling between associated metal ions as it reduces the ability of the bridge to mediate superexchange,201–203 which favors antiferromagnetic coupling of spins and contributes negatively to the total exchange coupling constant.204–208 The constrained Mn(IV)–O–Mn(IV) bond angle also disfavors the overlap of metal–ligand orbitals resulting in the damping of superexchange. This is reflected in the large positive value of the exchange coupling constants in the native OEC. However, the overall ferromagnetic coupling between the manganese sites leading to a S = 9/2 ground state in the synthetic cubane is consistent with the high-spin situation within the closed-cubane core of the OEC in the S2 and S3 states.71,82
The structural asymmetry induced by the external calcium ion in the complex of Mukherjee et al. has a strong effect on magnetic properties. Magnetic susceptibility data suggests strong ferromagnetic coupling among the manganese ions resulting a S = 9/2 spin ground state. The pairwise exchange coupling constants obtained both from fitting of the data and from theoretical calculations on the crystallographic model correspond to two moderately strong ferromagnetic and a weaker antiferromagnetic interaction (Table 1).
Both complexes serve to demonstrate that the Mn(IV)3CaO4 cube is an intrinsically high-spin unit. Christou and coworkers related the sign of J values to the Mn–O–Mn bond angles.195 As shown by Krewald et al.,196 the effect of the angle on superexchange can be visualized using the concept of corresponding orbitals209 between pairs of Mn ions. Although this is normally applied to dimeric systems, a pictorial view of the magnetic orbital interactions in the cubanes can be obtained by diamagnetic substitution, where one of the three Mn ions is replaced by the diamagnetic Ge4+. This allows the corresponding orbital analysis to be carried out for the two remaining Mn centers as in the case of a simple dimer, and hence to obtain orbital pairs such as those shown in Fig. 14. This analysis showed that the Mn–O–Mn angles within the cubane are such that the overlap between the magnetic orbitals is minimal, explaining the dominant ferromagnetic coupling.
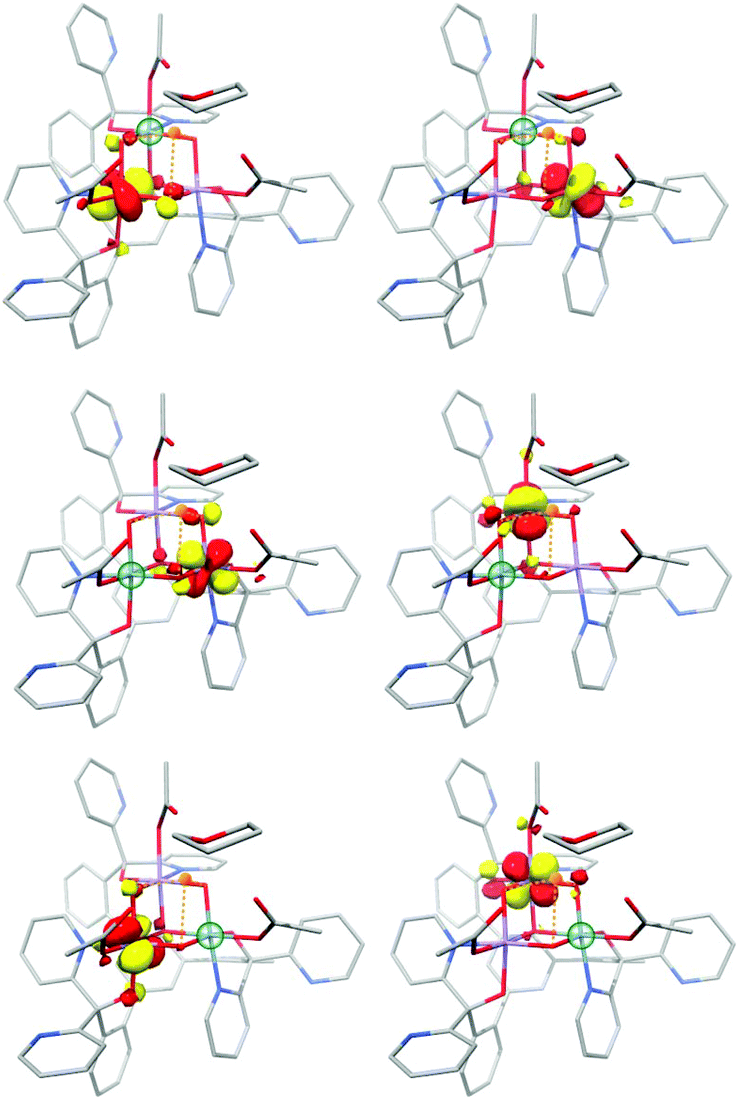 | ||
| Fig. 14 Selected corresponding orbital pairs between pairs of Mn ions, computed by the diamagnetic substitution approach; the green sphere indicates the manganese ion replaced by Ge4+ in each case. | ||
The magnetic coupling situation in these complexes can be described by “spin maps”, which indicate the ground state spin resulting from possible combinations of the three pairwise exchange coupling constants. Fig. 15 shows an example for a system of three coupled local S = 3/2 spins corresponding to three Mn(IV) ions. The two complexes discussed here fall into the S = 9/2 areas of the spin maps, although in one case (Mn3CaO4 cubane by Kanady et al.) all J couplings are weakly ferromagnetic and in the other (Mn3CaO4–Ca complex by Mukherjee et al.) there are two strong ferromagnetic and one weak antiferromagnetic coupling constants. In either case, for a complex of this type to fall outside the S = 9/2 region of the map would require significant asymmetry and hence structural distortions to an extent that in practice would probably mean disruption of the cubane framework.
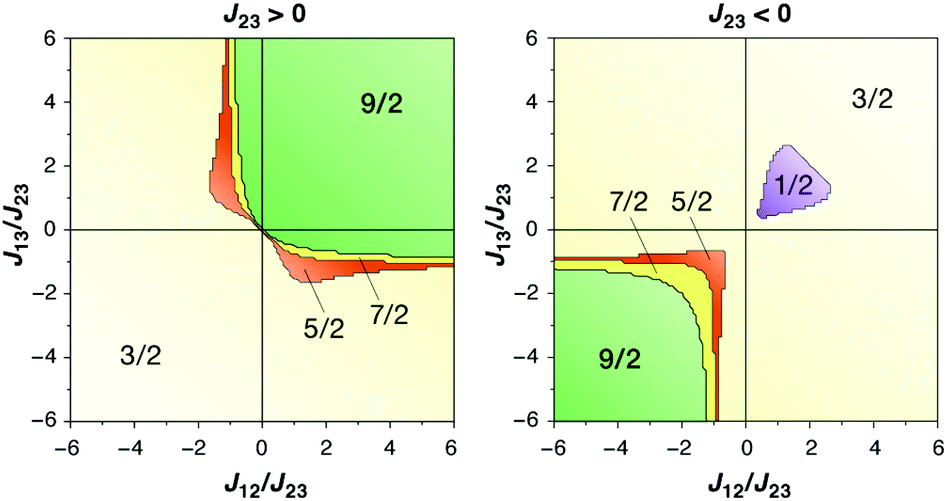 | ||
| Fig. 15 Spin maps indicating the ground spin state obtained by combinations of the three pairwise exchange coupling constants in a system of three local spins S = 3/2 (adapted with permission from Krewald et al.196 Copyright 2013 American Chemical Society). | ||
A relevant point here is that reduction of the cubane to produce a mixed valence Mn(III)Mn(IV)2 complex, which introduces axial Jahn–Teller elongation at the Mn(III) site, was suggested to be insufficient to open up the structure like in the S2 state of the OEC.196 This strongly suggests that the opening of the rigid framework of the cubane towards a more OEC-like topology is non trivial and depends on a number of factors beyond the Mn oxidation states.
The theoretical study further found the bulky tBu group in the case of the Mn3CaO4–Ca complex to be relevant for the magnetic topology of the complex,196 because in models where the tBu group was replaced by a methyl group, all three exchange coupling constants were predicted to be positive. The strong coupling sets the first excited S = 7/2 spin state much higher (57 cm−1 and 68 cm−1 from experimental fitting and computation respectively) above the S = 9/2 ground spin state, which is similar to the situation in the closed–cubane core of the OEC in the S2 and S3 states.71,82
The Mn3CaO4–Ca complex was also studied by EPR and ENDOR spectroscopies. Complicated hyperfine splitting is seen, as in other oligonuclear Mn(IV) containing complexes.14,105 To get a clear picture of the hyperfine splitting due to the three Mn nuclei, obscured in the inhomogeneously broadened EPR line, the 55Mn ENDOR spectrum was collected. Very low values for the anisotropic component of the hyperfine coupling constant were observed, which match well to other octahedral Mn(IV) systems, including the late Si states of the OEC.82,105 Experimental values of the isotropic hyperfine coupling constants are in agreement with computed values195,196 and are very similar for all Mn(IV) ions, with only small deviations consistent with the structural asymmetry of the cubane caused by the external Ca (Table 2).
Substituted Mn3AO4 cubanes
The role of calcium in the OEC has been a question of perennial interest that, in the absence of detailed understanding of the catalytic mechanism, remains without a convincing answer. Using their Mn3CaO4 cubane complexes as a platform, Agapie and coworkers contributed important results from synthetic models as they developed and studied a series of calcium-substituted Mn3AO4 complexes (A = Mn3+, Sr2+, Zn2+, Sc3+, Y3+, Gd3+, Ln3+).In all cubanes where Ca has been substituted with another cation, apart from the scandium complex Mn(III)Mn(IV)2ScO4, the manganese ions remain as Mn(IV), consistent with Mn–O bond distances of 1.821–1.913 Å. The Mn(III) center in the scandium complex shows an axial elongation in metal–ligand bond lengths (2.134–2.142 Å) and the reduced Gd3+ complex with Mn(IV)2Mn(III) composition also displays an axial elongation with Mn(III)–O distances of 2.169 and 2.163 Å, both cases typical of an axial Jahn–Teller distortion due to population of a metal–ligand σ-antibonding orbital of metal dz2 origin.182 The zinc complex shows similar structure as that of the Ca2+ and Sr2+ complexes but due to its smaller ionic radius the zinc center cannot bind solvent molecules. The symmetry of the complex has been systematically lowered by substituting the THF ligand with [ON4O]2−, which results in a broader range of Mn(IV)–Ca and Mn(IV)–Mn(IV) distances.181
Theoretical studies210 on these models similarly suggest that the Mn3O4 subunits of the Mn3AO4 series in both the oxidized and reduced forms show very limited variation, in contrast to the A–Mn and A–O distances which essentially reflect the differences in ionic radii of the cations An+. The geometries of the oxidized and reduced form also differ only in the Jahn–Teller elongation of the Mn(III) associated axis, while the others remain almost the same. This implies a predominantly ionic interaction between the redox–inactive An+ metal and the Mn3O4 framework.
Of relevance at this point is a Ca XAS study that detected significant change in the Ca K-pre-edge XAS data from Mn(IV)3 to Mn(IV)2Mn(III), which was related to the lengthening of the Mn(III)–O bonds.211 The longer bonds around the Jahn–Teller distorted Mn(III) ion in the reduced system imparts small changes in the Ca–bound bond distances and bond angles, which presumably lower the Ca 3d–2p mixing as reflected in ca. 23% lower pre-edge intensity. In terms of reactivity, the manganese-only cubane Mn4O4 was shown to be more reactive towards ligand exchange than the CaMn3O4 and ScMn3O4 complexes. This enhanced activity can be attributed to the presence of two Mn(III) sites in the Mn4O4 cubane, which imparts lability related to the presence of two Jahn–Teller axes. In the other two compounds all manganese ions are Mn(IV).44 Similarly, Mn(III)2Mn(IV)2O4 is more reactive in oxygen atom transfer compared with Mn(IV)3CaO4 because this requires dissociation of an acetate ligand that renders a Mn ion five-coordinate, a situation that is possible for Mn(III) but unfavorable for Mn(IV).212 Similar conclusions regarding the higher reactivity of the all-Mn cubane versus the Mn3CaO4 one were reached from computational studies that investigated hypothetical water oxidation pathways.213
The original Mn3CaO4 compound shows quasireversible reduction to Mn(IV)2Mn(III)CaO4 at −940 mV versus Fc/Fc+ in DMA, whereas the all-Mn cubane shows a quasireversible reduction at −700 mV in similar conditions. These data suggest that the presence of redox-inactive Ca may facilitate the formation of species with higher oxidation states at lower potential. However, this comparison is not entirely convincing because different redox couples are involved, i.e. Mn(IV)Mn(III)3O4/Mn(IV)2Mn(III)2O4 in case of the all-manganese cubane, and Mn(IV)2Mn(III)CaO4/Mn(IV)3CaO4 for the calcium-containing cubane. The reduction potentials of the Ca and Sr complexes are practically the same, whereas the reduction potential of the Zn complex is more positive (E1/2 = −630 mV), despite having the same total charge as that of the Ca and Sr complex. Similarly, despite the same charge, the reduction potentials of Sc and Y complexes differ by ca. 200 mV. The fact that the Ca and Sr compounds have the same redox potential has an obvious relevance for the OEC, which can function catalytically only with these two cations.44
A major outcome of the studies by the Agapie group was the identification of a linear correlation between the Lewis acidity of the redox-inactive cations (measured in terms of the pKa of their aquo complexes) and the reduction potential of the clusters (Fig. 16a). This points to a role of the redox-inactive metal cation in fine-tuning the redox potential of the OEC in biological water oxidation. The positive shift in redox potential with increasing Lewis acidity is interpreted as the increased electron-withdrawing effect upon the μ3-oxo ligands, which stabilizes the more reduced manganese oxidation state. On the other hand, increased number of oxo ligands per redox active metal site helps to stabilize higher oxidation states by providing an electron–rich environment.180,214 Additionally, in case of the Ln3+ cubanes the reduction potential is reduced with increase in the ionic radii and subsequent decrease in the Lewis acidity.182
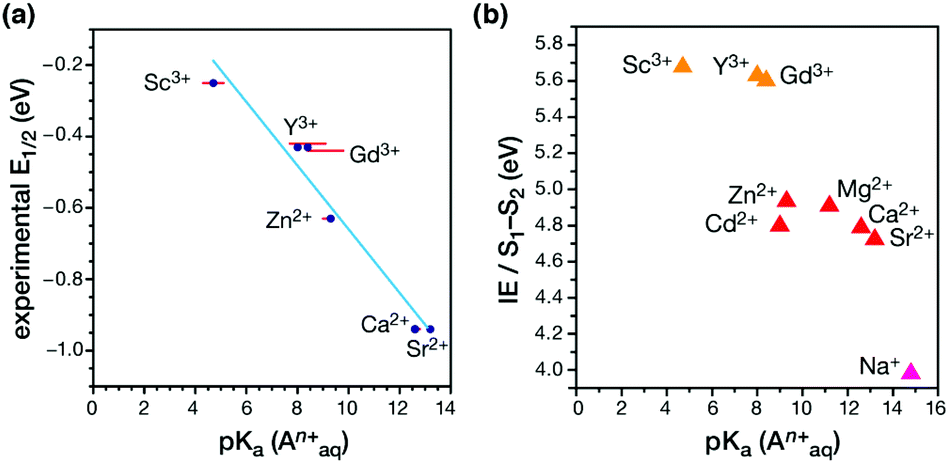 | ||
| Fig. 16 (a) Correlation between the experimental reduction potential E1/2 (eV) and the literature-known pKa values of the aquo-cations An+ in the Mn3AO4 cubane models.180,182 (b) Correlation of computed vertical ionization energies and pKa values of aquo-An+ cations for the S1–S2 transitions of the OEC210 (adapted from ref. 210 with permission from the PCCP Owner Societies). | ||
A recent computational study reproduced the correlation between Lewis acidity and reduction potential.210 However, no such correlation was found for models of the OEC in the S1–S2 (Fig. 16b) and 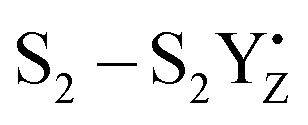 transitions. The theoretical analysis of the Mn3AO4 series of compounds and their comparison with the biological cluster reveals that the geometric relaxation energy for the cubane models upon ionization hardly varies across the series of An+ cations, whereas their vertical ionization energies vary systematically with the pKa values of the aquo-An+ cations. By contrast the OEC, modeled with the same range of An+ cations, shows a completely non-systematic variation in geometric relaxation energy from S1 to S2 or from
transitions. The theoretical analysis of the Mn3AO4 series of compounds and their comparison with the biological cluster reveals that the geometric relaxation energy for the cubane models upon ionization hardly varies across the series of An+ cations, whereas their vertical ionization energies vary systematically with the pKa values of the aquo-An+ cations. By contrast the OEC, modeled with the same range of An+ cations, shows a completely non-systematic variation in geometric relaxation energy from S1 to S2 or from 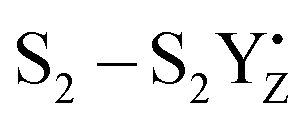 , while the change in vertical ionization energy depends entirely on the charge of An+ and not on the Lewis acidity (Fig. 17).
, while the change in vertical ionization energy depends entirely on the charge of An+ and not on the Lewis acidity (Fig. 17).
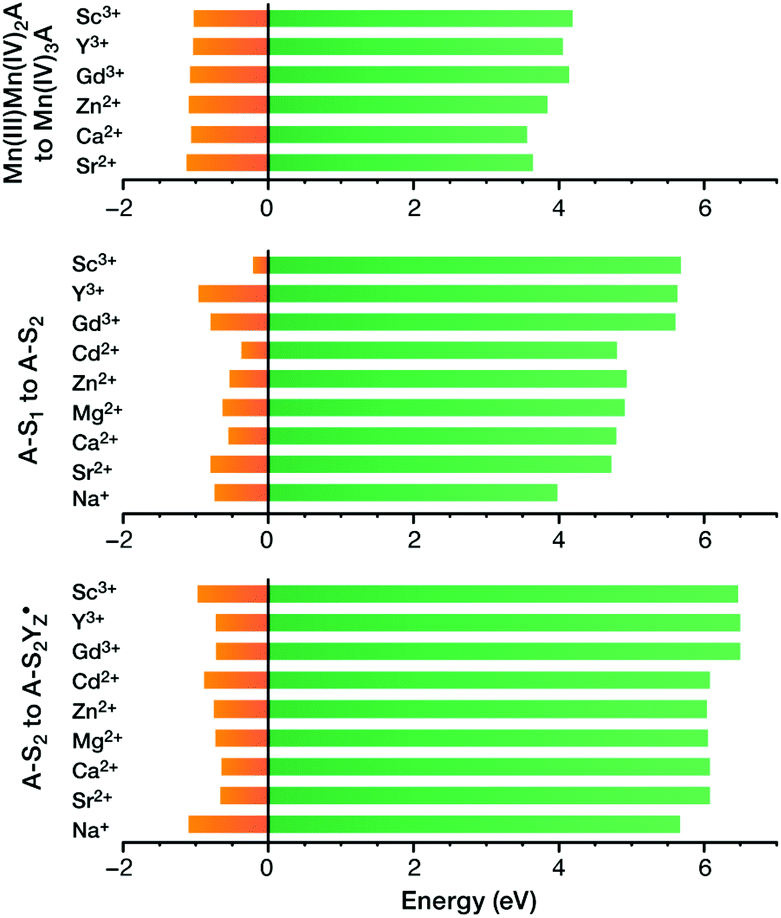 | ||
Fig. 17 Vertical ionization energies (eV, green) and geometry relaxation energies (eV, orange) for the systems studied here. Top to bottom: synthetic cubane models, OEC S1–S2 transition, OEC 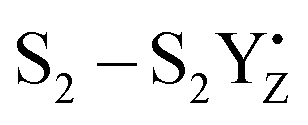 transition (adapted from ref. 210 with permission from the PCCP Owner Societies). transition (adapted from ref. 210 with permission from the PCCP Owner Societies). | ||
These results illustrate that the flexible core and first coordination sphere of the OEC can adopt a multitude of closely related geometries, for example by rearranging the hydrogen-bonding network of residues with structural waters, and/or adopting alternative orientations of water, amino acid side chains, etc. when exposed to structural perturbations induced by redox-inactive metal substitution. By contrast, the structurally rigid synthetic models show a markedly different response as a result of the structurally rigid hexadentate ligand scaffold, enabling “second-order correlations” to emerge.
In turn, this suggests that the attribution to the Ca2+ ion of a role as regulator of redox potential in the OEC is not straightforward. Other ideas have been advanced in this respect, for example in water delivery to the active site,72 in O2 release,215 and in fine-tuning the hydrogen bonding network that affects the properties of the redox-active tyrosine and its tyrosyl radical form.81
Mn3CaO4–Mn complex
An impressive step in the evolution of structural models of the OEC is the asymmetric complex [Mn4CaO4(ButCO2)8(ButCO2H)2(py)] (But: tert-butyl; py: pyridine) (Fig. 13), by Zhang and co-workers.197 The structural similarity between this complex and the OEC is portrayed through similar metal–metal and metal–ligand distances. All metal ions are pairwise connected by a bridging oxo group as well as a bridging carboxylate group, which matches the pattern in the biological system. Bond–valence sum analysis of the complex suggested a Mn1(III)–Mn2(IV)–Mn3(IV)–Mn4(III) valence distribution, in complete analogy with the OEC in its dark-stable S1 state. Mn–Mn distances also agree well with recent EXAFS36,63 and XFEL data on the OEC.70Nevertheless, this complex is still not a precise mimic of the OEC.70 A comparison of Fig. 2 and 13 reveals very clearly that the complex is instead an accurate synthetic realization of the Ferreira et al. model of the OEC.45 Like the older crystallographic model, the complex lacks the additional oxo bridge (O4 in the core of the OEC as depicted in Fig. 1 and 2) between the dangling Mn4 and the Mn3 ion; in the synthetic model this is substituted by a bridging carboxylate group. Furthermore, the bonds between the μ4-oxo and the associated metal ions are significantly shorter in the synthetic complex (1.85, 1.85 and 2.28 Å) relative to corresponding bond lengths in the S1 state of the OEC (2.3, 2.2 and 2.7 Å)70 despite the same oxidation states of the Mn ions. In addition, the number of peripheral carboxylate bridges is six in the synthetic complex compared to ten in the OEC.14 The distance between the terminal Mn ions, Mn1 and Mn4, is also significantly shorter in the model (3.59 Å) compared to the native OEC (4.89–4.97 Å).216 All of the above are different manifestations of a fundamental characteristic the distinguishes the synthetic model from the OEC: the fact that it is highly compact as a result of a rigid Mn3CaO4 cubane subunit, a part of the structure that is not formally present as a distinct structural unit in the S1 state of the OEC. Another difference is that the Jahn–Teller axes of the terminal Mn(III) ions are found to be oriented in a mutually perpendicular fashion, in contrast to the collinear orientation of Jahn–Teller axes in the S1 state of the OEC. Finally, we note the absence of any water or water derived ligands at the Ca and any of the Mn ions. However, exchangeable ligands at these sites do allow for the possibility of water coordination under appropriate conditions.
The structural relationship between the synthetic model and the native OEC inspired studies of the complex using quantum chemical methods.216–218 Computed exchange coupling constants from broken-symmetry DFT indicate that the ground spin state of the complex is S = 1, with low lying spin states within the range of 10 cm−1.217,218 This is in contrast to the S = 0 ground spin state of the OEC.42 This difference can be correlated with the much stronger antiferromagnetic coupling between the Mn3–Mn4 couple in the OEC, induced by the presence of the additional oxo bridge. The computational studies showed that the complex can access total oxidation states that correspond to the S0–S3 states of the OEC in terms of Mn oxidation states, Mn(III)3Mn(IV) to Mn(IV)4, with limited structural rearrangements.
More fascinating than geometric aspects are the various physicochemical properties of the complex that resemble those of the OEC much closer than any previous structural model. One of them is its electrochemical behavior. In its neutral form it resembles the S1 state of the OEC by requiring ca. 1 eV for one-electron oxidation. Most interestingly, the cyclic voltammogram of the complex in a mixture of organic solvents shows that the system can accumulate oxidation equivalents in a way similar to the OEC: reversible transitions were noted between oxidations states that might correspond to the S0–S3 states of the OEC. This type of extended redox behavior was not present in earlier Mn3CaO4 cubane complexes that did not contain the dangling Mn, which suggests that —in addition to the role of the ligands in potentially supporting a range of oxidation states— the fourth Mn playing an important role in controlling the redox behavior of the cluster.
The EPR results on the complex are particularly intriguing. A solution of the complex in its neutral form exhibits only a broad parallel mode EPR signal like that in the S1 state of OEC, however the effective g value of 12 in the synthetic complex is significantly different from the g ≈ 4.9 in the OEC. These differences likely stem from the distinctly different orientations of the Jahn–Teller axes of the Mn(III) ions in the two systems (perpendicular in the model and collinear in the OEC) which would result in the local d tensors contributing differently to the total D value of the system. The most striking aspect however is the simultaneous appearance of g ≈ 4.9 and g ≈ 2 EPR signals in the one-electron oxidized form, reminiscent of the S2 state of the native enzyme (Fig. 18).71 To obtain the EPR spectrum on the oxidized state of the complex, Zhang et al. employed [Fe(Phen)3]3+ as a chemical oxidant with a redox potential +1.1 V versus NHE. They observed two signals in perpendicular mode, a multiline signal centered at g ≈ 2 with a width of ca. 1600 G and more than 20 hyperfine peaks, and a signal centered at g ≈ 4.9, with a width of ca. 500 G without resolved hyperfine structure. Both EPR signals exhibit a linear Curie behavior suggesting that they are both ground-state signals. Such characteristic signals had never been observed before in other synthetic structural models of the OEC.219
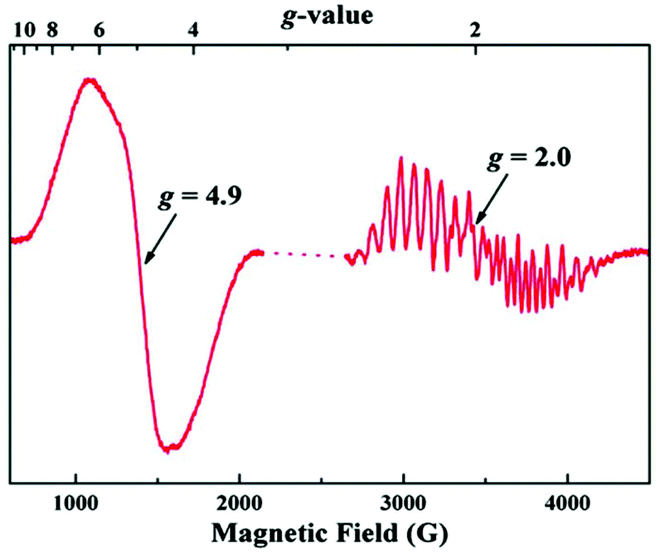 | ||
| Fig. 18 EPR spectrum of the oxidized Mn(IV)3Mn(III) form (“S2-state”) of the synthetic complex by Zhang et al. at 7 K197 (from Zhang et al., Science, 2015, 348, 690. Reprinted with permission from AAAS.). | ||
Zhang et al. could detect subtle structural differences, particularly in the Mn1–Mn4 and Ca–O4 distances between two monomers in the crystallographic unit cell of the neutral state. They assumed that the structural difference, possibly amplified during solvation or S1 to S2 state change, could affect the exchange coupling between Mn ions leading to two different spin states, which might explain the simultaneous appearance of two signals. The apparent similarities of the EPR of the oxidized form with the S2 state of the OEC, led also to the suggestion that the structural interpretation of the EPR of the OEC described by Pantazis et al.71 might also be valid here, i.e. that the two signals could arise by valence isomers of the oxidized synthetic complex.217 However, this hypothesis is not consistent with the experimental data. In stark contrast to the OEC, the two EPR signals of the oxidized synthetic model are non-interconvertible, suggesting that they are unlikely to arise from valence isomers as in the S2 state of the OEC. Given the rigid structural framework of the synthetic complex there is no need for extensive structural reorganization and hence electron hopping between Mn ions in the singly oxidized state of the complex should be more facile compared to the OEC instead of prohibited. This is a strong argument against the valence isomer hypothesis and indicates that the spectroscopic similarities with the S2 state of the OEC are superficial. This was recently confirmed with density functional theory and multireference wave function calculations by Paul et al.,218 who established that only one valence isomer is energetically accessible in the structurally intact one-electron oxidized form of the complex. This has a valence distribution that resembles the high-spin closed cubane form of the S2 state of the OEC, with the Mn(III) at the “dangler” position. It displays an S = 5/2 ground state and can readily explain the g = 4.9 signal. However, alternative valence isomers that might give rise to an S = 1/2 ground state are significantly higher in energy.218 These results demonstrate that despite geometric similarities the synthetic model does not mimic the valence isomerism of the OEC and suggest that the multiline EPR signal in the singly oxidized form may originate from an as yet unidentified rearrangement product.
A particularly interesting question is of course whether this complex can evolve oxygen by oxidizing water as a molecular catalyst. This is not clear from the available experimental data, but the answer is likely negative. Electrochemistry suggests that oxidation past the S3 state, i.e. to the equivalent of an oxygen-evolving S4 state, is irreversible. Consistent with this observation, Paul et al. showed that oxidation of the all-Mn(IV) S3 state would be ligand-centered.218 Besides, there are no water-derived ligands in the as-isolated complex that could serve as substrates. Indeed, a recent computational study220 suggested that a mechanism for water oxidation is conceivable only after extensive substitution of existing ligands by water molecules. It is unclear whether such a process should be expected to precipitate the chemically more plausible decomposition of the high-valent manganese cluster rather than enable catalytic water oxidation with an otherwise intact inorganic core.
Conclusions and perspectives
In this review we aimed to trace broadly, albeit selectively, the historical development of structural models for the oxygen-evolving complex and to highlight major achievements and insights. We singled out as three important milestones in this route the Mn3CaO4 cubane complex and related substituted systems by the Agapie group,151 the Mn3CaO4–Ca cubane complex by the Christou group,195 and the most recently reported Mn3CaO4–Mn cubane complex by the Zhang group.197 Although the structural parameters of the cubane units are very similar, these complexes define a series of increasing asymmetry induced by the presence of an external Ca2+ cation and a dangling redox-active manganese. They also define a series of increasing complexity in terms of behavior, with the Zhang complex displaying remarkable aspects of redox accumulation and spectroscopic properties.Obtaining insights from the synthetic models has often been inseparable from a theoretical analysis of their properties and has relied to a large extent on deciphering the fundamental electronic structure basis of geometry–property correlations using modern quantum chemistry that focuses on spectroscopic properties. These cubane-incorporating complexes have already served as sources of insight in this respect, in themselves and in comparison to models of the OEC, as showcased by numerous studies on magnetism, spectroscopy, and redox behavior.
However, we would also like to delineate some remaining challenges. Despite the enormous progress in ligand design, in controlling the meal stoichiometry and in incorporating asymmetry into the inorganic core, the precise stoichiometry (Mn4CaO5) and connectivity of the OEC (Fig. 1 and 2) have not been reproduced. Without exception, all models mimic the “closed cubane” forms of the OEC (or the older “London” model of the OEC). This motif first appears in a component of the S2 state but is not formally present in S0 and S1, or in one component of the S3 state.82 It is not considered likely to be involved in O–O bond formation according to existing mechanistic hypotheses,84 however closed cubane species were recently shown to be intimately involved in advancement to the S3 state,74 so this structural motif is certainly of high relevance to modeling the OEC. On the other hand, the rigidity of the cubane core itself in the synthetic models stands in contrast to the flexibility shown by the OEC. The exact Mn4CaO5 stoichiometry with the correct bridging modes, a more open connectivity pattern, and a potentially more flexible/bistable core constitute therefore part of future biomimetic challenges. To these, one might add the finer structure-based control of local and total spin, a crucial aspect of the OEC.78
Beyond these points, synthetic models that attempt to mimic the structure are still short of mimicking functionality, although it should be recognized that the structural determinants of function for the OEC itself remain ill-defined. Catalytic progression is tightly dependent on coupling, perhaps entirely, of substrate deprotonations with metal-centered oxidations preceding O–O bond formation. The function of the OEC depends critically on the protein matrix and on the tight control of water access, which allow the high-valent Mn cluster to perform water oxidation chemistry without damage and side reactions. Thus, eliciting function from geometric structure represents an entirely different level of challenge and would require exquisite architectural control of the second coordination sphere. For structural biomimetic chemistry the OEC still represents a peak to be conquered, not simply in terms of precisely reproducing the core topology and supporting ligand sphere in its stable states but also in targeting intermediates that might appear in the later catalytic stages, whose structural complexity has only very recently began to be fully appreciated.6,74
Acknowledgements
The authors acknowledge funding by the Max Planck Society and by the project MANGAN (03EK3545) funded by the Bundesministeriums für Bildung und Forschung (BMBF). Network support by the COST action CM1305 “Explicit Control Over Spin-states in Technology and Biochemistry (ECOSTBio)” is gratefully acknowledged. Open Access funding provided by the Max Planck Society.Notes and references
- R. E. Blankenship, Molecular Mechanisms of Photosynthesis, Blackwell, Oxford, 2001 Search PubMed.
- J. P. McEvoy and G. W. Brudvig, Chem. Rev., 2006, 106, 4455–4483 CrossRef CAS PubMed.
- J. Messinger, T. Noguchi and J. Yano, in Molecular Solar Fuels, ed. T. J. Wydrzynski and W. Hillier, The Royal Society of Chemistry, Cambridge, 2012, pp. 163–207 Search PubMed.
- V. Krewald, M. Retegan and D. A. Pantazis, Top. Curr. Chem., 2016, 371, 23–48 CrossRef PubMed.
- N. Cox, D. A. Pantazis, F. Neese and W. Lubitz, Acc. Chem. Res., 2013, 46, 1588–1596 CrossRef CAS PubMed.
- M. Pérez-Navarro, F. Neese, W. Lubitz, D. A. Pantazis and N. Cox, Curr. Opin. Chem. Biol., 2016, 31, 113–119 CrossRef PubMed.
- S. Mukhopadhyay, S. K. Mandal, S. Bhaduri and W. H. Armstrong, Chem. Rev., 2004, 104, 3981–4026 CrossRef CAS PubMed.
- W. Rüttinger and G. C. Dismukes, Chem. Rev., 1997, 97, 1–24 CrossRef.
- V. L. Pecoraro and W.-Y. Hsieh, Inorg. Chem., 2008, 47, 1765–1778 CrossRef CAS PubMed.
- C. S. Mullins and V. L. Pecoraro, Coord. Chem. Rev., 2008, 252, 416–443 CrossRef CAS PubMed.
- C. W. Cady, R. H. Crabtree and G. W. Brudvig, Coord. Chem. Rev., 2008, 252, 444–455 CrossRef CAS PubMed.
- K. Meelich, C. M. Zaleski and V. L. Pecoraro, Philos. Trans. R. Soc., B, 2008, 363, 1271–1281 CrossRef CAS PubMed.
- K. J. Young, B. J. Brennan, R. Tagore and G. W. Brudvig, Acc. Chem. Res., 2015, 48, 567–574 CrossRef CAS PubMed.
- B. Gerey, E. Gouré, J. Fortage, J. Pécaut and M.-N. Collomb, Coord. Chem. Rev., 2016, 319, 1–24 CrossRef CAS.
- J. D. Blakemore, R. H. Crabtree and G. W. Brudvig, Chem. Rev., 2015, 115, 12974–13005 CrossRef CAS PubMed.
- M.-N. Collomb and A. Deronzier, Eur. J. Inorg. Chem., 2009, 2009, 2025–2046 CrossRef.
- M. Wiechen, H.-M. Berends and P. Kurz, Dalton Trans., 2012, 41, 21–31 RSC.
- M. D. Kärkäs and B. Åkermark, Dalton Trans., 2016, 45, 14421–14461 RSC.
- X. Liu and F. Wang, Coord. Chem. Rev., 2012, 256, 1115–1136 CrossRef CAS.
- M. Hirahara, A. Shoji and M. Yagi, Eur. J. Inorg. Chem., 2014, 2014, 595–606 CrossRef CAS.
- M. D. Kärkäs, O. Verho, E. V. Johnston and B. Åkermark, Chem. Rev., 2014, 114, 11863–12001 CrossRef PubMed.
- J. Barber, Chem. Soc. Rev., 2009, 38, 185–196 RSC.
- D. G. Nocera, Acc. Chem. Res., 2012, 45, 767–776 CrossRef CAS PubMed.
- T. A. Faunce, W. Lubitz, A. W. Rutherford, D. MacFarlane, G. F. Moore, P. Yang, D. G. Nocera, T. A. Moore, D. H. Gregory, S. Fukuzumi, K. B. Yoon, F. A. Armstrong, M. R. Wasielewski and S. Styring, Energy Environ. Sci., 2013, 6, 695–698 Search PubMed.
- J. Messinger, W. Lubitz and J.-R. Shen, Phys. Chem. Chem. Phys., 2014, 16, 11810–11811 RSC.
- N. Cox, D. A. Pantazis, F. Neese and W. Lubitz, Interface Focus, 2015, 5, 20150009 CrossRef PubMed.
- D. Kim, K. K. Sakimoto, D. Hong and P. Yang, Angew. Chem., Int. Ed., 2015, 54, 3259–3266 CrossRef CAS PubMed.
- S. Berardi, S. Drouet, L. Francas, C. Gimbert-Surinach, M. Guttentag, C. Richmond, T. Stoll and A. Llobet, Chem. Soc. Rev., 2014, 43, 7501–7519 RSC.
- H. Dau, C. Limberg, T. Reier, M. Risch, S. Roggan and P. Strasser, ChemCatChem, 2010, 2, 724–761 CrossRef CAS.
- T. R. Cook, D. K. Dogutan, S. Y. Reece, Y. Surendranath, T. S. Teets and D. G. Nocera, Chem. Rev., 2010, 110, 6474–6502 CrossRef CAS PubMed.
- J. Barber, Biochemistry, 2016, 55, 5901–5906 CrossRef CAS PubMed.
- J.-R. Shen, Annu. Rev. Plant Biol., 2015, 66, 23–48 CrossRef CAS PubMed.
- B. Kok, B. Forbush and M. McGloin, Photochem. Photobiol., 1970, 11, 457–475 CrossRef CAS PubMed.
- P. Joliot, G. Barbieri and R. Chabaud, Photochem. Photobiol., 1969, 10, 309–329 CrossRef CAS.
- H. Dau and M. Haumann, Biochim. Biophys. Acta, Bioenerg., 2007, 1767, 472–483 CrossRef CAS PubMed.
- H. Dau and M. Haumann, Coord. Chem. Rev., 2008, 252, 273–295 CrossRef CAS.
- A. Klauss, M. Haumann and H. Dau, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 16035–16040 CrossRef CAS PubMed.
- A. Klauss, M. Haumann and H. Dau, J. Phys. Chem. B, 2015, 119, 2677–2689 CrossRef CAS PubMed.
- A. Zouni, H. T. Witt, J. Kern, P. Fromme, N. Krauss, W. Saenger and P. Orth, Nature, 2001, 409, 739–743 CrossRef CAS PubMed.
- K. Wieghardt, Angew. Chem., Int. Ed. Engl., 1989, 28, 1153–1172 CrossRef.
- Y. Umena, K. Kawakami, J.-R. Shen and N. Kamiya, Nature, 2011, 473, 55–60 CrossRef CAS PubMed.
- V. Krewald, M. Retegan, N. Cox, J. Messinger, W. Lubitz, S. DeBeer, F. Neese and D. A. Pantazis, Chem. Sci., 2015, 6, 1676–1695 RSC.
- G. Christou, Acc. Chem. Res., 1989, 22, 328–335 CrossRef CAS.
- E. Y. Tsui, J. S. Kanady and T. Agapie, Inorg. Chem., 2013, 52, 13833–13848 CrossRef CAS PubMed.
- K. N. Ferreira, T. M. Iverson, K. Maghlaoui, J. Barber and S. Iwata, Science, 2004, 303, 1831–1838 CrossRef CAS PubMed.
- P. E. M. Siegbahn and M. Lundberg, Photochem. Photobiol. Sci., 2005, 4, 1035–1043 CAS.
- E. M. Sproviero, J. A. Gascon, J. P. McEvoy, G. W. Brudvig and V. S. Batista, J. Chem. Theory Comput., 2006, 2, 1119–1134 CrossRef CAS PubMed.
- E. M. Sproviero, J. A. Gascon, J. P. McEvoy, G. W. Brudvig and V. S. Batista, Curr. Opin. Struct. Biol., 2007, 17, 173–180 CrossRef CAS PubMed.
- E. M. Sproviero, J. A. Gascon, J. P. McEvoy, G. W. Brudvig and V. S. Batista, J. Am. Chem. Soc., 2008, 130, 3428–3442 CrossRef CAS PubMed.
- E. M. Sproviero, J. A. Gascon, J. P. McEvoy, G. W. Brudvig and V. S. Batista, Coord. Chem. Rev., 2008, 252, 395–415 CrossRef CAS PubMed.
- B. Loll, J. Kern, W. Saenger, A. Zouni and J. Biesiadka, Nature, 2005, 438, 1040–1044 CrossRef CAS PubMed.
- P. E. M. Siegbahn, Chem. – Eur. J., 2008, 14, 8290–8302 CrossRef CAS PubMed.
- P. E. M. Siegbahn, Acc. Chem. Res., 2009, 42, 1871–1880 CrossRef CAS PubMed.
- P. E. M. Siegbahn, Dalton Trans., 2009, 10063–10068 RSC.
- G. N. George, R. C. Prince and S. P. Cramer, Science, 1989, 243, 789–791 CAS.
- V. K. Yachandra, K. Sauer and M. P. Klein, Chem. Rev., 1996, 96, 2927–2950 Search PubMed.
- V. K. Yachandra, V. J. DeRose, M. J. Latimer, I. Mukerji, K. Sauer and M. P. Klein, Science, 1993, 260, 675–679 CAS.
- J. Yano, J. Kern, K. Sauer, M. J. Latimer, Y. Pushkar, J. Biesiadka, B. Loll, W. Saenger, J. Messinger, A. Zouni and V. K. Yachandra, Science, 2006, 314, 821–825 CrossRef CAS PubMed.
- Y. Pushkar, J. Yano, P. Glatzel, J. Messinger, A. Lewis, K. Sauer, U. Bergmann and V. Yachandra, J. Biol. Chem., 2007, 282, 7198–7208 CrossRef CAS PubMed.
- H. Dau, A. Grundmeier, P. Loja and M. Haumann, Philos. Trans. R. Soc., B, 2008, 363, 1237–1243 CrossRef CAS PubMed.
- J. Yano, J. Kern, Y. Pushkar, K. Sauer, P. Glatzel, U. Bergmann, J. Messinger, A. Zouni and V. K. Yachandra, Philos. Trans. R. Soc., B, 2008, 363, 1139–1147 CrossRef CAS PubMed.
- H. Dau, I. Zaharieva and M. Haumann, Curr. Opin. Chem. Biol., 2012, 16, 3–10 CrossRef CAS PubMed.
- J. Yano and V. Yachandra, Chem. Rev., 2014, 114, 4175–4205 CrossRef CAS PubMed.
- A. Grundmeier and H. Dau, Biochim. Biophys. Acta, Bioenerg., 2012, 1817, 88–105 CrossRef CAS PubMed.
- H. Dau, P. Liebisch and M. Haumann, Phys. Chem. Chem. Phys., 2004, 6, 4781–4792 RSC.
- J. Yano, J. Kern, K.-D. Irrgang, M. J. Latimer, U. Bergmann, P. Glatzel, Y. Pushkar, J. Biesiadka, B. Loll, K. Sauer, J. Messinger, A. Zouni and V. K. Yachandra, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 12047–12052 CrossRef CAS PubMed.
- M. Grabolle, M. Haumann, C. Müller, P. Liebisch and H. Dau, J. Biol. Chem., 2006, 281, 4580–4588 CrossRef CAS PubMed.
- M. Askerka, D. J. Vinyard, J. Wang, G. W. Brudvig and V. S. Batista, Biochemistry, 2015, 54, 1713–1716 CrossRef CAS PubMed.
- P. Chernev, I. Zaharieva, E. Rossini, A. Galstyan, H. Dau and E.-W. Knapp, J. Phys. Chem. B, 2016, 120, 10899–10922 CrossRef CAS PubMed.
- M. Suga, F. Akita, K. Hirata, G. Ueno, H. Murakami, Y. Nakajima, T. Shimizu, K. Yamashita, M. Yamamoto, H. Ago and J.-R. Shen, Nature, 2015, 517, 99–103 CrossRef CAS PubMed.
- D. A. Pantazis, W. Ames, N. Cox, W. Lubitz and F. Neese, Angew. Chem., Int. Ed., 2012, 51, 9935–9940 CrossRef CAS PubMed.
- D. Bovi, D. Narzi and L. Guidoni, Angew. Chem., Int. Ed., 2013, 52, 11744–11749 CrossRef CAS PubMed.
- H. Isobe, M. Shoji, S. Yamanaka, Y. Umena, K. Kawakami, N. Kamiya, J. R. Shen and K. Yamaguchi, Dalton Trans., 2012, 41, 13727–13740 RSC.
- M. Retegan, V. Krewald, F. Mamedov, F. Neese, W. Lubitz, N. Cox and D. A. Pantazis, Chem. Sci., 2016, 7, 72–84 RSC.
- D. Narzi, D. Bovi and L. Guidoni, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 8723–8728 CrossRef CAS PubMed.
- M. Capone, D. Bovi, D. Narzi and L. Guidoni, Biochemistry, 2015, 54, 6439–6442 CrossRef CAS PubMed.
- M. Capone, D. Narzi, D. Bovi and L. Guidoni, J. Phys. Chem. Lett., 2016, 7, 592–596 CrossRef CAS PubMed.
- V. Krewald, M. Retegan, F. Neese, W. Lubitz, D. A. Pantazis and N. Cox, Inorg. Chem., 2016, 55, 488–501 CrossRef CAS PubMed.
- I. Zaharieva, H. Dau and M. Haumann, Biochemistry, 2016, 55, 6996–7004 CrossRef CAS PubMed.
- H. Sakamoto, T. Shimizu, R. Nagao and T. Noguchi, J. Am. Chem. Soc., 2017, 139, 2022–2029 CrossRef CAS PubMed.
- M. Retegan, N. Cox, W. Lubitz, F. Neese and D. A. Pantazis, Phys. Chem. Chem. Phys., 2014, 16, 11901–11910 RSC.
- N. Cox, M. Retegan, F. Neese, D. A. Pantazis, A. Boussac and W. Lubitz, Science, 2014, 345, 804–808 CrossRef CAS PubMed.
- A. Boussac, A. W. Rutherford and M. Sugiura, Biochim. Biophys. Acta, Bioenerg., 2015, 1847, 576–586 CrossRef CAS PubMed.
- P. E. M. Siegbahn, Biochim. Biophys. Acta, Bioenerg., 2013, 1827, 1003–1019 CrossRef CAS PubMed.
- M. Shoji, H. Isobe and K. Yamaguchi, Chem. Phys. Lett., 2015, 636, 172–179 CrossRef CAS.
- M. Askerka, J. Wang, D. J. Vinyard, G. W. Brudvig and V. S. Batista, Biochemistry, 2016, 55, 981–984 CrossRef CAS PubMed.
- M. Hatakeyama, K. Ogata, K. Fujii, V. K. Yachandra, J. Yano and S. Nakamura, Chem. Phys. Lett., 2016, 651, 243–250 CrossRef CAS.
- I. Ugur, A. W. Rutherford and V. R. I. Kaila, Biochim. Biophys. Acta, Bioenerg., 2016, 1857, 740–748 CrossRef CAS PubMed.
- H. Isobe, M. Shoji, J.-R. Shen and K. Yamaguchi, Inorg. Chem., 2016, 55, 502–511 CrossRef CAS PubMed.
- P. E. M. Siegbahn, Phys. Chem. Chem. Phys., 2012, 14, 4849–4856 RSC.
- P. E. M. Siegbahn, J. Am. Chem. Soc., 2013, 135, 9442–9449 CrossRef CAS PubMed.
- I. D. Young, M. Ibrahim, R. Chatterjee, S. Gul, F. D. Fuller, S. Koroidov, A. S. Brewster, R. Tran, R. Alonso-Mori, T. Kroll, T. Michels-Clark, H. Laksmono, R. G. Sierra, C. A. Stan, R. Hussein, M. Zhang, L. Douthit, M. Kubin, C. de Lichtenberg, L. Vo Pham, H. Nilsson, M. H. Cheah, D. Shevela, C. Saracini, M. A. Bean, I. Seuffert, D. Sokaras, T.-C. Weng, E. Pastor, C. Weninger, T. Fransson, L. Lassalle, P. Bräuer, P. Aller, P. T. Docker, B. Andi, A. M. Orville, J. M. Glownia, S. Nelson, M. Sikorski, D. Zhu, M. S. Hunter, T. J. Lane, A. Aquila, J. E. Koglin, J. Robinson, M. Liang, S. Boutet, A. Y. Lyubimov, M. Uervirojnangkoorn, N. W. Moriarty, D. Liebschner, P. V. Afonine, D. G. Waterman, G. Evans, P. Wernet, H. Dobbek, W. I. Weis, A. T. Brunger, P. H. Zwart, P. D. Adams, A. Zouni, J. Messinger, U. Bergmann, N. K. Sauter, J. Kern, V. K. Yachandra and J. Yano, Nature, 2016, 540, 453–457 CrossRef CAS PubMed.
- T. Ichino and Y. Yoshioka, Chem. Phys. Lett., 2014, 595–596, 237–241 CrossRef CAS.
- H. Bao and R. L. Burnap, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E6139–E6147 CrossRef CAS PubMed.
- X. Li, P. E. M. Siegbahn and U. Ryde, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 3979–3984 CrossRef CAS PubMed.
- H. Isobe, M. Shoji, J.-R. Shen and K. Yamaguchi, J. Phys. Chem. B, 2015, 119, 13922–13933 CrossRef CAS PubMed.
- I. Zaharieva, P. Chernev, G. Berggren, M. Anderlund, S. Styring, H. Dau and M. Haumann, Biochemistry, 2016, 55, 4197–4211 CrossRef CAS PubMed.
- M. Suga, F. Akita, M. Sugahara, M. Kubo, Y. Nakajima, T. Nakane, K. Yamashita, Y. Umena, M. Nakabayashi, T. Yamane, T. Nakano, M. Suzuki, T. Masuda, S. Inoue, T. Kimura, T. Nomura, S. Yonekura, L.-J. Yu, T. Sakamoto, T. Motomura, J.-H. Chen, Y. Kato, T. Noguchi, K. Tono, Y. Joti, T. Kameshima, T. Hatsui, E. Nango, R. Tanaka, H. Naitow, Y. Matsuura, A. Yamashita, M. Yamamoto, O. Nureki, M. Yabashi, T. Ishikawa, S. Iwata and J.-R. Shen, Nature, 2017, 543, 131–135 CrossRef PubMed.
- M. M. Najafpour, S. Heidari, S. E. Balaghi, M. Hołyńska, M. H. Sadr, B. Soltani, M. Khatamian, A. W. Larkum and S. I. Allakhverdiev, Biochim. Biophys. Acta, Bioenerg., 2017, 1858, 156–174 CrossRef CAS PubMed.
- X. Li and P. E. M. Siegbahn, Phys. Chem. Chem. Phys., 2015, 17, 12168–12174 RSC.
- S. Yamanaka, H. Isobe, K. Kanda, T. Saito, Y. Umena, K. Kawakami, J. R. Shen, N. Kamiya, M. Okumura, H. Nakamura and K. Yamaguchi, Chem. Phys. Lett., 2011, 511, 138–145 CrossRef CAS.
- N. Cox and J. Messinger, Biochim. Biophys. Acta, Bioenerg., 2013, 1827, 1020–1030 CrossRef CAS PubMed.
- J. Messinger, Phys. Chem. Chem. Phys., 2004, 6, 4764–4771 RSC.
- J. M. Peloquin, K. A. Campbell, D. W. Randall, M. A. Evanchik, V. L. Pecoraro, W. H. Armstrong and R. D. Britt, J. Am. Chem. Soc., 2000, 122, 10926–10942 CrossRef CAS.
- N. Cox, L. Rapatskiy, J.-H. Su, D. A. Pantazis, M. Sugiura, L. Kulik, P. Dorlet, A. W. Rutherford, F. Neese, A. Boussac, W. Lubitz and J. Messinger, J. Am. Chem. Soc., 2011, 133, 3635–3648 CrossRef CAS PubMed.
- J.-H. Su, N. Cox, W. Ames, D. A. Pantazis, L. Rapatskiy, T. Lohmiller, L. V. Kulik, P. Dorlet, A. W. Rutherford, F. Neese, A. Boussac, W. Lubitz and J. Messinger, Biochim. Biophys. Acta, Bioenerg., 2011, 1807, 829–840 CrossRef CAS PubMed.
- X. Wu, F. Li, B. Zhang and L. Sun, J. Photochem. Photobiol., C, 2015, 25, 71–89 CrossRef CAS.
- V. Artero and M. Fontecave, Chem. Soc. Rev., 2013, 42, 2338–2356 RSC.
- M. M. Najafpour, A. N. Moghaddam, H. Dau and I. Zaharieva, J. Am. Chem. Soc., 2014, 136, 7245–7248 CrossRef CAS PubMed.
- R. Tagore, H. Chen, H. Zhang, R. H. Crabtree and G. W. Brudvig, Inorg. Chim. Acta, 2007, 360, 2983–2989 CrossRef CAS PubMed.
- C. Baffert, S. Romain, A. Richardot, J.-C. Leprêtre, B. Lefebvre, A. Deronzier and M.-N. Collomb, J. Am. Chem. Soc., 2005, 127, 13694–13704 CrossRef CAS PubMed.
- H. Chen, R. Tagore, G. Olack, J. S. Vrettos, T.-C. Weng, J. Penner-Hahn, R. H. Crabtree and G. W. Brudvig, Inorg. Chem., 2007, 46, 34–43 CrossRef CAS PubMed.
- M. D. Kärkäs, E. V. Johnston, O. Verho and B. Åkermark, Acc. Chem. Res., 2014, 47, 100–111 CrossRef PubMed.
- M. Kondo and S. Masaoka, Chem. Lett., 2016, 45, 1220–1231 CrossRef CAS.
- A. R. Parent and K. Sakai, ChemSusChem, 2014, 7, 2070–2080 CrossRef CAS PubMed.
- M. M. Najafpour, G. Renger, M. Hołyńska, A. N. Moghaddam, E.-M. Aro, R. Carpentier, H. Nishihara, J. J. Eaton-Rye, J.-R. Shen and S. I. Allakhverdiev, Chem. Rev., 2016, 116, 2886–2936 CrossRef CAS PubMed.
- P. Kurz, Top. Curr. Chem., 2016, 371, 49–72 CrossRef PubMed.
- M. M. Najafpour, T. Ehrenberg, M. Wiechen and P. Kurz, Angew. Chem., Int. Ed., 2010, 49, 2233–2237 CrossRef CAS PubMed.
- D. Shevela, S. Koroidov, M. M. Najafpour, J. Messinger and P. Kurz, Chem. – Eur. J., 2011, 17, 5415–5423 CrossRef CAS PubMed.
- C. E. Frey, M. Wiechen and P. Kurz, Dalton Trans., 2014, 43, 4370–4379 RSC.
- C. E. Frey and P. Kurz, Chem. – Eur. J., 2015, 21, 14958–14968 CrossRef CAS PubMed.
- I. Zaharieva, M. M. Najafpour, M. Wiechen, M. Haumann, P. Kurz and H. Dau, Energy Environ. Sci., 2011, 4, 2400–2408 CAS.
- R. Manchanda, G. W. Brudvig and R. H. Crabtree, Coord. Chem. Rev., 1995, 144, 1–38 CrossRef CAS.
- K. Wieghardt, U. Bossek and W. Gebert, Angew. Chem., Int. Ed., 1983, 22, 328–329 CrossRef.
- K. S. Hagen, T. D. Westmoreland, M. J. Scott and W. H. Armstrong, J. Am. Chem. Soc., 1989, 111, 1907–1909 CrossRef CAS.
- C. E. Dubé, S. Mukhopadhyay, P. J. Bonitatebus, R. J. Staples and W. H. Armstrong, Inorg. Chem., 2005, 44, 5161–5175 CrossRef PubMed.
- W. F. Ruettinger, C. Campana and G. C. Dismukes, J. Am. Chem. Soc., 1997, 119, 6670–6671 CrossRef CAS.
- G. W. Brudvig and R. H. Crabtree, Proc. Natl. Acad. Sci. U. S. A., 1986, 83, 4586–4588 CrossRef CAS.
- J. B. Vincent and G. Christou, in Adv. Inorg. Chem, ed. A. G. Sykes, Academic Press, 1989, pp. 197–257 Search PubMed.
- J. S. Bashkin, H. R. Chang, W. E. Streib, J. C. Huffman, D. N. Hendrickson and G. Christou, J. Am. Chem. Soc., 1987, 109, 6502–6504 CrossRef CAS.
- D. N. Hendrickson, G. Christou, E. A. Schmitt, E. Libby, J. S. Bashkin, S. Wang, H. L. Tsai, J. B. Vincent and P. D. W. Boyd, J. Am. Chem. Soc., 1992, 114, 2455–2471 CrossRef CAS.
- Q. Li, J. B. Vincent, E. Libby, H.-R. Chang, J. C. Huffman, P. D. W. Boyd, G. Christou and D. N. Hendrickson, Angew. Chem., Int. Ed., 1988, 27, 1731–1733 Search PubMed.
- S. M. J. Aubin, N. R. Dilley, M. W. Wemple, M. B. Maple, G. Christou and D. N. Hendrickson, J. Am. Chem. Soc., 1998, 120, 839–840 CrossRef CAS.
- M. W. Wemple, D. M. Adams, K. S. Hagen, K. Folting, D. N. Hendrickson and G. Christou, J. Chem. Soc., Chem. Commun., 1995, 1591–1593 RSC.
- M. Brynda and R. D. Britt, in Metals in Biology: Applications of High-Resolution EPR to Metalloenzymes, ed. G. Hanson and L. Berliner, Springer New York, New York, NY, 2010, pp. 203–271 Search PubMed.
- G. Aromí, S. Bhaduri, P. Artús, K. Folting and G. Christou, Inorg. Chem., 2002, 41, 805–817 CrossRef.
- G. Aromí, M. W. Wemple, S. J. Aubin, K. Folting, D. N. Hendrickson and G. Christou, J. Am. Chem. Soc., 1998, 120, 5850–5851 Search PubMed.
- S. Wang, H.-L. Tsai, K. S. Hagen, D. N. Hendrickson and G. Christou, J. Am. Chem. Soc., 1994, 116, 8376–8377 CrossRef CAS.
- S. Wang, H.-L. Tsai, E. Libby, K. Folting, W. E. Streib, D. N. Hendrickson and G. Christou, Inorg. Chem., 1996, 35, 7578–7589 CrossRef CAS.
- S. Wang, M. S. Wemple, J. Yoo, K. Folting, J. C. Huffman, K. S. Hagen, D. N. Hendrickson and G. Christou, Inorg. Chem., 2000, 39, 1501–1513 CrossRef CAS PubMed.
- M. W. Wemple, D. M. Adams, K. Folting, D. N. Hendrickson and G. Christou, J. Am. Chem. Soc., 1995, 117, 7275–7276 CrossRef CAS.
- S. Wang, K. Folting, W. E. Streib, E. A. Schmitt, J. K. McCusker, D. N. Hendrickson and G. Christou, Angew. Chem., Int. Ed., 1991, 30, 305–306 CrossRef.
- S. Wang, H.-L. Tsai, W. E. Streib, G. Christou and D. N. Hendrickson, J. Chem. Soc., Chem. Commun., 1992, 677–679 RSC.
- W. Ruettinger, M. Yagi, K. Wolf, S. Bernasek and G. C. Dismukes, J. Am. Chem. Soc., 2000, 122, 10353–10357 CrossRef CAS.
- W. F. Ruettinger, D. M. Ho and G. C. Dismukes, Inorg. Chem., 1999, 38, 1036–1037 CrossRef CAS PubMed.
- W. F. Ruettinger and G. C. Dismukes, Inorg. Chem., 2000, 39, 1021–1027 CrossRef CAS PubMed.
- N. E. Chakov, K. A. Abboud, L. N. Zakharov, A. L. Rheingold, D. N. Hendrickson and G. Christou, Polyhedron, 2003, 22, 1759–1763 CrossRef CAS.
- J.-Z. Wu, E. Sellitto, G. P. A. Yap, J. Sheats and G. C. Dismukes, Inorg. Chem., 2004, 43, 5795–5797 CrossRef CAS PubMed.
- K. M. Van Allsburg, E. Anzenberg, W. S. Drisdell, J. Yano and T. D. Tilley, Chem. – Eur. J., 2015, 21, 4646–4654 CrossRef CAS PubMed.
- J.-Z. Wu, F. De Angelis, T. G. Carrell, G. P. A. Yap, J. Sheats, R. Car and G. C. Dismukes, Inorg. Chem., 2006, 45, 189–195 CrossRef CAS PubMed.
- J. S. Kanady, E. Y. Tsui, M. W. Day and T. Agapie, Science, 2011, 333, 733–736 CrossRef CAS PubMed.
- J. B. Vincent and G. Christou, Inorg. Chim. Acta, 1987, 136, L41–L43 CrossRef CAS.
- A. E. Kuznetsov, Y. V. Geletii, C. L. Hill and D. G. Musaev, J. Phys. Chem. A, 2010, 114, 11417–11424 CrossRef CAS PubMed.
- K. M. Davis, M. C. Palenik, L. Yan, P. F. Smith, G. T. Seidler, G. C. Dismukes and Y. N. Pushkar, J. Phys. Chem. C, 2016, 120, 3326–3333 CAS.
- R.-Z. Liao and P. E. M. Siegbahn, J. Photochem. Photobiol., B, 2015, 152, 162–172 CrossRef CAS PubMed.
- J. B. Vincent, C. Christmas, H. R. Chang, Q. Li, P. D. W. Boyd, J. C. Huffman, D. N. Hendrickson and G. Christou, J. Am. Chem. Soc., 1989, 111, 2086–2097 CrossRef CAS.
- J. B. Vincent, C. Christmas, J. C. Huffman, G. Christou, H.-R. Chang and D. N. Hendrickson, J. Chem. Soc., Chem. Commun., 1987, 236–238 RSC.
- C. Boskovic, K. Folting and G. Christou, Polyhedron, 2000, 19, 2111–2118 CrossRef CAS.
- E. Bouwman, M. A. Bolcar, E. Libby, J. C. Huffman, K. Folting and G. Christou, Inorg. Chem., 1992, 31, 5185–5192 CrossRef CAS.
- C. Cañada-Vilalta, J. C. Huffman and G. Christou, Polyhedron, 2001, 20, 1785–1793 CrossRef.
- E. Libby, K. Folting, C. J. Huffman, J. C. Huffman and G. Christou, Inorg. Chem., 1993, 32, 2549–2556 CrossRef CAS.
- E. Libby, J. K. McCusker, E. A. Schmitt, K. Folting, D. N. Hendrickson and G. Christou, Inorg. Chem., 1991, 30, 3486–3495 CrossRef CAS.
- J. Pistilli and R. H. Beer, Inorg. Chem. Commun., 2002, 5, 206–210 CrossRef CAS.
- S. Wang, H.-L. Tsai, K. Folting, J. D. Martin, D. N. Hendrickson and G. Christou, J. Chem. Soc., Chem. Commun., 1994, 671–673 RSC.
- M. K. Chan and W. H. Armstrong, J. Am. Chem. Soc., 1990, 112, 4985–4986 CrossRef CAS.
- M. K. Chan and W. H. Armstrong, J. Am. Chem. Soc., 1989, 111, 9121–9122 CrossRef CAS.
- G. Blondin, R. Davydov, C. Philouze, M.-F. Charlot, S. Styring, B. Åkermark, J.-J. Girerd and A. Boussac, J. Chem. Soc., Dalton Trans., 1997, 4069–4074 RSC.
- M. K. Chan and W. H. Armstrong, J. Am. Chem. Soc., 1991, 113, 5055–5057 CrossRef CAS.
- H. Chen, J. W. Faller, R. H. Crabtree and G. W. Brudvig, J. Am. Chem. Soc., 2004, 126, 7345–7349 CrossRef CAS PubMed.
- C. Philouze, G. Blondin, J.-J. Girerd, J. Guilhem, C. Pascard and D. Lexa, J. Am. Chem. Soc., 1994, 116, 8557–8565 CrossRef CAS.
- D. A. Pantazis, M. Orio, T. Petrenko, S. Zein, E. Bill, W. Lubitz, J. Messinger and F. Neese, Chem. – Eur. J., 2009, 15, 5108–5123 CrossRef CAS PubMed.
- C. F. Yocum, Coord. Chem. Rev., 2008, 252, 296–305 CrossRef CAS.
- R. M. Cinco, K. L. McFarlane Holman, J. H. Robblee, J. Yano, S. A. Pizarro, E. Bellacchio, K. Sauer and V. K. Yachandra, Biochemistry, 2002, 41, 12928–12933 CrossRef CAS PubMed.
- L. B. Jerzykiewicz, J. Utko, M. Duczmal and P. Sobota, Dalton Trans., 2007, 825–826 RSC.
- A. Mishra, W. Wernsdorfer, K. A. Abboud and G. Christou, Chem. Commun., 2005, 54–56 RSC.
- A. Mishra, J. Yano, Y. Pushkar, V. K. Yachandra, K. A. Abboud and G. Christou, Chem. Commun., 2007, 1538–1540 RSC.
- C. Chen, C. Zhang, H. Dong and J. Zhao, Dalton Trans., 2015, 44, 4431–4435 RSC.
- E. S. Koumousi, S. Mukherjee, C. M. Beavers, S. J. Teat, G. Christou and T. C. Stamatatos, Chem. Commun., 2011, 47, 11128–11130 RSC.
- I. J. Hewitt, J.-K. Tang, N. T. Madhu, R. Clerac, G. Buth, C. E. Anson and A. K. Powell, Chem. Commun., 2006, 2650–2652 RSC.
- E. Y. Tsui and T. Agapie, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 10084–10088 CrossRef CAS PubMed.
- J. S. Kanady, P.-H. Lin, K. M. Carsch, R. J. Nielsen, M. K. Takase, W. A. Goddard and T. Agapie, J. Am. Chem. Soc., 2014, 136, 14373–14376 Search PubMed.
- P.-H. Lin, M. K. Takase and T. Agapie, Inorg. Chem., 2015, 54, 59–64 CrossRef CAS PubMed.
- S. Singh, R. J. Debus, T. Wydrzynski and W. Hillier, Philos. Trans. R. Soc., B, 2008, 363, 1229–1235 CrossRef CAS PubMed.
- J. Dasgupta, G. M. Ananyev and G. C. Dismukes, Coord. Chem. Rev., 2008, 252, 347–360 CrossRef CAS PubMed.
- K. Becker, K. U. Cormann and M. M. Nowaczyk, J. Photochem. Photobiol. B, 2011, 104, 204–211 CrossRef CAS PubMed.
- M. Asada and H. Mino, J. Phys. Chem. B, 2015, 119, 10139–10144 CrossRef CAS PubMed.
- M. Retegan and D. A. Pantazis, Chem. Sci., 2016, 7, 6463–6476 RSC.
- S. Vassiliev, T. Zaraiskaya and D. Bruce, Biochim. Biophys. Acta, Bioenerg., 2012, 1817, 1671–1678 CrossRef CAS PubMed.
- S. Vassiliev, T. Zaraiskaya and D. Bruce, Biochim. Biophys. Acta, Bioenerg., 2013, 1827, 1148–1155 CrossRef CAS PubMed.
- M. Pérez Navarro, W. M. Ames, H. Nilsson, T. Lohmiller, D. A. Pantazis, L. Rapatskiy, M. M. Nowaczyk, F. Neese, A. Boussac, J. Messinger, W. Lubitz and N. Cox, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 15561–15566 CrossRef PubMed.
- J. Schraut and M. Kaupp, Chem. – Eur. J., 2014, 20, 7300–7308 CrossRef CAS PubMed.
- P. H. Oyala, T. A. Stich, R. J. Debus and R. D. Britt, J. Am. Chem. Soc., 2015, 137, 8829–8837 CrossRef CAS PubMed.
- T. Lohmiller, V. Krewald, M. Pérez Navarro, M. Retegan, L. Rapatskiy, M. M. Nowaczyk, A. Boussac, F. Neese, W. Lubitz, D. A. Pantazis and N. Cox, Phys. Chem. Chem. Phys., 2014, 16, 11877–11892 RSC.
- C. Chen, C. Zhang, H. Dong and J. Zhao, Chem. Commun., 2014, 50, 9263–9265 RSC.
- S. Mukherjee, J. A. Stull, J. Yano, T. C. Stamatatos, K. Pringouri, T. A. Stich, K. A. Abboud, R. D. Britt, V. K. Yachandra and G. Christou, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 2257–2262 CrossRef CAS PubMed.
- V. Krewald, F. Neese and D. A. Pantazis, J. Am. Chem. Soc., 2013, 135, 5726–5739 CrossRef CAS PubMed.
- C. Zhang, C. Chen, H. Dong, J.-R. Shen, H. Dau and J. Zhao, Science, 2015, 348, 690–693 CrossRef CAS PubMed.
- S. Romain, J. Rich, C. Sens, T. Stoll, J. Benet-Buchholz, A. Llobet, M. Rodriguez, I. Romero, R. Clérac, C. Mathonière, C. Duboc, A. Deronzier and M.-N. L. Collomb, Inorg. Chem., 2011, 50, 8427–8436 CrossRef CAS PubMed.
- V. Krewald and D. A. Pantazis, Dalton Trans., 2016, 45, 18900–18908 RSC.
- J. Yano, Y. Pushkar, P. Glatzel, A. Lewis, K. Sauer, J. Messinger, U. Bergmann and V. Yachandra, J. Am. Chem. Soc., 2005, 127, 14974–14975 CrossRef CAS PubMed.
- M. J. Baldwin, T. L. Stemmler, P. J. Riggs-Gelasco, M. L. Kirk, J. E. Penner-Hahn and V. L. Pecoraro, J. Am. Chem. Soc., 1994, 116, 11349–11356 CrossRef CAS.
- D. A. Pantazis, V. Krewald, M. Orio and F. Neese, Dalton Trans., 2010, 39, 4959–4967 RSC.
- V. Krewald, B. Lassalle-Kaiser, T. T. Boron, C. J. Pollock, J. Kern, M. A. Beckwith, V. K. Yachandra, V. L. Pecoraro, J. Yano, F. Neese and S. DeBeer, Inorg. Chem., 2013, 52, 12904–12914 CrossRef CAS PubMed.
- P. W. Anderson, Phys. Rev., 1950, 79, 350–356 CrossRef.
- P. W. Anderson, Phys. Rev., 1959, 115, 2–13 CrossRef CAS.
- P. J. Hay, J. C. Thibeault and R. Hoffmann, J. Am. Chem. Soc., 1975, 97, 4884–4899 CrossRef CAS.
- C. Kollmar, M. Couty and O. Kahn, J. Am. Chem. Soc., 1991, 113, 7994–8005 CrossRef CAS.
- S. Paul and A. Misra, J. Chem. Theory Comput., 2012, 8, 843–853 CrossRef CAS PubMed.
- F. Neese, J. Phys. Chem. Solids, 2004, 65, 781–785 CrossRef CAS.
- V. Krewald, F. Neese and D. A. Pantazis, Phys. Chem. Chem. Phys., 2016, 18, 10739–10750 RSC.
- V. Martin-Diaconescu, M. Gennari, B. Gerey, E. Tsui, J. Kanady, R. Tran, J. Pécaut, D. Maganas, V. Krewald, E. Gouré, C. Duboc, J. Yano, T. Agapie, M.-N. Collomb and S. DeBeer, Inorg. Chem., 2015, 54, 1283–1292 CrossRef CAS PubMed.
- J. S. Kanady, J. L. Mendoza-Cortes, E. Y. Tsui, R. J. Nielsen, W. A. Goddard and T. Agapie, J. Am. Chem. Soc., 2013, 135, 1073–1082 CrossRef CAS PubMed.
- C. Lee and C. M. Aikens, J. Phys. Chem. A, 2015, 119, 9325–9337 CrossRef CAS PubMed.
- E. Y. Tsui, R. Tran, J. Yano and T. Agapie, Nat. Chem., 2013, 5, 293–299 CrossRef CAS PubMed.
- S. Bang, Y.-M. Lee, S. Hong, K.-B. Cho, Y. Nishida, M. S. Seo, R. Sarangi, S. Fukuzumi and W. Nam, Nat. Chem., 2014, 6, 934–940 CrossRef CAS PubMed.
- M. Shoji, H. Isobe, T. Nakajima and K. Yamaguchi, Chem. Phys. Lett., 2015, 640, 23–30 CrossRef CAS.
- M. Shoji, H. Isobe, J. R. Shen and K. Yamaguchi, Phys. Chem. Chem. Phys., 2016, 18, 11330–11340 RSC.
- S. Paul, N. Cox and D. A. Pantazis, Inorg. Chem., 2017 DOI:10.1021/acs.inorgchem.6b02777.
- Y. Yu, C. Hu, X. Liu and J. Wang, ChemBioChem, 2015, 16, 1981–1983 CrossRef CAS PubMed.
- U. Raucci, I. Ciofini, C. Adamo and N. Rega, J. Phys. Chem. Lett., 2016, 7, 5015–5021 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |