
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sequential fractionation of feruloylated hemicelluloses and oligosaccharides from wheat bran using subcritical water and xylanolytic enzymes†
Andrea C.
Ruthes
a,
Antonio
Martínez-Abad
a,
Hwei-Ting
Tan
abc,
Vincent
Bulone
ab and
Francisco
Vilaplana
 *a
*a
aDivision of Glycoscience, School of Biotechnology, KTH Royal Institute of Technology, AlbaNova University Centre, SE-106 91 Stockholm, Sweden. E-mail: franvila@kth.se
bARC Centre of Excellence in Plant Cell Walls and School of Agriculture, Food and Wine, The University of Adelaide, Waite Campus, Urrbrae, SA 5064, Australia
cCentre for Tropical Crops and Biocommodities, Queensland University of Technology, Brisbane, QLD, Australia
First published on 28th March 2017
Abstract
Wheat bran is a major by-product of cereal production that still has limited use for advanced nutritional and material applications. A sequential process using subcritical water, membrane filtration and selective enzymatic treatments has been designed for the combined fractionation of functional high molar mass hemicelluloses (over 105 g mol−1) and oligosaccharides from wheat bran. This process not only offers increased total solid yield compared with conventional protocols based on alkaline extraction, but it also preserves the inherent functionalities of the phenolic groups that substitute the carbohydrate structures of the extracted hemicelluloses. Feruloylated arabinoxylans (F-AX) with high molar mass and significant radical scavenging activity can be isolated from the subcritical water extract. Structurally different oligosaccharides, including mixed-linkage β-D-glucan oligosaccharides (BGOs) and arabinoxylo-oligosaccharides (AXOs) can be recovered from the eluent after membrane filtration. The crosslinked residue after subcritical water extraction was further treated with xylanolytic enzymes to release valuable feruloylated arabinoxylo-oligosaccharides (FAXOs). The oligo- and polysaccharide fractions isolated from this sequential process show great potential for use as prebiotic or platform chemicals, and as polymeric matrices for carbohydrate-based materials with radical scavenging properties, respectively.
Introduction
Cereal processing generates large volumes of by-products that have not been exploited to their full potential for advanced nutritional and material applications.1 Each ton processed by the wheat milling industry generates up to 0.25 tons of wheat bran that corresponds to 14–16% of the cereal kernel.2 Due to its content of valuable biomacromolecules, such as polysaccharides (mainly hemicelluloses) and phenolic compounds, wheat bran has gained renewed interest as a raw material to be used in second generation biorefineries.2–4Xylan is one of the most abundant biopolymers in nature as a major non-cellulosic component of plant cell walls. In particular, arabinoxylan (AX) constitutes the main hemicellulose in grasses and cereal grains, with an average composition of 20–40% depending on the species and tissue considered.5,6 AX structure is based on a linear backbone of (1→4)-linked β-D-xylopyranose (Xylp) units, mainly 2-O-, 3-O- and/or 2,3-di-O-substituted by α-D-arabinofuranosyl (Araf) units. The xylan backbone can be 2-O-substituted to a lesser extent by α-D-glucuronic acid (GlcA) or its 4-O-methyl derivative (4-O-Me-GlcA). The extent and distribution of the glycosyl substitutions depend on the biological source and tissue.7 Phenolic compounds, mainly ferulic acid (FA), can further substitute the Araf residues in the C-5 position.5,8 These phenolic compounds can dimerize with other phenolic moieties from neighboring AX molecules, creating a complex covalent network structure. This heterogeneous and crosslinked molecular architecture is responsible for the recalcitrance of bran, and imposes major challenges for the biotechnological valorization of the constituent biomolecules.
Pretreatments using harsh alkaline conditions have been traditionally used for the extraction of hemicelluloses from cereal and other plant biomasses.9,10 However, these harsh conditions cause the loss of valuable functional groups, promoting the removal of acetyl, uronic acid and phenolic substitutions.9 Other methods have also been developed for the extraction and purification of AX from cereal by-products, including acid pretreatment,11 microwave assisted extraction,12 ultrasound assisted extraction,13,14 and steam explosion extraction.10,15 As an alternative, subcritical water extraction (SWE), also called pressurized hot water extraction (PHWE), appears as a promising green method for the isolation of hemicellulose fractions with preserved molecular functionalities and high molecular weight. The use of subcritical conditions (at temperature below critical point and pressure high enough to maintain liquid state) changes dramatically the properties of water, including density surface tension, polarity, viscosity and diffusion. Mass transfer during the extraction process is favored due to the decrease in the viscosity and an increase on diffusivity.16 Subcritical water has proven to be effective for the extraction of diverse bioactive and nutraceutical compounds, including polysaccharides from plants and food matrices.16–19 Moreover, subcritical water has been successfully used for the extraction of hemicelluloses from wood, with the possibility for scaling up.20–23 In parallel, the use of carbohydrate-active enzymes during biomass pretreatment further facilitates the extraction and fractionation of the polysaccharide components, showing good specificity.24 Xylanolytic enzymes that act on the xylan backbone and on the glycosyl decorations can be used to deconstruct xylan polysaccharides into mono- and oligosaccharides that can be further converted into bioethanol and platform chemicals.25–28 However, this process drastically reduces the molar mass of the hemicelluloses and restricts their use in biopolymeric material applications.
A sequential process that combines subcritical water and selective carbohydrate-active enzymes is here proposed to fractionate polymeric hemicelluloses and oligosaccharides from cereal by-products while preserving their native phenolic functionalities. The extraction conditions were optimized under subcritical water and compared to the alkaline processes. Finally, the effect of different carbohydrate-active enzymes was evaluated, as a mean to disrupt the crosslinked residues and further release of functional biomolecules.
Results and discussion
Composition of the defatted wheat bran
The summarized and extended compositions of the defatted wheat bran (wb) and destarched wheat bran (Dwb) are shown in Table 1 and ESI Table S1,† respectively. Polysaccharides represent the main constituents of wheat bran, with arabinoxylans (30%) and glucans (27%), including starch (17%), mixed-linkage β-D-glucan (6%) and cellulose (Fig. 1a), being the main populations, in agreement with previous reports.3,8,29,30 Residual presence of other hemicelluloses and pectins can be also detected by the relative abundance of other sugars in minor amounts (ESI Table S1†). Proteins and phenolic compounds represent the non-carbohydrate fractions of wb and Dwb. The protein content measured based on nitrogen quantification (Dumas method) was 19.7%, which is slightly higher compared to other studies (13.2–18.4%).3,29,31,32 The total hydroxycinnamic acid content was 2.2–2.4 mg g−1, with ferulic acid (FA) being the dominating compound. Similar or lower amounts of FA were reported for insoluble cell wall fractions of wheat endosperm33 and extracted AXs from wheat bran.8,34 Phytic acid (9.6 ± 0.5 mg g−1) and acetyl content (degree of acetylation, DA = 0.03) showed lower values than previously reported for wheat bran.8,35 Destarching effectively removed starch to below 3%, while enriching arabinoxylan (405.3 ± 0.1 mg per g DW) with an arabinose over xylose ratio (A/X) of 0.66, as previously reported for wheat bran (A/X ∼ 0.5–0.8).36,37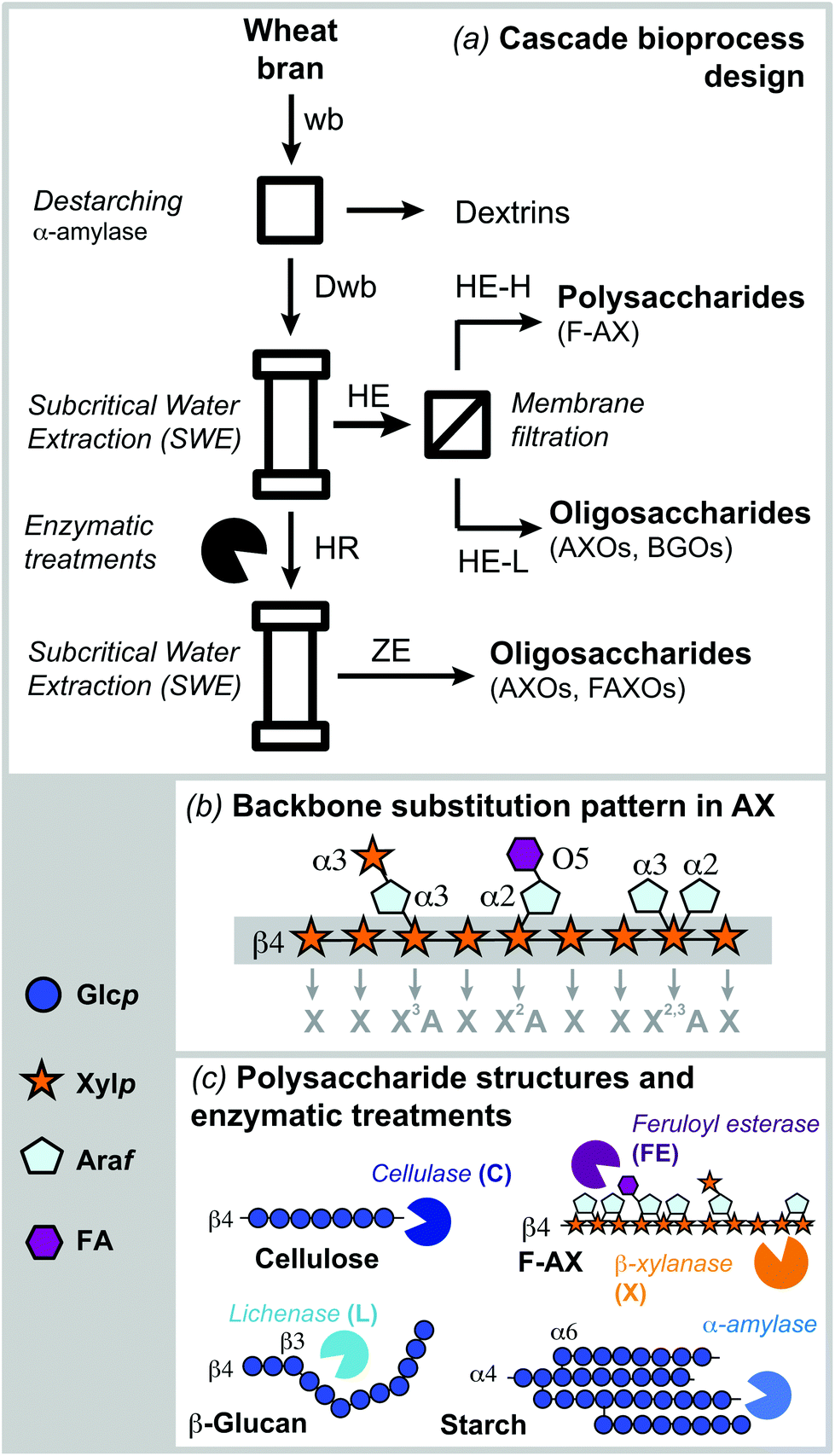 | ||
| Fig. 1 (a) Sequential fractionation process for the isolation of polymeric hemicelluloses (feruloylated arabinoxylan: F-AX) and oligosaccharides (arabino-xylooligosaccharides: AXOs, β-D-glucan oligosaccharides: BGOs, and feruloylated arabino-xylooligosaccharides: FAXOs) from wheat bran. (b) Nomenclature for the backbone substitution pattern in AX; (c) general structures of the main polysaccharides in wheat bran and summary of the enzymatic treatments used in the process. | ||
| wb | Dwb | |
|---|---|---|
| The values represent the average of triplicate measurements. The standard deviations are presented in parentheses.a Moisture determined gravimetrically after oven drying (110 °C, 24 h).b Carbohydrate content determined by phenol-sulfuric acid method.38c Determined from the monosaccharide composition (Table S1).d Starch and β-D-glucan content determined enzymatically (Megazyme).e Protein content determined using Dumas method.39f Hydroxycinnamic acid content determined by GC-MS.33g Klason lignin after acid hydrolysis (H2SO4).h Determined based on the difference between the sum of total carbohydrate, protein, phenolic and lignin and the initial dry weight. | ||
| Moisture contenta (%) | 22.0 (1.1) | 18.5 (1.1) |
| Total carbohydrateb (mg per g DW) | 595.9 (0.6) | 682.4 (0.1) |
| AX (as % of total carbohydrate)c | 48.6 (0.9) | 59.4 (0.4) |
| AXc (mg per g DW) | 289.6 (0.5) | 405.3 (0.1) |
| A/Xc | 0.59 | 0.66 |
| Glc (as % of total carbohydrate)c | 45.5 (0.7) | 35.7 (0.9) |
| Starchd (mg per g DW) | 169.4 (0.6) | 28.9 (2.9) |
| Mixed-linkage β-D-glucan (mg per g DW) | 57.0 (3.0) | 60.8 (1.0) |
| Other polysaccharidesc (mg per g DW) | 35.2 (0.4) | 33.5 (0.1) |
| Proteine (mg per g DW) | 202 | 197 |
| Hydroxycinnamic acidsf (mg per g DW) | 2.2 (0.1) | 2.4 (0.16) |
| Ligning (mg per g DW) | 90.0 (3.8) | 117.8 (1.4) |
| Other compoundsh (mg per g DW) | 109.9 (0.8) | 0.4 (0.05) |
Proposed scheme for the cascade fractionation of hemicelluloses and oligosaccharides from wheat bran
The sequential process combining subcritical water extraction, membrane filtration and xylanolytic enzymes was proposed in order to fractionate simultaneously feruloylated arabinoxylans in polymeric form and oligosaccharides from wheat bran (Fig. 1a). Before extraction, defatted wheat bran was destarched (Dwb) and no other pretreatments were required. The proteins were not removed from Dwb, since they may play important structural roles when preparing hybrid carbohydrate–protein biomaterials. Further enzymatic removal of proteins by proteases could be implemented in conjunction with destarching if required. Subcritical water (SWE) under pressurized conditions was selected as the extraction system due to its efficiency (short extraction times), process sustainability (green solvent system) and the expected ability to maintain the phenolic functionalities in the hemicellulose fractions. Membrane fractionation was introduced to separate the extracted biomolecules into high and low molar mass fractions. Finally, selective enzymatic treatments that target specific carbohydrate structures present in wheat bran (Fig. 1c) were used to increase the accessibility to the recalcitrant AX present in the residue and to release additional oligosaccharide fractions (Fig. 1c).Comparison between subcritical water and alkaline extraction
Alkaline and subcritical water extractions were compared in terms of total extraction yields, polysaccharide composition and phenolic content (Fig. 2 and ESI Table S2†). A monovalent (NaOH) and a divalent [Ca(OH)2] hydroxide were considered as alkaline agents. The use of monovalent hydroxides is reported to lead to higher yields, while divalent hydroxides have shown higher selectivity towards AX.40 Alkaline extractions using NaOH (NE) and Ca(OH)2 (CE), provided total solid yields of 31.3% and 12.4% (DW), respectively (Fig. 2a). Despite the lower yield, extracts from CE presented a higher content of AX (687 mg g−1) when compared to NE (612 mg g−1), confirming the selectivity of divalent hydroxides for AX extraction (Fig. 2b).40 High A/X ratios of 0.90 and 1.03 were obtained for NE and CE, respectively (ESI Table S2†). This may indicate that alkaline extraction is capable of releasing AXs from the outer layers of the wheat bran. Indeed, lower A/X values have been reported for AXs from the aleurone (0.44) and intermediate layers (0.42), while outer pericarp AXs are more substituted (1.16).36,37 The alkaline extracts and residues exhibited significantly lower phenolic acid contents (0.2–0.3 mg g−1) compared with the starting material (Fig. 2c), indicating the removal of ferulic acid.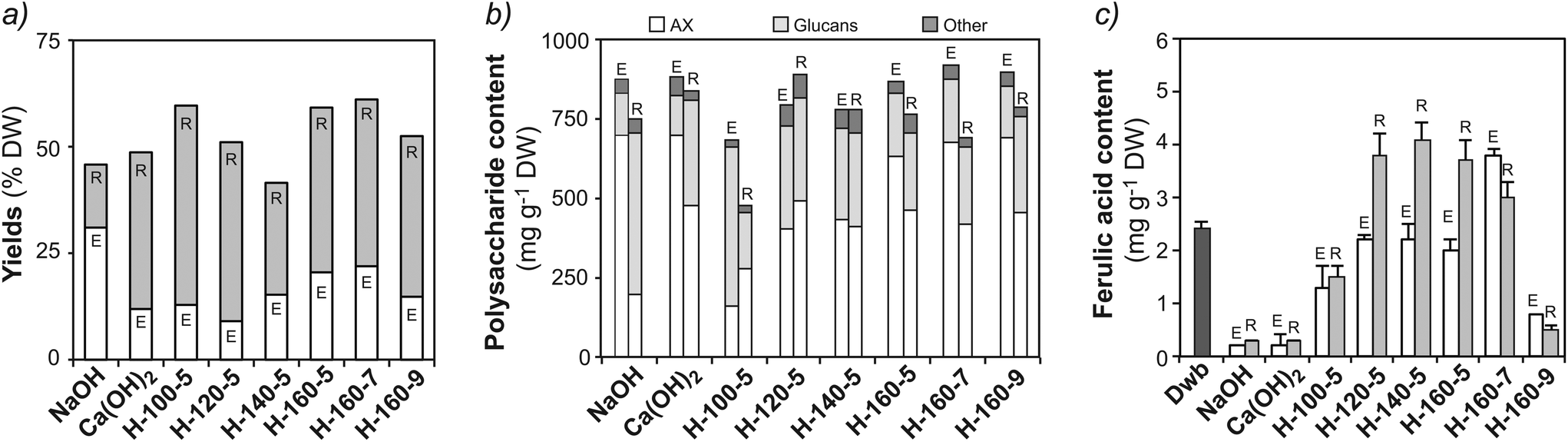 | ||
| Fig. 2 Comparison of alkaline (NaOH and Ca(OH)2) and subcritical water (SWE) extractions at different pH and temperature conditions: (a) total solid yields (based on % DW of Dwb); (b) polysaccharide composition (mg per g DW) of the extracts and residues in terms of arabinoxylan (AX), glucans and minor components. The composition was determined from the monosaccharide analysis after total acid hydrolysis (TFA – extracts and H2SO4 – residues) presented in ESI Table S2;† (c) ferulic acid content (mg per g DW). Note: H: subcritical water; E: extract; R: residue. | ||
SWE was optimized using pressurized buffered aqueous solutions under different temperature (100, 120, 140 and 160 °C) and pH (5.0, 7.0 and 9.0) conditions (Fig. 2a). The highest extraction yields were obtained at 160 °C and pH 7.0 (22.3% DW), with a high content of AX (670 mg per g DW) (Fig. 2b) and enriched ferulic acid content (2.8 mg g−1) (Fig. 2c). SWE offers somewhat lower extraction yields than NaOH extractions, but similar AX contents were obtained with preserved phenolic functionalities. As expected, higher temperatures result in increased total solid yields (Fig. 2a). However, extraction temperatures above 160–170 °C may result in hydrolysis of the polymeric fractions and additional costs to maintain the pressurized conditions.21,23 Temperature plays an important role in modulating the polysaccharide composition of the extracts, progressively increasing the AX content when temperature is increased from 100 °C at pH 5.0 (163 mg per g DW) to 160 °C at pH 7.0 (670 mg per g DW). Neutral pH conditions (7.0) offered optimal yields and AX content, with mild acidic conditions (pH 5.0) resulting in similar yields but lower AX content, and mild alkaline extractions (pH 9.0) leading to higher AX content but reduced ferulic acid content. The A/X ratio of the fractions extracted by SWE varied from 0.48–0.68 (ESI Table S2†) indicating that AXs from the aleurone and intermediate layers are preferentially extracted under subcritical water conditions.36,37
The morphology of the original wheat bran, extracts and residues is distinctively affected by alkaline and subcritical water extractions, as observed by fluorescence microscopy (Fig. 3). Auto-fluorescence emission attributed to ferulic acid can be used to indicate its presence and localization in wheat bran and other cereal kernels.36,41–43 The presence of ferulic acid can be clearly observed in the starting material (wb), as revealed by the selective blue DAPI filter (Fig. 3a, middle column), in order to minimize the possible interference due the presence of lignin.44,45 Ferulic acid appears to be localized in the aleurone and the hyaline layer, as it has been previously reported.35,43,46,47 These data suggest the presence of highly feruloylated crosslinked AXs in the wheat bran (wb and Dwb), and show that destarching did not promote the removal of ferulic acid (Fig. 3b). After alkaline extraction (NaOH), both the extract and residue exhibited no detectable auto-fluorescence (Fig. 3c and d), which demonstrates the cleavage of the ferulic acid moieties under such pH conditions.
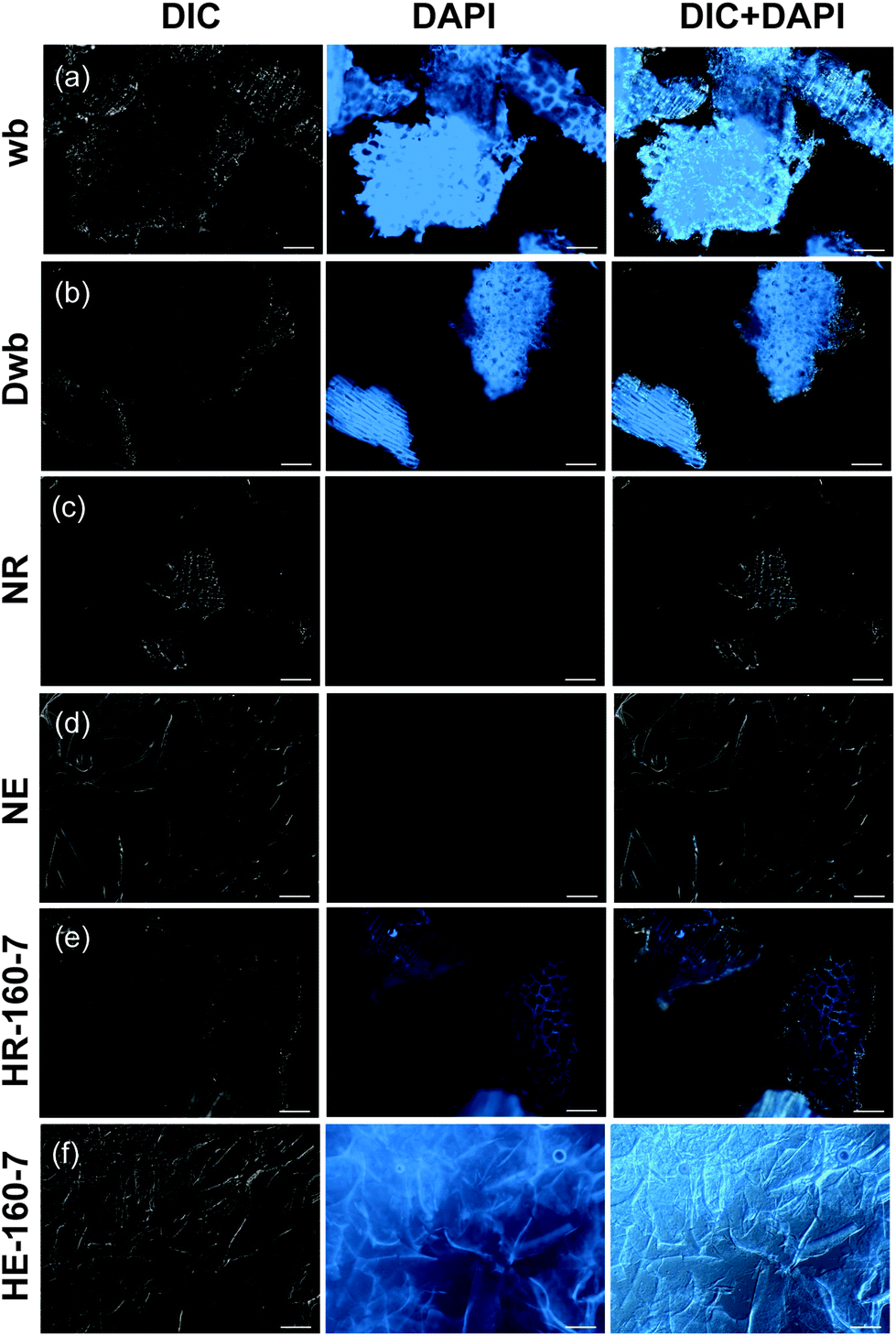 | ||
| Fig. 3 Fluorescence microscopy of wheat bran samples: (a) defatted wheat bran (wb); (b) destarched wheat bran (Dwb); (c) residue from NaOH extraction (NR); (d) extract from NaOH extraction (NE); (e) residue from SWE at 160 °C pH 7.0 (HR-160-7); (f) extract from SWE at 160 °C, pH 7.0 (HE-160-7). The images were obtained using differential interference contrast (DIC) (left column), DAPI filter (middle column) and DIC merged with DAPI filter (right column); optical objective 40×; scale bar 100 μm. | ||
Conversely, auto-fluorescence can be observed in both the extract and residue after SWE, indicating the preservation of ferulic acid while using this method (Fig. 3e and f). The extracts from SWE exhibit evenly distributed autofluorescence, with higher intensity along the observed fibrillar morphologies.
On the other hand, specific localization of the autofluorescence in the outer layers can still be observed in the residues after SWE (Fig. 3e), which indicates that non-extractable crosslinked populations with high ferulic acid densities remain in the outer bran layers. This suggests that subcritical water extracts preferentially AXs from the inner aleurone and intermediate layers, in agreement with the obtained A/X ratios. Such evidence is reinforced by the compositional analyses of the residues after SWE, which exhibit significant content of non-extractable AX (416 mg g−1; Fig. 2b) with high phenolic content (3.0 mg g−1 of FA; Fig. 2c and ESI Table S2†). These resilient AX populations remaining in the residue were the targets for the enzymatic treatments in the next steps.
Fractionation of oligo- and polysaccharide populations
The alkaline and subcritical water extracts exhibited bimodal molar mass distributions with two main (macro)molecular populations, a high molar mass polymeric fraction (105–106 g mol−1) and a low molar mass fraction (103–104 g mol−1) (Fig. 4a). The number-average molar mass (Mn), weight-average molar mass (Mw) and the dispersity index of the extracted fractions are presented in ESI Table S2.† Closed dialysis (Fig. 4b) and membrane ultrafiltration (Fig. 4c) were applied to fractionate the low molar mass (L) from the high molar mass (H) fractions. Both procedures efficiently separated the oligosaccharides from the polysaccharide populations. After membrane fractionation, the total solid yields; the polysaccharide composition and the molecular structure were evaluated for the polymeric and oligomeric fractions (Table 2). | ||
Fig. 4 Molecular properties of the hemicellulose fractions obtained from destarched wheat bran (Dwb). (a) Molar mass distributions w(log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) M) of the extracts after alkaline (NE and CE) and subcritical water extraction (HE); (b) molar mass distributions of the high molar mass fractions (H) after closed dialysis; (c) molar mass distributions of the polymeric (H) and oligomeric (L) fractions after membrane ultrafiltration of HE-160-7; (d) oligomeric mass profiling by ESI-MS of the polymeric fraction HE-160-7-H after enzymatic depolymerization with a xylanase (X) and a feruloyl esterase (FE); (e) oligosaccharide sequencing by ESI-MS/MS of selected feruloylated ions (m/z 745 and m/z 877) and proposed structures from the fragmentation pattern. The mass-to-charge (m/z) values of the fragments are presented in parenthesis. M) of the extracts after alkaline (NE and CE) and subcritical water extraction (HE); (b) molar mass distributions of the high molar mass fractions (H) after closed dialysis; (c) molar mass distributions of the polymeric (H) and oligomeric (L) fractions after membrane ultrafiltration of HE-160-7; (d) oligomeric mass profiling by ESI-MS of the polymeric fraction HE-160-7-H after enzymatic depolymerization with a xylanase (X) and a feruloyl esterase (FE); (e) oligosaccharide sequencing by ESI-MS/MS of selected feruloylated ions (m/z 745 and m/z 877) and proposed structures from the fragmentation pattern. The mass-to-charge (m/z) values of the fragments are presented in parenthesis. | ||
| NE | HE-120-5 | HE-140-5 | HE-160-5 | HE-160-7 | HE-160-9 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| H | L | H | L | H | L | H | L | H | L | H | L | |
| a Total yields (as % of initial DW of Dwb used for each extraction condition) determined gravimetrically after membrane filtration and freeze-drying. b Carbohydrate content determined by the phenol-sulfuric acid method. c Polysaccharide composition (as % of the total carbohydrate content) determined from the glycosidic linkage analysis (Tables S3 and S4). d Average backbone substitution pattern of AX. The values are normalized by the unsubstituted Xylp (X) units (see Fig. 1b for nomenclature). e Protein content determined using Dumas method. f Ferulic acid content determined by GC-MS. g Average molar masses determined by SEC-DRI. Note: The standard deviation is shown in parentheses. | ||||||||||||
| Yieldsa (% DW) | 79.1 | 9.2 | 68.8 | 28.8 | 78.3 | 19 | 58.6 | 40.6 | 65.9 | 32.9 | 76.8 | 21.8 |
| Carbohydrate contentb (mg per g DW) | 871.9 (3.4) | 750.0 (4.5) | 839.9 (24.9) | 616.3 (1.2) | 715.8 (7.6) | 743.3 (9.9) | 815.4 (1.8) | 564.2 (27.5) | 986.8 (5.2) | 598.1 (7.1) | 863.1 (0.4) | 660.6 (26.4) |
| AXc (%) | 92.0 | 54.6 | 77.5 | 50.4 | 77.0 | 73.6 | 76.4 | 70.5 | 85.6 | 80.4 | 85.9 | 75.2 |
| Xd | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| X2Ad | 14 | 23 | 9 | 8 | 10 | 11 | 11 | 11 | 15 | 12 | 10 | 14 |
| X3Ad | 30 | 45 | 19 | 14 | 16 | 25 | 22 | 24 | 24 | 19 | 20 | 30 |
| X2,3Ad | 45 | 40 | 32 | 28 | 27 | 41 | 25 | 34 | 27 | 26 | 23 | 37 |
| β-Glucanc (%) | 1.6 | 10.7 | 14.1 | 28.1 | 19 | 22.9 | 18.6 | 21.9 | 12.0 | 18.1 | 10.9 | 18.1 |
| (Arabino)galactanc (%) | 5.4 | 5.5 | 8.3 | 1.3 | 3.4 | 1.8 | 4.8 | 4.5 | 2.4 | 0.0 | 2.1 | 3.0 |
| Protein contente (mg per g DW) | 104.1 (2.2) | 51.6 (1.6) | 109.7 (32.5) | 155.1(24.9) | 88.9 (6.2) | 71.1 (10.1) | 78.0 (5.3) | 51.7 (2.4) | 81.0 (1.9) | 46.3 (14.8) | 71.2 (1.5) | 49.2 (3.4) |
| Ferulic acid contentf (mg per g DW) | 0.2 (0.04) | 0.2 (0.04) | 3.2 (0.14) | 2.2 (0.14) | 3.5 (0.13) | 1.0 (0.10) | 3.7 (0.14) | 1.8 (0.13) | 4.9 (0.13) | 2.2 (0.12) | 0.3 (0.1) | 0.2 (0.10) |
|
M
ng (g mol−1) |
135640 |
n.d. | 69622 |
1494 | 67330 |
1048 | 82419 |
1763 | 135040 |
1048 | 106100 |
1920 |
|
M
wg (g mol−1) |
458340 |
n.d. | 182170 |
1060 | 138410 |
833 | 133920 |
1314 | 193980 |
839 | 187840 |
1551 |
In general, the total yields for the high molar mass fractions were higher than for the oligomeric fractions, with ranges between 60–80% and 10–40%, respectively. The polysaccharide fractions were composed mainly of AX, with contents ranging between 70–90%. The alkaline (NE-H) fraction presented the highest AX content, whereas the AX content in the fractions extracted by subcritical water (HE-H) increased progressively with the extraction temperature, as reported earlier. The minor polysaccharide components were assigned to mixed-linkage β-D-glucan (12–20%) and arabinogalactan (AG) (5–8%), based on the results from glycosidic linkage analysis (ESI Table S3†). The presence of β-D-glucans and AG was higher for the fractions extracted at lower temperatures, which indicates that these populations are easier to extract than AX.
The A/X ratio and the substitution pattern determine the solubility, macromolecular conformation, physico-chemical and functional properties of these macromolecules.5,6 The A/X ratio for the NE-H AX fraction was 0.74, and on average 0.49 for the high molar mass fractions extracted by subcritical water. Again showing that the alkaline extracted AX is more substituted than the hydrothermal counterpart (ESI Table S3†). The average substitution pattern of the AX fractions was analyzed by glycosidic linkage analysis. Four basic building blocks can be seen in the backbone structure of AXs, which are responsible for determining the AX substitution pattern (Fig. 1b). The D-Xylp residues along the backbone can be unsubstituted (X), mono-substituted in the C2 (X2A), mono-substituted in the C3 (X3A), or di-substituted in both, C2 and C3 (X2,3A). The ratio of substituted D-Xylp units in the backbone was normalized towards the unsubstituted Xylp residues (X) and presented in Table 2. Di-substitutions (X2,3A) and unsubstituted D-Xylp residues (X) are more abundant than mono-substituted Xylp residues, whereas O-3 substitutions (X3A) are preferred over O-2 substitutions (X2A).5,6,48 Interesting differences can be observed in the substitution pattern of the polymeric extracts from subcritical water at different pH and temperature conditions. The presence of di-substituted X2,3A units progressively decreases and the relative presence of mono-substituted X3A and X2A units increases with higher extraction temperatures. Moreover, mild acidic (pH 5.0) and alkaline (pH 9.0) conditions result in AX fractions with lower substitution, compared to neutral (pH 7.0) conditions. These results suggest that it may be possible to modulate the substitution pattern of the AXs by tuning the pH and temperature conditions during SWE. Further pH and temperature conditions should be tested to verify this hypothesis.
The high molecular weight fractions showed a two-fold increase in the FA content compared to the native wheat bran. This indicates that the presence of feruloylated arabinoxylan (F-AX) populations was enriched during the extraction and fractionation processes. To confirm that the ferulic moieties are indeed covalently linked to the AX polysaccharide structures, the hydrothermal extract (HE-160-7-H) was enzymatically hydrolyzed with an endo-1,4-β-xylanase (X) alone and in combination with a feruloyl esterase (FE) and the resulting oligosaccharides were profiled by ESI-MS (Fig. 4d). The presence of FA in the oligosaccharide fragments was confirmed by the difference of m/z 176 between the non-feruloylated and the feruloylated sodium adducts ([M + Na]+). The ions assigned to the feruloylated oligosaccharides at m/z 745 and m/z 877 (corresponding to P4F and P5F, respectively) drastically decrease when treated with the FE, demonstrating the covalent linkage between the carbohydrate and phenolic moieties. The feruloylated motifs present in polymeric F-AX were further sequenced by tandem ESI-MS/MS. The fragments were assigned as proposed by Domon and Costello.49 The structures can be proposed as β-D-Xylp-(1→4)-[(5-O-trans-feruloyl)-α-L-Araf-(1→2)-β-D-Xylp-(1→4)-D-Xylp (XFAXX) and α-L-Araf-(1→3)-β-D-Xylp-(1→4)-[(5-O-trans-feruloyl)-α-L-Araf]-(1→2)-β-D-Xylp-(1→4)-D-Xylp (AXFAXX) (Fig. 4e). The presence of FA moieties in the polysaccharide structure is crucial for the scavenging activity of the polymeric fractions and for their further applicability, as discussed later.
The molar mass distributions and average molecular weights of the polymeric fractions were investigated by SEC analyses (Fig. 4b and Table 2). The polysaccharides in the alkaline extract exhibit higher molar mass (4.5 × 105 kg mol−1) than those in the subcritical water extracts (Mw between 1.3–1.8 × 105 g mol−1). These differences arise from the different extractability of the macromolecular populations under both processes. The harsh alkaline conditions disrupt the ferulic crosslinks in wheat bran, releasing AX populations with higher molar masses but without the covalently attached phenolic functionalities. Conversely, subcritical water is capable of extracting AX fractions from the most accessible parts of wheat bran with maintained feruloylated functionalities, but showing lower molar mass.
Another important issue is the potential degradation induced by subcritical water extraction at high temperatures. In a previous study on the extraction of hemicelluloses from wood (spruce), the combination of higher temperatures and acidic pH during subcritical water caused autohydrolysis and thus a reduction of the molar mass of the extracted fractions.21 In this study, a buffered aqueous solution was used to avoid such pH variations and to minimize degradation. The combination of an increased temperature (from 140 °C and 160 °C) and mild acidic conditions (pH 5.0) during SWE causes a slight reduction in the molecular weight of the polymeric fractions, which can be attributed to acid-catalyzed hydrolytic processes. Additionally, the acidic conditions during SWE may induce, as well, degradation of the Araf substitutions, which will affect the A/X ratio and the substitution pattern of the AX extracts. The fractions buffered at neutral (pH 7.0) and mild alkaline (pH 9.0) conditions at 160 °C exhibited the highest molar masses, comparable to the fractions extracted at 120 °C (Table 2). This demonstrates again the possibility of using pH and temperature to modulate the composition and substitution pattern of the AX fractions as well as the molecular weight.
The eluted fractions after fractionation are mainly composed of oligosaccharides with average molecular weights between 1000–2000 g mol−1 (Table 2). Arabinoxylan oligosaccharides (AXOs) and β-D-glucan oligosaccharides (BGOs) represent the main oligosaccharide populations, as evidenced by glycosidic linkage analyses (ESI Table S4†). In general the content of BGOs in these low molar mass fractions is enriched compared to the polymeric fractions. The AXOs are generally more substituted than their polymeric counterparts, with an A/X ratio of 0.94 and 0.45–0.65 for the alkaline and SWE fractions, respectively. The oligomeric mass profiling of these low molar mass fractions by ESI-MS showed the presence of BGOs and AXOs predominantly with 4–8 sugar units, although larger oligosaccharides can be detected as well (ESI Fig. S1†). This recovered oligosaccharide fractions could be used as prebiotic agents or platform chemicals, as discussed below.
Valorization of the residue by sequential enzymatic treatment and subcritical water extraction
The residues after SWE still exhibited approximately 50% of non-extractable AX with high ferulic acid content (Fig. 2B and C; ESI Table S2†). This crosslinked AX constitutes a valuable resource for further recovery using enzymatic treatments. The residue after extraction at 160 °C pH 7.0 (HR-160-7) was selected to develop the process methodology. In a first attempt, auxiliary enzymes, lichenase and cellulase, that degrade mixed-linked β-D-glucan and cellulose, respectively, were tested in order to release AX populations that could be in close contact with the β-D-glucans trapped in the hydrothermal residue. Although the activity of the auxiliary enzymes was verified by reducing sugar assay (ESI Fig. S2a†), the enzymatic treatments did not result in increased total yields after subsequent SWE (ESI Fig. S2b†). This could be due to the fact that the residual AX is not tightly associated with D-glucans (mixed-linkage β-D-glucan or cellulose), or due to the reduced enzymatic accessibility as a consequence of the presence of FA dehydrodimer crosslinks.50In a further attempt, a feruloyl esterase (FE, from family 1A) and an endo-1,4-β-D-xylanase (X, from family GH10) were used alone and in synergy to disrupt the phenolic crosslinked structures of the residue. The FE alone does not show significant activity towards the residue, but its activity increased after 16 h of incubation when combined with the xylanase (Fig. 5a). Additionally, the combination of both enzymes results in a significant increase of total solid yields after a subsequent cycle of subcritical water extraction (Fig. 5b). The polysaccharide composition and the phenolic acid content of the fractions after enzymatic treatments followed by SWE were analyzed (Fig. 5c and ESI Table S6†). The extracts after xylanase treatment (X), alone and in combination with feruloyl esterase (X + FE), present a higher A/X ratio (0.9–1.2) in comparison to HE-160-7, indicating the release of highly branched F-AX populations. Ferulic acid could be determined only in the samples that were treated with FE, alone or in combination with X (Fig. 5d).
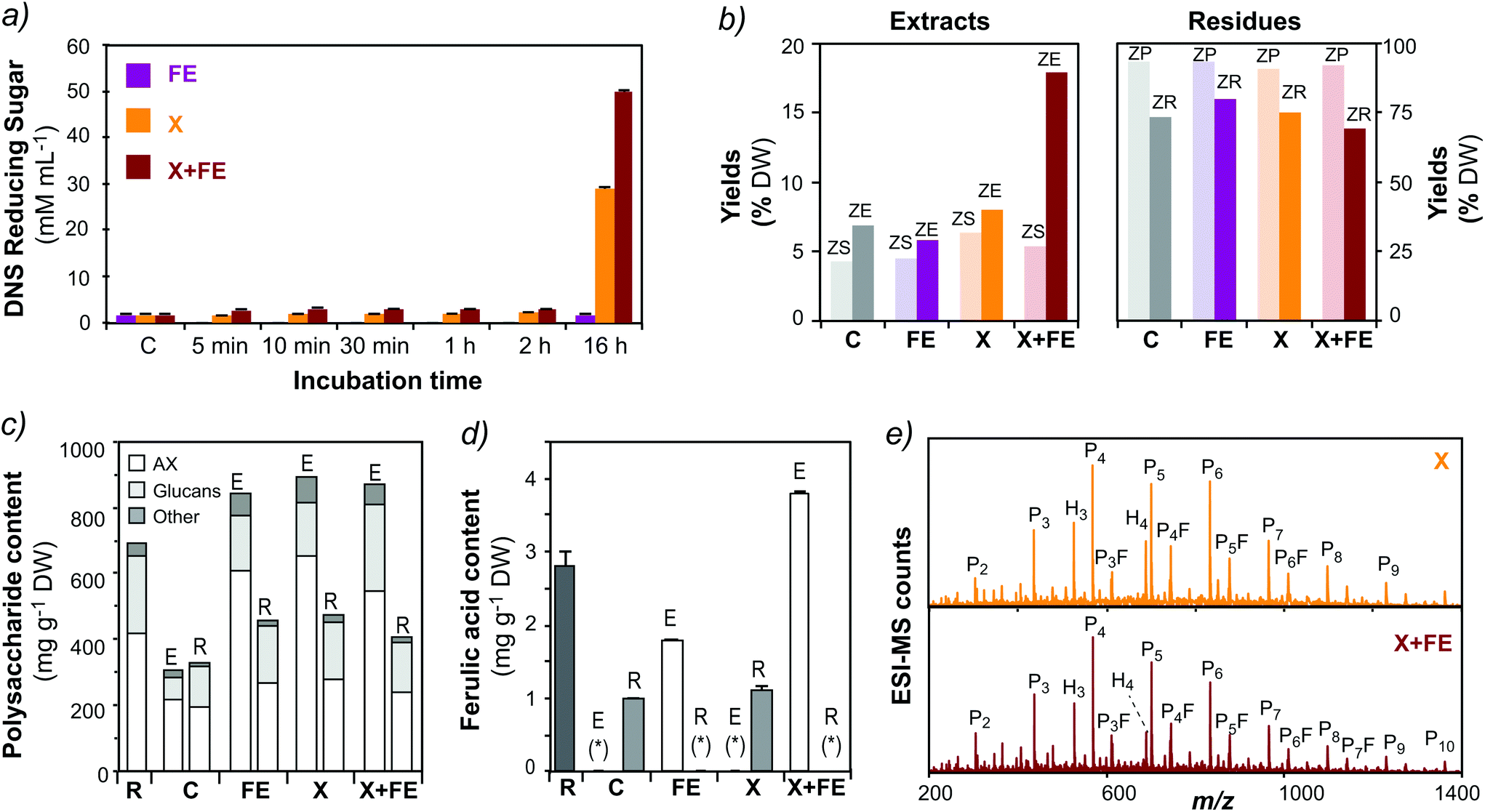 | ||
| Fig. 5 Valorization of the crosslinked residue by enzymatic treatments and subsequent subcritical water extraction. (a) Activity of the enzymatic treatments evaluated by reducing sugar assay: C: control, FE: feruloyl esterase; X: β-xylanase; (b) total yields of the extracts and the residue after enzymatic treatment (ZS: supernatant; ZP: precipitate) and subsequent subcritical water extraction (ZE: extract; ZR: residue). (c) Polysaccharide content and (d) ferulic acid content in the extracts (E) and residues (R). Note: (*) not detected. (e) Mass profiling of the oligosaccharides released after enzymatic treatments by ESI-MS. Note: H: hexoses; P: pentoses; F: ferulic acid. Peak assignment as reported in ESI Table S5.† | ||
The polysaccharide composition and the phenolic acid content of the fractions after enzymatic treatments followed by SWE were analyzed (Fig. 5c and ESI Table S6†). The extracts after xylanase treatment (X), alone and in combination with feruloyl esterase (X + FE), present a higher A/X ratio (0.9–1.2) in comparison to HE-160-7, indicating the release of highly branched F-AX populations. Ferulic acid could be determined only in the samples that were treated with FE, alone or in combination with X (Fig. 5d). The oligosaccharide fractions released after the combined enzymatic treatment and SWE were characterized by mass spectrometric techniques. The oligomeric mass profiling of the extracts treated with X and X + FE shows a range of AXOs from 2 to 12 pentose units, together with a profile of F-AXOs containing 4–7 pentose (P) units plus ferulic acid (F) (Fig. 5e). The structural diversity of the released oligosaccharides will be the subject of further investigations. This is a proof of concept of the effectiveness of the enzymatic treatment using xylanolytic enzymes to disrupt the crosslinked feruloylated network and to valorize the non-extractable residue into valuable functional biomolecules. This procedure opens the possibility to test a wide variety of FE from different families, alone or in combination with other xylanolytic enzymes (β-xylanases, α-L-arabinofuranosidases) to release AX populations from the crosslinked residues. Indeed, FEs constitutes a heterogeneous family of carbohydrate esterases (CEs) based on protein homology, their specificity towards phenolic substrates, and the ability to release phenolic dimers.51–53 Few studies have focused on the specificity and substrate recognition of such FE, alone or in combination with other xylanolytic enzymes, towards complex crosslinked substrates as the ones we encounter in wheat bran.54 Different enzyme families may show distinct activity and selectivity towards the ferulic crosslinked network, which may allow further optimization of the yields of released biomolecules and tuning of their molecular structures.
Antioxidant activities of the AX fractions
The radical scavenging activity of the alkaline and the SWE fractions was assessed against 2,2-diphenyl-1-picrylhydrazyl (DPPH˙) and 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulphonic acid) (ABTS˙+) radicals. Both assays are based on electron transfer, involving the reduction of a colored oxidant. Ascorbic acid and ferulic acid were used as positive controls, whereas wheat endosperm AX (WE-AX; Megazyme) was used as a negative control.55 The radical scavenging activity of selected samples is presented in Fig. 6, whereas the results on all the samples together with the half maximal effective concentration (EC50) values are presented in the ESI Table S7.†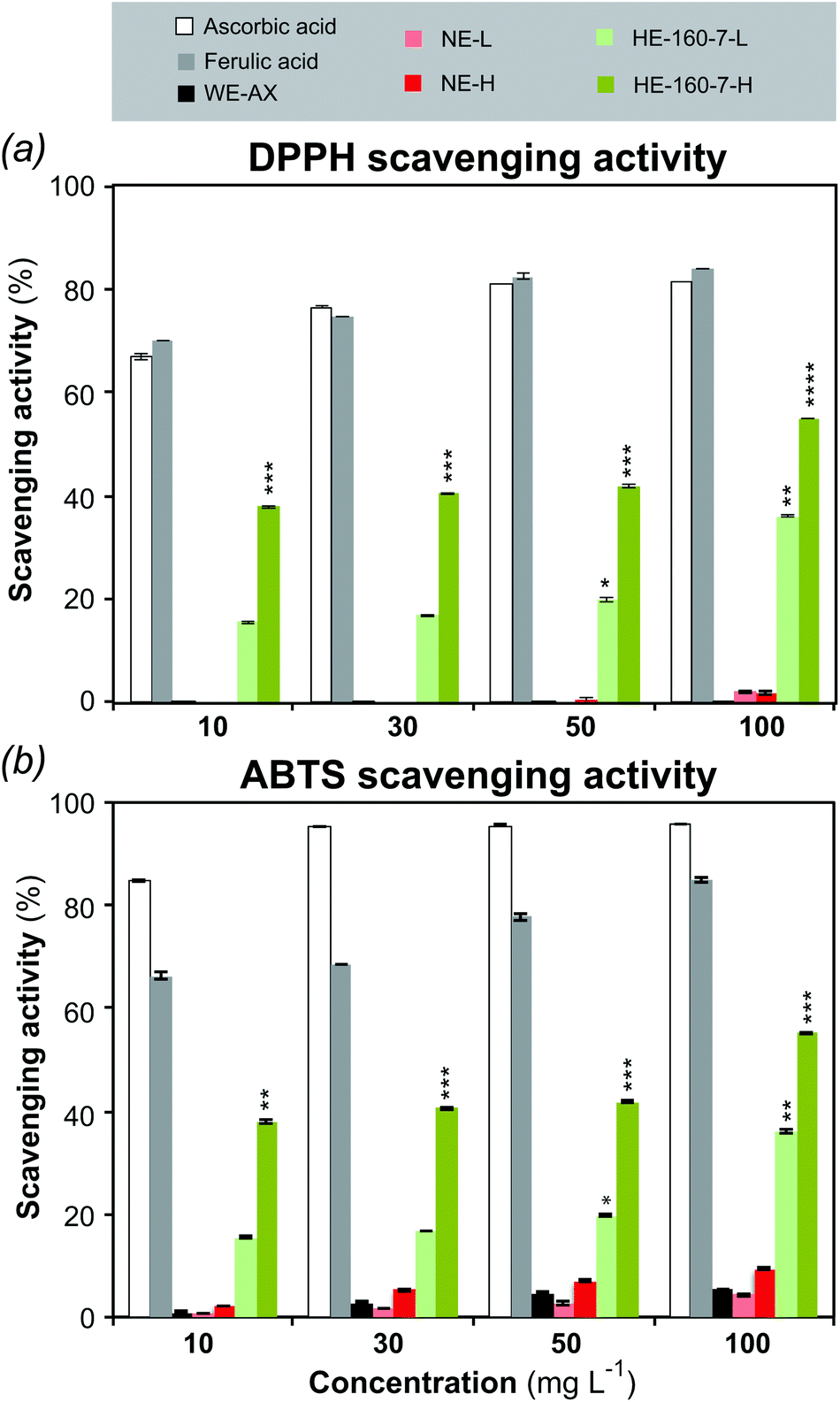 | ||
| Fig. 6 Radical scavenging activity of the alkaline and SWE high and low molar mass fractions from wheat bran. (a) DPPH (b) ABTS. Ascorbic acid and ferulic acid were used as positive control; wheat endosperm arabinoxylan (WE-AX) was used as negative control. Values were expressed as mean ± SD of triplicate measurements. Asterisks indicate a significant difference (*P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.005) relative to control. | ||
The alkaline extract did not show statistically significant scavenging activity compared with the controls. Contrarily, both the low molar mass (HE-160-7-L) and the high molar mass (HE-160-7-H) fractions from SWE showed significant and increasing scavenging activity with higher doses, with this effect being more marked in the high molar mass fraction. HE-160-7-H shows the highest scavenging activity against DPPH radicals (55.1 ± 0.06%) with an EC50 value of 53.6 ± 1.34 μg ml−1 for the highest concentration (100 μg ml−1). At the same concentration, the EC50 value for the alkaline fraction NE-H is 294.9 ± 1.34 μg ml−1, which represents a 6-fold lower potency. For the ABTS˙+ scavenging assay, the whole range of concentrations was tested only for the positive and negative controls (ascorbic acid, ferulic acid and WE-AX) and for the polymeric fractions (H) of the NE and HE-160-7, whereas only the highest concentration (100 μg ml−1) was tested for all the different fractions (ESI Table S7†). A good quantitative correlation is obtained between the antioxidant capacities measured by the ABTS and DPPH assays for the positive controls. The qualitative trends between both methods are maintained for the alkaline and SWE extracts, with fraction HE-160-7-H showing the highest ABTS scavenging activity (Fig. 6b).
As the commercial wheat endosperm AX and the alkaline fractions did not show significant scavenging activity in both assays, the ferulic acid present in the SWEs can be considered as the main contributor to the antioxidant properties.46,56–59 Phenolic compounds are reported to have strong antioxidant activity that can be associated to their ability to destroy oxygen-derived free radicals, break radical chain reaction and/or chelate metals.60–62 The DPPH free radical scavenging of FA probably occurs due to its hydrogen-donating ability.56,61 The DPPH or any reactive radical colliding with FA phenolic nucleus can easily abstract a hydrogen atom to form a resonance-stabilized phenoxy radical, accounting for its antioxidant activity.59 Many publications have reported the antioxidant activity of free FA or conducted studies to evaluate the free radical scavenging capacity of wheat, wheat bran or other cereal extractives and oligosaccharides.47,61,63–66 However, just a handful number of studies have demonstrated the scavenging activity of isolated feruloylated AXs in polymeric form. Hydroxycinnamic acid bound AXs extracted using mild alkali conditions from different sources of millet bran,67 maize fiber59 and sorghum bran60 showed antioxidant activity. These extractions exhibit a lower total phenolic content (between 1–2.4 mg g−1) resulting in a lower potency of the scavenging activity when compared to the high molar mass subcritical water extracts obtained in the present study. The high efficiency of the polymeric AX fractions with bound ferulic acid as radical scavenging agents aligns with a recent re-evaluation of the antioxidant activity of complex carbohydrates, which indicate the important role of minor phenolic components in the antioxidant properties.55 However, the nature of the correlation between ferulic acid content and antioxidant properties is still not fully understood. Further studies of the antioxidant activity of F-AXs with a range of ferulic acid content should cast more light on this issue.
Final mass balances and potential of the hemicellulose and oligosaccharide fractions from wheat bran
The proposed cascade bioprocess generates enriched hemicellulose polymeric populations (mainly F-AX) and oligosaccharides from wheat bran. Table 3 presents the total mass balance of the designed process in comparison with the alkaline extraction, in terms of total solid, total AX and ferulic acid yields. The proposed bioprocess offers higher total yields and similar AX content compared to traditional alkaline extraction. As an advantage, it only uses mild aqueous solvents combined with biocatalysis (carbohydrate-active enzymes) preserving the phenolic substitutions (ferulic acid) attached to the polymeric and oligomeric hemicellulose fractions. The phenolic functionalities confer valuable antioxidant and radical scavenging properties to these fractions, which may be extremely valuable for food applications and in the design of functional materials and platform chemicals.| Fraction | Total yields (%) | AX yields (%) | FA yields (%) | |||
|---|---|---|---|---|---|---|
| NE | HE-160-7 | NE | HE-160-7 | NE | HE-160-7 | |
| NE: alkaline Na(OH) extraction; HE-160-7: optimized SWE at 160 °C and pH 7; H: high molar mass fraction; L: low molar mass fraction; EZ + SWE: fraction after enzymatic treatment (FE + X) and subsequent SWE at 160 °C pH 7. | ||||||
| H | 24.8 | 14.7 | 68.6 | 42.9 | 2.3 | 32.7 |
| L | 2.9 | 7.3 | 4.1 | 12.2 | 0.3 | 7.3 |
| ZS + ZE | n.d. | 9.2 | n.d. | 17.3 | n.d. | 15.8 |
| Total | 27.6 | 31.2 | 72.6 | 72.3 | 2.5 | 55.9 |
Arabinoxylans and their derived AXOs are important from a nutritional point of view, since they both have been associated with a great variety of health benefits as dietary fibers, including prebiotic and immunomodulatory activities.58 The mechanisms of dietary fiber polysaccharides, and of arabinoxylans in particular, in modulating the gut microbiome are starting to be understood.68,69 However, further research is required to better correlate the fine structure–function relationships that regulate the health-promoting mechanisms of dietary fibers.70 From a technological point of view, AXs have shown great potential as biomaterials due to their biocompatibility, biodegradability, and barrier properties to oxygen.71,72 The extracted polymeric F-AX fractions show excellent potential for the design of bio-based materials, combining the good rheological and viscoelastic properties of the polysaccharide core with the inherent antioxidant properties of the phenolic moieties. Moreover, the FA could offer further interesting functionalities, such as UV absorbance73 and the possibility for enzymatic crosslinking.74,75 This opens new routes for the development of multifunctional films, hydrogels, and texturizing agents to be used in food packaging, drug delivery, and other biomedical applications. These inherent radical scavenging materials may contribute to an extended shelf life of unstable foods and drugs (preventing oxidation of less stable components) and to mitigate the presence of reactive oxygen species (ROS) in inflammatory processes.76
Conclusions
We have designed a cascade bioprocess using subcritical water extraction, membrane filtration, and enzymatic treatments for the isolation of functional biomolecules from cereal byproducts. With this approach, we do not only fractionate hemicelluloses with high molecular weight and intact phenolic functionalities, but we also produce valuable oligosaccharides from the non-extractable residues. The use of both subcritical water and xylanolytic enzymes offers evident environmental benefits as green solvents and biocatalysts avoiding the use of hazardous substances. The purity, substitution pattern, and molecular weight of the polymeric extracts can be modulated by controlling the temperature and pH conditions during subcritical water extraction. The SW extracts show significant radical scavenging activity compared to the alkaline extracts, which can be attributed to the presence of ferulic acid attached to the carbohydrate backbones. Moreover, xylanolytic enzymes release functional oligosaccharides from the unextractable residue, increasing the total AX and phenolic yields. This study opens up the possibility of exploring the use of different families of xylan-degrading enzymes to disrupt the recalcitrant crosslinked network, in order to maximize and tune the yields, molecular structure and properties of the released oligosaccharides. These polysaccharide and oligosaccharide fractions show great potential for their utilization in advanced nutritional (prebiotic) and material applications in the food and biomedical sectors, combining the macroscopic properties of the carbohydrates with the antioxidant activity of the ferulic moieties. This sequential procedure can be extended to basically any cereal side stream and opens the possibility to an improved utilization of plant biomass.Experimental
Materials
Fine granulometry defatted wheat bran (wb) was provided by Lantmännen (Stockholm, Sweden). All chemicals, analytical standards and reagents were from Sigma-Aldrich (Stockholm Sweden). Folin Ciocalteau Phenol reagent was from Merck (Solna, Sweden). Spectra/Por 1 and 3 Dialysis Membrane, 6–8 kDa and 3.5 kDa MWCO, respectively and Biotech CE dialysis tubing, 0.1–0.5 kDa and 20 kDa MWCO were purchased from SpectrumLabs (Breda, The Netherlands). Centramate cassete 30 kDa MWCO was purchased from VWR. Feruloyl esterase 1A (Ruminococcus albus) was purchased from NZytech (Lisbon, Portugal). endo-1,4-β-Xylanase (Cellvibrio mixtus), cellulase (Talaromyces emersonii) and lichenase (Bacilus subtilis), and wheat endosperm high viscosity arabinoxylan were purchased from Megazyme (Wicklow, Ireland).Bioprocess design
:10 (w/v) and boiled for 5 min to gelatinize the starch granules. The samples were equilibrated at 40 °C and incubated with α-amylase (2 U mg−1 of carbohydrate) for 1 h at 40 °C; a second incubation was performed for further 30 min. The polysaccharides were precipitated by addition of four volumes of cold absolute ethanol at −8 °C overnight and centrifuged (1500g, 5 min). The precipitate was washed with cold absolute ethanol (×3) and water, centrifuging between washes, prior to freeze-drying. Starch removal was verified under the microscope by iodine staining. The total starch content was determined gravimetrically and enzymatically before and after the α-amylase treatment.
:8 (w/v). The recovered supernatants (NE and CE) and the residues (NR and CR) were neutralized with acetic acid, dialyzed for 48 h using a 3.5 kDa MWCO dialysis membrane and freeze-dried.
Characterization of the extracted fractions
:1 v/v, 100 μL) at 100 °C for 30 min.78 The alditol acetates were extracted with ethyl acetate and analyzed by GC-MS using a HP-6890 gas chromatographer and an HP-5973 electron-impact mass spectrometer (Agilent Technologies, Santa Clara, CA).
:1:4 v/v), 100 °C, 60 min) into amino acid methyl esters (aaMAs).79
Klason lignin was determined gravimetrically from the residues after two-step total sulfuric acid hydrolysis (72 wt% H2SO4, 3 h, R.T.; 1 M H2SO4, 100 °C, 3 h).80
:1 v/v, 200 μL) at 100 °C for 30 min. The per-O-methylated alditol acetates were extracted with ethyl acetate and quantified in triplicate by GC-MS, by comparison with typical polysaccharide standards.
000 analytical columns (PSS, Mainz, Germany). Calibration of the SEC separation was performed using pullulan standards provided by Polymer Standards Services (PSS, Mainz, Germany).
Acknowledgements
This work was funded by the Lantmännen Research Foundation (project 2014F1-0001) and the Swedish Research Council Formas (project 942-2016-119).References
- O. O. Onipe, A. I. O. Jideani and D. Beswa, Int. J. Food Sci. Technol., 2015, 50, 2509–2518 CrossRef CAS.
- L. Stevenson, F. Phillips, K. O'Sullivan and J. Walton, Int. J. Food Sci. Nutr., 2012, 63, 1001–1013 CrossRef CAS PubMed.
- S. Apprich, Ö. Tirpanalan, J. Hell, M. Reisinger, S. Börhmdorfer, S. Siebenhandl-Ehn, S. Novalin and W. Kneifel, LWT – Food Sci. Technol., 2014, 56, 222–231 CrossRef CAS.
- J. Hell, L. Donaldson, H. Michlmayr, M. Kraler, W. Kneifel, A. Potthast, T. Rosenau and S. Böhmdorfer, Carbohydr. Polym., 2015, 121, 18–26 CrossRef CAS PubMed.
- M. S. Izydorczyk and C. G. Biliaderis, Carbohydr. Polym., 1995, 28, 33–48 CrossRef CAS.
- L. Saulnier, F. Guillon, P.-E. Sado and X. Rouau, in Comprehensive Glycoscience, ed. H. Kamerling, Elsevier, France, 2007, pp. 653–689, DOI:10.1016/B978-044451967-2/00147-1.
- A. Ebringerova, Z. Hromadkova and T. Heinze, in Polysaccharides I, ed. T. Heinze, 2005, ch. 136816, vol. 186, pp. 1–67 Search PubMed.
- M. Aguedo, C. Fougnies, M. Dermience and A. Richel, Carbohydr. Polym., 2014, 105, 317–324 CrossRef CAS PubMed.
- I. Egüés, A. M. Stepan, A. Eceiza, G. Toriz, P. Gatenholm and J. Labidi, Carbohydr. Polym., 2014, 102, 12–20 CrossRef PubMed.
- Z. Zhang, C. Smith and W. Li, Food Res. Int., 2014, 65, 423–436 CrossRef CAS.
- A. Höije, M. Gröndahl, K. Tommeraas and P. Gatenholm, Carbohydr. Polym., 2005, 61, 266–275 CrossRef.
- D. J. Rose and G. E. Inglett, Food Chem., 2010, 119, 1613–1618 CrossRef CAS.
- A. Ebringerová, Z. Hromádková, J. Alfödi and V. Hríbalová, Carbohydr. Polym., 1998, 37, 231–239 CrossRef.
- Z. Hromádková, J. Kováciková and A. Ebringerová, Ind. Crops Prod., 1999, 9, 101–109 CrossRef.
- S. G. Allen, D. Schulman, J. Lichwa, M. J. J. Antal, M. Laser and L. R. Lynd, Ind. Eng. Chem. Res., 2001, 40, 2934–2941 CrossRef CAS.
- Ó. Benito-Róman, E. Alonso and M. J. Cocero, presented in part at the III Iberoamerican Conference on Supercritical Fluids, Cartagena de Indias, 2013.
- Ó. Benito-Román, E. Alonso, M. J. Cocero and M. Goto, Food Bioprod. Process., 2016, 98, 21–28 CrossRef.
- A. Mustafa and C. Turner, Anal. Chim. Acta, 2011, 703, 8–18 CrossRef CAS PubMed.
- C. C. Teo, S. N. Tan, J. W. H. Yong, C. S. Hew and E. S. Ong, J. Chromatogr., A, 2010, 1217, 2484–2494 CrossRef CAS PubMed.
- O. Hosseinaei, S. Wang, T. G. Rials, C. Xing and Y. Zhang, Cellulose, 2011, 18, 841–850 CrossRef CAS.
- J. V. Rissanen, H. Grénaman, C. Xu, S. Willför, D. Y. Murzin and T. Salmi, ChemSusChem, 2014, 7, 2947–2953 CrossRef CAS PubMed.
- T. Song, A. Pranovich and B. Holmbom, Holzforschung, 2011, 65, 35–42 CrossRef CAS.
- P. O. Kilpeläinen, S. S. Hautala, O. O. Byman, L. J. Tanner, R. I. Korpinen, M. K. J. Lillandt, A. V. Pranovich, V. H. Kitunen, S. M. Willför and H. S. Ilvesniemi, Green Chem., 2014, 16, 3186–3194 RSC.
- A. Martínez-Abad, A. C. Ruthes and F. Vilaplana, J. Appl. Polym. Sci., 2016, 133, 42523 Search PubMed.
- R. Deutschmann and R. F. H. Dekker, Biotechnol. Adv., 2012, 30, 1627–1640 CrossRef CAS PubMed.
- R. Faryar, J. A. Linares-Pastén, P. Immerzeel, G. Mamo, M. Andersson, H. Stålbrand, B. Mattiasson and E. N. Karlsson, Food Bioprod. Process., 2015, 93, 1–10 CrossRef CAS.
- I. Finore, A. Poli, P. Di Donato, L. Lama, A. Trincone, M. Fagnano, M. Mori, B. Nicolaus and A. Tramice, Green Chem., 2016, 18, 2460–2472 RSC.
- L. S. McKee, H. Sunner, G. E. Anasontzis, G. Toriz, P. Gatenholm, V. Bulone, F. Vilaplana and L. Olsson, Biotechnol. Biofuels, 2016, 9, 1–13 CrossRef PubMed.
- E. Dornez, K. Gerbruers, S. Wiame, J. A. Delcour and C. M. Courtin, J. Agric. Food Chem., 2006, 54, 8521–8529 CrossRef CAS PubMed.
- Y. Hemery, X. Rouau, V. Lullien-Pellerin, C. Barron and J. Abecassis, J. Cereal Sci., 2007, 46, 327–347 CrossRef CAS.
- G. Di Lena, V. Vivanti and G. B. Quaglia, Food, 1997, 41, 285–288 CAS.
- M. Reisinger, Ö. Tirpanalan, M. Prückler, F. Huber, W. Kneifel and S. Novalin, Bioresour. Technol., 2013, 144, 179–185 CrossRef CAS PubMed.
- P. Comino, H. Collins, J. Lahnstein, C. Beahan and M. J. Gidley, Food Hydrocolloids, 2014, 41, 219–226 CrossRef CAS.
- S. Zhou, X. Liu, Y. Guo, Q. Wang, D. Peng and L. Cao, Carbohydr. Polym., 2010, 81, 784–789 CrossRef CAS.
- A. Fadet, Nutr. Res. Rev., 2010, 23, 65–134 CrossRef PubMed.
- C. Barron, A. Surget and X. Rouau, J. Cereal Sci., 2007, 45, 88–96 CrossRef CAS.
- K. Gebruers, E. Dornez, D. Boros, A. Fras, W. Dynkowska, Z. Bedõ, M. Rakszegi, J. A. Delcour and C. M. Courtin, J. Agric. Food Chem., 2008, 56, 9740–9749 CrossRef CAS PubMed.
- M. Dubois, K. A. Gilles, J. K. Hamilton, P. A. Rebers and F. Smith, Anal. Chem., 1956, 28, 350–356 CrossRef CAS.
- J. B. A. Dumas, Ann. Chim. Phys., 1831, 2, 198–213 Search PubMed.
- M. E. F. Schooneveld-Bergmans, A. M. C. P. Hopman, G. Beldman and A. G. J. Voragen, Carbohydr. Polym., 1998, 35, 39–47 CrossRef CAS.
- G. B. Fincher, J. Inst. Brew., 1976, 82, 347–349 CrossRef CAS.
- H. K. Lichtenthaler and J. Schweiger, J. Plant Physiol., 1998, 152, 272–282 CrossRef CAS.
- V. Pussayanawin, D. L. Wetzel and R. G. Fulcher, J. Agric. Food Chem., 1988, 36, 515–520 CrossRef CAS.
- B. Albinsson, S. Li, K. Lundqvist and R. Stomberg, J. Mol. Struct., 1999, 508, 1–3 CrossRef.
- L. Donaldson, IAWA J., 2013, 34, 3–19 CrossRef.
- B. Laddomada, S. Caretto and G. Mita, Molecules, 2015, 20, 15666–15685 CrossRef CAS PubMed.
- X. Yuan, J. Wang and H. Yao, Food Chem., 2005, 90, 759–764 CrossRef CAS.
- Y. Sun, S. W. Cui, X. Gu and J. Zhang, Carbohydr. Polym., 2011, 85, 615–621 CrossRef CAS.
- B. Domon and C. E. Costello, Glycoconjugate J., 1988, 5, 397–409 CrossRef CAS.
- D. M. de Oliveira, A. Finger-Teixeira, T. R. Mota, V. H. Salvador, F. C. Moreira-Vilar, H. B. C. Molinari, R. A. C. Miitchell, R. Mearchiosi, O. Ferrarese-Filho and W. D. dos Santos, Plant Biotechnol. J., 2015, 13, 1224–1232 CrossRef CAS PubMed.
- V. F. Crepin, C. B. Faulds and I. F. Connerton, Appl. Microbiol. Biotechnol., 2004, 63, 647–652 CrossRef CAS PubMed.
- I. Benoit, E. G. J. Danchin, R.-J. Bleichrodt and R. P. de Vries, Biotechnol. Lett., 2008, 30, 387–396 CrossRef CAS PubMed.
- D. B. R. K. G. Udatha, I. Kouskoumvekaki, L. Olsson and G. Panagiotou, Biotechnol. Adv., 2011, 29, 94–110 CrossRef CAS PubMed.
- C. B. Faulds, D. Zanichelli, V. F. Crepin, I. F. Connerton, N. Juge, M. K. Bhat and K. W. Waldron, J. Cereal Sci., 2003, 38, 281–288 CrossRef CAS.
- S. Hu, J. Yin, S. Nie, J. Wang, G. O. Phillips, M. Xie and S. W. Cui, Bioact. Carbohydr. Diet. Fibre, 2016, 7, 19–27 CrossRef CAS.
- H. Kikuzaki, M. Hisamoto, K. Hirose, K. Akiyama and H. Taniguchi, J. Agric. Food Chem., 2002, 50, 2161–2168 CrossRef CAS PubMed.
- K. Masisi, T. Beta and M. H. Moghadasian, Food Chem., 2016, 196, 90–97 CrossRef CAS PubMed.
- M. Mendis and S. Simsek, Food Hydrocolloids, 2014, 42, 239–243 CrossRef CAS.
- M. Srinivasan, A. R. Sudheer and V. P. Menon, J. Clin. Biochem. Nutr., 2007, 40, 92–100 CrossRef CAS PubMed.
- G. G. Duthie, S. J. Duthie and J. A. M. Kyle, Nutr. Res. Rev., 2000, 13, 79–106 CrossRef CAS PubMed.
- J. Wang, X. Yuan, B. Sun, Y. Tian and Y. Cao, Food Technol. Biotechnol., 2009, 47, 39–46 CAS.
- S. Itagaki, T. Kurokawa, C. Nakata, Y. Saito, S. Oikawa, M. Kobayashi, T. Hirano and K. Iseki, Food Chem., 2009, 114, 466–471 CrossRef CAS.
- P. Katapodis, M. Vardakou, E. Kalogeris, D. Kekos, B. J. Macris and P. Christakopoulos, Eur. J. Nutr., 2003, 42, 55–60 CrossRef CAS PubMed.
- A. Mpofu, H. D. Spirstein and T. Beta, J. Agric. Food Chem., 2006, 54, 1265–1270 CrossRef CAS PubMed.
- D. Povilaitis, V. Sulniute, P. R. Venskutonis and V. Kraujaliene, J. Cereal Sci., 2015, 62, 117–123 CrossRef CAS.
- B. R. Veenashri and G. Muralikrishna, Food Chem., 2011, 126, 1475–1481 CrossRef CAS.
- V. Bijalwan, U. Ali, A. K. Kesarwani, K. Yadav and K. Mazunder, Int. J. Biol. Macromol., 2016, 88, 296–305 CrossRef CAS PubMed.
- A. Rogowski, J. A. Briggs, J. C. Mortimer, T. Tryfona, N. Terrapon, E. C. Lowe, A. Basle, C. Morland, A. M. Day, H. Zheng, T. E. Rogers, P. Thompson, A. R. Hawkins, M. P. Yadav, B. Henrissat, E. C. Martens, P. Dupree, H. J. Gilbert and D. N. Bolam, Nat. Commun., 2015, 6, 7481 CrossRef CAS PubMed.
- E. C. Martens, A. G. Kelly, A. S. Tauzin and H. Brumer, J. Mol. Biol., 2014, 426, 3851–3865 CrossRef CAS PubMed.
- B. R. Hamaker and Y. E. Tuncil, J. Mol. Biol., 2014, 426, 3838–3850 CrossRef CAS PubMed.
- G.-G. Chen, X.-M. Qi, Y. Guan, F. Peng, C.-L. Yao and R.-C. Sun, ACS Sustainable Chem. Eng., 2016, 4, 1985–1993 CrossRef CAS.
- M. J. Costa, M. A. Cerqueira, H. A. Ruiz, C. Fougnies, A. Richel, A. A. Vicente, J. A. Teixeira and M. Aguedo, Ind. Crops Prod., 2015, 66, 305–311 CrossRef CAS.
- N. Kumar and V. Pruthi, Biotechnol. Rep., 2014, 4, 86–93 CrossRef.
- G. Oudgenoeg, R. Hilhorst, S. R. Piersma, C. G. Boeriu, H. Gruppen, M. Hessing, A. G. J. Voragen and C. Laane, J. Agric. Food Chem., 2001, 49, 2503–2510 CrossRef CAS PubMed.
- E. Carvajal-Millan, V. Landillon, M.-H. Morel, X. Rouau, J.-L. Doublier and V. Micard, Biomacromolecules, 2005, 6, 309–317 CrossRef CAS PubMed.
- Y.-H. Cheng, S.-H. Yang and F.-H. Lin, Biomaterials, 2011, 32, 6953–6961 CrossRef CAS PubMed.
- J. F. Saeman, W. E. Moore, R. L. Mitchell and M. A. Millett, Tappi J., 1954, 37, 336–343 CAS.
- A. B. Blakeney, P. J. Harris, R. J. Henry and B. A. Stone, Carbohydr. Res., 1983, 113, 291–299 CrossRef CAS.
- G. L. Sassaki, L. M. Souza, R. V. Serrato, T. R. Cipriani, P. A. J. Gorin and M. Iacomini, J. Chromatogr., A, 2008, 1208, 215–222 CrossRef CAS PubMed.
- J. B. Sluiter, R. O. Ruiz, C. J. Scarlata, A. D. Sluiter and D. W. Templeton, J. Agric. Food Chem., 2010, 58, 9043–9053 CrossRef CAS PubMed.
- C. Xu, A. S. Leppänen, P. Eklund, P. Holmlund, R. Sjöholm, K. Sundberg and S. Willför, Carbohydr. Res., 2010, 345, 810–816 CrossRef CAS PubMed.
- F. A. Pettolino, C. Walsh, G. B. Fincher and A. Basic, Nat. Protoc., 2012, 7, 1590–1606 CrossRef CAS PubMed.
- G. L. Miller, Anal. Chem., 1959, 31, 426–428 CrossRef CAS.
- W. Brand-Williams, M. E. Cuvelier and C. Berset, LWT – Food Sci. Technol., 1995, 28, 25–30 CrossRef CAS.
- R. Re, N. Pellegrini, A. Proteggente, A. Pannala, M. Yang and C. Rice-Evans, Free Radical Biol. Med., 1999, 26, 1231–1237 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6gc03473j |
| This journal is © The Royal Society of Chemistry 2017 |