
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFuture perspectives for formaldehyde: pathways for reductive synthesis and energy storage
Leo E.
Heim†
,
Hannelore
Konnerth†
and
Martin H. G.
Prechtl
 *
*
Universität zu Köln, Department of Chemistry, Greinstr. 6, 50939 Köln, Germany. E-mail: martin.prechtl@uni-koeln.de; Web: http://www.h2.bio Web: http://www.catalysis.bio Web: http://www.catalysislab.de
First published on 16th December 2016
Abstract
Formaldehyde has been a key platform reagent in the chemical industry for many decades in a large number of bulk scale industrial processes. Thus, the annual global demand reached 30 megatons per year, and currently it is solely produced under oxidative, energy intensive conditions, using high-temperature approaches for the methanol oxidation. In recent years, new fields of application beyond the use of formaldehyde and its derivatives as i.e. a synthetic reagent or disinfectant have been suggested. For example dialkoxymethane could be envisioned as a direct fuel for combustion engines or aqueous formaldehyde and paraformaldehyde may act as a liquid organic hydrogen carrier molecule (LOHC) for hydrogen generation to be used for hydrogen fuel cells. To turn these new perspectives in feasible approaches, it requires also new less energy-intensive technologies for the synthesis of formaldehyde. This perspective article spreads light on the recent directions towards the low-temperature reductive synthesis of formaldehyde and its derivatives and low-temperature formaldehyde reforming for hydrogen generation. These aspects are important for the future demands on modern societies’ renewable energy management, in the form of a methanol and hydrogen economy, and the required formaldehyde-feedstock for the manufacture of many formaldehyde-based daily products.
 Leo E. Heim | Leo Heim is a PhD student at the Prechtl Lab and has been researching since his Master's thesis on catalytic formaldehyde decomposition. Since 2012 he has served as a contracted student research assistant in the same lab. Previously he worked on his Bachelor's thesis with Axel Jacobi von Wangelin and served as chair of the Student Representatives of the Department of Chemistry. He is a co-inventor for a patent on hydrogen generation, and the first and co-author of six articles published for example in Nat. Commun. and Angew. Chem. |
 Hannelore Konnerth | Hannelore Konnerth is a PhD student as a Manchot-Fellow at the Prechtl Lab and works on catalytic hydrogenation in multiphase-systems. She performed research on biomass conversion in water for her Master's thesis at the NU Singapore in collaboration with Ning Yan. She has been granted with a competitive Contact-Singapore/A-Star-Fellowship for her research at the NUS. Since 2012 she has served as a contracted research assistant in the Prechtl Lab. Previously she worked on her Bachelor's thesis with Axel Jacobi von Wangelin. She is the first and co-author of six articles and published her most recent results in Chem. Commun. (Emerging Investigator Issue 2016). |
 Martin H. G. Prechtl | Martin Prechtl (FRSC), named Humboldtian and Heisenbergian, received the NRW Returnee Award 2009 (1.25 M€) and works at the University of Cologne as Privatdozent. He is the editor for Wiley, RSC, DeGruyter and Elsevier. Besides other awards, he holds the Ernst-Haage-Prize 2014 (MPI-CEC) for his contribution to hydrogen storage (Nat. Commun., 2014, PCT-patent). His group develops molecular and nanoscale (de)hydrogenation catalysts and is known for H2-generation from methanol/formaldehyde at room temperature. Before Cologne he performed research in Wuppertal, Bridgetown, Sao Paulo, Aachen, Muelheim/Ruhr, Porto Alegre and Berlin. |
Introduction
Formaldehyde is widely abundant in nature and the anthropogenic environment owing to multiple natural and artificial decomposition pathways of both biological and artificial organic matter.1–3 Currently, these formaldehyde production pathways still play a minor role, in terms of its valorisation in industry. However, this will change when vegetal biomass processing based on renewable sources will take on full ride for the future substitution of fossil based processes which will consequently lead to a biomass-derived formaldehyde production.2 For many decades, formaldehyde has been used in over 50 industrial processes as a crucial building block (i.e. cross-linker) for daily life commodities such as germicides/disinfectants in hospitals, pharmaceuticals, paints/inks, cosmetics, resins, polymers/adhesives, besides many others.4–6 The commercial importance of all these branches sums up to a globally multi-billion dollar market with the demands of over 30 mega tons per year of formaldehyde,7 respectively paraformaldehyde and the forecast predicts an annual growth of 4%. However, so far formaldehyde as a platform compound in the global energy storage sector does not play a significant role, although it provides interesting properties in comparison to other alternative solutions. For example, oxymethylene ethers (OME), synthesised from formaldehyde and alcohols, are investigated as potential candidates for combustion fuels and fuel additives.8–10 It could be envisioned to extend this approach using long-chain bio-derived alcohols, thus resulting in long-chain OMEs suitable to complement biofuels such as biodiesel (Scheme 1).8 Worth mentioning is also the long established use of solid formaldehyde derivatives as solid fuel tablets for direct combustion commonly used for small camping stoves especially in military equipment since the 1930s. These ashless burning fuel tablets consist of 1,3,5-trioxane, or more commonly of hexamethylenetetramine (also known as urotropine), and the latter ones are commonly sold and known under the commercial name Esbit®. Additionally, formaldehyde can be harvested in the form of OME during the lignin depolymerisation processing using ethanol as a scavenger for formaldehyde, thus OME would be a valuable by-product during the production of aromatics.11 In another context, formaldehyde impurities also play a role in terms of safety and storage issues for aqueous nuclear waste treatment owing to the observation that formaldehyde-containing nuclear waste generates flammable mixtures of H2 and N2O.12 In this regard, novel formaldehyde decontamination technologies would be desirable for the safe pre-treatment of contaminated and potential flammable waste previous to storage or further processing.1,13 In another perspective, formaldehyde itself is a hydrogen-rich molecule containing 6.7 wt% H2 in the gaseous phase, and in aqueous media this value rises to 8.4 wt% owing to the favourable equilibrium towards methanediol.14 Thus, it provides potential to become suitable as a liquid organic hydrogen carrier (LOHC) molecule for the delivery of hydrogen in high-purity for hydrogen fuel cell technologies. In this regard, it might be even more attractive than the hydrogen-richer methanol (12.5 wt%) of the C1-family owing to the different thermodynamic properties. In vast contrast to methanol which needs a larger energy input during the endergonic methanol reforming process, the formaldehyde reforming process is exergonic, thus more feasible for a self-sustaining hydrogen delivery pathway. In combination with a low-temperature formaldehyde synthesis, the formaldehyde reforming would result in a rechargeable hydrogen battery suitable for hydrogen fuel cell technologies (Scheme 1).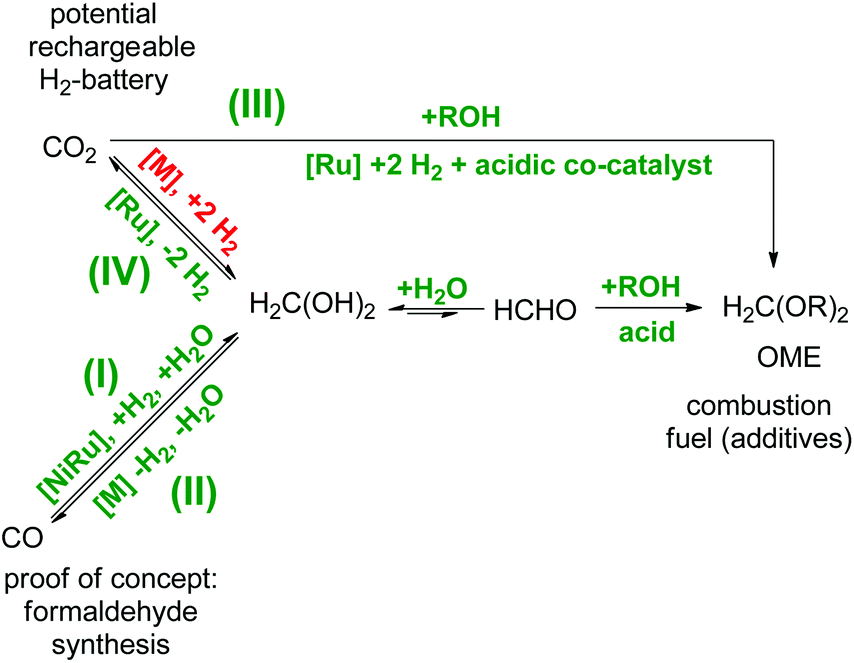 | ||
| Scheme 1 C1-Interconversion network of aqueous formaldehyde in relation to: (I) a reductive low-temperature formaldehyde synthesis,2,5,7 (II) the decomposition of formaldehyde to syngas,4 (III) OME as potential combustion fuel (additives)8,10 and (IV) the application as LOHC for hydrogen fuel cells.1,14,15 The pathways in green are realized and the pathway in red remains the missing puzzle piece in this network. | ||
In this perspective article the recent progress and efforts towards more sustainable formaldehyde synthesis5,7,16 and its derivatives8,17–21via the reduction of CO2 and CO are described, as well as the approaches for low-temperature formaldehyde reforming.1,14,15,22 New formaldehyde production methods are also required to feed the potential future application in the energy sector on a large scale because the currently produced formaldehyde is already reserved for the established synthetic applications. Likewise the current annual global methanol production capacity (∼110 Mt) cannot serve solely the energy sector owing to the requirements for methanol (∼80–90 Mt per year) in the chemical and pharmaceutical industries.
Direct formaldehyde synthesis
Since the early commercialisation of formaldehyde its production is based on an energy-intensive gas phase process via partial oxidation and/or dehydrogenation of methanol (i.e. Formox process).3,14,23 Interestingly already in the late 19th century, at least since the 1870s there are early reports about chemical, photochemical, and especially electrochemical and high-temperature attempts described to reduce CO and CO2 to formaldehyde; the high-temperature decomposition of formaldehyde to syngas (CO/H2) can be found in the literature.24–29 These early attempts have been fallen in oblivion for about 80 years owing to thermodynamic disfavours.30,31 Still nowadays, when methanol production from CO2 has been already realised, and Carbon Recycling International (CRI) installed even a CO2 methanol plant in Iceland (covering about 3% of Iceland's energy demands),32 the Formox process and related methanol oxidation processes remain the technical state of the art for the formaldehyde production. Thus, even so the more sustainable production of methanol can be realised, the production of formaldehyde remains energy (300–400 °C) and cost intensive on a large scale. Currently >35% of the world methanol production is used to cover the global formaldehyde demands, which turns formaldehyde to the number one product directly derived from methanol. The conventional formaldehyde production includes a highly energetic three-step process line to formaldehyde consisting of steam reforming of natural gas (700–1100 °C) to syngas, followed by methanol synthesis (200–300 °C) and finally partial oxidation/dehydrogenation of methanol (300–400 °C) to yield formaldehyde as the major product (Scheme 2).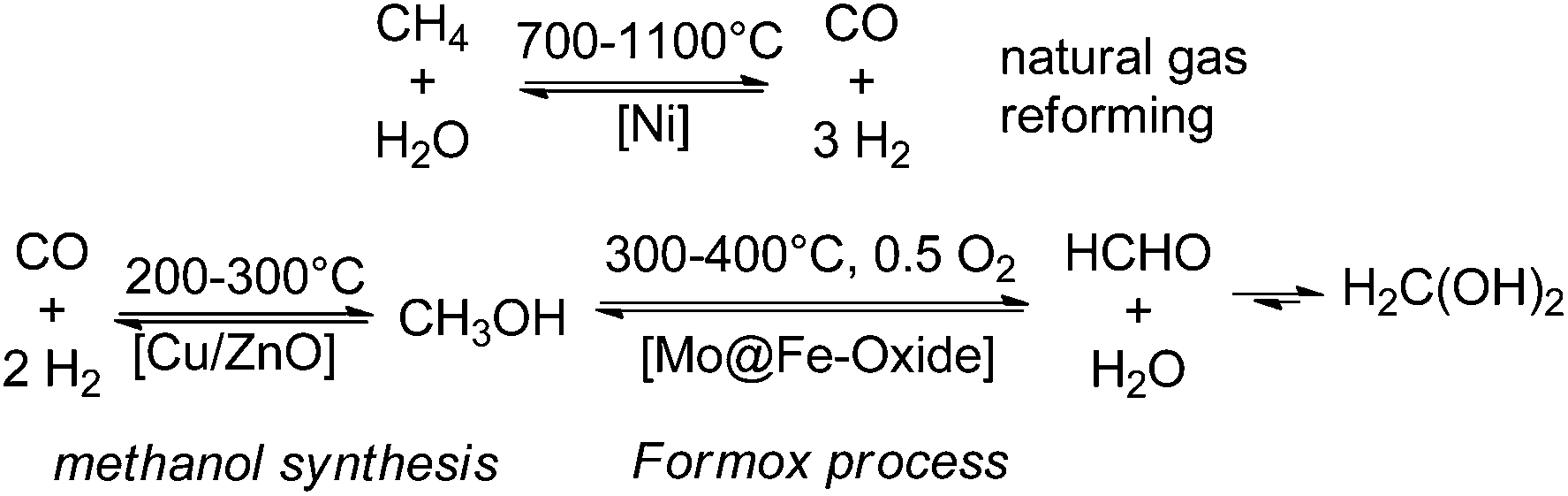 | ||
| Scheme 2 Classical high temperature gas phase process line to formaldehyde: (I) natural gas reforming, (II) methanol synthesis, and (III) partial methanol oxidation. | ||
The mentioned energy-related drawbacks owing to the high temperature required for this process line are accompanied by further energy-intensive compression and purification steps which results in economic and ecological deficiencies.3 Today's modern societies are largely based on C1-molecules and their derivatives in many daily life products.2,4,5,7 To sustain the status quo of modern life, it urgently requires more sustainable processes for the interconversion of those molecules. Owing to the massive global demands of methanol, the efforts focus on the substitution of the traditional oxidative methanol synthesis from natural gas by a reductive process utilizing abundant CO2.33–35 Taking into account studies about the life-cycle assessment for the conversion of CO2 to methanol, this approach is promising in the case that the required hydrogen is delivered from renewables or plain water.36 In vast contrast, conventional hydrogen derived from fossil sources leads to the so-called CO2-paradoxon owing to the fact that the total CO2-production is superior in comparison to the required hydrogen for the CO2 reduction to methanol.37 Interestingly, the research about improved methanol syntheses is much more advanced than the progress for the formaldehyde synthesis, even so that most of the methanol (35%) is simply produced to produce formaldehyde. Over 80 years the valorisation of syngas has quite developed and is widely used for example for hydroformylation,4,38 or synthetic fuels (Fischer–Tropsch synthesis).39,40 However, it was not considered for the formaldehyde synthesis because of thermodynamic disfavours in gas phase reactions and low yields (<0.2%).30,31 In comparison to the classical formaldehyde synthesis, a direct conversion of syngas to formaldehyde would eliminate two energy intensive steps (methanol synthesis and oxidation) and most recently this has been proven as an elegant alternative.5 Surprisingly, the direct catalytic hydrogenation of carbon dioxide to formaldehyde or formalin has rarely been reported. In vast contrast, many reports on CO2 hydrogenation to formic acid and methanol are available, likewise reports where methanol and formic acid are described as a hydrogen source.1,14,15,34,35,37,41–51 However, there are reports of trapping formaldehyde in the form of derivatives such as acetals. An overview about those chemical transformations is given in another section of this article. Moreover, a more general review article by Klankermayer and Leitner is of interest for those who want to gain a general overview about the interconversion of C1-molecules in general.32
In the mid-1980s Gambarotta and co-workers reported a stoichiometric stepwise reduction of carbon dioxide to formaldehyde and methanol using the zirconium metallocene [Cp2Zr(H)(Cl)]n as a reducing agent.52 The authors fully characterised the corresponding complexes formed in the course of the metal hydride transfer to CO2. In the first step an oxo-bridged dimeric complex [Cp2Zr(Cl)]2O is formed, indicating a C–O bond cleavage of CO2. In a second step [Cp2Zr(H)(Cl)] reacts with formaldehyde, yielding [Cp2Zr(OMe)(Cl)] (Scheme 3).
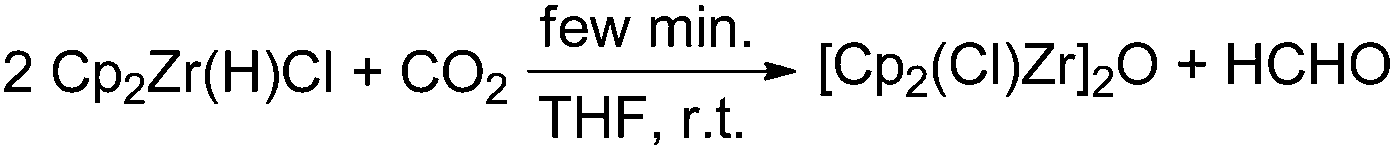 | ||
| Scheme 3 Stoichiometric reaction of CO2 to formaldehyde at room temperature.52 | ||
Corriu and co-workers reported a few years later the insertion of CO2 in the Si–H bonds of a hypervalent organosilane in the presence of a tertiary amine (Scheme 4).53 In the first step a silyl formate is formed which is stable at room temperature. The silyl ester decomposes at 65–85 °C under argon within two hours yielding a trimeric siloxane and formaldehyde. The formaldehyde was trapped and characterised as a hydrazone using 2,4-dinitrophenylhydrazine.
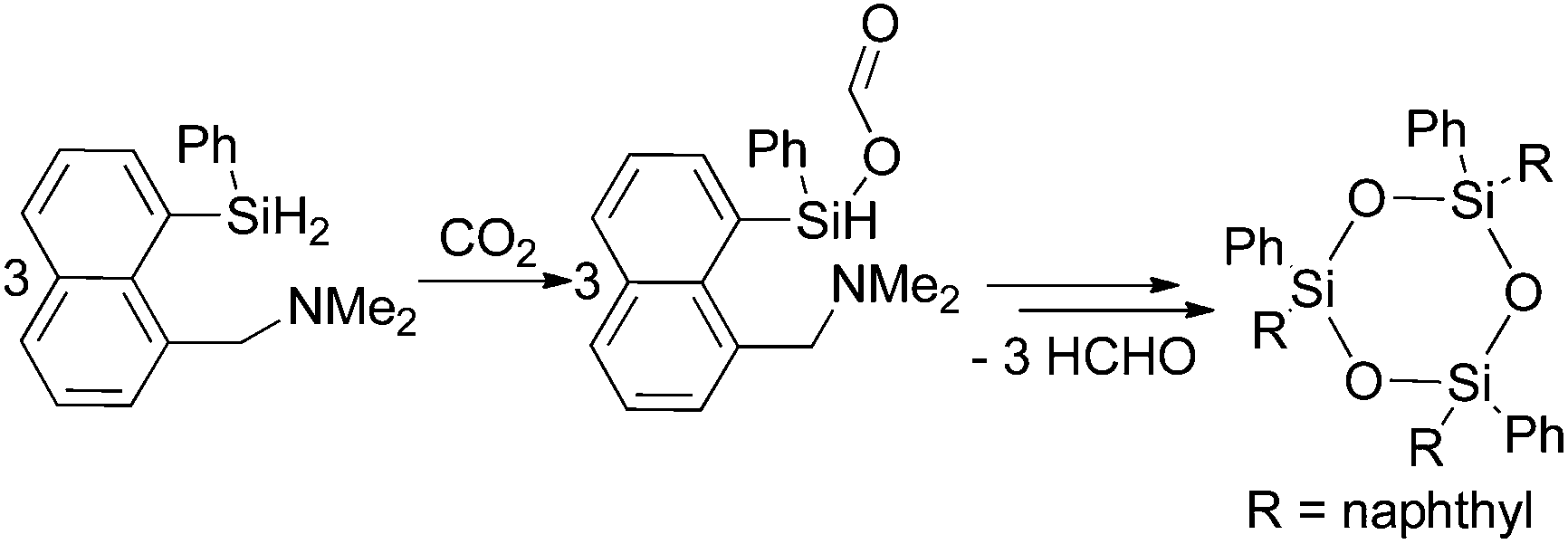 | ||
| Scheme 4 Stoichiometric reaction of CO2 to formaldehyde via decomposition of a silyl formate.53 | ||
In an enzymatic approach Liu and co-workers demonstrated the NADH-coupled reduction of CO2 to formaldehyde using formate dehydrogenase (FDH) and formaldehyde dehydrogenase (FADH) as catalysts (Scheme 5).54 The optimised reactions have been performed at 37 °C in phosphate buffer (pH 6) with 1 mL of 100 mmol L−1 NADH solution under 5 bar of CO2 yielding about 0.901 mmol L−1 HCHO for example after 12 h.
 | ||
| Scheme 5 Sequential reduction of CO2 by means of FDH and FADH enzymes and NADH co-factor.54 | ||
In contrast to the above mentioned molecular approaches, there is a report on the direct formation of free formaldehyde starting from CO2 using solid catalyst materials. Lee and co-workers reported a pronounced selectivity even for the hydrogenation of CO2 to formaldehyde at 150 °C under a total pressure of 6 bar with a H2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CO2 ratio of 20:1. They used a PtCu/SiO2 (Pt/Cu = 0.03) catalyst and obtained 0.87 × 10−4 mol min−1 of formaldehyde per gram catalyst, and 0.20 × 10−4 mol min−1 of methanol per gram catalyst.55 At a H2:CO2 ratio of 3:1 the selectivity inverted to 0.2:0.8 (HCHO:MeOH; Scheme 6).
:CO2 ratio of 20:1. They used a PtCu/SiO2 (Pt/Cu = 0.03) catalyst and obtained 0.87 × 10−4 mol min−1 of formaldehyde per gram catalyst, and 0.20 × 10−4 mol min−1 of methanol per gram catalyst.55 At a H2:CO2 ratio of 3:1 the selectivity inverted to 0.2:0.8 (HCHO:MeOH; Scheme 6).
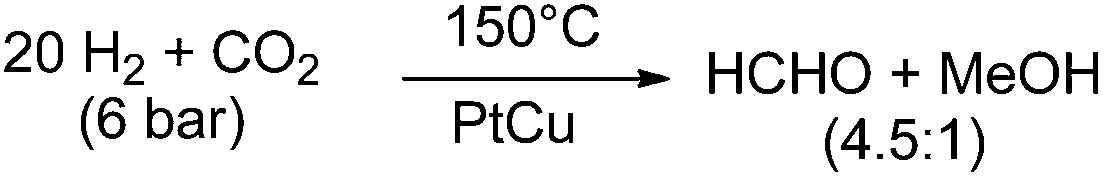 | ||
| Scheme 6 Hydrogenation of CO2 with a PtCu catalyst.55 | ||
In a photocatalytic approach using ruthenium(II)-complex/carbon nitride hybrid photocatalysts (with ∼8 μmol g−1 Ru: [Ru(bipy)2Cl2(CO)2] as a precursor), the authors reported the formation of HCHO (380 nmol) in the course of the conversion of CO2 to CO and formic acid.56 In another photocatalytic method, the authors used a dye-sensitized TiO2 film with bis(tetrabutylammonium)-cis-bis(isothiocyanato)-bis(2,2′′-bipyridyl-4,4′′-dicarboxylato)-ruthenium(II) for the reduction of CO2 to formic acid and methanol.57 At pH 10 they observed for example the formation of 0.0835 mmol cm−2 formic acid, 0.1292 mmol cm−2 formaldehyde and 0.1781 mmol cm−2 methanol after visible light illumination for 5 h (Scheme 7). In other reports about photocatalytic reduction of CO2 typically a mixture of formic acid, formaldehyde, methanol or even methane has been observed on either a low conversion level and/or with low selectivity for formaldehyde.58–63
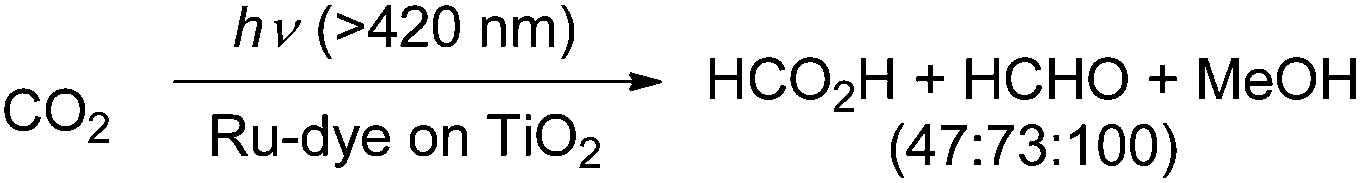 | ||
| Scheme 7 Photocatalytic conversion of CO2 into HCHO.57 | ||
In 2014 a promising electrocatalytic method converting CO2 into formaldehyde even in seawater has been presented by Nakata and Einaga (Scheme 8).16 The authors used boron-doped diamond (BDD) electrodes with p-type surfaces and a platinum counter electrode in the range from −1.0 V to −1.9 V vs. Ag/Ag+ in a methanol electrolyte. They reported a maximum Faradaic efficiency for formaldehyde of 74% at −1.7 V vs. Ag/Ag+. The efficiency is maintained between 1 h and 20 h. Formic acid is formed with just 15% Faradaic efficiency at −1.5 V, and H2 is formed with 1.1% below −1.7 V. The reduction of formic acid, instead of CO2 as the starting material, yielded formaldehyde with 85% at −1.5 V. After optimization of the set up with artificial electrolytes they tested seawater as a natural electrolyte successfully yielding 36% formaldehyde, and 7.5 × 10−3 M per hour. As a reference they tested also 0.1 M aq. NaCl yielding 62% formaldehyde. The lower activity in seawater is attributed to natural impurities. Other electrochemical approaches gave Faradaic efficiencies between 0.08% and 25% for formaldehyde derived from CO2 using polyoxometalate as a catalyst material.64
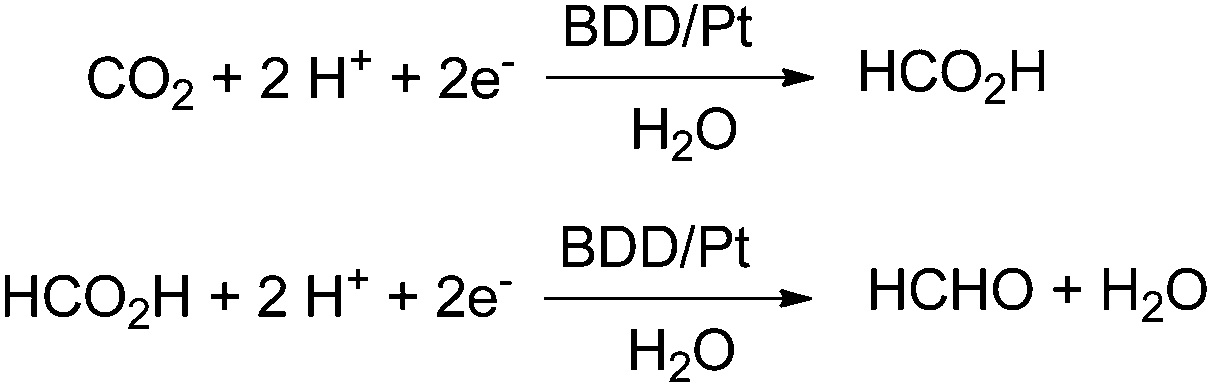 | ||
| Scheme 8 Electrocatalytic reduction with boron doped-diamond electrodes with highest efficiency for formaldehyde at −1.7 V.16 | ||
Again in 2014, the first direct conversion of syngas (CO:H2 = 1:1) to formaldehyde has been realized in aqueous media (Scheme 9, patent filed).5 The driving force to push the equilibrium into the desired direction was the performance of the reaction in the liquid rather than in the gaseous phase. Indeed, this reaction requires a protic environment to stabilise formaldehyde in the form of methanediol or (hemi)acetals and prevent over-reduction to methanol. In contrast to the performance in the aqueous phase, the gas phase hydrogenation of CO to formaldehyde with a solid catalyst is thermodynamically very limited (vide infra).5 In consequence to these observations, considering the reactivity of formaldehyde towards water,14 water has been used as a solvent. Formaldehyde is known to form methanediol as the major product in aqueous solution.14 This low-temperature approach has the potential to save costs and energy owing to the elimination of two high-temperature reaction steps.
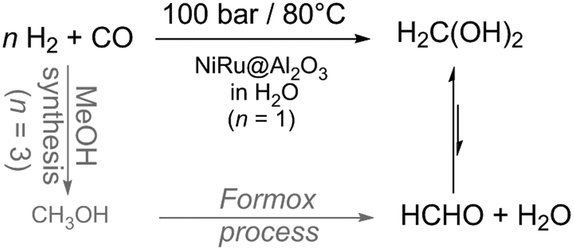 | ||
| Scheme 9 Elimination of two energy and cost intensive synthesis steps towards low temperature formaldehyde synthesis from syngas in aqueous solution.5 | ||
The hydrogenation of CO to methanediol in water was then thermodynamically favourable and highly selective (100%) for methanediol at a conversion level of 19% in a slurry reactor, pointing out that formic acid, methanol or methane was not detectable. The importance of water is underlined by comparison with the gas phase reaction with the same catalyst in a fixed bed reactor resulting in a conversion of ∼10−4%. The used catalyst is bimetallic nanoscale NiRu on Al2O3 applied at 80 °C under 100 bar syngas (CO:H2 = 1:1). The thermodynamic analysis supports experimental observations reflected by the equilibrium constant for the gas phase reaction (8.8 × 10−7 mol−1) which differs a lot from the constant (17.3 mol−1) for the aqueous phase reaction at 298 K. Moreover, the Gibbs free energy (ΔG) is positive for the gas phase reaction and negative for the aqueous phase reaction (Table 1). Most likely, a rigorous mixing of the gases into the liquid phase is beneficial for efficient conversion. Additionally, it has been proven that high temperatures (≫80 °C) are not beneficial for the product yield which stays again in agreement with the thermodynamics (Table 1) and Le Chatelier's principle. The limitation of the Tanksale-reaction is the syngas solubility in water. In 2016, Tanksale reported on solvent effects (solvent mixtures) and four different reaction pathways for the formalin formation from CO have been proposed.7 They determined methanol to be the best solvent under their conditions yielding 15.6 mmol L−1 gcat−1 HCHO at 90 °C and a pressure of 100 bar with 100% selectivity and without any CO2 formation. This yield corresponds to a yield four times higher than the best in the previous report.
| T [K] | Gas phase | Aqueous phase | ||
|---|---|---|---|---|
| X [%] | ΔG [kJ mol−1] (K [mol−1]) | X [%] | ΔG [kJ mol−1] (K [mol−1]) | |
| 298 | 4.38 × 10−3 | 34.565 (8.760 × 10−7) | 31.40 | −7.071 (17.332) |
| 323 | 4.67 × 10−3 | 37.297 (9.348 × 10−7) | 18.42 | −5.648 (8.186) |
| 373 | 4.88 × 10−3 | 42.933 (9.764 × 10−7) | 3.86 | −1.297 (1.519) |
| 423 | 4.86 × 10−3 | 48.705 (9.711 × 10−7) | 5.84 × 10−1 | 4.853 (2.52 × 10−1) |
Formaldehyde derivatives
In 2013 Sabo-Etienne and Bontemps trapped formaldehyde as bis(boryl)methylene acetal in the course of the CO2 reduction with pinacol borane with [RuH2(η-H2)2(PCy3)2] (Cy = cyclohexyl; 10 mol%) as the catalyst precursor (Scheme 10). The key step for the CO2 reduction goes via the insertion of CO2 into the ruthenium hydride bond. Later the authors showed that an iron catalysed pathway involving a metal hydride catalyst is also feasible for the generation of borylmethylene derivatives with good selectivities.65 Likewise most recently in 2016, Cantat and Berthet showed that besides iron, also copper and cobalt phosphine complexes are active for the borylation of CO2 yielding mixtures of bisboryl acetal and methoxy borane with a broad range of selectivity for these two products.66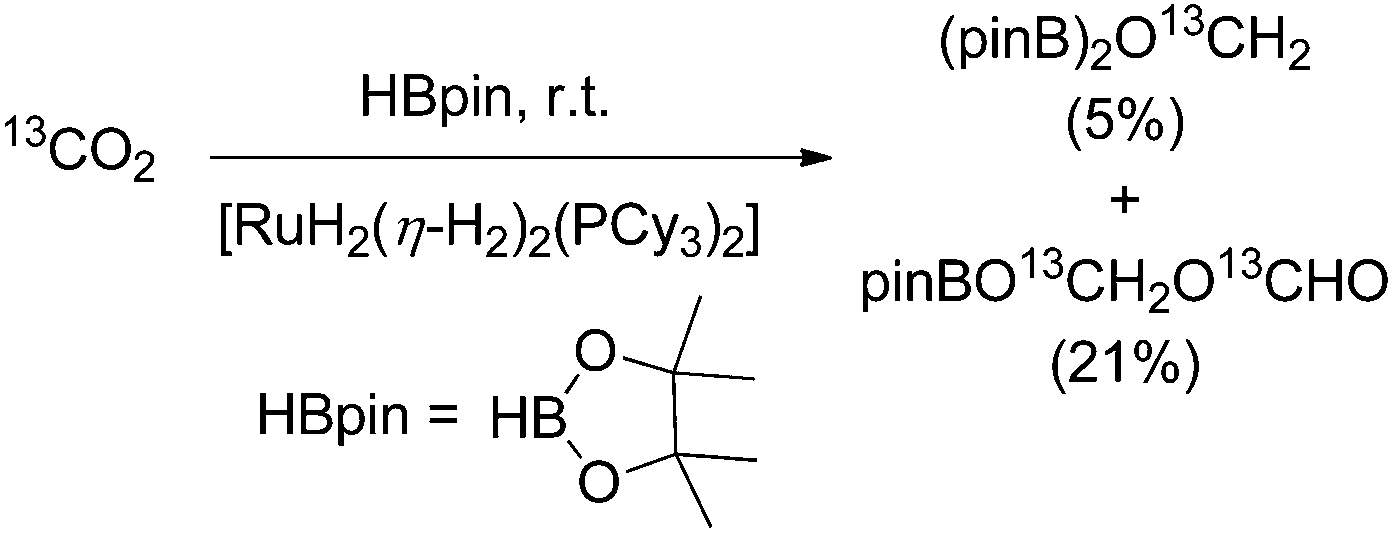 | ||
| Scheme 10 Reduction of CO2 with pinacol borane yielding boryl methylene acetal.67 | ||
In their ongoing research they focussed on the generation of free formaldehyde and achieved this with a slightly modified protocol.68 Using this catalyst precursor [RuH2(η-H2)2(PCyp3)2] (Cyp = cyclopentyl; 1–10 mol%) they achieved the formation of 22% formaldehyde by reduction of CO2 with pinacol borane besides the above mentioned boryl methylene acetals (6 and 12%). For further improvement of the formaldehyde yield, they added bulky diisopropyl aniline to the starting material resulting in the corresponding formyl imine (74%) and upon addition of aqueous methanol, the imine is hydrolysed yielding aqueous formaldehyde and paraformaldehyde (Scheme 11).68 Interestingly, in a study with phosphine-borane catalysts about hydroboration of CO2 yielding methoxyboranes, formaldehyde borane adducts have been postulated as catalytically active species.18
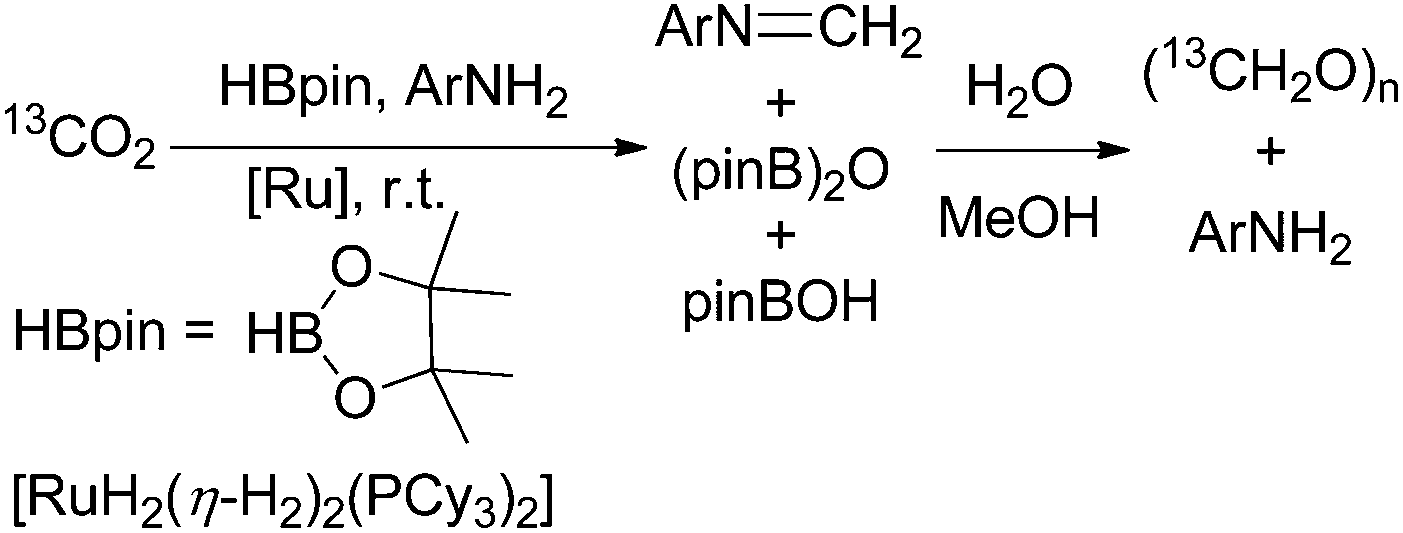 | ||
| Scheme 11 Reduction of CO2 with pinacol borane in the presence of aniline, followed by hydrolysis yielding aqueous formaldehyde and paraformaldehyde.68 | ||
Oestreich and Metsänen reported a ruthenium-catalysed hydrosilylation of CO2 with Et3SiH yielding silylated formaldehyde or methanol.19 Here the key step is the formation of the bis(silyl)methylene acetal which gives high selectivities (81–99%) at moderate to very high conversions (33 to >99%; Scheme 12) at 80 °C. Silylated methanol can be obtained after 7 days at 150 °C with 75% selectivity and 69% conversion.
 | ||
| Scheme 12 Metal catalysed reduction of CO2 with Et3SiH yielding bis(silyl)methylene acetal.17,19–21 | ||
Under similar conditions, Serrano and Rodriguez evaluated a nickel PBP-complex for the formation of bis(silyl)methylene acetal from CO2 in the presence of Et3SiH with high selectivities (78 to >99%) and moderate to high conversion (41–91%) at 70 °C.17 Already in 2013, Berke and co-workers reported the hydrosilylation of CO2 using a Frustrated Lewis Pair (FLP) consisting for example of a rhenium hydride and B(C6F5)3 (Scheme 12) at 80 °C.21 Here the authors reported yields of ∼90%. Piers and co-workers reported a scandium complex for the above described reaction.20 In general, it should be underlined that the interest in the synthesis of boryl and silyl methylene acetals from CO2 is of fundamental academic nature, and this might stimulate interest in the preparation of inorganic/organic hybrid materials incorporating formaldehyde derived from CO2. Contrarily, the above described approaches seem unsuitable for their application to generate organic combustion fuels or liquid organic hydrogen carrier molecules for energy storage and hydrogen storage systems based on CO2-derived formaldehyde.
Following the above described efforts on trapping formaldehyde in the form of boryl and silyl methylene acetals (Schemes 11 and 12), most recently in 2016, Klankermayer et al. demonstrated for the first time the catalytic conversion of CO2 (20 bar) and H2 (60 bar) in the presence of alcohols into dialkoxymethane ethers (oxymethylene ethers) in a multistep reaction using a molecular ruthenium-triphos catalyst and aluminium triflate as a Lewis acidic co-catalyst (Scheme 13).8 The reaction has been validated for a wide range of linear and branched aliphatic alcohols (C1–C10) and benzyl alcohol. The achieved turnovers reached about 200 TONs for methanol and varied for the higher aliphatic alcohols between 50 and 120, showing a dependency of the chain-length, with an activity increase for long-chain alcohols. The Klankermayer protocol gives direct access to dimethoxymethane and higher oxymethylene ethers which are considered to be potential fuels or fuel additives10 using abundant CO2 and renewable hydrogen.
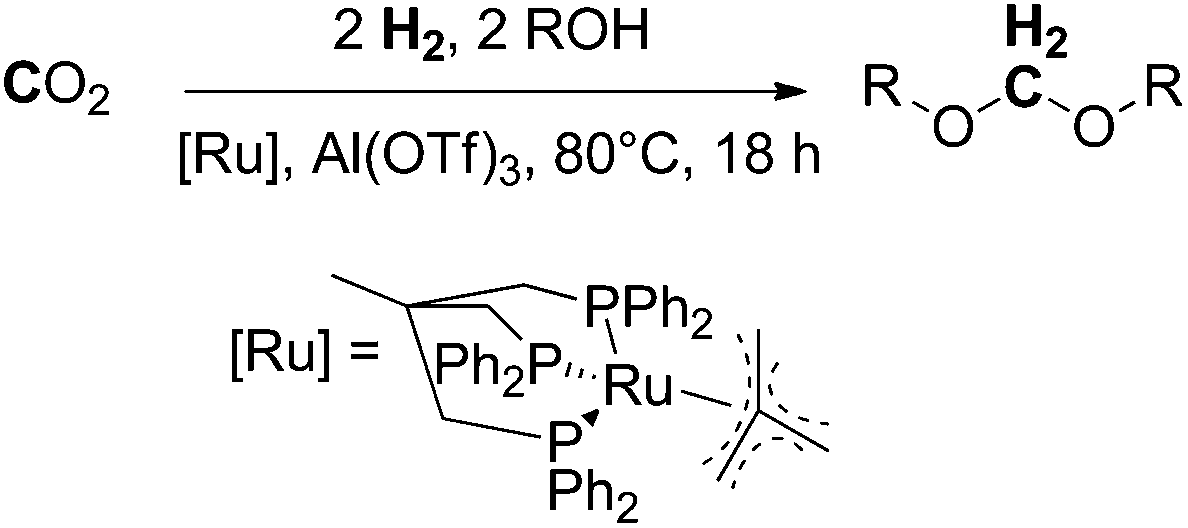 | ||
| Scheme 13 Ruthenium and Lewis acid catalysed reductive conversion of CO2 with H2 in the presence of alcohol into dialkoxymethanes.8 | ||
Formaldehyde as hydrogen source
In contrast to the above described obstacles and challenges for the synthesis of formaldehyde under reductive conditions starting from CO2 and CO, the dehydrogenative decomposition of aqueous formaldehyde and paraformaldehyde to H2 and CO2 is much more straightforward,14 even so, in contrast to long-investigated hydrogen evolution from formic acid or methanol,69,70 it has been rarely considered and investigated.Recently, we showed that water in the presence of C1-entities like (para)formaldehyde (6.7 wt% per unit) is suitable for molecular hydrogen-storage as these molecules can be easily and selectively dehydrogenated forming pure H2 and CO2 (Scheme 14).14 A theoretical efficiency of 8.4 wt% of H2 considering 1 equiv. H2O and H2CO is possible owing to the favourable equilibrium constant for methanediol in aqueous solution. In this regard, the incorporation of water into the formaldehyde molecule yielding methanediol can be interpreted as a chemical indirect water-splitting reaction, where water acts as a proton source and formaldehyde as a hydride source. The H2-content is higher than for formic acid (4.4 wt%), even when technical aqueous H2CO is used, the solution has a minimum efficiency of 5.0 wt%. This catalytic decomposition of H2CO can be envisioned as a novel approach for simultaneous H2 production and decontamination treatment of wastewater with formaldehyde impurities as a waste to value approach.1,13
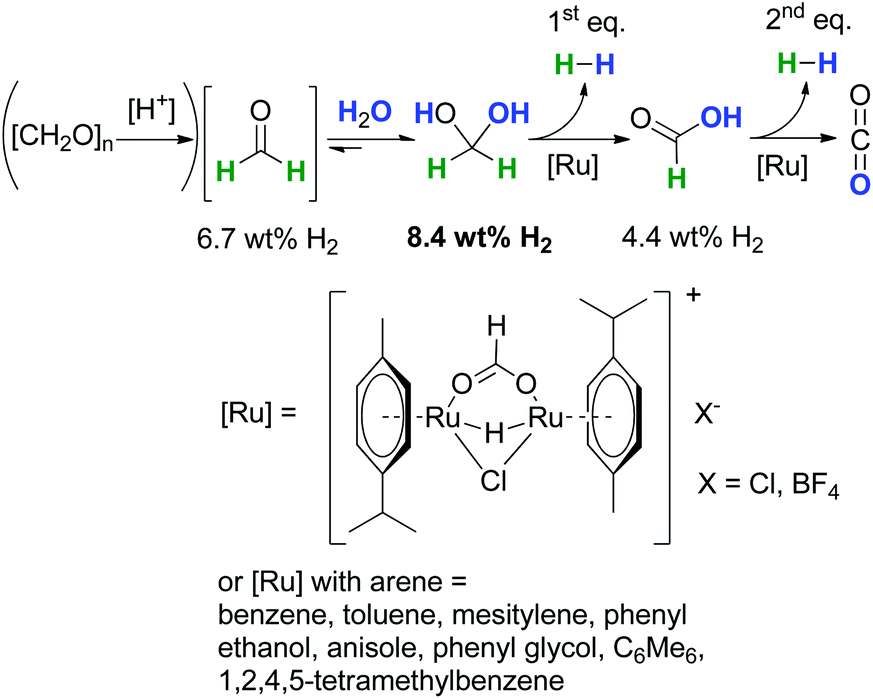 | ||
| Scheme 14 Low-temperature formaldehyde reforming between 25 °C and 95 °C.1,14,15 | ||
The dehydrogenation reaction of aqueous formaldehyde proceeds under mild conditions in the temperature range between room temperature and 95 °C using a molecular ruthenium catalyst under base-free, respectively pH neutral conditions, without any prevention to exclude air.1,14,15 As catalyst precursors a wide range of [Ru(arene)Cl2]2 complexes are suitable (arene = benzene, toluene, p-cymene, etc.). The catalyst is in situ transformed into a cationic ruthenium hydride formiato species [Ru(arene)(μ-H)(μ-Cl)(μ-HCO2)Ru(arene)]+X− (X: Cl, BF4) which plays a major role in the catalytic cycle.1 The hydrogen evolution is solely accompanied by CO2 formation without any carbon monoxide present. In regard to the hydrogen production yields of 85% of pure H2 using concentrated formaldehyde solutions have been obtained within 60 minutes of reaction time. Additionally, the capability for the wastewater purification has been proven with diluted formaldehyde solutions. Those tests gave conversions of ≫99.95%, and the residual amount of formaldehyde in solution was as low as 10–40 ppm. These values are in the range for the tolerance of formaldehyde in wastewater, where the highest reported TOF was 3142 h−1 using [Ru(p-cymene)Cl2]2 as the precatalyst.1
An extension of aqueous formaldehyde as LOHC has been further investigated with regard to room-temperature methanol reforming.15 The room temperature reforming of methanol follows a bio-inspired approach using a multi-catalytic system consisting of enzymes and a biomimetic formaldehyde dehydrogenase (Scheme 15). The limitations of this approach are related to the compatibility of the metal complex and the enzymes which might be overcome with metal–protein technologies,71 and it is also related to the reaction conditions.72 The catalyst modification and also slightly modified reaction conditions turn the catalytic formaldehyde dehydrogenation into an efficient biomimetic dismutase reaction pathway.72
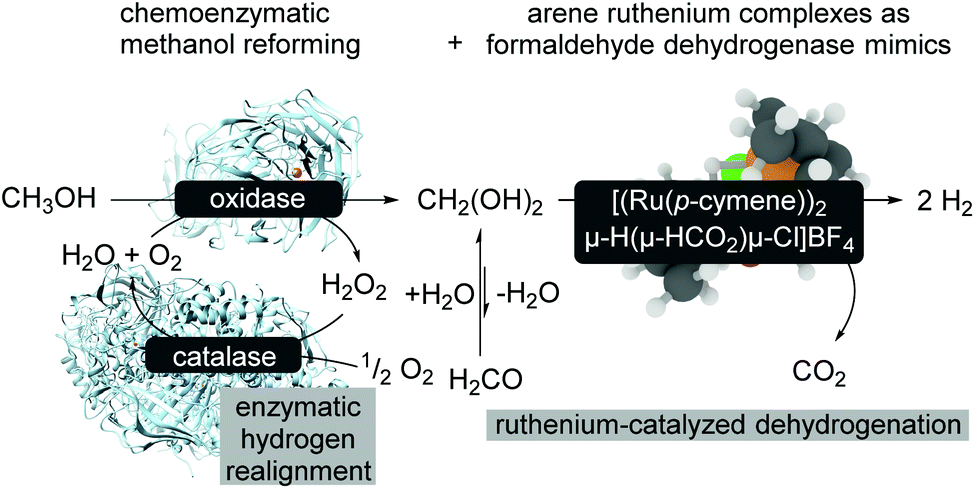 | ||
| Scheme 15 Bioinduced methanol reforming at room-temperature.15 | ||
Besides the above described ruthenium catalysed hydrogen generation from formaldehyde in water, Fukuzumi and co-workers showed that a water-soluble iridium complex is suitable for hydrogen generation from paraformaldehyde in water at room-temperature, however so far with TONs <100 depending on the basicity (pH 6–10) of the solutions (Scheme 16).22 Yamaguchi and colleagues demonstrated the formaldehyde dehydrogenation with an iridium complex carrying a cooperative ligand-site, resulting in conversions up to 89% (TON = 178).73 In both cases it has been shown that pH has an influence on the catalyst structure in solution and on the activity and yield. Both reactions show better performance under basic conditions.
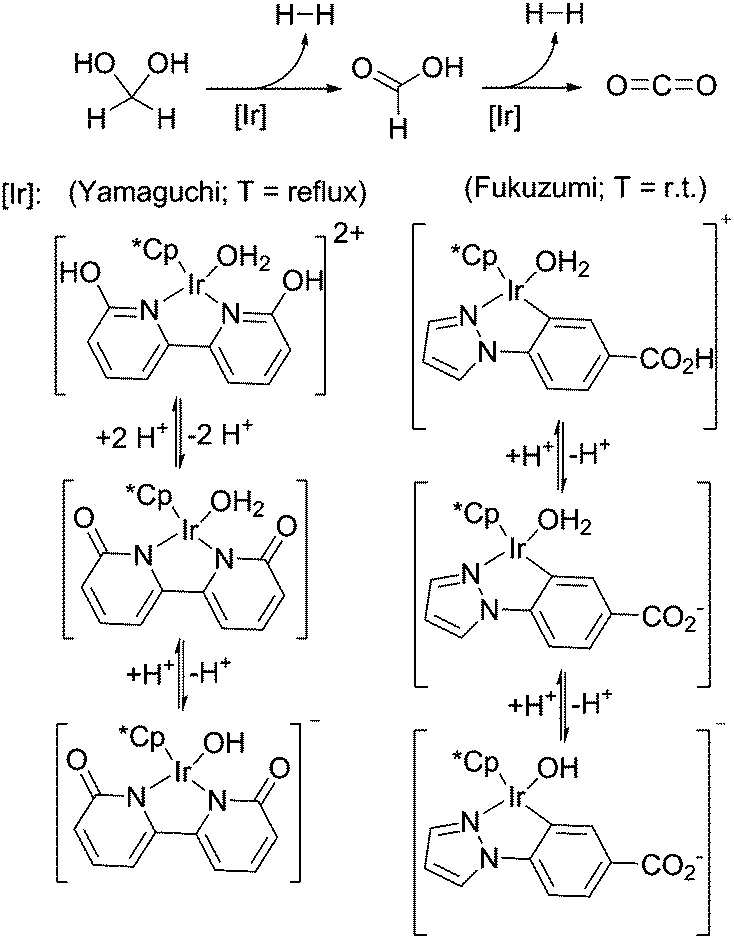 | ||
| Scheme 16 Low-temperature formaldehyde reforming with iridium catalysts.22,73 | ||
Summary & outlook
In this decade, major progress has been achieved towards the low-temperature synthesis of formaldehyde and its derivatives starting from CO and CO2. For the first time a promising direct formaldehyde synthesis from syngas has been realized. Likewise the first synthesis of dialkoxymethane via catalytic reduction of CO2 has been demonstrated. Moreover an electrocatalytic conversion of CO2 to formaldehyde using sea water as the electrolyte shows a further promising direction. These novel pathways in the C1-chemistry puzzle open new windows of opportunities towards more sustainable cost and energy saving production chains with formaldehyde as the platform reagent. The methods may become independent of fossil sources if the future hydrogen production processes will use renewables or plain water. The same efforts will be required for the future production of syngas using biomass or organic waste as the feedstock. The scenario of the stabilization of formaldehyde in solution with water and alcohol will play a key role to further improve these reductive syntheses because the thermodynamic parameters caused by the reactive protic solvents have a tremendous influence on the all over reaction pathways and the reactivity of all solute species and intermediates: methanediol, (di)alkoxymethane in the formaldehyde case or carbonic acid and carbonates in the CO2 case as dominant species in solution. Other approaches for the stabilization of formaldehyde in solution included the formation of bis(silyl) or bis(boryl)methylene acetals, which might generate further interest for inorganic materials with formaldehyde as a cross-linker. Still the selective hydrogenation of CO2 to formaldehyde is a missing piece of the C1-puzzle. In analogy to the CO hydrogenation in water, the key for a selective CO2 hydrogenation will be probably the application of water as the solvent to trap formaldehyde in its form as methanediol, hence formalin solution. Notably, free formaldehyde can be also obtained in sequential enzyme catalysis applying FDH and FADH in the presence of NADH as a co-factor. This clearly shows that also the biotechnology pathway might be promising one day to complement the global demands for the large amounts of formaldehyde already required nowadays for established application.Considering new potential fields for the application of formaldehyde in the energy sector for new combustion fuels or as liquid hydrogen carrier molecules for hydrogen fuel cell technologies, consequently this will require further efforts to enhance significantly the global production of formaldehyde. Thus, one needs to consider also biomass- and waste-based formaldehyde production. In this regard also the hydrogen production from wastewater containing residual formaldehyde might become an interesting aspect to complement the hydrogen production. The realization of a formaldehyde-based hydrogen battery requires the immobilization of the catalyst. In terms of long-term stability the system seems quite promising, also taking into account the usual limitations of battery life-time issues and the superior energy content of hydrogen and LOHC in comparison to conventional batteries.
Acknowledgements
We gratefully acknowledge financial support provided by the Ministerium für Innovation, Wissenschaft und Forschung (NRW-returnee award 2009 to M. H. G. P.), the Heisenberg-Program (Deutsche Forschungsgemeinschaft), the COST Action “Catalytic Routines for Small Molecule Activation (CARISMA)” and the Ernst-Haage-Prize 2014 of the Max-Planck Institute for Chemical Energy Conversion. H. K. acknowledges the Jürgen-Manchot-Stiftung for a PhD scholarship.Notes and references
- L. E. Heim, S. Vallazza, D. van der Waals and M. H. G. Prechtl, Green Chem., 2016, 18, 1469–1474 RSC.
- A. M. Bahmanpour, A. Hoadley and A. Tanksale, Rev. Chem. Eng., 2014, 30, 583–604 CAS.
- G. Reuss, W. Disteldorf, A. O. Gamer and A. Hilt, Ullmann's Encyclopedia of Industrial Chemistry, Wiley, Weinheim, 2003 Search PubMed.
- B. Sam, B. Breit and M. J. Krische, Angew. Chem., Int. Ed., 2015, 54, 3267–3274 CrossRef CAS PubMed.
- A. M. Bahmanpour, A. Hoadley and A. Tanksale, Green Chem., 2015, 17, 3500–3507 RSC.
- D. Roeda and C. Crouzel, Appl. Radiat. Isot., 2001, 54, 935–939 CrossRef CAS PubMed.
- A. M. Bahmanpour, A. Hoadley, S. H. Mushrif and A. Tanksale, ACS Sustainable Chem. Eng., 2016, 4, 3970–3977 CrossRef CAS.
- K. Thenert, K. Beydoun, J. Wiesenthal, W. Leitner and J. Klankermayer, Angew. Chem., Int. Ed., 2016, 55, 12266–12269 CrossRef CAS PubMed.
- X. L. Zhang, A. O. Oyedun, A. Kumar, D. Oestreich, U. Arnold and J. Sauer, Biomass Bioenergy, 2016, 90, 7–14 CrossRef CAS.
- J. Burger, M. Siegert, E. Strofer and H. Hasse, Fuel, 2010, 89, 3315–3319 CrossRef CAS.
- X. M. Huang, T. I. Koranyi, M. D. Boot and E. J. M. Hensen, Green Chem., 2015, 17, 4941–4950 RSC.
- S. Kapoor, F. A. Barnabas, M. C. Sauer, D. Meisel and C. D. Jonah, J. Phys. Chem., 1995, 99, 6857–6863 CrossRef CAS.
- M. H. G. Prechtl, L. E. Heim and N. E. Schloerer, Wastewater treatment and hydrogen production (patent), 2013, WO2015/003680A003681 and PCT/DE002014/000344 Search PubMed.
- L. E. Heim, N. E. Schloerer, J. H. Choi and M. H. G. Prechtl, Nat. Commun., 2014, 5, 3621, DOI:10.1038/ncomms4621.
- L. E. Heim, D. Thiel, C. Gedig, J. Deska and M. H. G. Prechtl, Angew. Chem., Int. Ed., 2015, 54, 10308–10312 CrossRef CAS PubMed.
- K. Nakata, T. Ozaki, C. Terashima, A. Fujishima and Y. Einaga, Angew. Chem., Int. Ed., 2014, 53, 871–874 CrossRef CAS PubMed.
- P. Rios, N. Curado, J. Lopez-Serrano and A. Rodriguez, Chem. Commun., 2016, 52, 2114–2117 RSC.
- R. Declercq, G. Bouhadir, D. Bourissou, M. A. Legare, M. A. Courtemanche, K. S. Nahi, N. Bouchard, F. G. Fontaine and L. Maron, ACS Catal., 2015, 5, 2513–2520 CrossRef CAS.
- T. T. Metsänen and M. Oestreich, Organometallics, 2015, 34, 543–546 CrossRef.
- F. A. LeBlanc, W. E. Piers and M. Parvez, Angew. Chem., Int. Ed., 2014, 53, 789–792 CrossRef CAS PubMed.
- Y. F. Jiang, O. Blacque, T. Fox and H. Berke, J. Am. Chem. Soc., 2013, 135, 7751–7760 CrossRef CAS PubMed.
- T. Suenobu, Y. Isaka, S. Shibata and S. Fukuzumi, Chem. Commun., 2015, 51, 1670–1672 RSC.
- Chem. Process Eng., 1970, 51, 100–101.
- K. Jahn, Ber. Dtsch. Chem. Ges., 1889, 22, 989–989 CrossRef.
- W. A. Bone and H. L. Smith, J. Chem. Soc., Dalton Trans., 1905, 910–916 RSC.
- D. L. Chapman and A. Holt, J. Chem. Soc., Dalton Trans., 1905, 916–921 RSC.
- H. J. H. Fenton, J. Chem. Soc., Dalton Trans., 1907, 687–693 RSC.
- A. P. Mathews, Science, 1910, 32, 926 CAS.
- B. C. Brodie, Proc. R. Soc. London, 1873, 22, 171–172 CrossRef.
- G. T. Morgan, D. V. N. Hardy and R. A. Proctor, J. Soc. Chem. Ind., 1932, 51, 1–7T CrossRef CAS.
- R. H. Newton and B. F. Dodge, J. Am. Chem. Soc., 1933, 55, 4747–4759 CrossRef CAS.
- J. Klankermayer, S. Wesselbaum, K. Beydoun and W. Leitner, Angew. Chem., Int. Ed., 2016, 55, 7296–7343 CrossRef CAS PubMed.
- M. Behrens, F. Studt, I. Kasatkin, S. Kuhl, M. Havecker, F. Abild-Pedersen, S. Zander, F. Girgsdies, P. Kurr, B. L. Kniep, M. Tovar, R. W. Fischer, J. K. Norskov and R. Schlogl, Science, 2012, 336, 893–897 CrossRef CAS PubMed.
- M. Nielsen, E. Alberico, W. Baumann, H. J. Drexler, H. Junge, S. Gladiali and M. Beller, Nature, 2013, 495, 85–89 CrossRef CAS PubMed.
- S. Wesselbaum, T. vom Stein, J. Klankermayer and W. Leitner, Angew. Chem., Int. Ed., 2012, 51, 7499–7502 CrossRef CAS PubMed.
- M. Aresta, A. Caroppo, A. Dibenedetto and M. Narracci, Environmental Challenges and Greenhouse Gas Control for Fossil Fuel Utilization in the 21st Century, Springer, 2002, pp. 331–347 Search PubMed.
- P. G. Jessop, T. Ikariya and R. Noyori, Chem. Rev., 1995, 95, 259–272 CrossRef CAS.
- R. Franke, D. Selent and A. Borner, Chem. Rev., 2012, 112, 5675–5732 CrossRef CAS PubMed.
- M. Scariot, D. O. Silva, J. D. Scholten, G. Machado, S. R. Teixeira, M. A. Novak, G. Ebeling and J. Dupont, Angew. Chem., Int. Ed., 2008, 47, 9075–9078 CrossRef CAS PubMed.
- D. O. Silva, J. D. Scholten, M. A. Gelesky, S. R. Teixeira, A. C. B. Dos Santos, E. F. Souza-Aguiar and J. Dupont, ChemSusChem, 2008, 1, 291–294 CrossRef CAS PubMed.
- J. D. Scholten, M. H. G. Prechtl and J. Dupont, ChemCatChem, 2010, 2, 1265–1270 CrossRef CAS.
- G. A. Olah, Angew. Chem., Int. Ed., 2005, 44, 2636–2639 CrossRef CAS PubMed.
- T. Zell and D. Milstein, Acc. Chem. Res., 2015, 48, 1979–1994 CrossRef CAS PubMed.
- X.-L. Du, Z. Jiang, D. S. Su and J.-Q. Wang, ChemSusChem, 2016, 9, 322–332 CrossRef CAS PubMed.
- W.-H. Wang, Y. Himeda, J. T. Muckerman, G. F. Manbeck and E. Fujita, Chem. Rev., 2015, 115, 12936–12973 CrossRef CAS PubMed.
- W. Wang, S. Wang, X. Ma and J. Gong, Chem. Soc. Rev., 2011, 40, 3703–3727 RSC.
- W. Leitner, Coord. Chem. Rev., 1996, 153, 257–284 CrossRef CAS.
- P. G. Jessop, F. Joo and C. C. Tai, Coord. Chem. Rev., 2004, 248, 2425–2442 CrossRef CAS.
- P. G. Jessop, T. Ikariya and R. Noyori, Nature, 1994, 368, 231–233 CrossRef CAS.
- M. Grasemann and G. Laurenczy, Energy Environ. Sci., 2012, 5, 8171–8181 CAS.
- A. Behr and K. Nowakowski, in CO2 Chemistry, 2014, vol. 66, pp. 223–258 Search PubMed.
- S. Gambarotta, S. Strologo, C. Floriani, A. Chiesivilla and C. Guastini, J. Am. Chem. Soc., 1985, 107, 6278–6282 CrossRef CAS.
- P. Arya, J. Boyer, R. J. P. Corriu, G. F. Lanneau and M. Perrot, J. Organomet. Chem., 1988, 346, C11–C14 CrossRef CAS.
- W. F. Liu, Y. H. Hou, B. X. Hou and Z. P. Zhao, Chin. J. Chem. Eng., 2014, 22, 1328–1332 CrossRef CAS.
- D. K. Lee, D. S. Kim and S. W. Kim, Appl. Organomet. Chem., 2001, 15, 148–150 CrossRef CAS.
- R. Kuriki, O. Ishitani and K. Maeda, ACS Appl. Mater. Interfaces, 2016, 8, 6011–6018 CAS.
- G. H. Qin, Y. Zhang, X. B. Ke, X. L. Tong, Z. Sun, M. Liang and S. Xue, Appl. Catal., B, 2013, 129, 599–605 CrossRef CAS.
- L. Jia, J. Li and W. Fang, Catal. Commun., 2009, 11, 87–90 CrossRef CAS.
- K. Tennakone, A. H. Jayatissa and S. Punchihewa, J. Photochem. Photobiol., A, 1989, 49, 369–375 CrossRef CAS.
- Y. Matsumoto, M. Obata and J. Hombo, J. Phys. Chem., 1994, 98, 2950–2951 CrossRef CAS.
- T. Inoue, A. Fujishima, S. Konishi and K. Honda, Nature, 1979, 277, 637–638 CrossRef CAS.
- K. Li, X. An, K. H. Park, M. Khraisheh and J. Tang, Catal. Today, 2014, 224, 3–12 CrossRef CAS.
- B. Aurianblajeni, M. Halmann and J. Manassen, Sol. Energy, 1980, 25, 165–170 CrossRef CAS.
- M. Girardi, S. Blanchard, S. Griveau, P. Simon, M. Fontecave, F. Bedioui and A. Proust, Eur. J. Inorg. Chem., 2015, 3642–3648 CrossRef CAS.
- G. H. Jin, C. G. Werncke, Y. Escudiee, S. Sabo-Etienne and S. Bontemps, J. Am. Chem. Soc., 2015, 137, 9563–9566 CrossRef CAS PubMed.
- A. Aloisi, J.-C. Berthet, C. Genre, P. Thuery and T. Cantat, Dalton Trans., 2016, 45, 14774–14788 RSC.
- S. Bontemps and S. Sabo-Etienne, Angew. Chem., Int. Ed., 2013, 52, 10253–10255 CrossRef CAS PubMed.
- S. Bontemps, L. Vendier and S. Sabo-Etienne, J. Am. Chem. Soc., 2014, 136, 4419–4425 CrossRef CAS PubMed.
- P. Mars, J. J. F. Scholten and P. Zwietering, in Advances in Catalysis, ed. H. P. D. D. Eley and P. B. Weisz, Elsevier, Amsterdam, 1963, vol. 14, p. 35 Search PubMed.
- G. M. Meaburn, F. W. Mellows and A. Reiffste, Nature, 1964, 204, 1301–1302 CrossRef CAS.
- T. R. Ward, Acc. Chem. Res., 2011, 44, 47–57 CrossRef CAS PubMed.
- D. van der Waals, L. E. Heim, S. Vallazza, C. Gedig, J. Deska and M. H. G. Prechtl, Chem. – Eur. J., 2016, 22, 11568–11573 CrossRef CAS PubMed.
- K. Fujita, R. Kawahara, T. Aikawa and R. Yamaguchi, Angew. Chem., Int. Ed., 2015, 54, 9057–9060 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this article. |
| This journal is © The Royal Society of Chemistry 2017 |