
Practical molecular thermodynamics for greener solution chemistry†
Steven
Abbott
ab,
Jonathan J.
Booth‡
c and
Seishi
Shimizu
 *d
*d
aSteven Abbott TCNF Ltd., 7 Elsmere Road, Ipswich, Suffolk IP1 3SZ, UK
bSchool of Mechanical Engineering, University of Leeds, Leeds LS2 9JT, UK
cSchool of Chemistry, University of Leeds, Leeds LS2 9JT, UK
dYork Structural Biology Laboratory, Department of Chemistry, University of York, Heslington, York YO10 5DD, UK. E-mail: seishi.shimizu@york.ac.uk
First published on 14th December 2016
Abstract
We all know that to enhance solubility using greener chemistry we should harness sound principles of molecular-based thermodynamics. The problem is that even for simple systems it can be hard to know how to use fundamental tools for formulation benefit, and for the more complex systems that we must often use, calculations required for molecular thermodynamics can often be quite involved. In this paper we show that a fundamental, assumption-free statistical thermodynamics approach, the Kirkwood–Buff theory, can be used in practical, complex aqueous systems to provide the insights we need to optimise formulations. The theory itself is not that difficult, but its implementation, which requires many steps of thermodynamic calculations, has up to now not been straightforward. Taking full advantage of an interactive approach, here we review what the Kirkwood–Buff theory can provide for formulators; we use the power of modern web browsers to provide open-source, user-friendly, responsive-design apps to do the hard work of data analysis, leaving formulators to focus on the interpretation of the results for their specific optimisation task. Indeed the apps are intended to be used by researchers and formulators for specific systems of interest to them.
 Steven Abbott | Steven Abbott has a chemistry PhD from Oxford and Harvard and has worked for many years in the chemicals and coating industry. He is now an independent scientist and consultant. He is Visiting Professor at the School of Mechanical Engineering, University of Leeds. His Practical Adhesion, Practical Solubility and Practical Surfactants websites represent his attempt to “appify” as much science as possible in these areas, allowing the science to become alive and usable by non-experts. His most recent apps are designed for Virtual Reality. |
 Jonathan J. Booth | Jonathan Booth studied Chemistry at the University of York where, under the supervision of Dr Seishi Shimizu, he carried out an MChem research project of solubility enhancement. This led to the first rational theory of how hydrotropes work, and the development of a thermodynamic toolkit for explaining many areas of solubility and formulation science. After graduating Jonathan completed a PhD in computer modelling and algorithm development at the University of Leeds under the supervision of Prof. Dmitry Shalashilin. Jonathan now works for Croda as a formulation scientist. |
 Seishi Shimizu | Seishi Shimizu is a Senior Lecturer in the York Structural Biology Laboratory, Department of Chemistry, University of York. He has received a BSc in Physics, MSc in Physics and PhD in Biotechnology from the University of Tokyo, prior to his postdoctoral training in the Faculty of Medicine, University of Toronto. As a theorist specializing in statistical thermodynamics, he has worked in wide-ranging fields and applications including solvation, protein folding and stability, protein–ligand interaction, food science, formulation science and hydrotropy. |
Introduction
When water is chosen as a principal solvent for green chemistry,1–5 the challenge is to find green “solubilizers” (using a deliberately vague word) to help bring hydrophobic solutes into solution.6–11 If we are lucky we can use conventional “cosolvents” such as ethanol and we need little new theory to guide us in the right choice. Similarly, it is relatively straightforward to find surfactant systems (such as Tweens) that increase solubility when present at concentrations above their critical micelle concentration (CMC) by bringing the solute somewhere within the micelle structure (tail, head or intermediate region, depending on the solute).6–11 Although the theory of such systems is complex,12,13 in many situations a formulator can find adequate formulations via simple design of experiment approaches.Our concern here is with solubilizers that fall between these two extremes. Here there is a confusing mix of terminologies such as “hydrotrope”, “solvosurfactant”, “solubilizer”, “cosolvent” and even “pre-ouzo” formulations.6–11 Although such systems offer much promise, it is hard to formulate rationally because of the confusing terminology and the obscurity of the mechanisms by which solubility is increased.14–17 The confusion leads directly to a waste of intellectual and developmental resources, as well as sub-optimal formulations. The aim of this paper is to provide a set of practical tools to counter this wastefulness.
We can illustrate the problems through the use of three classical hydrotropes: urea,18 nicotinamide19 and sodium cumene sulfonate, SCS.20 In each case, the hydrotrope has little effect on solubility at low concentrations, then above a Minimum Hydrotrope Concentration (MHC)21 the solubility of the solute increases. Because of the striking analogy between the MHC and CMC it has been natural to say that the hydrotrope forms clusters within which the solute is soluble.6–10,21 With SCS, for example, which looks a little like a typical ionic surfactant, the temptation to accept the analogy is obvious. Another example, nicotinamide, is well-known to self-associate in water so the temptation persists.22 However, urea has no significant tendency to self-associate in water, so for urea at least, the analogy cannot be correct.23 As we shall see, the analogy is positively wrong.14–17
Another temptation has been to invoke the phrase “water structure” to suggest that the hydrotrope breaks up the structure, allowing the solute to dissolve.24,25 However, invoking the phrase seldom brings helpful formulation understanding and we shall show that water structure is thermodynamically irrelevant for hydrotrope solubilization.14–17
A final temptation was to think in terms of “complexes” between solute and hydrotrope.26,27 There are three obvious problems with this idea. The first is that the term “complex” brings images of stoichiometry and chemists’ inclinations to think in terms of specific interaction.15 The second is that there has seldom been convincing evidence for such complexes.15 The third is that no reasonable theory of complex formation could reproduce a typical hydrotrope solubility curve.14–17
Resolving the debate via fundamental molecular thermodynamics
For decades the debate about the general mechanism of hydrotropes could not be resolved because there was no objective way to look at the thermodynamics of this complex ternary system.14–17 Classical thermodynamic values such as enthalpy and entropy are useful, yet are not the most straightforward entry into a molecular understanding, as will be shown below.By using a fundamental, assumption-free molecular (statistical) thermodynamics technique called the Kirkwood–Buff (KB) theory,28–36 the present authors were able to show conclusively that none of the above hypotheses had any merit.14–17 The structure of the water could be shown to change negligibly across the entire range of hydrotrope concentrations.14 The self-association of a hydrotrope (e.g. nicotinamide) could be shown unambiguously to reduce its capability to act as a hydrotrope, for the reason, obvious in hindsight, that any hydrotrope molecules that were self-associating were not associating with the solute.17 And the idea of a “complex” such as 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 or 2:1 was demonstrated to be unrealistic.15 Indeed, stoichiometric complexation, which assumes a strong, specific interaction, contradicts the weak, non-specific interactions that take place in aqueous hydrotrope solutions.14–17
:1 or 2:1 was demonstrated to be unrealistic.15 Indeed, stoichiometric complexation, which assumes a strong, specific interaction, contradicts the weak, non-specific interactions that take place in aqueous hydrotrope solutions.14–17
Instead it was shown that on average the solute and hydrotrope were mutually attracted, reducing the overall free energy of the system.14–17 Interestingly, the MHC was shown to be due to solute-induced clustering of the hydrotrope – in other words, once a hydrotrope started to become associated with the solute, that attracted more hydrotrope molecules.16
By looking at different solutes, some patterns emerged. Nicotinamide was generally more likely to want to associate with aromatic solutes but sometimes urea (which was not especially associated to the solute) was the more effective hydrotrope because none of it was wasted in hydrotrope clusters.17
The numerical analysis of these effects (discussed properly below) requires a modest amount of algebraic analysis of some raw data. The necessary data are simple and basic:14–17
1. The solubility curve versus hydrotrope concentration (of course);
2. The density dependence of the aqueous hydrotrope solutions;
3. The vapour pressure (VP) or (equivalently) osmotic pressure of the hydrotrope solutions.
No complex experimental equipment is required (though it is possible to derive some of the data via SAXS/SANS after much effort and cost) and modern high throughput machines make it trivial to obtain the necessary data.
This means that the green formulator merely has to add a few rather routine measurements (density & VP) to their solubility measurements to get an assumption-free, fundamental thermodynamic analysis of all the relevant effects within their solubilization experiments. The community gains a universal tool for comparing/contrasting the effects of different hydrotropes on different solutes and once a sufficient body of data exists, the technique will allow prediction and optimization. As a community we can transition from rather vague (and often confusing) terms such as “hydrotrope” to a few fundamental thermodynamic numbers which describe what is really going on.
A friendly guide to Kirkwood–Buff theory
KB starts with a thought experiment that is natural to any formulator.33 Imagine one of the molecules in the solution, for example the solute. Look around and count how many molecules of another component (e.g. the hydrotrope) are present at various distances. In the far distance the numbers will be the bulk statistical average. Close up the numbers might be much less than the average (if there are unfavourable interactions) or much more (favourable interactions).33 From this counting one can create the radial distribution function (RDF) as shown in top part of Fig. 1 which comes from the first app used in this paper (found at http://www.stevenabbott.co.uk/practical-solubility/rdf.php). Note that screenshot images are provided as a visual impression – readers should click on the link to be able to examine the data properly. The apps run on all modern platforms and are open source, written in standard HTML5/CSS3/Javascript allowing those interested to examine the code in detail.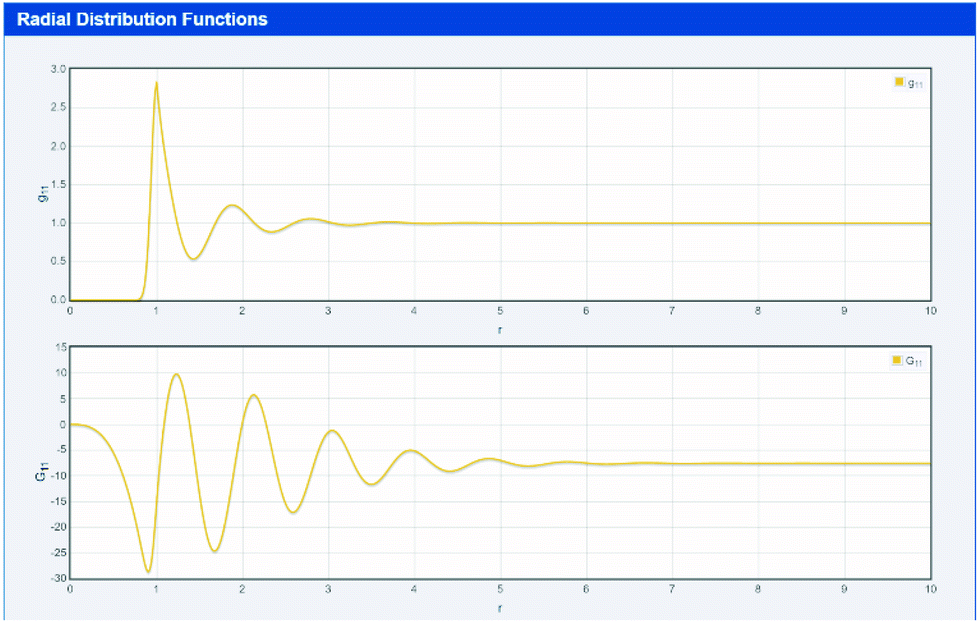 | ||
| Fig. 1 A simple simulation of an RDF (gij(r)) and 4πr2[gij(r) − 1] as a function of intermolecular separation, r. | ||
The RDF between species i and j is designated gij(r), which is a function of the intermolecular separation, r. By integrating the gij(r) − 1 over the whole solution space (via the function 4πr2[gij(r) − 1] shown in the bottom part of Fig. 1) one obtains a number which would be 0 if the distribution were entirely average (no special interactions), positive for typical small molecules if the molecules have fairly strong interactions and negative for systems with unfavourable interactions.14–17,28–36 The values for systems of interest are readily found from the hydrotrope app discussed below. These numbers are called the Kirkwood–Buff Integrals (KBI) and the free energy of the system can be calculated on a rigorous, assumption-free basis from the KBI.14–17,28–36 For two components i and j the KBI is written as Gij. It is important to note that a positive (negative) KBI implies an increase (decrease) in the measured density of the solution, i.e. microscopic phenomena are reflected in macroscopic effects, as discussed below.
By definition, Gij = Gji so to completely specify the pair-wise interactions in a water (1), hydrotrope (2) and solute (u) system we need G11, G12, G1u, G22, G2u and Guu, though for the purposes of this paper the low concentration of the solute allows Guu to be ignored.14–17 These numbers change as the relative concentrations change so we get a full picture of how the pair-wise interactions vary as the relative proportions vary in the formulation. Crucially, the Gij values are numbers that make sense to a formulator. A large positive Gij can be imagined as a local clustering of the species j around i. The details of the clustering will be vague for two reasons. The first is that this is statistical thermodynamics so the clusters are statistical – we are not talking about complexes.14–17 The second is, regrettably, a fundamental downside of KB theory. It can be shown that many different RDFs can produce the same KBI. In other words, we have no knowledge from the KBI whether a large Gij is due to a very compact clustering within the first solvation shell or a very loose cluster in the first shell with a steadily decreasing longer-range interaction.28–36 This RDF problem also means that we have to do the experiments to get the data rather than rely simply on molecular dynamics simulations where it is trivially easy to calculate KBIs from the simulation but extremely hard to avoid small errors at long ranges which can cause massive changes to the calculated KBI values.37
A simple 2D pseudo-molecular dynamics app (based upon an interactive molecular dynamics programme by Schroeder38) allows the rapid development of an intuitive feel for RDFs and the resulting Gij values. The app is http://www.stevenabbott.co.uk/practical-solubility/rdf-demo.php and the screen shot (Fig. 2) gives some idea of the capabilities of the app.
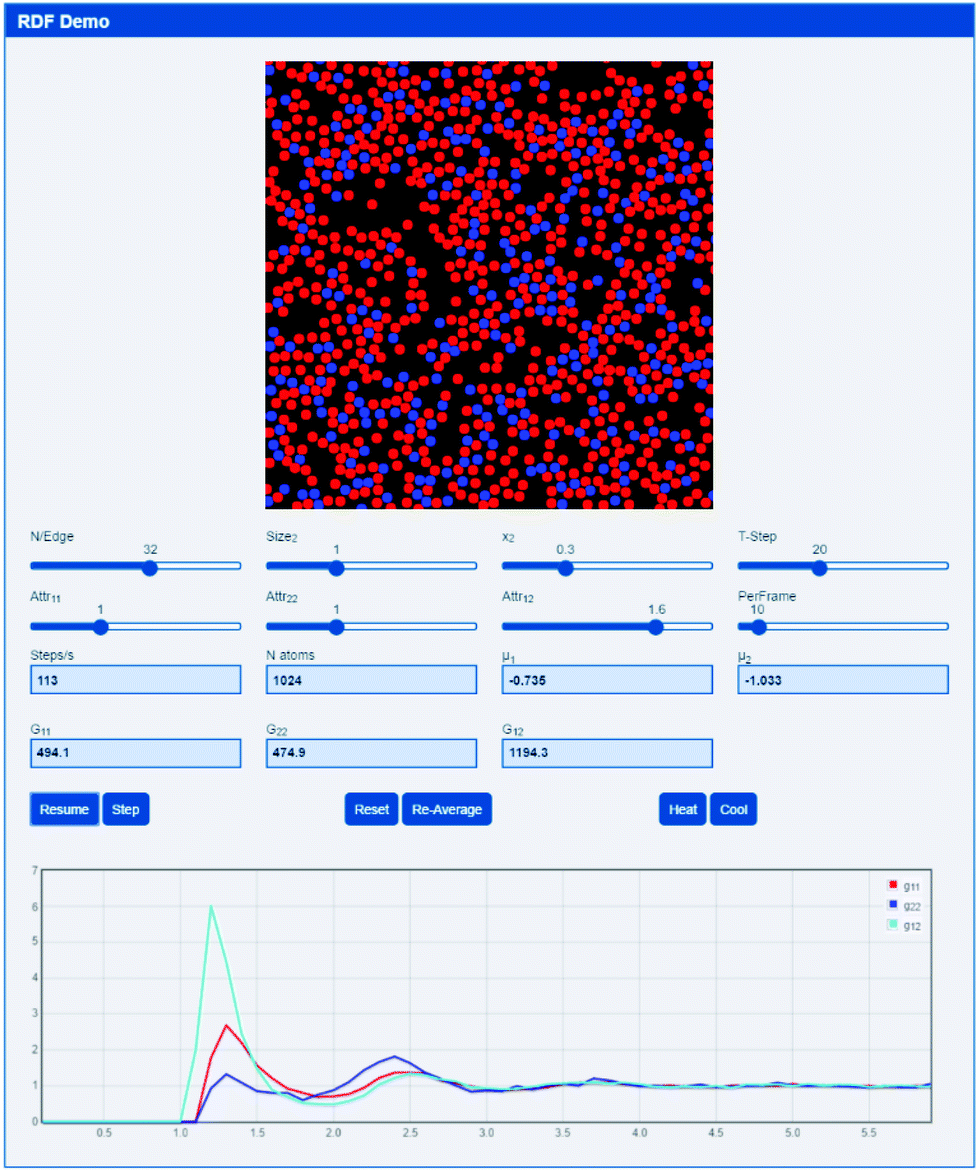 | ||
| Fig. 2 A 2D simulation of simple Lennard-Jones fluid mixtures from which an RDF is calculated. In this example, the 1-2 “attraction” is 50% greater than that of 1-1 and 2-2 so the G12 values are higher. | ||
Density and exclusion
Few formulators are interested in measuring densities – they seem such an insignificant parameter. It is easy to see, however, that densities are far from insignificant. The units of KBI are cc mol−1. These are the same units as those of molar volume (MVol) i.e. MWt/density. If two liquids that have very similar shapes and interactions are mixed together, then the density of the mixture is simply the average.39 But if there are disparities in interactions between the molecules then the density will increase (attractive) or decrease (repulsive) relative to the average.39 It follows that it is impossible to have a large, positive KBI without an increase in density over the ideal value. So we already know that by measuring densities of mixtures we are gaining some access to KBI information. It is a key strength of the KB approach that something as simple as measuring density to 4 or 5 decimal places using a few μl of solution in a modern automated densitometer takes you a long way to deriving the KBIs.40Another advantage of KBIs is in their ability to rationalize the formulators’ general preference for smaller solvents to larger ones for dissolution. The KBI is the integral of the radius r from 0 to infinity of the RDF.28–36 As shown in Fig. 1, the RDF is exactly 0 in the early part of the integral because other molecules are excluded from the volume of whichever molecule we are taking to be the reference for our distribution. This means that larger molecules have a 0 value of an RDF for a longer distance than smaller ones.30–33 So, other things being equal, KBIs for larger molecules will always be smaller (more negative) than those for smaller molecules. And because KBIs are a measure of the strengths of interactions, larger molecules always start with a disadvantage. There are many ways to talk of this “excluded volume” effect and it is common to describe it in terms of entropy. But by thinking of it in terms of the KBI, an idea that can be slippery and confusing becomes concrete and intuitive.30–33 An app (http://www.stevenabbott.co.uk/practical-solubility/ev-demo.php, not shown) allows exploration of this effect by omitting all other confusing variables.
So two simple concepts, density and excluded volume are key to understanding many formulation issues, yet are used surprisingly little.
Vapour pressure, osmotic pressure
Most formulators are comfortable with the idea of “activity coefficient”.39 If, for example, volatile species i is strongly attracted to species j at a given concentration in an i, j mixture then the vapour pressure of i above the mixture will be lower than expected from an ideal mixture, and will be higher if i is repelled by j at that concentration.39 In the first case the activity coefficient is less than 1 and in the second it is greater than 1. If measuring vapour pressure is not convenient then the same information can be found using an osmometer.39KBIs are directly related to activity coefficients, provided that density changes are also taken into account.14–17,28–37 Because this paper is deliberately app-based, the reader is urged to explore the relationship between activity coefficients, densities and molecular weights (which affect molar volumes and excluded volumes) by using http://www.stevenabbott.co.uk/practical-solubility/kbgs.php.
The default settings, shown in Fig. 3, use constant densities, identical molecular weights and (via pseudo Wilson parameters) a system with modestly large activity coefficients. The three KBI (G11, G12 and G22) are plotted across the mole fraction range of 0 to 1 and will quickly make intuitive sense as the relative activity coefficients are changed. By changing molecular weight, simple excluded volume effects become apparent, then by adding a polynomial density plot of any shape that seems of interest, i.e. adding the effects of internal interactions, the full system can be explored.
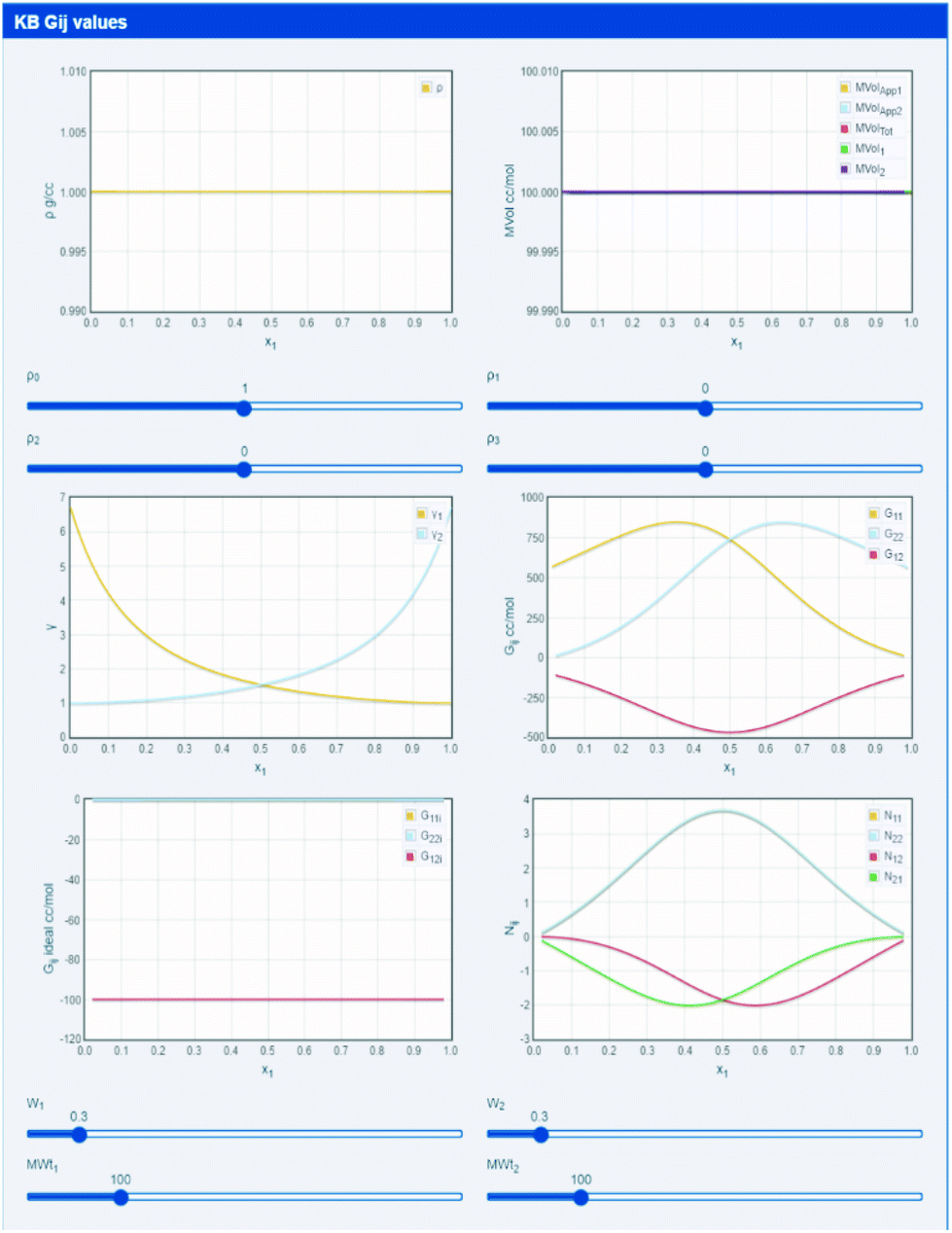 | ||
| Fig. 3 KBIs derived directly from MWt, density and activity coefficient data. Extra parameters such as “excess numbers” are also calculated. | ||
The app introduces two other concepts. We know that a large positive Gij means attraction and a large negative value means repulsion.14–17 So the Gij of an ideal mix might be expected to equal 0. But excluded volume effects mean that ideal Gij are always negative. With some elementary arithmetic, by subtracting the ideal Gij from the real value, it is possible to calculate Nij the “excess number”, i.e. how many more molecules of j are around i than would be found in the ideal case. They give an indication of the scale of the influence of molecules on each other.14–17,30–32 If Nij = 2 that does not mean that there is a trimer, it just means that somewhere in the vicinity of i there are, on average, 2 more j than expected. Note that there are 4 excess numbers because although Gij = Gji, Nij ≠ Nji.14–17,30–32
Applying the theory
The general ideas set us up for the only equation to be used in this paper. KB theory shows that the derivative of the solute's molar solubility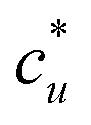 with respect to the hydrotrope concentration c2 is given by:14–17,31,32
with respect to the hydrotrope concentration c2 is given by:14–17,31,32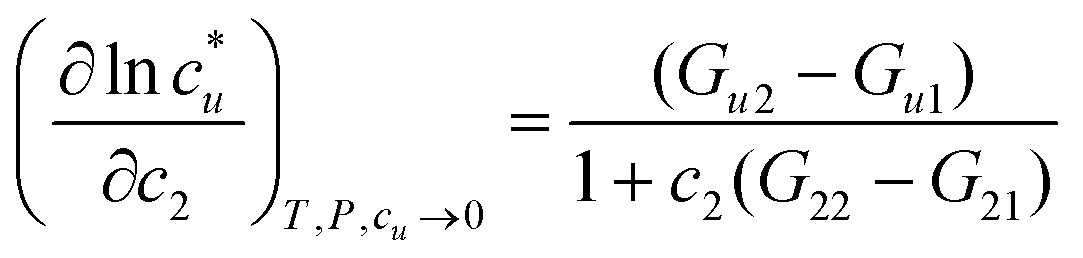 | (1) |
The derivative on the left-hand side is simply obtained from the curve of solubility versus hydrotrope concentration, with a large value implying a large increase in concentration.14–17,31,32 So, fundamental, assumption-free thermodynamics tells us that for a large increase in solubility we need a large difference between Gu2 and Gu1 and/or a small difference between G22 and G21. This already tells us that water structure, measured by G11 is irrelevant as it does not appear in the rigorous equation.14,17 It also says that self-clustering of the hydrotrope, G22 reduces the solubility by increasing the value of the denominator.14–17 We instinctively know that Gu1 is small because if it were large then without the help of the hydrotrope the solubility would be large. So, to increase solubility a large Gu2 is required – the hydrotrope and solute should show a strong positive interaction.14–17
All that remains for the formulator to have a deep understanding of any given system is to see how the various Gij values change over the concentration range of interest and then, to find optimum systems, compare and contrast how changes to the system change the key terms in the equation.
The equations for obtaining the various Gij data from the experimental data are not particularly complex and the procedure is not especially difficult. They are fully described in our previous papers.14–17 Nevertheless, most formulators, including the present authors, would struggle to get every detail correct and to avoid classic pitfalls in a plethora of unit conversions. The (open-source) KB-hydrotrope app, http://www.stevenabbott.co.uk/practical-solubility/kb-hydrotropes.php, does all the hard work (see ESI† for instructions on how to use it).
The app comes with 16 datasets created by the authors from raw data found in the literature, but users can readily load their own data. The user provides a simple tab-separated (or comma-separated) file containing the key data for the analysis:
1. Molar solubility of the solute versus molar concentration of the hydrotrope
2. Density values (g cc−1) for a series of molar hydrotrope concentrations
3. Osmolality versus molality for a series of hydrotrope concentrations (the same samples can be used for density measurements, but this is not a necessary requirement).
Outputs of the Gij values are visual (with mouse read-out), but a comprehensive data set is also provided within a text box. Via a copy/paste from this box into Excel, the user can analyse the data more deeply. This rather crude way of extracting data is a consequence of the natural security of HTML5/Javascript apps – they are not allowed to directly do anything to the user's file system or Clipboard.
As indicated in eqn (1) the example shown in the app, butyl acetate with urea as hydrotrope, the solubilisation is due to the high Gu2, peaking at (from the mouse readout) 2300 (all values in cc per mole). In this case, the low self-association of urea is shown by a G22 of −40. The comparative efficacies of sodium benzoate and sodium salicylate, and their root causes can equally be studied in the app and can be compared to published analyses.14 It is interesting to compare (easily done in the app, not shown here) the cases of p-aminobenzoic acid solubilised by urea and nicotinamide. With urea the Gu2 is relatively small, 760, and the same modest G22 of −40, but with nicotinamide Gu2 is higher 1450, but also with a significant G22 of 1200 which somewhat reduces its efficacy. As before, the app data can be compared to the published analysis.17
Again, the expectation is that from such analyses of many datasets the reasons for the success or failure of various solute/hydrotrope pairs will become apparent.
Limitations and alternatives
The current app is limited to situations where the solute is at relatively low concentrations.14–17 At higher concentrations the KB theory still remains assumption-free but the methodology for extracting the Gij values becomes more complex and is not included in the current version. An alternative app based upon a more general theory36 for handling a full ternary phase diagram shows that KB theory has no intrinsic limitation. However, the ternary app deals only with fully miscible liquids and is currently not so useful for the green formulator.Another limitation is that the KB equations are not good at critical points of phase separation.28–38 So a ternary system that has regions of immiscibility can only give Gij numbers surrounding the phase boundaries. KB can probably handle the fascinating “pre-ouzo” region (which has attractive green solubilization potential) but certainly cannot handle the phase separation domain.8,11 Nevertheless, a good delineation of phase boundaries still provides a lot of key information that maps to a formulator's intuition.
In the present app, an alternative approach, which can describe the cooperative (sigmoidal) solubility increase, has also been implemented.41 This operative solubilisation theory attributes the sigmoidal solubility increase to the enhancement of n-body hydrotrope association when the solute comes into the solution.41 As can be demonstrated in the app featured in Fig. 4, the overall sigmoidal shape can be reproduced using only three parameters described in the reference, including n. This theory is approximate, and does not contain all the KBIs, but may be useful for an overall understanding of hydrotrope action.41
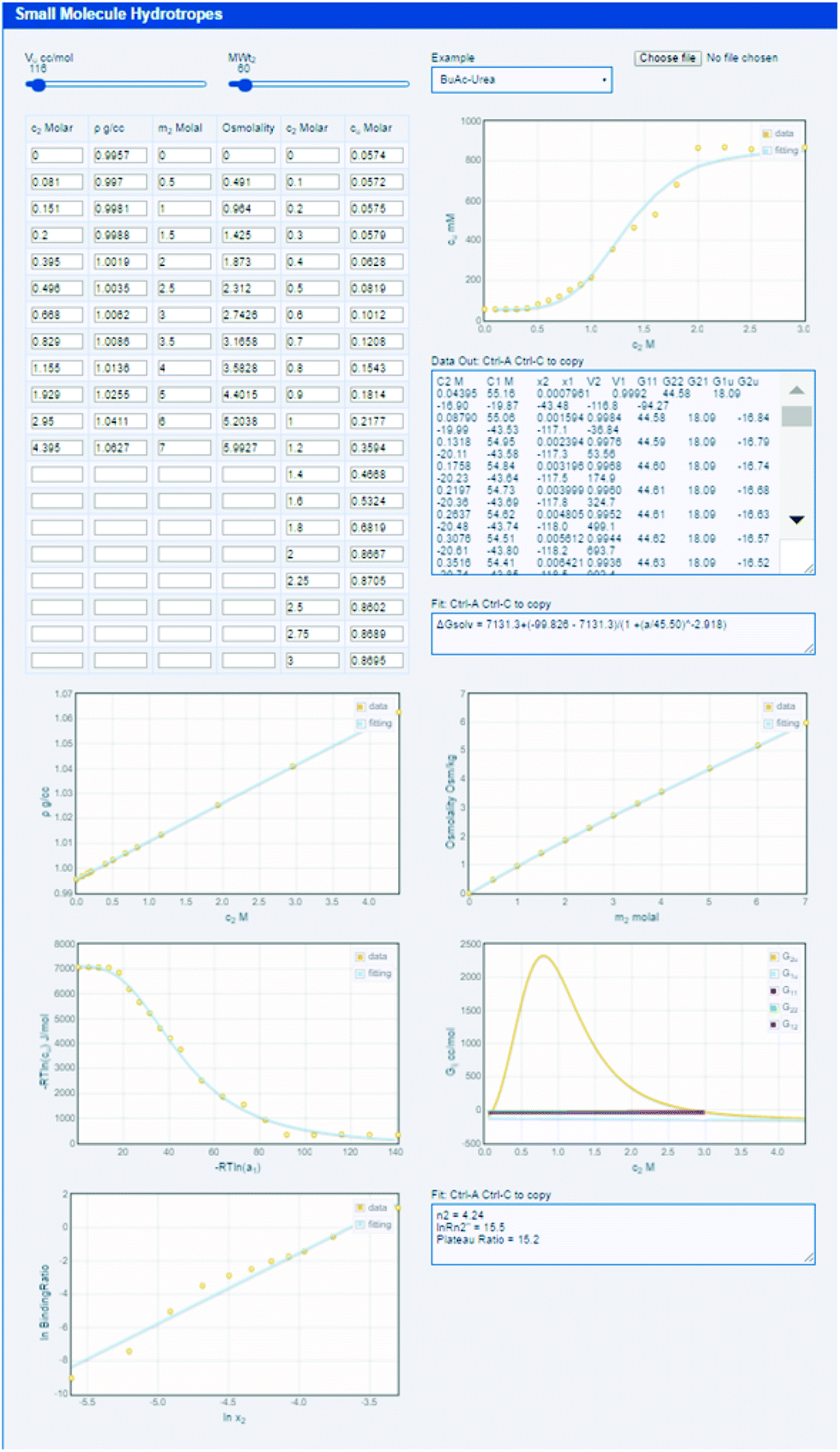 | ||
| Fig. 4 The KB-hydrotrope app does all the hard work of fitting the raw data to the fundamental theory, outputting graphs and data for further processing. In addition to 16 datasets from the literature, the user can load their own datasets for analysis. | ||
One of the many beautiful aspects of thermodynamics is that the same problem can be viewed from several different perspectives. Those who prefer to work with enthalpies and entropies (and the issue of entropy/enthalpy compensation) could, in principle, take the same experimental data and derive thermodynamic values that are equally valid. The problem is that there is no direct, rigorous and tractable link between solubility and solution structure from an entropy/enthalpy point of view,42–44 nor is there an appified version of such an approach that allows the formulator to easily gain a deep, intuitive understanding of what is going on. It would be very useful if such alternatives existed because the different ways of looking at the same problem can provide insights that neither approach on its own can give. For those who are interested in such aspects, it is indeed possible to go from KBIs to entropy/enthalpy values but the present authors have not provided this functionality in the apps.
What about prediction? As mentioned above, even with advanced molecular dynamics simulations it happens to be extremely difficult to go from computed RDFs to reliable KBIs.38 Even if this were possible, molecular dynamics will show the same complicated picture of multiple, statistical interactions that are somewhat stronger or weaker depending on the system. So prediction, as opposed to understanding, is currently not possible via the KB approach.
This is, to say the least, unfortunate. But the present authors believe that with a sufficient corpus of datasets, formulators will be able to generate rules of thumb or more sophisticated algorithms to provide the necessary prediction. Already it is possible to see why urea may or may not be a better hydrotrope than nicotinamide.17 For solutes where, by intuition, nicotinamide is not going to be strongly attracted to the solute, urea would tend to win because the rather large G22 value of nicotinamide gives it a significant disadvantage. With a proper KB analysis of the cases where SCS (or the similar sodium xylene sulfonate, SXS) are used on an industrial scale it should be possible to intuit why they are so successful in these specific applications, and to suggest alternatives for cases where they do not work.
Conclusion
The KB molecular thermodynamics approach described here links the macroscopic world of solubilities, densities and vapour pressures to the molecular realm in a fundamental manner.14–17 For issues of solubilization in the area between the comparatively well-understood zones of cosolvency and micellar solvency, it offers a fundamental, assumption-free way for the green formulator to grasp what is going on in a complex aqueous system.14–17 Because KB uses radial distribution functions,28–38 the formulator can gain an intuitive insight, via KB integrals, into what is going on at the molecular level. Because the key values can be obtained from a set of rather simple measurements (solubility, density, vapour pressure) which, in turn, can be obtained by modern high-throughput techniques,14–17,28–38 the green community has the opportunity to build up a wide dataset of KB values for different solutes and solubilizers. From a wider dataset, a more general understanding of what works and what does not should emerge.For those who are comfortable with the algebra of statistical thermodynamics, the approach adopted here is described in rigorous detail in our previous papers.14–17 For those who are less comfortable, the KB theory itself can be explained via a series of apps that allow the formulator to build up an understanding of what the theory really means.
While the extraction of the KB parameters from experimental data requires a series of tedious calculations, a set of general-purpose, open source apps allows the green community to obtain the key KB values easily and reliably from their own experimental data. By being open source, the apps can be challenged, modified and developed collaboratively by the hydrotrope community.
Alternative app-based approaches via different, complementary thermodynamic methods would be welcomed. The present authors have chosen KB because it seems to handle these complex systems with a welcome clarity and precision and provides numbers (KBIs) that map onto chemists’ understanding of radial distribution functions. But clearly, other approaches have their own merits.
Acknowledgements
We thank Joshua Reid and Shuntaro Chiba for insightful discussions. S. S. acknowledges the support from the Gen Foundation.References
- A. F. M. Cláudio, M. C. Neves, K. Shimizu, J. N. Canongia Lopes, M. G. Freire and J. A. P. Coutinho, Green Chem., 2015, 17, 3948–3963 RSC.
- A. Lavergne, L. Moity, V. Molinier and J.-M. Aubry, RSC Adv., 2013, 3, 5997–6007 RSC.
- V. Molinier and J.-M. Aubry, Sugar-based hydrotropes: preparation, properties and applications, in Carbohydrate Chemistry, ed. A. P. Rauter, T. Lindhorst and Y. Queneau, RSC, London, 2014, vol. 40 Search PubMed.
- K. B. Ansari and V. G. Gaikar, Chem. Eng. Sci., 2014, 115, 157–166 CrossRef CAS.
- M. L. Klossek, D. Touraud and W. Kunz, Phys. Chem. Chem. Phys., 2013, 15, 10971–10977 RSC.
- C. Neuberg, Biochem. Z., 1916, 76, 107–176 CAS.
- A. M. Saleh and L. K. Elkhordagui, Int. J. Pharm., 1985, 24, 231–238 CrossRef CAS.
- P. Bauduin, A. Renoncourt, A. Kopf, D. Touraud and W. Kunz, Langmuir, 2005, 21, 6769–6775 CrossRef CAS PubMed.
- T. K. Hodgdon and E. W. Kaler, Curr. Opin. Colloid Interface Sci., 2007, 12, 121–128 CrossRef CAS.
- C. V. Subbarao, I. P. K. Chackravarthy, A. V. S. L. Sai Bharadwaj and K. M. M. Prasad, Chem. Eng. Technol., 2012, 35, 225–237 CrossRef CAS.
- W. Kunz, K. Holmberg and T. Zemb, Curr. Opin. Colloid Interface Sci., 2016, 22, 99–107 CrossRef CAS.
- A. M. Troncoso and E. Acosta, J. Phys. Chem. B, 2012, 116, 14051–14061 CrossRef CAS PubMed.
- A. M. Troncoso and E. Acosta, J. Colloid Interface Sci., 2016, 466, 400–412 CrossRef PubMed.
- J. J. Booth, S. Abbott and S. Shimizu, J. Phys. Chem. B, 2012, 116, 14915–14921 CrossRef CAS PubMed.
- S. Shimizu, J. J. Booth and S. Abbott, Phys. Chem. Chem. Phys., 2013, 15, 20625–20632 RSC.
- S. Shimizu and N. Matubayasi, J. Phys. Chem. B, 2014, 118, 10515–10524 CrossRef CAS PubMed.
- J. J. Booth, M. Omar, S. Abbott and S. Shimizu, Phys. Chem. Chem. Phys., 2015, 17, 8028–8037 RSC.
- N. N. Gandhi, M. D. Kumar and N. Sathyamurthy, J. Chem. Eng. Data, 1998, 43, 695–699 CrossRef CAS.
- S. T. Kumar and N. N. Gandhi, Int. J. Pharm. Pharm. Sci., 2012, 4, 324–330 Search PubMed.
- Y. P. Koparkar and V. G. Gaikar, J. Chem. Eng. Data, 2004, 49, 800–803 CrossRef CAS.
- D. Balasubramanian, V. Srinivas, V. G. Gaikar and M. M. Sharma, J. Phys. Chem., 1989, 93, 3865–3870 CrossRef CAS.
- R. E. Coffman and D. O. Kildsig, Pharm. Res., 1996, 13, 1460–1463 CrossRef CAS.
- R. Chitra and P. E. Smith, J. Phys. Chem. B, 2002, 106, 1491–1500 CrossRef CAS.
- H. S. Frank and F. Franks, J. Chem. Phys., 1968, 48, 4746–4757 CrossRef CAS.
- R. E. Coffman and D. O. Kildsig, J. Pharm. Sci., 1996, 85, 951–954 CrossRef CAS PubMed.
- R. Sanghi, D. Evans and S. Yalkowsky, Int. J. Pharm., 2007, 336, 35–41 CrossRef PubMed.
- Y. Cui, Int. J. Pharm., 2010, 397, 36–43 CrossRef CAS PubMed.
- J. G. Kirkwood and B. F. Buff, J. Chem. Phys., 1951, 19, 774–777 CrossRef CAS.
- A. Ben-Naim, Molecular Theory of Solutions, Oxford University Press, New York, 2006 Search PubMed.
- S. Shimizu, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 1195–1199 CrossRef CAS PubMed.
- S. Shimizu and C. L. Boon, J. Chem. Phys., 2004, 121, 9147–9155 CrossRef CAS PubMed.
- S. Shimizu and N. Matubayasi, J. Phys. Chem. B, 2014, 118, 3922–3930 CrossRef CAS PubMed.
- S. Shimizu, R. Stenner and N. Matubayasi, Food Hydrocolloids, 2017, 62, 128–139 CrossRef CAS.
- P. E. Smith, J. Phys. Chem. B, 2004, 108, 18716–18724 CrossRef CAS.
- I. L. Shulgin and E. Ruckenstein, J. Phys. Chem. B, 2006, 110, 12707–12713 CrossRef CAS PubMed.
- http://www.stevenabbott.co.uk/practical-solubility/kb-ternary.php based on. E. Matteoli, P. Gianni and L. Lepori, Kirkwood-Buff integrals in fully miscible ternary systems: thermodynamic data, calculation, and interpretation, in Fluctuation Theory of Solutions, ed. P. E. Smith, E. Matteoli and J. P. O'Connell, CRC Press, Boca Raton, FL, 2013 Search PubMed.
- S. Chiba, T. Furuta and S. Shimizu, J. Phys. Chem. B, 2016, 31, 7714–7723 CrossRef PubMed.
- D. V. Schroeder, Am. J. Physiol., 2015, 83, 210–218 CAS.
- P. Atkins and J. de Paula, Atkins’ Physical Chemistry, Oxford University Press, Oxford, 10th edn, 2014 Search PubMed.
- T. V. Chalikian, Annu. Rev. Biophys. Biomol. Struct., 2003, 32, 207–235 CrossRef CAS PubMed.
- S. Shimizu and N. Matubayasi, Phys. Chem. Chem. Phys., 2016, 18, 25621–25628 RSC.
- N. Matubayasi, L. H. Reed and R. M. Levy, J. Phys. Chem., 1995, 98, 10640–10649 CrossRef.
- N. Matubayasi and R. M. Levy, J. Phys. Chem., 1996, 100, 2681–2688 CrossRef CAS.
- N. Matubayasi, E. Gallicchio and R. M. Levy, J. Chem. Phys., 1998, 109, 4864–4872 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6gc03002e |
| ‡ Present address: Croda Europe, Cowick Hall, Snaith, Goole, East Yorkshire, DN14 9AA, UK. |
| This journal is © The Royal Society of Chemistry 2017 |