
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Furfurylamines from biomass: transaminase catalysed upgrading of furfurals†
Alice
Dunbabin
a,
Fabiana
Subrizi
a,
John M.
Ward
b,
Tom D.
Sheppard
 a and
Helen C.
Hailes
*a
a and
Helen C.
Hailes
*a
aDepartment of Chemistry, University College London, 20 Gordon Street, London WC1H 0AJ, UK. E-mail: h.c.hailes@ucl.ac.uk; Tel: +44 (0)20 7679 4654
bDepartment of Biochemical Engineering, University College London, Bernard Katz Building, Gordon Street, London, WC1H 0AH, UK
First published on 20th September 2016
Abstract
Furfural is recognised as an attractive platform molecule for the production of solvents, plastics, resins and fuel additives. Furfurylamines have many applications as monomers in biopolymer synthesis and for the preparation of pharmacologically active compounds, although preparation via traditional synthetic routes is not straightforward due to by-product formation and sensitivity of the furan ring to reductive conditions. In this work transaminases (TAms) have been investigated as a mild sustainable method for the amination of furfural and derivatives to access furfurylamines. Preliminary screening with a recently reported colorimetric assay highlighted that a range of furfurals were readily accepted by several transaminases and the use of different amine donors was then investigated. Multistep synthetic routes were required to synthesise furfurylamine derivatives for use as analytical standards, highlighting the benefits of using a one step biocatalytic route. To demonstrate the potential of using TAms for the production of furfurals, the amination of selected compounds was then investigated on a preparative scale.
Introduction
Furfurals have attracted significant interest in recent years as renewable feedstocks for the production of biofuels and chemicals.1 In particular, furfural 1a and 5-hydroxymethylfurfural (5-HMF) 2a are valuable platform chemicals, prepared via the acid-catalysed dehydration of pentoses and hexoses obtained by the hydrolysis of cellulosic biomass waste including cornstalks, corncobs and rice waste.2 Due to improved catalytic processes furfurals are becoming more available and their production has been integrated into a biorefinery context.3 Indeed, renewable 5-HMF is being manufactured commercially.4 As a consequence, several processes have been developed for the conversion of furfurals into a number of valuable chemicals and fuels such as furfuryl alcohols, THFs, furfurylamine 1b, 1,5-pentanediol and functionalised aromatics.3,5 Among these, the production of furfurylamines by the selective reductive amination of furfurals has received interest due to many potential applications, including as intermediates in the synthesis of pharmaceuticals such as antiseptic agents, antihypertensives, and diuretics (e.g. Furosemide).6 The 5-HMF 2a derived amine 2b also has potential as a curing agent in epoxy resins. Selective synthesis of these primary amines from carbonyl compounds is still challenging. Indeed, the use of traditional synthetic routes is not straightforward due to the sensitivity of the furan ring to reductive conditions and the tendency to form secondary or tertiary amine by-products.7 Moreover, the waste generated from using such reducing agents has to be considered especially within the green chemistry agenda.Transaminase (TAm) enzymes have been investigated in recent years for the transformation of pro-chiral ketones and aldehydes into the corresponding chiral secondary, or primary amines.8 These transferases can provide a sustainable high yielding, selective route to amines under mild aqueous conditions. The use of TAms with furfural analogues in the literature has surprisingly received very little attention with only two previous reports: a kinetic resolution of racemic 1-(2-furfuryl)ethylamine using commercially available TAms;9 use of 5-HMF 2a in an enzyme cascade incorporating the Vibrio fluvialis TAm with alanine dehydrogenase to shift the TAm equilibrium.10
Here we describe the use of TAms for the amination of furfural derivatives 1a–9a and ketone 10a to access amines 1b–10b (Scheme 1). High conversions were observed with a range of substrates. The amination of selected compounds was then investigated for the production of value-added chemicals on a preparative scale.
 | ||
| Scheme 1 Use of TAms with co-factor pyridoxal-5-phosphate (PLP), aldehydes and ketone 1a–10a to generate amines 1b–10b. | ||
Results and discussion
Initial substrate screening
Ten furan-based substrates were screened including furfural 1a, 5-HMF 2a, halogenated furfurals 3a–6a, 5-methylfurfural 7a, 5-formyl-2-furancarboxylic acid 8a, 2,5-furandicarboxaldehyde 9a, and 2-acetylfuran 10a (Fig. 1). TAms selected for screening included the (S)-ω-TAm Chromobacterium violaceum DSM30191 (CV-TAm), which has been used with a range of cyclic and aromatic substrates and demonstrated tolerance towards the low cost amine donor isopropylamine (IPA) 11.11–14 The (R)-selective TAms Arthrobacter sp. variant ArRMut1115 and Mycobacterium vanbaalenii (Mv-TAm),16 were also selected due to their complementary enantioselectivities compared to CV-TAm for use with the ketone 10a. In addition, ArRMut11 has been used with a wide range of substrates and also has good tolerance towards IPA 11 as an amine donor.15–19 | ||
| Fig. 1 Furan-based substrates 1a–10a screened against TAms, and amine donors IPA 11 and MBA 13. | ||
Initial screening for the conversion of aldehydes and ketone 1a–10a, was carried out using crude cell extract and our recently reported rapid and sensitive TAm colorimetric assay (Scheme 2).20 It uses an inexpensive amine donor 12 that forms the corresponding aldehyde shown when a TAm reaction occurs: the aldehyde generated can then react with the major amine present (12) to form an imine which tautomerises to give a red precipitate, indicating that the TAm reaction has occurred.20 The results of the colorimetric assay are shown in Fig. 2 against benzaldehyde (+) as a positive control. When using CV-TAm a strong coloration was observed with all aldehyde substrates. Notably, furfural 1a, 5-HMF 2a and acid 8a were readily converted showing a deep red coloration. In addition, aldehydes were accepted by both ArRMut11 and Mv-TAm while ketone 10a showed little coloration with all TAms perhaps due to imine formation with 12.
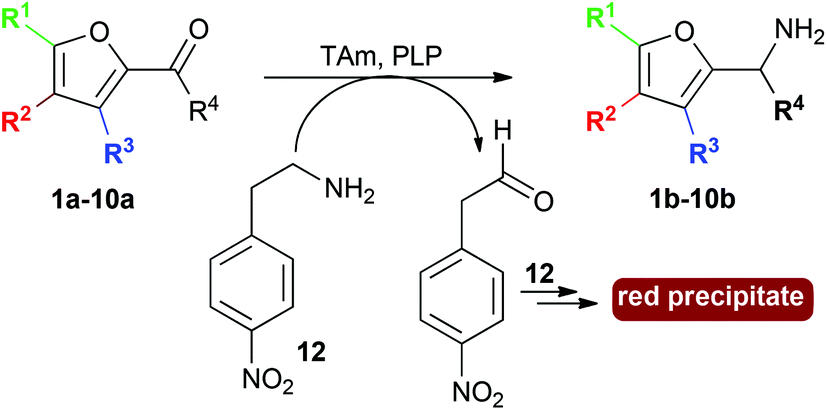 | ||
| Scheme 2 Colorimetric TAm assay using 12. | ||
 | ||
| Fig. 2 Colorimetric assay (in duplicate) using 2-(4-nitrophenyl)ethan-1-amine 12 as amine donor: 12 (25 mM), amino acceptor (10 mM), PLP (0.2 mM), KPi buffer pH 7.5 (100 mM) and enzyme crude lysate, 24 h, 30 °C, 500 rpm. Benzaldehyde was used as a positive control (+) and water as a negative control (−). | ||
Activity of the TAms towards 1a–10a (with benzaldehyde as a positive control) was also confirmed using either (S)- or (R)-α-methylbenzylamine (MBA) 13 as amine donors, depending on the selectivity of the enzyme (Fig. 3). Notably furfural 1a and 5-HMF 2a were readily accepted by all three enzymes and in comparable conversions to benzaldehyde with CV-TAm and Mv-TAm (see Fig. 3 caption). Also, the 3- and 4-bromo derivatives 3a and 4a gave 50–60% conversions with all the selected transaminases. Within this series, the 5-substituted furfurals 5a and 7a were particularly well accepted by Mv-TAm (82% and 79% conversions respectively), while significantly lower conversions were achieved with 5a and both CV-TAm and ArRMut11 and 7a with CV-TAm. However, the less polar 5-methylfurfural 7a showed a 60% conversion with ArRMut11.
 | ||
| Fig. 3 Screening results for substrates 1a–10a (5 mM) using 25 mM (S)-13 (CV-TAm) or 25 mM (R)-13 (ArRMut11 and Mv-TAm) as amine donors, 24 h, 30 °C. The product acetophenone was detected by HPLC analysis (at 254 nm) and used to determine % conversions. Background levels of acetophenone production were subtracted from the assay results in control reactions, and all reactions were performed in triplicate (standard deviations were less than 10%). Benzaldehyde (conversion of 68% with CV-TAm, 34% with ArRMut11, and 64% with Mv-TAm) was used as a positive control. *For 9a product formation was determined using a synthesised diamine standard 9b (see below). | ||
Notably, the acid 8a was accepted by CV-TAm in approximately 50% conversion, while very low conversions were achieved with ArRMut11. By comparison, 2,5-furan dicarboxaldehyde 9a performed well with all the selected enzymes. Due to complexities arising from the potential formation of monoamine or diamine products when using 9a, product yields were determined using the diamine 9b (see Scheme 3 for synthesis), which was formed in good yield with all the TAms (56–70%). Finally, 2-acetylfuran 10a was accepted by all three TAms, with Mv-TAm giving the highest conversions. Overall, following the initial screening hits from the colorimetric assay, the MBA-screen highlighted that the presence of more polar groups at C-5 reduced conversions for ArRmut11, but this had less effect with CV-TAm and Mv-TAm.
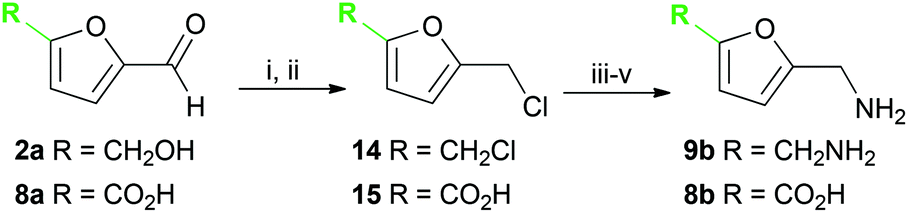 | ||
| Scheme 3 (i) NaBH4; (ii) HCl; (iii) NaN3; (iv) PPh3 then Boc2O; (v) HCl. | ||
Amine donor selection and synthesis of 2b, 8b and 9b
Several methods are known to shift the TAm equilibrium towards the amine product, including the use of excess amine donor and enzyme coupled systems. One of the most operationally straightforward and cost-effective is the use of IPA 11 as the amine donor. Since IPA and the co-product acetone are both volatile, the scale-up and isolation of products is also more facile. Furfural 1a, 5-HMF 2a, acid 8a and dialdehyde 9a were taken forward for reaction optimisation using IPA 11 as the amine donor. The reaction with 2-acetylfuran 10a was also explored further to establish stereoselectivities. Since furfurylamines 2b, 8b, and 9b were not readily available commercially but required as analytical standards they were synthesised (Scheme 3). As the furan ring is sensitive to reductive conditions, and traditional reductive amination routes resulted in mixtures of di- and trisubstituted amines,7 a multi-step route for the preparation of 9b and 8b was established through chlorination to 14 and 15 and azide formation followed by reduction with triphenylphosphine (PPh3). Derivatisation of the amine with tert-butoxycarbonyl anhydride (Boc2O) was necessary to enable more straightforward product isolation and purification from side-products. The amine standards were then used as chemical standards to determine TAm reaction yields by HPLC.Efforts to prepare 5-hydroxymethylfurfurylamine 2b starting from 5-HMF 2a following a similar strategy were unsuccessful. Moreover, attempts to reduce the carboxylic acid 8b or the corresponding azide with a range of reducing agents including LiAlH4 and BH3-THF resulted in complex mixtures of products that could not be purified. Instead, an enzymatic reaction using CV-TAm and IPA as amine donor was the most efficient method of preparing 2b, which was isolated after scale-up (see below) and used as a standard.
It is worth noting that procedures reported to afford 2b are typically very low yielding (4% yield) or use unsustainable metal-based catalysts.21,22 This example, together with the generally low yields of the multi-step synthesis, emphasises problems with such traditional synthetic approaches and the benefits of a one step biocatalytic route to compounds such as the amino acid 8b.
Reaction product yields when using amine donors 11 and 13 are shown in Table 1 using typical reaction conditions previously utilised in such TAm reactions.14 For IPA 11 (10 equiv.) higher yields were generally observed than when using MBA 13 (5 equiv.), highlighting the benefits of using excess amine donor and possibly the slightly higher reaction temperature. Yields of up to 92% were obtained for furfurylamine 1b with CV-TAm, but again lower yields (34%) were observed when using ArRMut11. The same trend was observed using 2a, which was readily accepted by CV-TAm and Mv-TAm, but showed a lower product yield with ArRMut11. Notably, with CV-TAm the amino acid 8b was produced in 88% yield with IPA, compared to 47% with MBA, and again ArRMut11 gave 8b in very low yield. For the dialdehyde 9a, when using IPA, yields of the diamine 9b were slightly lower than for MBA. However, since a double transamination was required the use of twice as much enzyme was explored with IPA 11. Yields of the diamine increased up to 60% for CV-TAm (Table 1). It was not possible to determine the yield of the mono-aminated product since it was unstable during synthetic investigations, forming complex polymerised mixtures via imine formation: this may also account for why the TAm yields observed did not increase markedly when using twice as much enzyme with 9a. The product yields for 2-acetylfuran 10a with MBA were confirmed using a commercial sample of 10b and HPLC, again highlighting Mv-TAm (54%) as the most productive transaminase, giving (R)-10b in 78% ee (absolute stereochemistry based on the reported stereoselectivity of this TAm). The reaction with CV-TAm was lower yielding so the ee was not investigated.
| Product |
|
|
|
|
|
||||
|---|---|---|---|---|---|---|---|---|---|
| Amine donor | MBA 13 | IPA 11 | MBA 13 | IPA 11 | MBA 13 | IPA 11 | MBA 13 | IPA 11 | MBA 13 |
| a Reactions were performed in triplicate and conversions were determined using HPLC against product standards. b Assays were performed on a 200 μL total reaction volume containing MBA 13 (25 mM) or IPA 11 (100 mM), PLP (1 mM), potassium phosphate buffer (100 mM, pH 7.5 for MBA and pH 8 for IPA), amine acceptor (5 mM when MBA was used or 10 mM when IPA was used) and crude cell lysate (20 μL) at 30 °C with MBA and 35 °C with IPA, for 24 h. (S)-13 or (R)-13 was used depending on enzyme selectivity. c Yields obtained using double the amount of TAm enzyme. d IPA was used as an amine donor except for compound 10b; ee determined using chiral HPLC; n.d. not determined. | |||||||||
| CV-TAm | 80% | 92% | 75% | 89% | 47% | 88% | 70% | 48% (60%c) | 18% [ee n.d.] |
| ArRMut11 | 59% | 34% | 69% | 44% | 4% | 3% | 57% | 45% (53%c) | 1% [ee n.d.] |
| Mv-TAm | 75% | 78% | 53% | 66% | 32% | 59% | 56% | 46% (52%c) | 54% [78% ee (R)] |



Preparative scale reaction
Furfurylamines are valuable biomass-derived products and direct syntheses were then investigated on a preparative scale using CV-TAm, due to its high yields using IPA 11 as the amine donor (Table 1), together with aldehydes 1a, 2a, and 8a. The reactions were performed on a 20 mM scale (in 50 mL) for 24 h after which no starting materials remained. HPLC analysis established product yields of 83%, 58%, and 40% for 1b, 2b, and 8b respectively. The lower yields for 2b and 8b may have been due to some product inhibition at higher substrate concentrations. Furfurylamine 1b was not isolated as it is commercially available. As mentioned above, 2b was required as a product standard for analytical purposes in this work and was readily generated using this one-step enzymatic route from 5-HMF 2a. The product was isolated in 54% yield, highlighting the potential and benefits of this synthetic strategy compared to traditional chemical approaches. The preparative scale reaction using acid 8a was also of particular interest as amino acid 8b is a cyclic analogue of γ-aminobutyric acid (GABA) and has activity as an inhibitor of GABA aminotransferase, with Km values similar to GABA.23 Moreover, 8b belongs to the class of furanoid sugar amino acids which has been incorporated into novel anticancer peptides.24 The product 8b of the biotransformation reaction was successfully isolated as Boc-8b for ease of isolation purposes, and directly deprotected to give 8b in 31% overall isolated yield and high purity.Conclusions
Amines derived from furfural have diverse applications, including the synthesis of furan containing polymers from the amine products of 5-HMF and 2,5-furandicarboxaldehyde, which are a renewable alternative to conventional polymers.4 Three TAms have been investigated for the transamination of a range of furan-based aldehydes and a ketone. The initial colorimetric assay highlighted that all the furfurals 1a–9a were readily accepted by the selected TAms using 2-(4-nitrophenyl)ethan-1-amine hydrochloride 12 as the donor, and this was validated using an MBA screen. The use of IPA was then investigated as an alternative amine donor with 1a, 2a, 8a, and 9a. In most cases higher yields were obtained with some yields in excess of 90%, while 2-acetylfuran 10a was more readily converted to the corresponding amine when MBA was used as the amine donor. Procedures using IPA allowed reactions to be performed on a preparative scale in one-step and good yields. This approach uses a sustainable feedstock for the preparation of known and novel fine chemicals. The biocatalytic process for the production of furfurylamine and derivatives has significant potential in applications for manufacturing drug synthons and monomers for use in polymer production with high-performance25 and biodegradable26 properties.Experimental
General
All chemicals were obtained from commercial suppliers and used as received unless otherwise stated. Thin layer chromatography (TLC) analysis was performed on Merck Kieselgel precoated aluminium-backed silica gel plates and compounds visualised by exposure to UV light, potassium permanganate or ninhydrin stains. Flash column chromatography was carried out using silica gel (particle size 40–60 μm). NMR: 1H and 13C NMR spectra were recorded at 298 K at the field indicated using Bruker spectrometers AMX400, Avance 500, and Bruker Avance III 600. Coupling constants (J) are measured in Hertz (Hz) and multiplicities for 1H NMR couplings are shown as s (singlet), d (doublet), and m (multiplet). Chemical shifts (in ppm) are given relative to tetramethylsilane and referenced to residual protonated solvent. Infrared spectra were recorded on a Perkin Elmer Spectrum 100 FTIR spectrometer. Mass spectrometry analyses were performed at the UCL Chemistry Mass Spectrometry Facility using a Finnigan MAT 900 XP mass spectrometer and the EPSRC UK National Mass Spectrometry Facility at Swansea University using a Thermo scientific LTQ Orbitaltrap XL. Melting points were recorded on an Electrothermal IA9000 Series melting point apparatus and are uncorrected.Analytical HPLC
Analyses of the reactions were performed using an Agilent 1260 Infinity HPLC with an Ace 5 C18 column (150 × 4.6 mm). Elution was carried out at 1 mL min−1 with a linear gradient of acetonitrile/H2O containing 0.1% TFA, with detection at 250 or 210 nm, injection volume of 10 μL and column temperature of 30 °C.Transaminase expression and cell crude extract preparation
Selected TAm glycerol stocks of Chromobacterium violaceum DSM3019111 (CV-TAm), Arthrobacter sp. variant ArRMut1115 and Mycobacterium vanbaalenii (Mv-TAm),16 from the UCL TAm library were used to inoculate 2TY broth (5 mL) containing kanamycin (50 μg mL−1) and incubated at 37 °C for 16–18 h. This pre-inoculum was then used to inoculate a larger culture (500 mL) containing the same antibiotic which was incubated at 37 °C for 3 h until an OD600 of 0.5–0.7 was reached. Enzyme expression was induced by the addition of isopropyl-β-D-thiogalactopyranoside (1 mM), and the temperature was reduced to 30 °C.14 After 5 h cells were harvested by centrifugation (8000 rpm, 20 min) and the pellet suspended in potassium phosphate buffer (100 mM, pH 7.5) containing pyridoxal-5-phosphate (PLP) (1 mM) and freeze dried. The freeze dried cells were used fresh or stored at −20 °C for up to six months. To prepare crude cell extract, freeze dried cells (25 mg) were suspended in potassium phosphate buffer (1 mL, 100 mM, pH 7.5), lysed by sonication on ice (10 s on and 10 s off for 5 cycles) and used as a crude lysate (10% v/v).TAm concentrations were determined by SDS-PAGE densitometry: densitometry of samples electrophoresed on a NuPAGE 10% Bis–Tris gel (Novex) was used. The Coomassie stained gel was imaged using LabWork image software to calculate the TAm band density. A range of concentrations of commercial BSA was run in each SDS-PAGE gel (ESI Fig. 1†), and used to calculate a standard curve based on integrated optical density for calibration of the enzyme concentration. Total protein was determined using a standard Bradford assay. TAm concentrations in the crude lysates were determined as 5.7 mg mL−1 CV-TAm, 4.0 mg mL−1 ArRMut11 and 2.5 mg mL−1 Mv-TAm.
Colorimetric screening
The assay was performed in a 96 well-plate with a total volume of 200 μL containing 2-(4-nitrophenyl)ethan-1-amine hydrochloride 12 (25 mM) as amine donor, amine acceptor (10 mM), PLP (0.2 mM) and potassium phosphate buffer (100 mM, pH 7.5). The reaction was started by the addition of E. coli crude cell extract (20 μL) containing the overexpressed TAm and the reaction was incubated at 30 °C and 500 rpm for 24 h. Two negative controls were also performed, one without amine acceptor and another without enzyme. An orange/red coloration indicated that the TAms were active towards the selected furfurals (Fig. 2).MBA (13) screening
The assay was performed in an Eppendorf tube (200 μL total volume) containing (R)- or (S)-MBA (13) (25 mM), PLP (1 mM), potassium phosphate buffer (100 mM, pH 7.5), amine acceptor (5 mM) and crude cell lysate (20 μL). After incubation at 30 °C and 300 rpm for 24 h, the reaction was stopped by the addition of 10% trifluoroacetic acid (TFA) in water (10 μL). Denatured protein was removed by centrifugation (4 °C, 3000 rpm, 4 min) and the supernatant diluted and analysed by analytical HPLC.Isopropylamine (11) assay
The assay was performed in an Eppendorf tube containing isopropylamine (11) (100 mM, pH 8), PLP (1 mM), potassium phosphate buffer (100 mM, pH 8), amine acceptor (10 mM) and crude cell lysate (20 μL). After incubation at 35 °C and 300 rpm for 24 h, the reaction was stopped by the addition of 10% TFA in water (10 μL). Denatured protein was removed by centrifugation (4 °C, 3000 rpm, 4 min) and the supernatant diluted and analysed by analytical HPLC.5-Hydroxymethyl-2-furancarboxylic acid
To 5-formyl-2-furancarboxylic acid 8a (140 mg, 1.00 mmol) in methanol (5 mL) at 0 °C, NaBH4 (57 mg, 1.5 mmol) was added in portions and the reaction was stirred for 4 h. The reaction was quenched with brine (5 mL) and the methanol removed under reduced pressure. The aqueous residue was acidified and extracted with ethyl acetate (3 × 30 mL), washed with brine (2 × 30 mL), dried (Na2SO4) and the solvent removed under reduced pressure to give 5-hydroxymethyl-2-furancarboxylic acid27 as a colourless solid (115 mg, 81%). M.p. 157–158 °C (EtOAc), lit. 163–164 °C;28vmax (film) 3236, 2414 br, 1650, 1595 cm−1; 1H NMR (MeOH-d4; 500 MHz) 4.56 (2H, s, CH2OH), 6.45 (1H, d, J 3.4 Hz, 4-H), 7.15 (1H, d, J 3.4 Hz, 3-H); 13C NMR (MeOH-d4; 125 MHz) 57.5, 110.2, 120.0, 145.7, 160.7, 161.8; m/z (EI) 142 ([M]+, 28%), 123 (22), 97 (100), 69 (50).5-Chloromethyl-2-furancarboxylic acid (15)
To 5-hydroxymethyl-2-furancarboxylic acid (91 mg, 0.64 mmol) in CH2Cl2 (6 mL), conc. HCl (37%, 2 mL) was added. The reaction was stirred at room temperature for 24 h. Water (10 mL) was added and the reaction was extracted with CH2Cl2 (3 × 15 mL), washed with brine (2 × 15 mL), dried (Na2SO4) and the solvent removed under reduced pressure to give 15 as a colourless solid (45 mg, 44%). M.p. 123–124 °C (CH2Cl2); vmax (film) 3121, 2929, 2800 br, 1675, 1590 cm−1; 1H NMR (MeOH-d4; 600 MHz) 4.70 (2H, s, CH2Cl), 6.59 (1H, d, J 3.5 Hz, 3-H), 7.15 (1H, d, J 3.5 Hz, 4-H); 13C NMR (CDCl3; 125 MHz) 36.6, 111.8, 121.1, 144.0, 155.5, 163.1; m/z MS (EI) 162 ([M37Cl]+, 6%), 160 ([M35Cl]+, 16), 125 (100), 79 (33); HRMS (FTMS) found [M − H]− 158.9858; C6H435ClO3 requires 158.9854.5-Aminomethyl-2-furancarboxylic acid (8b)
Sodium azide (53 mg, 0.82 mmol) was added to 15 (44 mg, 0.27 mmol) in dry DMF (5 mL) under argon, and the reaction was heated at 65 °C for 16 h. The DMF was removed under reduced pressure and the residue dissolved in methanol (8 mL). PPh3 (212 mg, 0.809 mmol) was then added and the reaction was stirred at room temperature. After 3 h Boc2O (118 mg, 0.541 mmol) was added and the reaction was stirred for 16 h. The methanol was removed under reduced pressure, sat. NaHCO3 was added (30 mL) and side-products removed with ethyl acetate (3 × 30 mL). The aqueous layer was then acidified (HCl) and extracted with ethyl acetate (3 × 30 mL). The combined organic extracts were dried (Na2SO4) and the solvent removed under reduced pressure. The product was then stirred in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of methanol and 4 M HCl (6 mL) for 2 h, and the solvent removed under reduced pressure to give 5-aminomethyl-2-furancarboxylic acid hydrochloride salt 8b·HCl23 (29 mg, 60%) as a colourless solid. M.p. 250 °C (decomp.; H2O); vmax (film) 3150, 3011, 2791 br, 1685, 1586 cm−1; 1H NMR (MeOH-d4; 600 MHz) 4.26 (2H, s, CH2NH2), 6.71 (1H, d, J 3.5 Hz, 4-H), 7.22 (1H, d, J 3.5 Hz, 3-H); 13C NMR (MeOH-d4; 600 MHz) 36.8, 113.8, 119.9, 147.4, 152.1, 161.2; HRMS (CI) found [M + H]+ 142.0499; C6H7NO3 requires 142.0499.
:1 mixture of methanol and 4 M HCl (6 mL) for 2 h, and the solvent removed under reduced pressure to give 5-aminomethyl-2-furancarboxylic acid hydrochloride salt 8b·HCl23 (29 mg, 60%) as a colourless solid. M.p. 250 °C (decomp.; H2O); vmax (film) 3150, 3011, 2791 br, 1685, 1586 cm−1; 1H NMR (MeOH-d4; 600 MHz) 4.26 (2H, s, CH2NH2), 6.71 (1H, d, J 3.5 Hz, 4-H), 7.22 (1H, d, J 3.5 Hz, 3-H); 13C NMR (MeOH-d4; 600 MHz) 36.8, 113.8, 119.9, 147.4, 152.1, 161.2; HRMS (CI) found [M + H]+ 142.0499; C6H7NO3 requires 142.0499.
2,5-Bis(hydroxymethyl)furan
NaBH4 (170 mg, 4.49 mmol) was added in portions to 5-HMF 2a (378 mg, 3.00 mmol) in methanol (10 mL) at 0 °C. The reaction was stirred for 4 h, brine (5 mL) was added and the methanol removed under reduced pressure. The aqueous residue was extracted with ethyl acetate (3 × 30 mL), washed with brine (2 × 30 mL), dried (Na2SO4) and the solvent removed under reduced pressure to give 2,5-bis(hydroxymethyl)furan29 as a colourless oil (296 mg, 77%). vmax (film) 3281, 2924, 2866, 1631 cm−1; 1H NMR (CDCl3; 400 MHz) 2.45 (2H, br s, 2 × OH), 4.56 (4H, s, 2 × CH2OH), 6.22 (2H, s, 3-H, 4-H); 13C NMR (CDCl3; 150 MHz) 57.5, 108.7, 154.2; m/z (CI) 256 ([2M]+, 100%), 191 (38), 173 (15), 146 ([MH + NH3]+ 35), 128 (5).2,5-Bis(aminomethyl)furan hydrochloric salt (9b)
To 2,5-bis(hydroxymethyl)furan (256 mg, 2.00 mmol) in CH2Cl2 (6 mL), conc. HCl (37%; 2 mL) was added. The reaction was stirred at room temperature for 16 h. Water (10 mL) was added and the product was extracted with CH2Cl2 (3 × 15 mL), washed with brine (2 × 15 mL), dried (Na2SO4) and the solvent removed under reduced pressure to give 1430 as a brown oil (82 mg, 25%) which was taken directly through to the next step. 1H NMR (CDCl3; 600 MHz) 4.58 (4H, s, CH2Cl), 6.34 (2H, s, 3-H, 4-H); 13C NMR (CDCl3; 150 MHz) 37.4, 110.9, 151.1.Sodium azide (130 mg, 2.00 mmol) was added to 14 (83 mg, 0.50 mmol) in dry DMF (8 mL) under argon, and the reaction was heated at 65 °C for 16 h. The DMF was removed under reduced pressure and the residue was dissolved in methanol (8 mL). PPh3 (525 mg, 2.00 mmol) was added and the reaction was stirred at room temperature. After 2 h Boc2O (436 mg, 2.00 mmol) was added and the reaction was stirred for 16 h. The methanol was removed under reduced pressure, sat. NaHCO3 added (30 mL) and the product was extracted with ethyl acetate (3 × 30 mL), dried (Na2SO4) and the solvent removed under reduced pressure. 2,5-Bis(Boc-aminomethyl)furan was purified by column chromatography (ethyl acetate/petroleum ether (40–60), 1:4) to give a brown solid (63 mg, 39%). It was then directly deprotected by stirring in a 1:1 mixture of methanol and 4 M HCl (6 mL) for 1 h, and the solvent removed under reduced pressure to give 9b·2HCl31 as a brown solid (34 mg, 34% from 14). M.p. 240 °C (decomp.; H2O); vmax (film) 3090, 2851 br, 1596 cm−1; 1H NMR (MeOH-d4; 600 MHz) 4.21 (4H, s, 2 × CH2NH2), 6.61 (2H, s, 3-H, 4-H); 13C NMR (MeOH-d4; 150 MHz) 36.8, 113.2, 149.6; m/z (EI) 126 ([M]+, 30%), 96 (100); HRMS (EI) found [M]+ 126.0788; C6H10N2O requires 126.0788.
1-Furan-2-ethylamine ee determination for Mv-Tam
The assay was performed (800 μL total volume) containing (R)-(13) (25 mM), PLP (1 mM), potassium phosphate buffer (100 mM, pH 7.5), amine acceptor (5 mM) and crude cell lysate (80 μL). After incubation at 30 °C and 300 rpm for 24 h, the reaction was stopped by the addition of 10% trifluoroacetic acid (TFA) in water (40 μL). Denatured protein was removed by centrifugation (4 °C, 3000 rpm, 4 min), the supernatant extracted with diethyl ether (1 mL) and the solvent evaporated. The residue was dissolved in THF (1 mL), 2 M NaOH (50 μL) and benzyl chloroformate (100 μL) were added and the reaction shaken at room temperature for 16 h. Solvents were removed under reduced pressure and the residue dissolved in water (500 μL), extracted with diethyl ether (1 mL) and the solvent evaporated. The product was dissolved in EtOH (100 μL) and analysed by chiral HPLC to give the ee (78% (R)-assigned on the basis of the reported selectivity for Mv-TAm). Retention times: (R)-isomer 27.5 min, (S)-isomer 35.1 min.Preparative scale biocatalytic reactions
The TAm reaction was scaled up (50 mL) with substrate (20 mM), isopropylamine (200 mM, pH 8), potassium phosphate buffer (100 mM, pH 8), PLP (1 mM) and CV-TAm crude cell lysate (10 mL). The reaction was incubated at 37 °C and 200 rpm for 24 h.Furfurylamine (1b)
Furfural 1a (83 μL, 1.0 mmol) was subjected to the preparative scale reaction conditions, and after removal of the denatured protein by centrifugation (4000 rpm, 4 °C, 20 min) was analysed by HPLC, to give the product in 83% yield.5-Hydroxymethylfurfurylamine (2b)
5-HMF 2a (126 mg, 1.00 mmol) was subjected to the preparative scale biocatalytic reaction conditions, quenched with MeOH (100 mL) and the denatured protein removed by centrifugation (4000 rpm, 4 °C, 20 min). Volatile organics were removed under reduced pressure and the remaining aqueous solution extracted with ethyl acetate (3 × 50 mL) to remove any remaining starting material. The pH was changed to pH 10 by addition of 2 M NaOH and the aqueous layer extracted with ethyl acetate (10 × 30 mL) and dried (Na2SO4). The solvent was removed under reduced pressure to give 2b21 as a yellow oil (product yield 58% by HPLC; isolated yield 69 mg, 54%). vmax (film) 3288, 2926, 1650 cm−1; 1H NMR (MeOH-d4; 600 MHz) 3.76 (2H, s, CH2NH2), 4.47 (2H, s, CH2OH), 6.17 (1H, d J = 3.2 Hz, 3-H), 6.22 (1H, d, J = 3.2 Hz, 4-H); 13CNMR (MeOH-d4; 150 MHz) 39.2, 57.4, 107.7, 109.2, 155.3, 156.2; m/z MS (EI) 127 ([M]+, 20%), 96 (100).5-Aminomethyl-2-furancarboxylic acid (8b)
5-Formyl-2-furancarboxylic acid 8a (140 mg, 1.00 mmol) was subjected to the preparative scale reaction conditions, quenched with MeOH (100 mL) and the denatured protein removed by centrifugation (4000 rpm, 4 °C, 20 min). The solution was evaporated to dryness under reduced pressure and the residue dissolved in MeOH (15 mL) (product yield by HPLC 40%). To this solution was added Boc2O (874 mg, 4.00 mmol) and the reaction was stirred at room temperature for 3 h. Methanol was removed under reduced pressure and the residue dissolved in water (20 mL) and extracted with ethyl acetate (2 × 30 mL) to remove side products. The aqueous solution was acidified and extracted with ethyl acetate (3 × 30 mL), dried (Na2SO4) and the solvent removed under reduced pressure to give Boc-8b. This was directly deprotected in a 1:1 mixture of methanol and 4 M HCl (6 mL) for 2 h, and the solvent removed under reduced pressure to give 5-aminomethyl-2-furancarboxylic acid hydrochloride salt 8b·HCl23 (55 mg, 31%). The characterisation data was identical to 8b·HCl synthesised above.
Acknowledgements
The authors would like to thank the UK Engineering and Physical Sciences Research Council (EPSRC) for financial support of this work to A. D. and for F. S. (EP/K014897/1), as part of their Sustainable Chemical Feedstocks programme. Also the Department of Chemistry UCL for part-funding A. D. Furthermore, we gratefully acknowledge the UCL Mass Spectrometry and NMR Facilities in the Department of Chemistry UCL and the EPSRC UK National Mass Spectrometry Facility at Swansea University.Notes and references
- For example:
(a)
T. Werpy and G. Peterson, Top Value Added Chemicals from Biomass, U.S. Department of Energy, Washington, DC, 2004, vol. 1 Search PubMed
; (b) J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539–554 RSC
- For example:
(a) A. A. Rosatella, S. P. Simeonov, R. F. M. Frade and C. A. M. Afonso, Green Chem., 2011, 13, 754–794 RSC
- For example:
(a) O. O. James, S. Maity, L. A. Usman, K. O. Ajanaku, O. O. Ajani, T. O. Siyanbola, S. Sahu and R. Chaubey, Energy Environ. Sci., 2010, 3, 1833–1850 RSC
- AVA Biochem 2014. First Industrial Production for Renewable 5-HMF. Available at http://www.ava-biochem.com/pages/en/downloads/press-releases.php.
- For example:
(a) J. J. Pacheco and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 8363–8367 CrossRef CAS PubMed
-
(a)
T. Ayusawa, S. Mori, T. Aoki and R. Hamana, US4598159, 1986 Search PubMed
-
(a) N. Kise and N. Ueda, Tetrahedron Lett., 2001, 42, 2365–2368 CrossRef CAS
- For example:
(a) D. Koszelewski, K. Tauber, K. Faber and W. Kroutil, Trends Biotechnol., 2010, 28, 324–332 CrossRef CAS PubMed
- F. Blume, M. H. Albeiruty and J. Deska, Synthesis, 2015, 2093–2099 CAS
-
T. Haas, J. C. Pfeffer, K. Faber and M. Fuchs, WO2012171666, 2012 Search PubMed
- U. Kaulmann, K. Smithies, M. E. B. Smith, H. C. Hailes and J. M. Ward, Enzyme Microb. Technol., 2007, 41, 628–637 CrossRef CAS
- M. E. B. Smith, B. H. Chen, E. G. Hibbert, U. Kaulmann, K. Smithies, J. L. Galman, F. Baganz, P. A. Dalby, H. C. Hailes, G. J. Lye, J. M. Ward, J. M. Woodley and M. Micheletti, Org. Process Res. Dev., 2010, 14, 99–107 CrossRef CAS
- T. Sehl, H. C. Hailes, J. M. Ward, U. Menyes, M. Pohl and D. Rother, Green Chem., 2014, 16, 3341–3348 RSC
- N. Richter, R. C. Simon, H. Lechner, W. Kroutil, J. M. Ward and H. C. Hailes, Org. Biomol. Chem., 2015, 13, 8843–8851 CAS
- C. K. Savile, J. M. Janey, E. C. Mundorff, J. C. Moore, S. Tam, W. R. Jarvis, J. C. Colbeck, A. Krebber, F. J. Fleitz, J. Brands, P. N. Devine, G. W. Huisman and G. J. Hughes, Science, 2010, 329, 305–309 CrossRef CAS PubMed
- M. Höhne, S. Schätzle, H. Jochens, K. Robins and U. T. Bornscheuer, Nat. Chem. Biol., 2010, 6, 807–813 CrossRef PubMed
- D. Pressnitz, C. S. Fuchs, J. H. Sattler, T. Knaus, P. Macheroux, F. G. Mutti and W. Kroutil, ACS Catal., 2013, 3, 555–559 CrossRef CAS
- E. Busto, R. C. Simon, B. Grischek, V. Gotor-Fernandez and W. Kroutil, Adv. Synth. Catal., 2014, 356, 1937–1942 CrossRef CAS
- S. Schätzle, F. Steffen-Munsberg, A. Thontowi, M. Höhne, K. Robins and U. T. Bornscheuer, Adv. Synth. Catal., 2011, 353, 2439–2445 CrossRef
- D. Baud, N. Ladkau, T. S. Moody, J. M. Ward and H. C. Hailes, Chem. Commun., 2015, 51, 17225–17228 RSC
- M. S. Holfinger, A. H. Conner, D. R. Holm and C. G. Hill, J. Org. Chem., 1995, 60, 1595–1598 CrossRef CAS
- M. Chatterjee, T. Ishizaka and H. Kawanami, Green Chem., 2016, 18, 487–496 RSC
- D. D. Hawker and R. B. Silverman, Bioorg. Med. Chem., 2012, 20, 5763–5773 CrossRef CAS PubMed
-
S. Prasad, T. K. Chakraborty, A. Mathur, M. Jaggi, A. Kunwar, R. Mukherjee and A. Burman, US20050032707, 2005 Search PubMed
- L. B. Maktouf, I. Ghorbel, A. Afli, S. Abid and A. Gandini, Polym. Bull., 2011, 67, 1111–1122 CrossRef
- H. L. Wei, K. Yao, H. J. Chu, Z. C. Li, J. Zhu, Y. M. Shen, Z. X. Zhao and Y. L. Feng, J. Mater. Sci., 2012, 47, 332–340 CrossRef CAS
- T. Matsui, A. Kudo, S. Tokuda, K. Matsumoto and H. Hosoyama, J. Agric. Food Chem., 2010, 58, 10876–10879 CrossRef CAS PubMed
- S. Nielek and T. Leslak, J. Prakt. Chem., 1988, 330, 825–829 CrossRef CAS
- J. Ohyama, A. Esaki, Y. Yamamoto, S. Arai and A. Satsuma, RSC Adv., 2013, 3, 1033–1036 RSC
- J. M. Timko, S. S. Moore, D. M. Walba, P. C. Hiberty and D. J. Cram, J. Am. Chem. Soc., 1977, 99, 4207–4219 CrossRef CAS
- T. El Hajj, A. Masroua, J. C. Martin and G. Descotes, Bull. Soc. Chim. Fr., 1987, 855–860 CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6gc02241c |
| This journal is © The Royal Society of Chemistry 2017 |