
The E factor 25 years on: the rise of green chemistry and sustainability
Roger A.
Sheldon
ab
aMolecular Sciences Institute, School of Chemistry, University of the Witwatersrand, Wits 2050, Johannesburg, South Africa. E-mail: roger.sheldon@wits.ac.za
bBiocatalysis & Organic Chemistry, Delft University of Technology, 2628 BL Delft, Netherlands. E-mail: r.a.sheldon@.tudelft.nl
First published on 30th September 2016
Abstract
The global impact, over the last 25 years, of the principles of green chemistry and sustainability, and the pivotal role of the E factor concept in driving resource efficiency and waste minimisation, in the chemical and allied industries, is reviewed. Following an introduction to the origins of green chemistry and the E factor concept, the various metrics for measuring greenness are discussed. It is emphasised that mass-based metrics such as atom economy, E factors and process mass intensity (PMI) need to be supplemented by metrics, in particular life cycle assessment, which measure the environmental impact of waste and, in order to assess sustainability, by metrics which measure economic viability. The role of catalysis in waste minimisation is discussed and illustrated with examples of green catalytic processes such as aerobic oxidations of alcohols, catalytic C–C bond formation and olefin metathesis. Solvent losses are a major source of waste in the pharmaceutical and fine chemical industries and solvent reduction and replacement strategies, including the possible use of neoteric solvents, such as ionic liquids and deep eutectic solvents, are reviewed. Biocatalysis has many benefits in the context of green and sustainable chemistry and this is illustrated with recent examples in the synthesis of active pharmaceutical ingredients. The importance of the transition from an unsustainable economy based on fossil resources to a sustainable bio-based economy is delineated, as part of the overarching transition from an unsustainable linear economy to a truly green and sustainable circular economy based on resource efficiency and waste minimisation by design.
 Roger A. Sheldon | Roger Sheldon (http://www.sheldon.nl) is currently Distinguished Professor of Biocatalysis Engineering at the University of the Witwatersrand in Johannesburg. He is a recognised authority on Green Chemistry and widely known for developing the E factor concept for assessing the environmental impact of chemical processes. He authored several books on catalysis, >470 papers and 55 granted patents. Current interests are in biocatalysis, enzyme immobilisation and biomass conversion. He received the RSC 2010 Green Chemistry Award and was elected a Fellow of the Royal Society in 2015. He has a Ph.D. in organic chemistry from Leicester University and was Professor at Delft University (1991–2007), CEO of CLEA Technologies (2006–2015), VP R&D at DSM-Andeno (1980–1990) and Shell Research Amsterdam (1969–1980). |
1. Introduction: origins of the E factor concept
The first publication on the E factor concept appeared in 1992.1 Now, twenty-five years later, it seemed a good time for an assessment of its impact in the context of waste minimisation and resource efficiency in chemicals manufacture. In the early 1980s our attention was drawn to the problem of waste in the fine chemicals industry by our experience with the industrial production of the pharmaceutical intermediate, phloroglucinol (1,3,5-trihydroxybenzene) in three steps from 2,4,6-trinitrotoluene (TNT).2 The first step was an oxidation with potassium dichromate in fuming sulfuric acid (oleum). Overall, 40 kgs of solid chromium-containing waste were produced for every kg of phloroglucinol. In short, the process involved a hazardous starting material (TNT), a known human carcinogen (hexavalent chromium) as a reagent and generated 40 kgs of hazardous waste per kg of product. Perhaps not surprisingly, the plant was shut down in the mid-1980s when the cost of waste disposal approached the revenues generated by the product.Since we were curious to know if this amount of waste was typical for fine chemicals manufacture, we conducted an inventory of the amounts of waste generated in processes for other fine chemicals, pharmaceutical intermediates and some bulk chemicals. This soon revealed that tens of kgs waste per kg product were no exception in the fine chemicals industry. We concluded that a new paradigm was needed for efficiency in organic synthesis, from the traditional concept based on chemical yield to one that assigns value to maximising resource efficiency, minimising waste and avoiding the use of hazardous and toxic chemicals. Clearly, an environmental factor was missing and this led us, in the late 1980s, to propose the E(nvironmental) factor – mass of waste/mass of product, usually expressed as kgs/kg – for assessing the environmental impact of manufacturing processes. The E factors collated in Table 1 emphasised the enormity of the waste problem in different segments of the chemical industry.1
| Industry segment | Tonnes per annum | E factor (kg waste per kg product) |
|---|---|---|
| Oil refining | 106–108 | <0.1 |
| Bulk chemicals | 104–106 | <1–5 |
| Fine chemicals | 102–104 | 5–50 |
| Pharmaceuticals | 10–103 | 25 – >100 |
In the late 1980s, as a result of increasing environmental awareness, emphasis gradually switched to waste prevention at source as opposed to waste remediation and pollution control by end-of-pipe solutions. The US Pollution Prevention Act of 1990![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 3 focused attention on the need to reduce environmental pollution and recognised that waste prevention at source not only eliminates the cost of waste treatment but actually strengthens economic competitiveness through more efficient use of raw materials. It led to a fundamental shift in the blueprint of the US Environmental Protection Agency (EPA) for environmental protection – from “end of pipe” waste treatment to waste prevention – and culminated in the introduction of the term Green Chemistry at the EPA in the early 1990s.
3 focused attention on the need to reduce environmental pollution and recognised that waste prevention at source not only eliminates the cost of waste treatment but actually strengthens economic competitiveness through more efficient use of raw materials. It led to a fundamental shift in the blueprint of the US Environmental Protection Agency (EPA) for environmental protection – from “end of pipe” waste treatment to waste prevention – and culminated in the introduction of the term Green Chemistry at the EPA in the early 1990s.
The term gained formal recognition with the publication of the 12 principles of green chemistry, by Anastas and Warner in 1998,4 in which the overall guiding element is ‘benign by design’. A succinct definition is: Green chemistry efficiently utilises (preferably renewable) raw materials, eliminates waste and avoids the use of toxic and/or hazardous reagents and solvents in the manufacture and application of chemical products. Green chemistry is primary pollution prevention rather than waste remediation. In the last twenty-five years the concept has become firmly entrenched in both industry and academia.
2. Green metrics: atom economy, E factors and beyond
As Lord Kelvin so astutely observed, “To measure is to know”. If we want to know how green a process is then we need metrics to measure greenness. The two oldest green metrics are the above-mentioned E factor and Trost's atom economy, first proposed in 1991.5 Atom economy (AE) is calculated by dividing the molecular weight of the product by the sum total of the molecular weights of all substances formed in the stoichiometric equation for the reaction involved. AE is a theoretical number which assumes the use of exact stoichiometric quantities of starting materials and a chemical yield of 100%, and disregards substances, such as solvents and chemicals used in the work-up of the reaction mixture, which do not appear in the stoichiometric equation. Nonetheless, it is an extremely useful tool for rapid evaluation, before any experiments are performed, of the resource efficiency and, by the same token, the amounts of waste that will be generated in alternative processes.The E factor, in contrast, is the actual amount of waste produced in the process and differs from AE in two important ways. First, it takes the product yield into account and waste from all of the auxiliary components, e.g. solvent losses, chemicals used in work-up, which are disregarded by AE. Second, AE is applied to individual steps but the E factor can easily be applied to a multi-step process thus facilitating a holistic assessment of a complete process. In our original publication we defined waste as “everything but the desired product”, with the exception of water. Our rationale for excluding water was that including it could lead to a skewing of E factors and render meaningful comparison of processes difficult. However, the current trend in the pharmaceutical industry is to include water in the E factor and in an assessment of a biocatalytic process for atoravastatin (Lipitor) we calculated the E factor both with and without water.6 Energy was implicitly included in the E factor since its input results in the formation of waste, mainly in the form of carbon dioxide. However, in practice it is often not included in E factors for the manufacture of fine chemicals and active pharmaceutical ingredients (APIs), where energy usage is often not allocated to individual products in multipurpose production facilities. In contrast, energy efficiency and CO2 generation can't be disregarded in the production of commodity chemicals in the bio-based economy (see section 9).
A higher E factor means more waste and, consequently, greater negative environmental impact. The ideal E factor is zero. It can also be calculated, for a particular product, production site, or even a whole company, from knowledge of the number of tons of raw materials purchased and the number of tons of product sold. Importantly, lower E factors have been shown7 to correlate well with reduced manufacturing costs of APIs which is a reflection of lower process materials input and output, reduced costs of hazardous and toxic waste disposal, improved capacity utilisation and reduced energy demand. In short, there are strong economic incentives for the pharmaceutical industry to integrate green chemistry into the entire process research, development and manufacturing lifecycle. The concepts of atom economy and E factors have motivated industrial and academic chemists worldwide to explicitly consider waste generation, in addition to the common criteria such as synthetic convergence, chemical yield and cost of goods, when designing a synthesis of a target molecule. Moreover, in the last decade the E factor and atom economy concepts have been incorporated into chemistry textbooks and curricula at both university and high school level.8
Various alternative metrics have been proposed9 for measuring the environmental footprint of processes and attempts have been made to unify the different green metrics.10 Constable and coworkers of GSK11 proposed the use of reaction mass efficiency, a refinement of atom economy which takes yield and use of excess reagents into account, and mass intensity (MI), defined as the total mass (including water) used in a process divided by the mass of product, i.e. MI = E factor + 1. The Green Chemistry Institute Pharmaceutical Round Table adopted this metric, renaming it Process Mass Intensity (PMI), to benchmark the environmental footprint of processes for APIs, and to use this data to drive the greening of the pharmaceutical industry.12 In our opinion none of these alternative metrics offer any particular advantage over the E factor for describing how wasteful a process is. The ideal PMI is 1, whereas the ideal E factor is 0, which more clearly reflects the ultimate goal of zero waste. The E factor also has the advantage that, in evaluating a multi-step process, E factors of individual steps are additive but PMIs are not because PMI doesn't discount step products from the mass balance.7
3. System boundaries and intrinsic E factors
A major source of waste in the pharmaceutical industry is solvent losses (see section 7). If solvent losses were not known, in order to calculate an E factor we generally assumed that 90% of the solvent could be recovered and re-used. However, this may have been too optimistic. More recently, we have suggested7 the use of simple E factors (sEF) and complete E factors (cEF), depending on the stage of development of the process. The sEF doesn't take solvents and water into account and is more appropriate for early route scouting activities, whereas the cEF accounts for all process materials including solvents and water, assuming no recycling, and is more appropriate for total waste stream analysis. However, the true commercial E factor will fall somewhere between the sEF and cEF and can be calculated when reliable data for solvent losses are available.| sEF = ∑m(raw materials) + ∑m(reagents) − m(product)/m(product) |
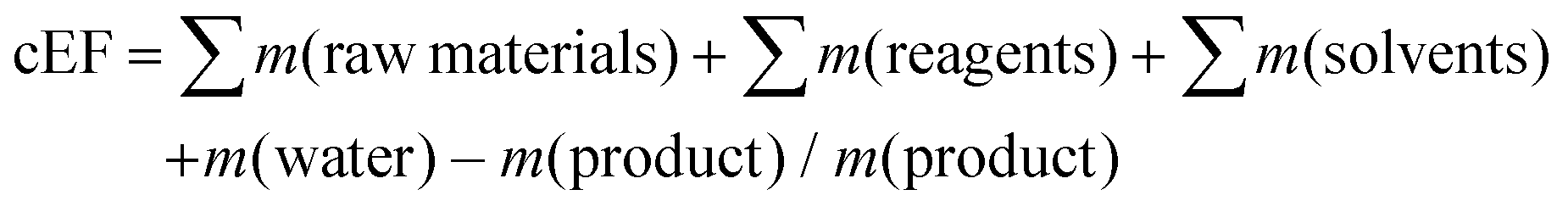
E factors, indeed all mass-based metrics, are very much dependent on the starting point of the synthesis, i.e. it is necessary to define the boundary conditions for calculation of E factors.7 In our original development of the concept, E factors were calculated on a gate-to-gate basis,1i.e. the starting point is the raw material entering the factory gate and the end point is the product leaving it. The E factor includes only those processes conducted at the manufacturing site of, for example, an API. However, we have always recognised that a raw material used in the synthesis of an API may itself be an advanced intermediate, prepared in a multi-step process from readily available raw materials. Indeed, one way to dramatically reduce your E factor overnight is to purchase an intermediate rather than produce it in-house. This can lead to inconsistencies in the application of green metrics to pharmaceuticals. It is important, therefore, to consider the intrinsic E factors associated with procured raw materials and one possibility7 is to define the starting point as a commodity-type, commercially available raw material. For example, in an E factor calculation for the Pfizer process for manufacture of sildenafil citrate (Viagra™) shown in Scheme 1.
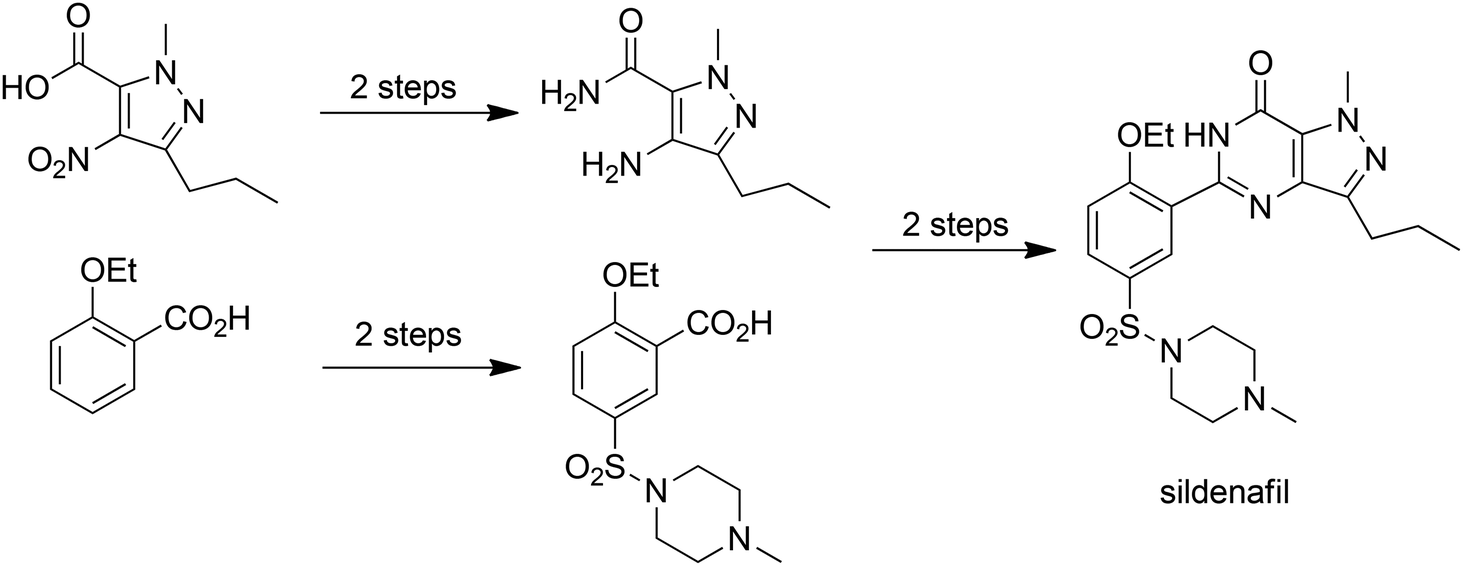 | ||
| Scheme 1 Pfizer's commercial process for sildenafil (Viagra™). | ||
Analysis of this process7 afforded a traditional E factor, including 10% of solvents used and excluding water, of 6.4 kg kg−1 which corresponds very well with Pfizer's reported 6 kg kg−1.13 The sEF (excluding solvents and water) is 3.9 kg kg−1 and the cEF is 50.3 kg kg−1. However, one of the primary raw materials, 1-methyl-4-nitro-3-propyl-1H-pyrazole-5-carboxylic acid (1) doesn't meet the starting point criterion of a readily available commodity-type chemical. Hence, we argued7 that the intrinsic E factor of (1) should be included in the calculation of the E factor for Sildenafil citrate production. Inclusion of the intrinsic E factor of (1), which is derived from readily available diethyl oxalate and 2-pentanone in a five-step process (Scheme 2), results in significant increases in the overall E factors: the sEF increases from 3.9 to 9.9 kg kg−1, the cEF from 50.3 to 85.5 kg kg−1 and the E factor from 6.4 to 13.8 kg kg−1. This example serves to stress the importance of implementing an industry-wide standardised starting point concept for green process analysis.
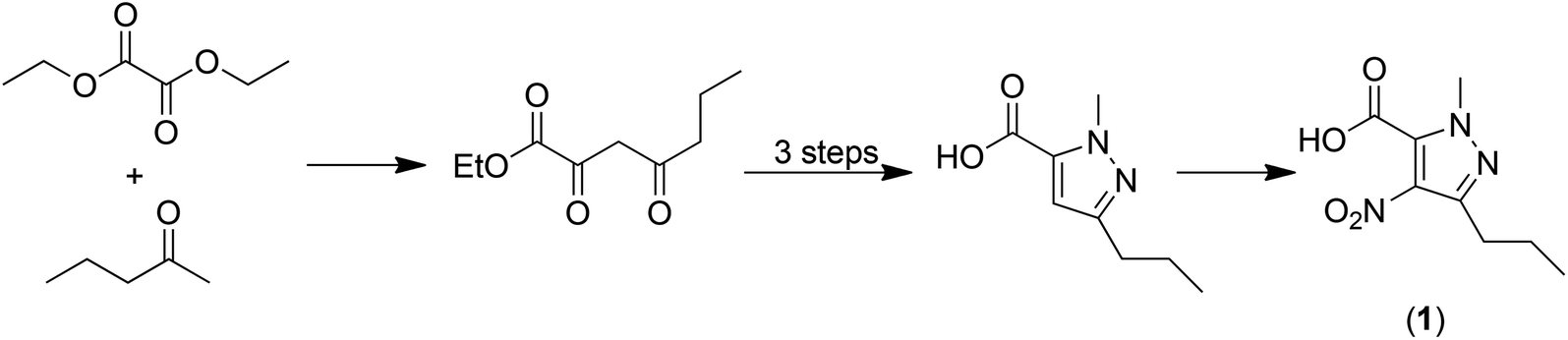 | ||
| Scheme 2 Synthesis of 1-methyl-4-nitro-3-propyl-1H-pyrazole-5-carboxylic acid (1). | ||
A primary cause of the high E factors of processes in the pharmaceutical industry is the high molecular complexity of APIs and the correspondingly large number of chemical steps needed to assemble APIs from commercially available starting materials. Hence, one way to reduce waste is to develop step economic processes as advocated by Wender and coworkers.14 Indeed, Wender's step economy is a very useful metric, perhaps more useful than AE, in driving the greenness of an overall process. The molecular complexity is also taken into account in the Green Aspiration Level (GAL) concept7 as a novel performance metric for quantifying the environmental impact of API manufacturing processes.
However, the larger E factors in the fine chemical and pharmaceutical industries, compared to bulk chemicals and oil refining, are also due to the widespread use of organic solvents and classical stoichiometric reagents rather than catalysts. Hence, the solution to the waste problem is the development of alternative atom and step economic, catalytic technologies, preferably in solvent-free processes.
4. Environmental impact of waste: life cycle assessment
All of the metrics discussed above take only the mass of waste generated into account. A shortcoming of simple mass-based metrics is that they assign the same weighting to all types of waste. However, it is worth pointing out that the original intention of the E factor and other mass-based metrics was to draw attention to the inefficiency of many batch chemical processes and the fact that this is the direct cause of the generation of copious amounts of waste in, for example, the fine chemical and pharmaceutical industries. Nonetheless, the environmental impact of the waste must be considered. One kg of sodium chloride doesn't have the same impact on the environment as 1 kg of a chromium salt or 1 kg of dichloromethane solvent. We recognised this when we introduced the E factor in 1992 and, therefore, we subsequently introduced15 the term ‘environmental quotient’, EQ, where Q is an arbitrarily assigned unfriendliness multiplier. For example, one could arbitrarily assign a Q value of 1 to NaCl and, say, 100–1000 to a heavy metal salt, such as chromium, depending on its toxicity, ease of recycling, etc. Although the values arbitrarily assigned to Q are debatable it is clear that (monetary) values can be assigned to waste streams and much attention has been devoted in the last two decades to developing methodologies for quantification of Q values.For example, Eissen and Metzger16 developed the EATOS (Environmental Assessment Tool for Organic Synthesis) software in which metrics related to health hazards, persistence, bioaccumulation and ecotoxicity were used to determine the environmental index of the input (substrates, reagents, solvents) and output (product and waste). The outcome is equivalent to EQ in that it constitutes an integration of the amount of waste with quantifiable environmental indicators based on the nature of the waste. It also introduced the cost of raw materials into the assessment. Similarly, van Aken and coworkers17 introduced the EcoScale, a semi-quantitative method to evaluate both economic and environmental impact factors of organic syntheses based on yield, cost, safety hazards, conditions and ease of down-stream processing.
Life Cycle Assessment (LCA)18 is a methodology specifically designed to assess the environmental impact of all stages of a product's life from raw materials extraction through materials processing to distribution, use and disposal or recycling, so-called from cradle-to-grave analysis. The idea was originally conceived by Teasley at The Coca Cola company in 1969 and developed together with the Midwest Research Institute (MRI) in Kansas City.19 Their goal was to quantify the energy, material and environmental consequences of the entire life cycle of beverage cans from raw materials extraction to disposal and the possibility of using a plastic bottle was being considered, a revolutionary idea at the time. The methodology was referred to as Resource and Environmental Profile Analysis (REPA). The term LCA was coined at the first international workshop sponsored by the Society of Environmental Technology and Chemistry (SETAC) in 1990.20
LCA is conducted on the basis of quantifiable environmental impact indicators, such as energy usage, global warming, ozone depletion, acidification, eutrophication, smog formation, and human- and eco-toxicity, in addition to waste generated. The methodology is defined by the ISO 14040:2006 standard21 and is divided into 4 phases: (i) goal and scope definition, (ii) inventory analysis, (iii) impact assessment, and (iv) interpretation. Depending on the goal, an LCA can have different system boundaries. It could cover the entire supply chain (cradle-to-grave), raw materials extraction and processing (cradle-to-gate) or just the manufacturing domain (gate-to-gate). It is also worth noting that the relative importance of environmental indicators can vary depending on, inter alia, industry segment, geographical location and even when it is conducted. Whether or not renewable resources are used is probably a moot point in the pharmaceutical industry, for example. Similarly, an LCA conducted today would probably attach more importance to the global warming potential (greenhouse gas emissions) whereas not so long ago ozone depletion would have topped the list.
Around the turn of the century, it was recognised that LCA could be used to compare the environmental footprints of conventional organic syntheses with green alternatives. In 1999 Graedel broke a lance for extending green chemistry from merely green synthesis to the greening of entire life cycles, noting that “adding a life-cycle perspective to green chemistry enlarges its scope and enhances its environmental benefits”.22 Subsequently, integration of green chemistry with quantitative assessment of environmental impact using LCA was subsequently described by several authors.23 Jessop and coworkers,24 for example, used a combination of nine LCA environmental impact indicators – acidification, ozone depletion, smog formation, global warming, human toxicity by ingestion and inhalation, persistence, bioaccumulation, and abiotic resource depletion – in a gate-to-gate assessment of the greenness of alternative routes to a particular product. GREENSCOPE (Gauging Reaction Effectiveness for the Environmental Sustainability of Chemistries with a multi-Objective Process Evaluator) was introduced by the EPA for evaluating and designing more sustainable processes. It is based on the four Es: environment, energy, efficiency, and economics which are used for a direct comparison between two similar processes with differing reaction chemistries or process technologies.25
Some companies developed their own LCA methodologies modified according to their own goals. Glaxo Smith Kline (GSK), for example, has been particularly active in using LCA-based techniques. A modular, cradle-to-gate LCA for the evaluation of processes for API manufacture was reported by Jiménez-González and coworkers at GSK in 2004.26 They concluded that solvent usage is a major contributor to the cradle-to-gate life cycle impacts of APIs. The same group subsequently developed the FLASC (Fast Life Cycle Assessment of Synthetic Chemistry) tool27 for the evaluation of synthetic routes to APIs and has recently described the evolution of LCA in pharmaceutical and chemical applications.28 BASF developed Eco-efficiency Analysis29 which took both economic and environmental aspects into account, including energy, raw materials, emissions, toxicity, hazards and land use and aims to identify the most economically efficient environmental improvements.
Chimex, a subsidiary of L'Oreal producing primarily cosmetic ingredients, introduced Eco-footprint, a new tool for assessing “Made in Chimex” processes.30 The Eco-footprint covers the supply chain from the supplier's gate to the product leaving the Chimex gate and consists of a manufacturing footprint and an eco-design footprint. The manufacturing footprint is based on five indicators: water consumption, carbon footprint related to transport of raw materials, aqueous waste valorisation, used solvents valorisation and energy consumption. The eco-design footprint also consists of five indicators: the E factor of the process, synthetic pathway efficiency which combines number of steps with yields, raw materials of renewable origin, and potential environmental impacts of raw materials and waste.
The flavour and fragrance company, Mane, introduced Green Motion™ as a green metric tool to evaluate health, safety and environmental impacts of their manufacturing processes on a 0–100 scale.31 The starting point was to group the twelve principles of green chemistry into seven fundamental concepts: raw material, solvent, hazard and toxicity of the reagents, reaction, process, hazard and toxicity of the final product and the waste. Penalty points are then allocated within each category based on well-defined criteria, such as renewable or synthetic origin of raw materials, yield, number of steps and solvents in the process and amount of waste as expressed as the E factor. The authors claim that a full assessment can be made in only half an hour.
Full-scale cradle-to-grave or cradle-to-cradle LCAs are useful for comparing products and processes which have already been commercialised but conducting a full scale LCA in the design or development phase is generally too difficult and time consuming.32 The application of LCA to the synthesis of fine chemicals and APIs is particularly challenging owing to the acute lack of life cycle inventory data33 and the absence of a coherent framework for characterizing the toxicological impacts of chemicals.34 One approach to bridging the data gap is to make use of molecular structure-based models, such as the Finechem tool developed by Wernet.35 The latter is based on artificial neural networks and can estimate key inventory parameters and environmental impacts, such as cumulative energy demand and global warming potential using only molecular features as input data. The lack of a coherent framework for evaluating the toxicological impact of chemicals is a consequence of the fact that LCA metrics are wholly dependent on emissions and do not capture potential risks posed by inherently hazardous chemicals. This led Eckelman36 to propose the adoption of life cycle inherent toxicity, i*, as a novel metric that adapts the computational framework of LCA by attaching measures of inherent hazard to intermediate chemical flows, rather than considering only emissions, thereby providing additional information that can be used in conjunction with current green chemistry metrics.
In the final analysis, for an assessment of the “greenness” of a process or product, a multivariate approach that includes impacts across all the different metrics is needed. The assessment tool for process industries can be simplified by splitting the system boundaries into two domains: the raw materials production and supply domain and the gate-to-gate manufacturing domain.37
5. Sustainable development and the circular economy
The integration of mass-based green metrics with LCA affords an extremely useful tool for evaluating the environmental impact of processes for the manufacture of bulk and fine chemicals and APIs and the methodology continues to be further refined.38 However, the fact that there is no economic component implicit in green chemistry is seen as a major shortcoming by industry which has a preference, therefore, for the concept of sustainable development. The concept was first introduced in 1987 in ‘Our Common Future’, a report of The World Commission on Environmental Development.39 It is defined as development that meets the needs of the present generation without compromising the ability of future generations to meet their own needs. The underpinning philosophy is that in order to retain enough of the planet's resources, to satisfy the needs of future generations, the current rate of resource extraction has to be restrained. A balance needs to be found between economic development, environmental impact and societal equity, often referred to as the triple bottom line.This is reflected in the three pillars of sustainability: societal, ecological and economic, otherwise referred to as the three Ps: people, planet and profit. This can be illustrated with a Venn diagram containing three overlapping circles (Scheme 3) where each circle represents one of the three pillars of sustainability and has its own metrics, often known as indicators which are, by definition, one-dimensional (1-D).40 Three types of 2-D metrics – eco-efficiency, socio-economic and socio-ecological – are obtained at the intersections of two circles and a technology is fully sustainable when all three aspects of sustainability are fulfilled, as measured by 3-D sustainability indicators in the space where three circles overlap.
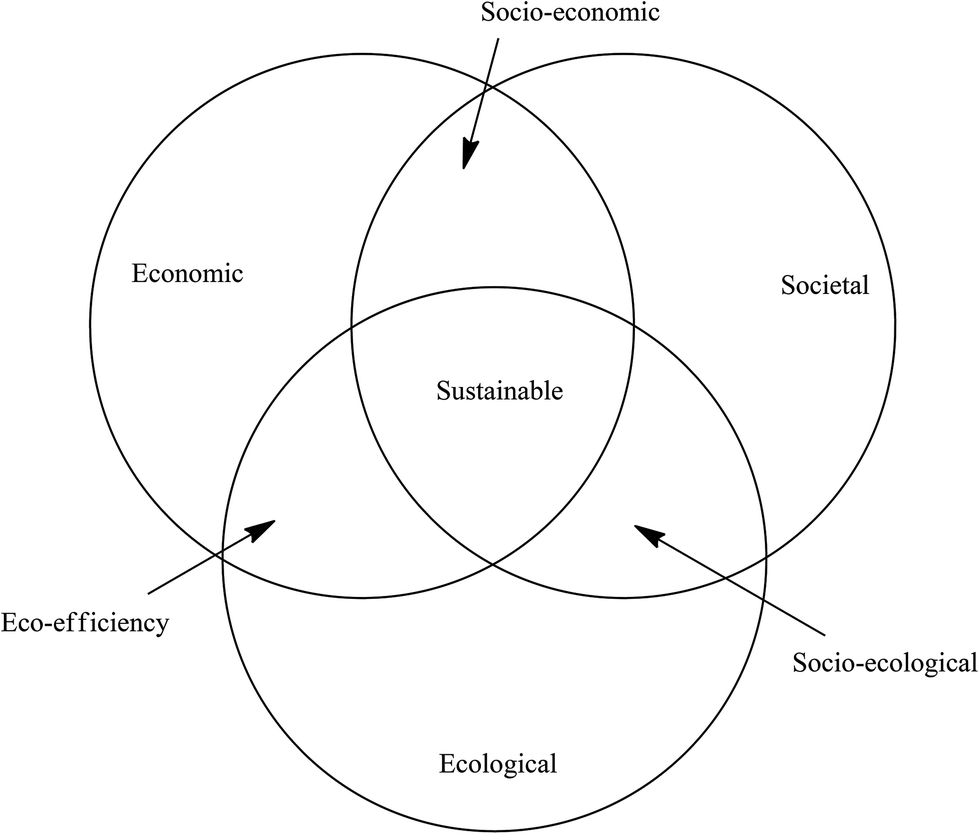 | ||
| Scheme 3 Sustainability metrics Venn diagram. | ||
The integration of environmental and economic metrics for assessing fine chemical processes was already described in 1998 by Heinzle and coworkers.41 It similarly underlies BASF eco-efficiency analysis and, more recently, the eight criteria for good chemical manufacturing processes defined by Roschangar and coworkers at Boehringer Ingelheim.42
As Graedel observed,43 sustainability is dependent on the rate of resource utilisation and the rate of waste generation. In order to be sustainable a technology must fulfill two conditions: (i) natural resources should be used at rates that do not unacceptably deplete supplies over the long term and (ii) residues should be generated at rates no higher than can be assimilated readily by the natural environment. With regard to (i), it is abundantly clear, for example, that non-renewable fossil resources – oil, coal and natural gas – are being used at a much higher rate than they are replaced by natural geological processes and their use is, therefore, not sustainable in the long term. Furthermore, the use of fossil resources is generating carbon dioxide at rates that can't be assimilated by the natural environment and this is widely accepted to be a root cause of climate change.44 This marked discrepancy between the time scale of formation of natural resources and their exploitation is referred to45 as the “ecological time-scale violation”.
5.1. The circular economy
Increased interest in green and sustainable growth, coupled with growing concern for climate change, has focused attention on resource efficiency and is driving transition from a traditional linear flow of materials in a ‘take-make-use-dispose’ economy, to a greener, circular economy.46 The latter seeks to eliminate waste through deliberate design of products and processes with resource efficiency and recycling in mind, the underlying philosophy of the European Commission's “Roadmap to a resource efficient Europe”.47Barry Commoner, a pioneer of industrial ecology, already recognised the linear vs. circular economy issue in the 1960s in observing:48 “We have broken out of the circle of life, converting its endless cycles into man-made linear events: oil is taken from the ground, distilled into fuel, burned in an engine, converted thereby into noxious fumes which are emitted into the air”. However, the transition from an unsustainable linear economy to a greener circular one is seriously hampered by the fact that economic comparisons are not being conducted on a level playing field. The true costs of the established ‘take-make-use-dispose’ production chains have to include the externalised costs of resource depletion, waste management and environmental pollution. These need to be internalised and new economic indicators are needed that take resource efficiency and circularity into account. We need to rethink how to close the loops of production chains and optimise resource efficiency.
6. The role of catalysis in waste minimisation
A major cause of waste, particularly in fine chemicals and pharmaceuticals manufacture, is the use of stoichiometric, mainly inorganic, reagents in organic synthesis. Pregnant examples are reductions with metals (Na, Mg, Zn, Fe) and metal hydride reagents (LiAlH4, NaBH4) and oxidations with permanganate, manganese dioxide and chromium(VI) reagents. Similarly, mineral acids (H2SO4, HF, H3PO4) and Lewis acids (AlCl3, ZnCl2, BF3), employed as reagents in the reaction or in downstream processing, are major sources of waste. The solution is evident: substitution of archaic stoichiometric methodologies with atom economic catalytic alternatives. Relevant examples of high atom economy processes are catalytic hydrogenation, oxidation, carbonylation and hydroformylation (Scheme 4). The generation of copious amounts of inorganic salts can similarly be largely circumvented by replacing stoichiometric mineral and Lewis acids and stoichiometric bases, such as NaOH, KOH, with recyclable solid acids and bases, preferably in catalytic amounts.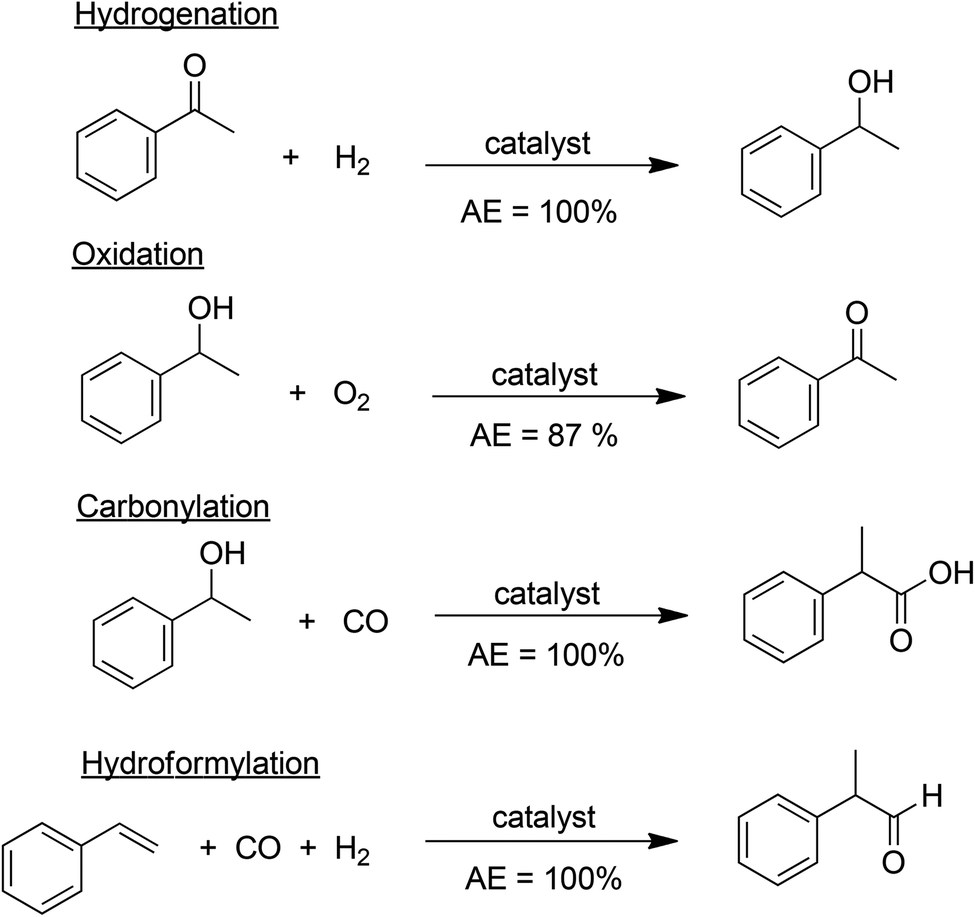 | ||
| Scheme 4 Atom economic catalytic processes. | ||
In short, the key to green and sustainable chemistry is the use of catalysis – heterogeneous, homogeneous, organocatalysis and biocatalysis – in organic synthesis.49 Since the solution is so simple why have catalytic processes not been as widely used in pharmaceuticals and fine chemicals manufacture as in bulk chemicals? One reason is that the volumes involved are much smaller and the need to minimise waste was less acute than in bulk chemicals manufacture. A second reason is that the economics of bulk chemicals manufacture dictate the use of the least expensive reagent, which was generally the most atom economical: O2 or H2O2 for oxidation H2 for reduction and CO for C–C bond formation. Two additional reasons are the broad applicability of time-honoured, classical technologies and the shorter development time compared with that of cleaner, catalytic alternatives. Consequently, environmentally (and economically) inferior technologies are often used to meet stringent market deadlines and subsequent process changes are prohibitive owing to problems associated with regulatory approval. Yet another reason is that, synthetic organic chemists were traditionally not raised on a ‘catalytic diet’, perhaps with the exception of catalytic hydrogenation, and, hence, catalytic methodologies were only sporadically applied in organic synthesis. However, motivated by the need to reduce waste, in the last two decades more emphasis has been placed on the use of catalytic methods in organic synthesis.
6.1. Alcohol oxidations
Pertinent examples where there are pressing needs for catalytic methodologies are provided by redox reactions which, in addition to often having significant scale-up problems, have serious environmental issues. Indeed, the concept of redox economy50 teaches that redox steps should be avoided in organic synthesis unless they are involved in skeleton forming or setting the stereochemistry. The best oxidation (or reduction) is no oxidation (or reduction) and ideally there should be no change in oxidation state on going from substrate to product.Oxidations of primary and secondary alcohols, for example, are pivotal reactions in organic synthesis. The recommended textbook method for the conversion of a secondary alcohol to a ketone (Scheme 5) involves oxidation with the Jones reagent, which consists of stoichiometric quantities of chromium trioxide in sulfuric acid and affords chromium sulfate as the coproduct. The atom economy of this reaction, using α-methylbenzyl alcohol as a model, is 44% which would translate to an E factor of ca. 1.3. In practice the E factor is often more than 3 owing to the use of an excess of reagents and a yield of less than 100%. Other stoichiometric oxidants that are popular with medicinal chemists are the Swern reagent51 and the Dess–Martin periodinane.52 The former generates toxic byproducts and the stench of dimethyl sulfide while the latter is shock sensitive and prohibitively expensive and both reagents have very poor atom economies (Scheme 5a). This should be compared with catalytic aerobic oxidation (Scheme 5b) which has an atom economy of 91%, water as the coproduct and a theoretical E factor of ca. 0.1 (zero if we exclude water).
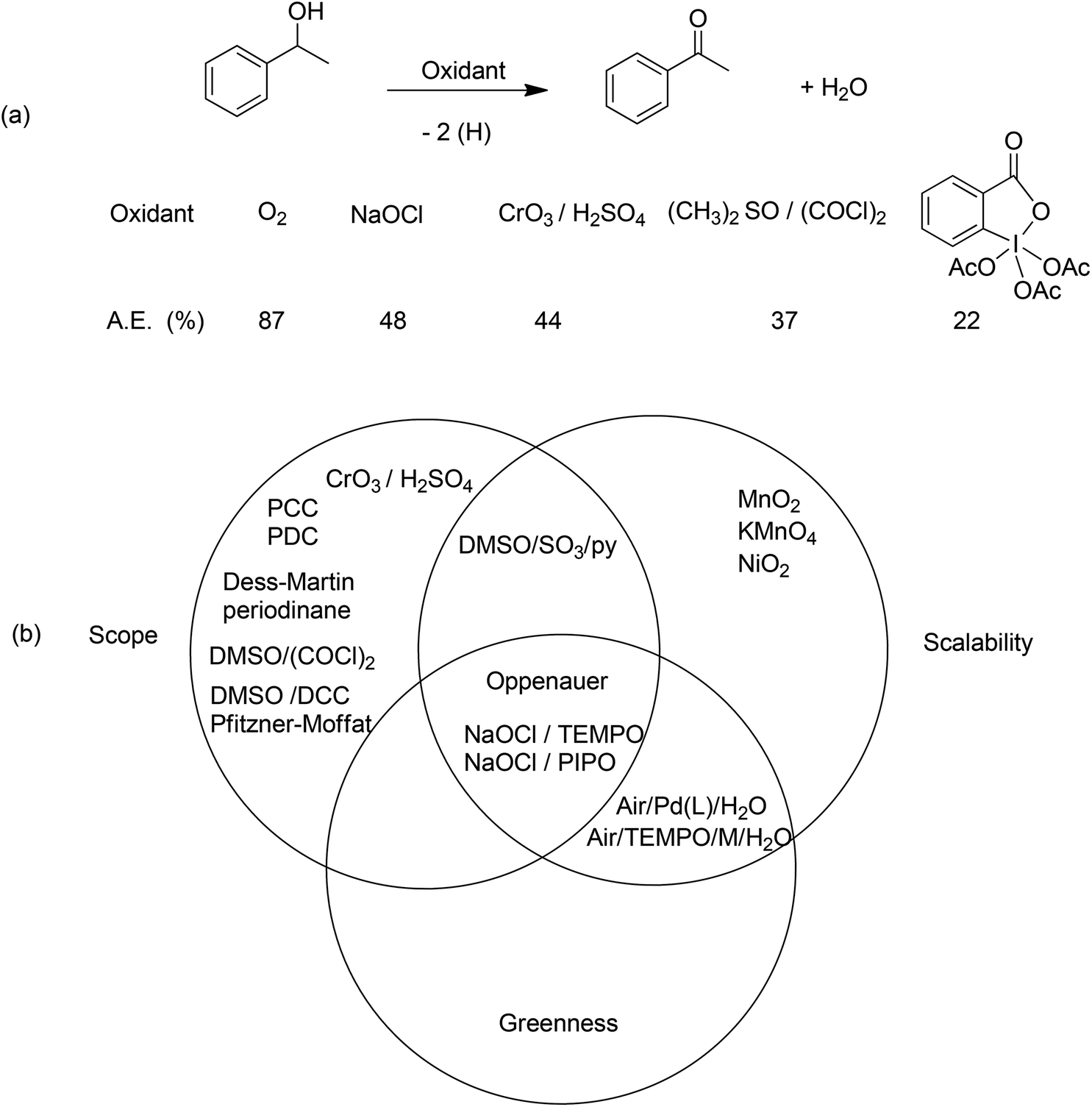 | ||
| Scheme 5 Oxidation of secondary alcohols. | ||
There is clearly a need in the fine chemical and pharmaceutical industry for systems that are green, scalable and have broad synthetic utility. A more environmentally friendly method, for example, is oxidation with household bleach (NaOCl) catalysed by stable nitroxyl radicals such as TEMPO53 or PIPO.54 Although the use of NaOCl as a stoichiometric oxidant affords an equivalent of NaCl as the coproduct, and perhaps the possibility of forming chlorinated impurities, it constitutes a dramatic improvement compared to chromium(VI) and other reagents referred to above. Moreover, because of the scale of pharmaceuticals manufacture, the volumes of NaCl produced are not an issue.
The suitability of various methods for the oxidation of secondary alcohols in the synthesis of fine chemicals and APIs can be depicted by incorporating them in a Venn diagram (Scheme 5b).55 Technologies which fall in the zone representing all three desirable characteristics – broad utility (scope), scalability and greenness – are sustainable. Oxidations with NaOCl fall into this category. One might expect to also find catalytic aerobic oxidations in this category but the authors noted that the use of molecular oxygen (air) presents significant safety issues in connection with the explosion hazards of mixtures of oxygen and volatile organic solvents in the vapour phase. Moreover, the cost benefits of using air may be outweighed by the extra capital investment involved in conducting reactions under pressure in autoclaves compared to NaOCl oxidations which are conducted in standard reactors. Another issue with NaOCl oxidations is the use of dichloromethane (see section 7) as the solvent for the organic phase in the standard protocol but it was recently shown56 that there are several greener alternatives, e.g. the use of ethyl acetate or isopropyl acetate gave similar or even better results than dichloromethane.
The safety profile of aerobic oxidations is substantially improved by conducting them in water which, because of its inert character, reduces the flammability of the vapour phase. A variety of methods are available for catalytic oxidation of alcohols, with dioxygen or hydrogen peroxide, in aqueous media.57 For example primary and secondary alcohols can be selectively oxidized with air as the oxidant and a water-soluble palladium complex as the catalyst in an organic-solvent-free, aqueous biphasic system, affording the corresponding aldehyde or ketone, respectively, in high yield.58 Furthermore, the catalyst, contained in the water phase, could be recovered by simple phase separation and recycled several times without loss of activity.
A system comprising a mixture of TEMPO or PIPO and a bipyridyl-Cu(II) complex, together with a base, also formed an excellent catalyst for the selective aerobic oxidation of primary alcohols in an aqueous medium.59 The copper-dependent oxidase, laccase (E.C. 1.10.3.2), in combination with TEMPO as a cocatalyst, also catalyses the selective aerobic oxidation of primary benzylic alcohols,60via an oxoammonium cation as the active oxidant, analogous to the TEMPO catalysed oxidations with NaOCl.61
6.2. Catalytic C–C and C–N bond formation
C–C bond formations are key transformations in organic synthesis and an atom efficient method for generating C–C bonds is catalytic carbonylation. An early and elegant example of this is the Hoechst-Celanese process for the manufacture of the analgesic, ibuprofen in two catalytic steps (hydrogenation and carbonylation) from p-isobutylactophenone (Scheme 6) with 100% atom economy.62 This process replaced a classical route involving more steps and a much higher E factor. A drawback of this reaction, and palladium catalysed reactions in general (see below), in the context of API synthesis is the regulatory requirement to reduce Pd contamination of the product to <10 ppm, which translates to a need for a costly purification step.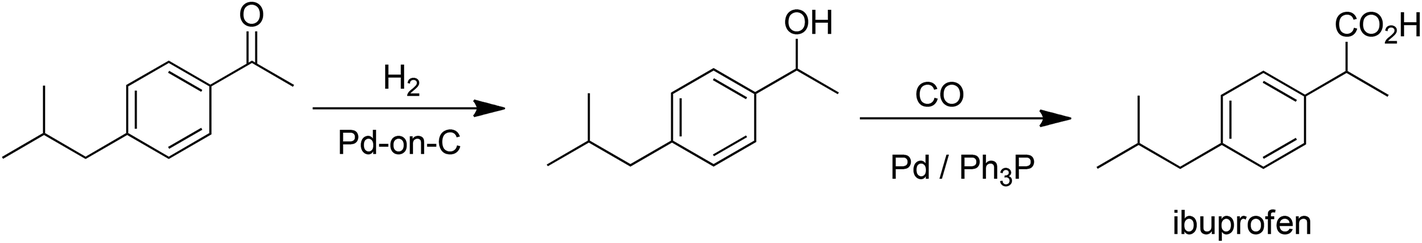 | ||
| Scheme 6 Ibuprofen synthesis by catalytic carbonylation. | ||
In addition to carbonylation, and hydroformylation many other transition metal (particularly palladium) catalysed C–C and C–N coupling reactions have been reduced to industrial practice in the pharmaceutical and fine chemical industries, in the last 25 years.63 These include the well-known Heck–Mizoroki64 and Suzuki–Miyaura65 C–C coupling and the Buchwald–Hartwig C–N coupling (amination)66 reactions (Scheme 7).
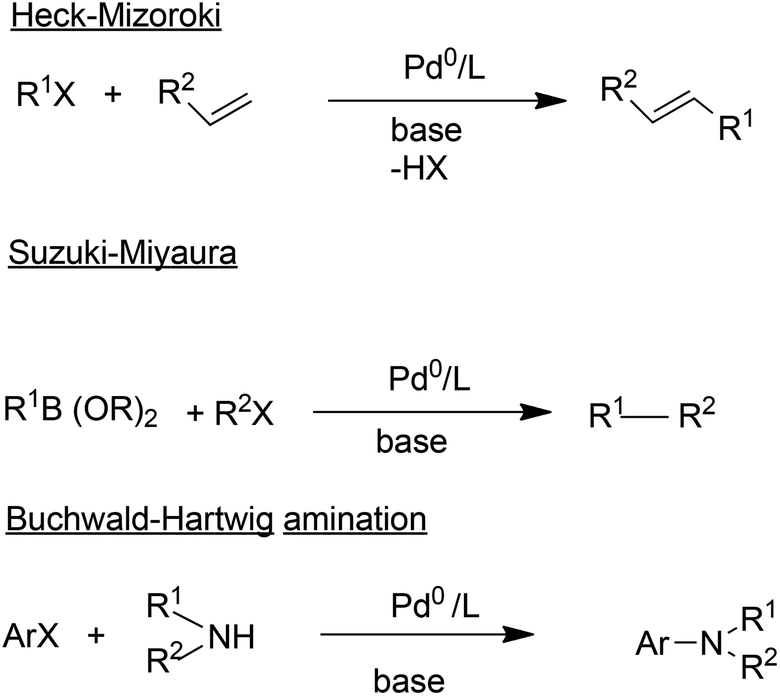 | ||
| Scheme 7 Palladium catalysed coupling reactions. | ||
Another elegant C–C bond forming process which has received considerable attention in the last two decades is olefin metathesis. Following its discovery in the 1960s, olefin metathesis was applied to bulk chemicals manufacture, a prominent example being the Shell Higher Olefins Process (SHOP).67 In the succeeding decades the advent of more versatile catalysts, in particular Grubbs’ development of robust ruthenium-based catalysts which perform well in the presence of most functional groups, paved the way for widespread application of olefin metathesis in the synthesis of complex organic molecules. In awarding the 2005 Nobel Prize in Chemistry to Chauvin, Grubbs and Schrock for the development of olefin metathesis, the Swedish academy noted that it was “a great step forward for green chemistry”. Olefin metathesis is actually a family of reactions and the two sub classes which are important in the context of API and specialty chemicals manufacture are ring-closing metathesis (RCM) and cross metathesis (CM), respectively (Scheme 8).68
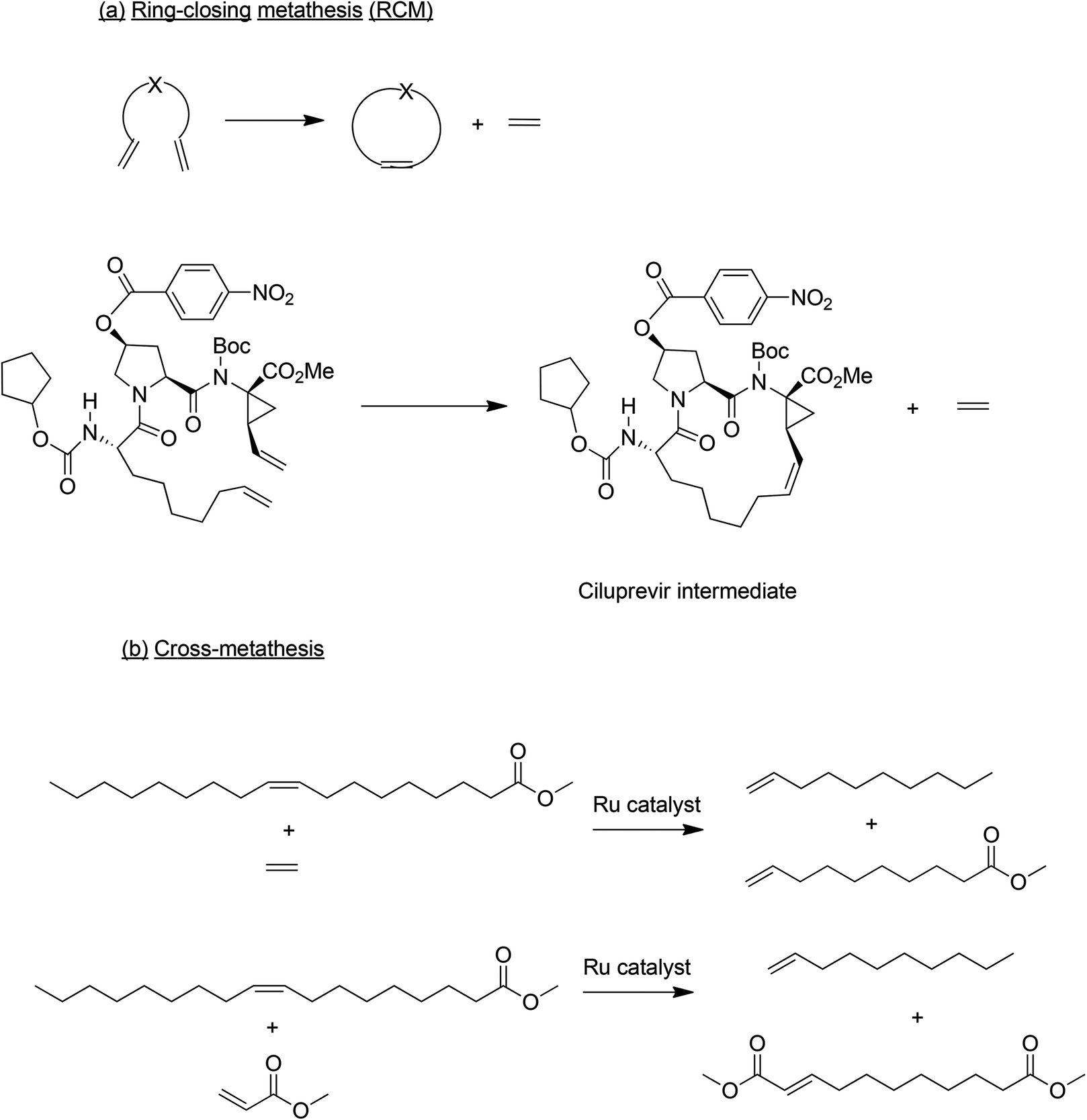 | ||
| Scheme 8 Olefin metathesis reactions. | ||
RCM transforms relatively small linear substrates to cyclic olefins and is, therefore, used to prepare medium and large ring molecules which are otherwise difficult to synthesise. The first large scale use of RCM was in the production of Ciluprevir,69 used in the treatment of hepatitis C virus (HCV), in which the key step was a ruthenium catalysed RCM (Scheme 8a). Subsequently, other VCH drug candidates have been prepared using ruthenium catalysed RCM.64 It should be noted, however, that in order to comply with regulations the ruthenium content of the product needs to be <5 ppm which could be a challenge.
Cross metathesis is used in the conversion of renewable unsaturated plant oils – palm, soy bean and rape seed – to specialty olefin building blocks.70 This innovation is driven by the increasing demand for products from renewable resources and advances in olefin metathesis catalysts over the last two decades have enabled the green catalytic conversion of renewable oils to specialty chemicals. Both ruthenium- and molybdenum-based catalysts can be used. Cross metathesis of methyl oleate with ethylene, for example, affords methyl-9-decenoate and 1-decene (Scheme 8b), both important platform chemicals for polymers and surfactants. Similarly, cross metathesis of fatty acid esters with methyl acrylate affords α,ω-diesters (Scheme 8b) as precursors to polyesters and polyamides.71
6.3. One-pot reactions with multifunctional catalysts
Corma and coworkers72 described a novel approach to catalytic generation of C–C bonds. It involves a one-pot, aldol type condensation and in situ reduction using a heterogeneous bifunctional catalyst, resulting in an order of magnitude reduction in E factors compared with existing, conventional processes. Palladium supported on nanocrystalline magnesium oxide was used, for example, in the one-pot, solvent-free synthesis of the aroma chemical, 4-(4-methoxyphenyl)-2-butanone (raspberry ketone), from 4-methoxybenzaldehyde and acetone (Scheme 9a) in a yield of 99%. The E factor for this process was 0.01 compared with 9.69 for the conventional process. When the intrinsic E factor for the synthesis of 4-methoxybenzaldehyde from p-cresol is included the overall E factors are 0.99 and 10.67, respectively. Similarly, 2-cyclopentylcyclopentanone, an aroma chemical with a jasmine-like odour, was synthesised in 91% selectivity at 98% conversion in a one-pot self-condensation and subsequent hydrogenation of cyclopentanone with Pd-on-MgO/H2 (Scheme 9b). The E factor was 0.10 compared with 1.16 for the conventional two-step process.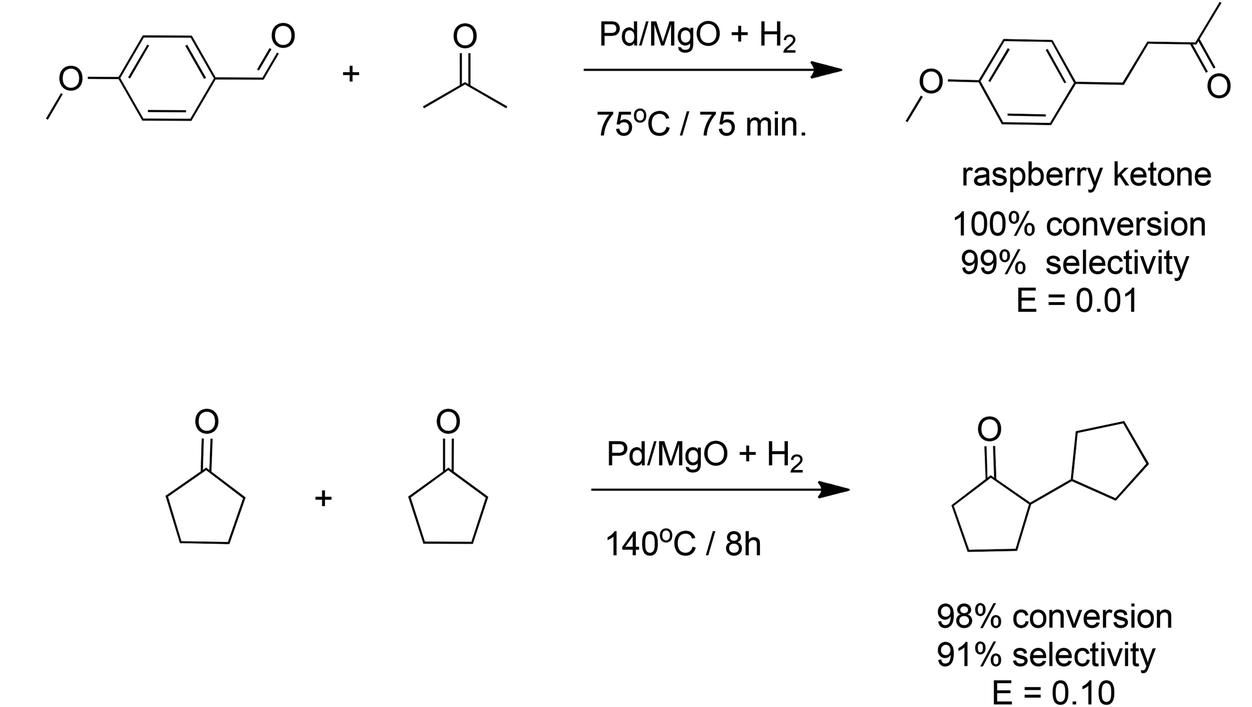 | ||
| Scheme 9 One-pot synthesis of aroma chemicals with multifunctional catalyst. | ||
7. The medium is the message: the best solvent is no solvent
Another important source of waste in fine chemicals manufacture derives from the use of organic solvents. Moreover, so many of the favourite organic solvents of synthetic chemists, such as chlorinated hydrocarbons, have been blacklisted that the whole question of solvent use requires rethinking and has become a primary focus in the pharmaceutical industry.73 According to one report,74 solvents constitute 80–90% of the non-aqueous mass of materials used to make APIs. Another survey,12a revealed that 56% of materials used in API manufacture are organic solvents, with water contributing a further 32%, and it has been estimated that solvents account for 50% of green-house gas emissions from pharmaceutical manufacture.75More recently, a survey of 388 publications in the journal Organic Process Research and Development in the period 1997–2012 concluded76 that there was much room for improvement across the global pharmaceutical industry. The solvents were divided into three categories: (i) solvents of concern, (ii) dipolar aprotic solvents and (iii) neoteric solvents. The eight most commonly used solvents of concern, for reasons of toxicity or fire and explosion hazard, were dichloromethane, n-hexane, diisopropyl ether, 1,2-dimethoxyethane, 1,4-dioxane, diethyl ether, 1,2-dichloroethane and chloroform. Some encouraging trends were observed, such as the reduction in n-hexane and chloroform use after 2001 and the fact that Pfizer has not transferred a process which used a chlorinated hydrocarbon solvent to its manufacturing division in the last eight years. However, the general conclusion was that overall the global industry was not making significant progress and that many of these solvents of concern are still being selected even though greener alternatives are at hand.
Dipolar aprotic solvents are often deemed to be indispensable to the pharmaceutical industry where they are widely used as reaction media for, inter alia, nucleophilic substitutions. The five most commonly used solvents in this class are acetonitrile, dimethylformamide (DMF), dimethyl sulfoxide (DMSO), N-methyl pyrrolidone (NMP) and dimethylacetamide (DMAC). All of these solvents have significant toxicity or safety (DMSO) issues and three of them (DMF, DMAC and NMP) are on the REACH list of substances of very high concern (SVHC).77 A broader screening of solvents was recommended as it is not always necessary to use polar aprotic solvents for nucleophilic substitutions. Eight examples in the category neoteric solvents, which included ionic liquids, supercritical fluids, fluorous solvents, water and solvents from renewable raw materials, such as ethyl lactate and 2-methyl tetrahydrofuran (2-MeTHF), were examined. Only the use of 2-MeTHF grew significantly in the period 1997–2012. Reluctance by the industry to adopt new solvents was attributed to cost and availability, consistency of purity, lack of regulatory guidance and the fact that subsequent environmental studies often do not support initial greenness claims.78
The clear need for replacement and/or reduction in the use of organic solvents in API manufacture led pharmaceutical companies to develop solvent guides to promote this process. GSK published its first solvent guide in 199979 which was subsequently expanded to include LCA in solvent selection in 2005.80 Further modifications and expansions were published in 201181 and in 2016.82 The overall methodology remained the same, namely ranking of solvents relative to each other based on waste disposal, environment, health and safety. A score is assigned for each of these areas, which in turn consist of multiple sub-categories. For example, the waste score comprises scores for incineration, recycling, biotreatment and volatile organic chemical (VOC) emissions.
Each solvent is then given a composite score, defined as the geometric mean of waste, environmental impact, health and safety scores. Traffic light inspired colour designations are then used to categorise solvents: green, amber and red. Certain LCA categories, such as energy usage, water consumption, amount of fossil fuels as feedstock, acidification and eutrophication equivalents, were not included because of significant gaps in the available data bases. A single page view of the solvents most widely used at GSK, as well as greener alternatives, was prepared from the data to provide an at-a-glance comparison of solvents in single chemical classes and across multiple classes. It is immediately apparent that members of certain chemical classes, e.g. alcohols, esters and carbonates, tend to be greener than structurally similar solvents from other classes.
Other pharmaceutical companies, including Pfizer55 and Sanofi,83 have published their own solvent selection guides based on similar scoring systems and colour highlighting and less comprehensive solvent guides have also been published for specific reactions84 which are widely used in practice, e.g. amide bond formation,85 reductive amination86 and olefin metathesis.87 Sanofi classified 96 solvents into four categories: recommended, substitution advisable, substitution requested and banned (highlighted in green, amber, red and brown, respectively). A recent survey of solvent selection guides88 concluded that there is reasonable agreement between the guides, minor differences being largely a reflection of culture and policy. Pena-Pereira and coworkers89 recently published a solvent selection guide where 151 solvents were assessed using chemometrics and multi-criteria decision analysis. The procedure involved grouping of solvents according to their physicochemical parameters and ranking within clusters according to their toxicological and hazard parameters. Hellweg and coworkers90 suggested the use of a relatively simple Environmental, Health and Safety (EHS) screening tool to identify potential hazards in early stage process design. Additional indicators – persistence and spatial range – that describe the exposure potential of chemicals, were suggested for use in the process development stage and a full blown LCA after commercialisation of the process.
As noted in section 3, in our original studies of E factors we assumed, in the absence of data, that solvents would be recycled by distillation and that this would involve a 10% loss. However, the organic chemist's penchant for using different solvents for the various steps in multistep syntheses makes recycling difficult owing to cross contamination. Hence, it is not only better to substitute undesirable solvents with green alternatives, the number of solvents used needs to be reduced. A striking example is the redesign of Pfizer's sertraline manufacturing process.91 Among other improvements, a three step sequence was streamlined by employing ethanol as the sole solvent, thus eliminating the need to use, distil and recover four solvents (methylene chloride, tetrahydrofuran, toluene and hexane). Pfizer workers also reported13 impressive reductions in solvent usage in the process for sildenafil (Viagra) manufacture (see in section 3), from 1700 ltr per kg product used in the medicinal chemistry route to 7 ltr per kg in the commercial process and a future target of 4 ltr per kg.
There is currently a marked trend away from hydrocarbons and chlorinated hydrocarbons as solvents towards lower alcohols, esters and, in some cases, ethers. More recently, attention has been focused on the use of solvents derived from renewable biomass (Scheme 10).92 Inexpensive fermentation products such as ethanol have the added advantage of being readily biodegradable and ethyl lactate, produced from two innocuous fermentation products, is an environmentally attractive solvent for chemical reactions. Other examples include glycerol, a by-product of biodiesel production, which is a good solvent for biocatalytic processes and its derivative, glycerol carbonate. Similarly, γ-valerolactone and 2-Me-THF, are obtained from lignocellulose via 5-hydroxymethyl furfural (HMF) and furfural, respectively, and isosorbide is derived from glucose via sorbitol.
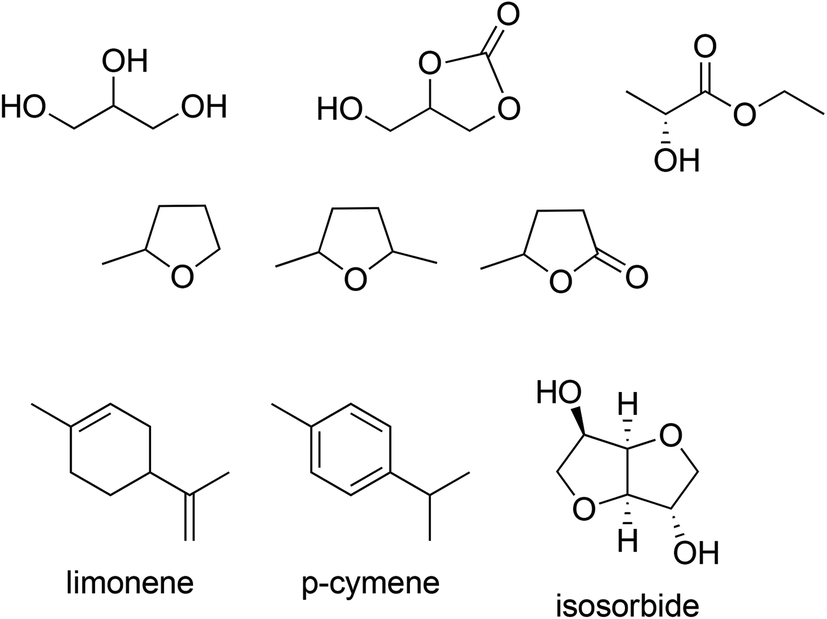 | ||
| Scheme 10 Examples of bio-based solvents. | ||
7.1. Alternative reaction media
The problem with organic solvents is not so much their use but the seemingly inherent inefficiencies associated with their containment, recovery and reuse. Alternative solvents should, therefore, be readily removed from the product. Another important issue is recovery and reuse of the catalyst in (bio)catalytic processes. In pharmaceuticals manufacture separation of noble metal catalyst residues from the product, for example, is mandatory. An insoluble solid, i.e. a heterogeneous catalyst, is easily separated by centrifugation or filtration but recovery and recycling of homogeneous catalysts is challenging. Alternatively, the advantages of homogeneous catalysts can be maintained by employing liquid–liquid biphasic catalysis.93 The catalyst is dissolved in one phase and the reactants and product(s) in a second liquid phase and the catalyst is recovered and recycled by simple phase separation. Preferably, the catalyst solution remains in the reactor and is reused with a fresh batch of reactants without further treatment or, ideally, it is adapted to continuous operation.The best solvent is no solvent but if a solvent (diluent) is needed then water has much to offer: it is non-toxic, non-inflammable, abundantly available and inexpensive. Hence, catalytic conversions in aqueous media have been extensively studied.94 Furthermore, performing catalytic reactions in an aqueous biphasic system95 allows for recovery and recycling of the catalyst by simple phase separation. The water phase, containing the catalyst, can remain in the reactor after separation of the product which significantly limits the amount of water needed. A classic example of large scale application of aqueous biphasic catalysis is the Ruhrchemie/Rhône Poulenc process for the hydroformylation of propylene to n-butanal which employs a water-soluble rhodium(I) complex of trisulfonated triphenylphosphine (tppts) as the catalyst and has an E factor of 0.1 compared to 0.6–0.9 for conventional monophasic hydroformylation processes.96
Another innovative approach, is to use small amounts of environmentally benign designer amphiphiles (surfactants) containing a lipophilic portion which spontaneously self-assembles in water into micelles. In essence, nano-micelle reactors are formed and function as the reaction vessel in which a desired reaction occurs between otherwise water-insoluble substrates and catalysts. The choice of amphiphile is crucial, just as the choice of solvent is crucial to the performance of standard reactions in organic media. Lipshutz and coworkers used 2–5 wt% of the commercially available designer amphiphiles PTS,97 TPGS-750-M,98 based on vitamin E and, more recently SPGS-550-M99 based on β-sitosterol (see Scheme 11 for structures), dissolved in organic-solvent-free water, to perform a variety of palladium catalysed coupling reactions (Scheme 11).100,101 The product can be separated by extraction with a minimum amount of an environmentally acceptable solvent, such as ethyl acetate, while the amphiphile remains in the water phase. Alternatively, solid products that are insoluble in water can precipitate and be separated by filtration. The E factors (calculated including the total volume of organic solvent used and with and without water) of reactions with 2–5% amphiphile were an order of magnitude less than the same reactions conducted in the traditional way in an organic solvent. Similarly, many other metal catalysed reactions, e.g. rhodium catalyzed hydroformylation,102 can be performed in water in the presence of designer amphiphiles.
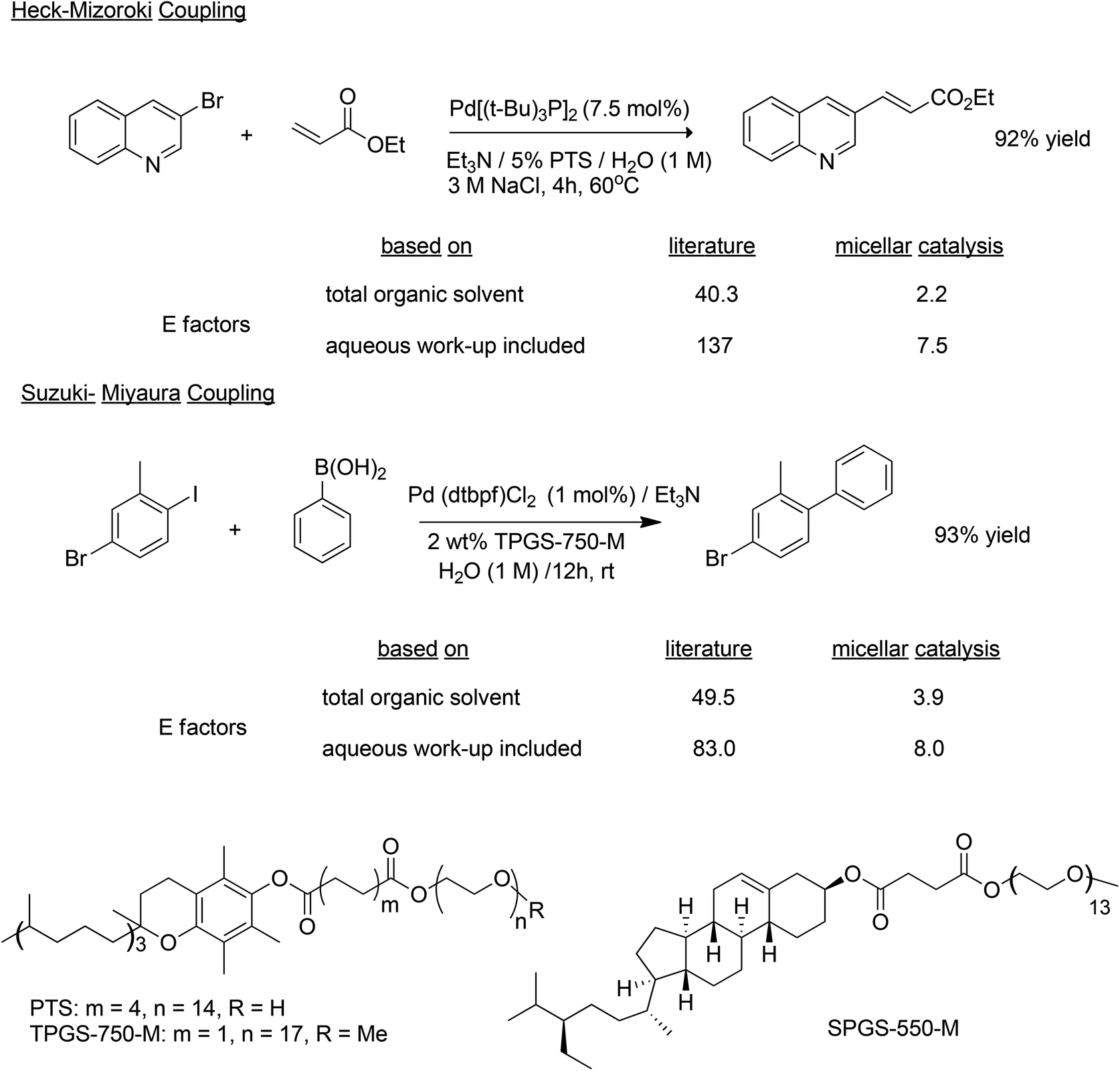 | ||
| Scheme 11 Comparison of E factors for Pd catalysed coupling reactions. | ||
An aqueous biphasic system may not be the answer in all cases, however, and a number of so-called neoteric solvents, such as fluorous biphasic systems,103 supercritical carbon dioxide104 and ionic liquids (ILs),105 as well as biphasic mixtures thereof,106 have been extensively studied. Based on their low flammability and negligible vapour pressure, ILs could be alternatives for volatile organic solvents. However, ILs have significant solubility in water and many first generation ILs are poorly biodegradable107 and exhibit aquatic ecotoxicity.108 Furthermore, their preparation often involves circuitous, high E factor processes, making them prohibitively expensive.109 ILs cannot be generalised as being green or not green; their environmental impact is strongly dependent on the structure of the cation and anion.110 More recently, second generation ILs containing more biocompatible cations and anions, derived from relatively inexpensive, eco-friendly natural products, such as carbohydrates and amino acids have emerged.111 Cholinium ILs, for example, are prepared by reaction of inexpensive choline hydroxide with a wide variety of, mostly naturally occurring, carboxylic acids, affording the corresponding carboxylate salt and water as the only byproduct.112 Such cholinium ILs combine low toxicity with high biodegradability.113 Protic ionic liquids (PILs) also exhibit reduced ecotoxicity and biodegradability and their simple preparation, by mixing a tertiary amine with an acid, such as a carboxylic acid114,115 or sulfuric acid,116 makes them relatively inexpensive (Scheme 12). 2-Hydroxyethylammonium lactates are examples of PILs which are easily prepared from commodity mono-, di- and tri-ethanolamines and renewable lactic acid.117
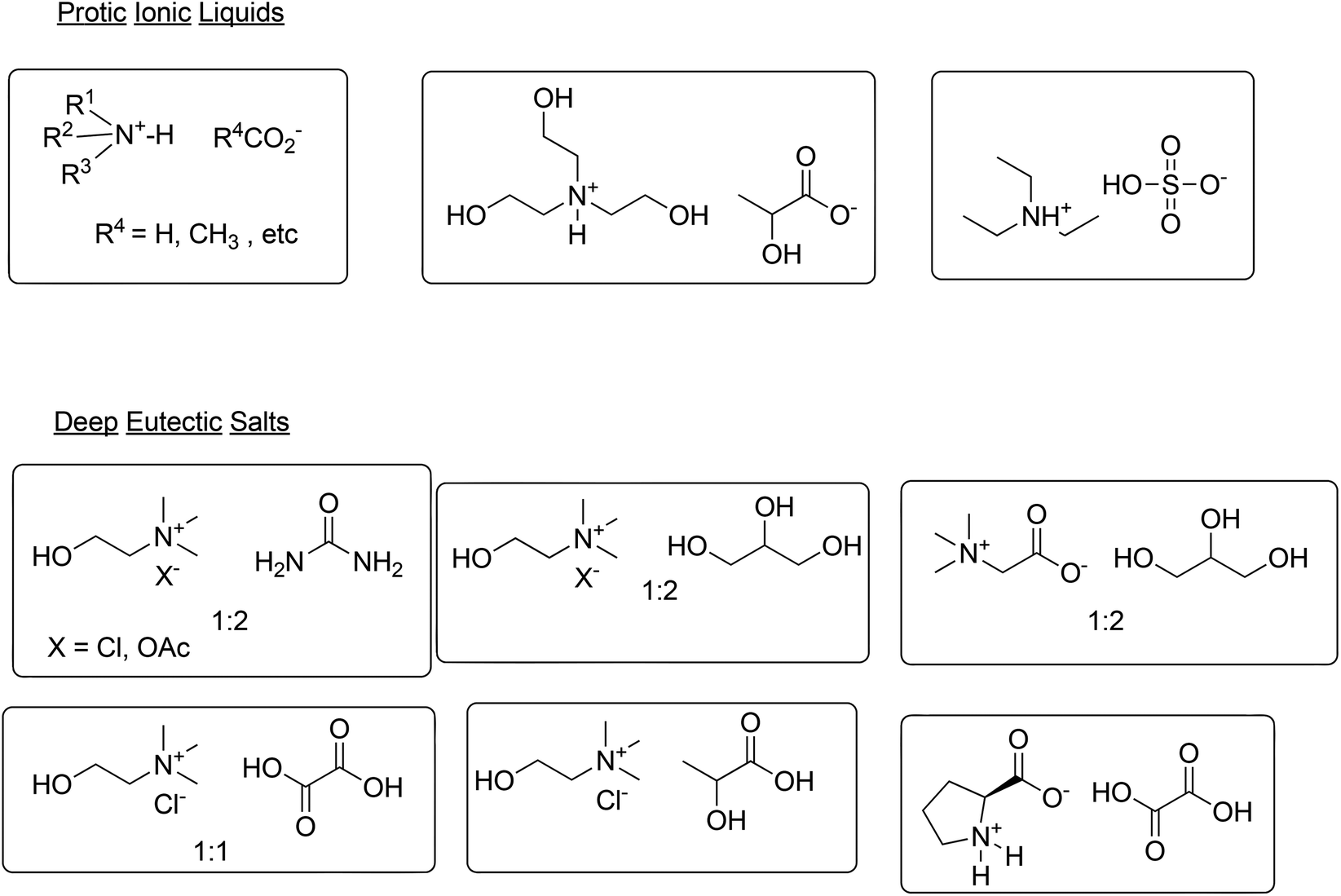 | ||
| Scheme 12 Examples of PILs and DESs. | ||
Another class of alternative reaction media has emerged in recent years: deep eutectic solvents (DESs),118 otherwise known as low-transition temperature mixtures (LTTMs),119 formed by mixing, for example, ammonium or phosphonium salts with a hydrogen bond donor (HBD) such as alcohols, polyols, carboxylic acids and amides. The HBD interacts with the anion of the salt, thereby increasing its effective size and causing a decrease in melting point. For example, mixing choline chloride (m.p. 302 °C) with urea (m.p. 132 °C), in a 1:2 molar ratio, affords a DES with a m.p. of 12 °C. DESs have properties in common with ILs, such as low volatility and high thermal stability, but are easier to synthesise, by mixing the two components and gently heating the mixture. Because they are generally made from naturally occurring, biocompatible substrates, they are also non-toxic and biodegradable. Choline chloride (ChCl), for example, is a readily available, inexpensive feed additive produced in bulk quantities, urea is a common fertiliser and glycerol is a byproduct of biodiesel manufacture. Hence, mixtures of ChCl with urea120 and glycerol,121 in molar ratios of 1:2, are readily available, inexpensive, biocompatible and biodegradable (Scheme 12).
DESs have also been prepared from ChCl and a variety of carbohydrates122 and certain natural deep eutectic solvents (NADES)123 derived from metabolites, such as sugars, amino acids, choline, and natural organic acids, may actually function as reaction media for the in vitro synthesis of sparingly water insoluble compounds such as flavonoids and steroids in living cells. Indeed, NADES have been referred to as ‘solvents for the 21st century’.124
A variety of synthetic transformations have been performed in DESs.125 A quite remarkable example is the recently described126 use of ChCl-based DESs as solvents for reactions with highly polar organometallic reagents, such as Grignard and organolithium reagents at room temperature in the presence of air. This contrasts with existing procedures which generally involve reactions at low temperatures in dry ethereal solvents in an inert atmosphere and opens up new avenues for organometallic chemistry under environmentally friendly conditions (green solvents, room temperature in the presence of air). DESs are also ideal solvents for conducting biocatalytic transformations which we shall address in the next section.
8. Biocatalysis is green and sustainable
Organic synthesis with enzymes has been known for a long time but up until fairly recently was only sporadically used in industrial organic synthesis. However, in the second half of the 1980s the application of enzymes in industrial organic synthesis was triggered by a confluence of two developments.127 First, the landmark 1984 paper by Zaks and Klibanov,128 describing enzymatic catalysis at 100 °C in organic media, demonstrated that many enzymes were actually more thermally stable in organic solvents, such as toluene, than in water. This was a revelation for most synthetic organic chemists. It led to the realisation that biocatalysis had a much broader scope in organic synthesis than was previously imagined. Second, at about the same time recognition of the importance of stereoisomerism in drug action signaled the advent of regulations by, for example, the FDA which required that chiral drugs be marketed as the single, active enantiomer. This created a need for cost-effective methods for enantioselective synthesis and, since enzymes are usually highly enantioselective, biocatalytic methods were obvious candidates.Hence, biocatalysis evolved from an academic curiosity to an industrially attractive technology for the enantioselective synthesis of APIs.129 However, twenty-five years ago a major obstacle to widespread use was the limited number of commercially available enzymes. The enzymes available to process chemists were largely limited to those already in use in the food and beverages and detergent industries, which consisted mainly of hydrolases such as lipases, esterases and glycosidases, often from animal origin. Over the last two decades this situation has changed dramatically thanks to modern biotechnology. As a result of high throughput DNA sequencing, around 20000 whole genome sequences have become publicly available. Thanks to advances in recombinant DNA technology it is now possible to identify a target gene, by in silico analysis of a genome sequence data base, have the gene synthesised chemically within two weeks, ready for cloning into a host production organism, typically at a cost of ca. $ 1000. Consequently, more enzymes are available and they can be produced for commercially acceptable prices. Moreover, protein engineering techniques, such as directed (in vitro) evolution,130 have made it possible to re-engineer enzymes such that they exhibit pre-defined properties with regard to, inter alia, substrate specificity, activity, selectivity, stability and pH optimum.131 Twenty five years ago it was necessary to modify the process to fit the available enzyme, often resulting in a nightmare process. Now it is eminently feasible to optimise the enzyme to fit a pre-defined optimum process, i.e., genuine benign by design. Furthermore, the development of effective immobilisation techniques has paved the way for optimising their storage and operational stability and their cost-effectiveness by facilitating recovery and recycling as free-flowing solids.132
Consequently, in the last decade biocatalysis has been integrated into mainstream organic synthesis,133,134 particularly in the pharmaceutical industry.135 Indeed, Turner and O'Reilly136 proposed that guidelines and rules for biocatalytic retrosynthesis be introduced to aid synthetic chemists in identifying where biocatalysts could be applied to the synthesis of target molecules. Its broad application can also be attributed to its numerous environmental and economic benefits. The catalyst (an enzyme) is derived from renewable resources and is biocompatible, biodegradable, essentially nonhazardous and nontoxic. Biocatalysis avoids the use of scarce precious metals and the associated, often prohibitive, costs of removing traces of noble metals, to an acceptable ppm level, from end products (see earlier). Enzymatic reactions are performed under mild conditions (physiological pH and ambient temperature and pressure) in water, often without the need for functional-group activation and protection and deprotection steps required in conventional organic syntheses. This translates to more step economic syntheses, higher selectivities, and purer products from processes that are more resource and energy efficient and generate less waste than conventional routes. Enzymatic processes (but not fermentations) can be conducted in standard multi-purpose batch reactors and, hence, do not require any extra investment, e.g. for high-pressure equipment. Lastly, most enzymatic reactions are conducted under roughly the same conditions of temperature and pressure and, hence, it is relatively easy to integrate multiple reactions into eco-efficient catalytic cascade processes.137 In short, biocatalysis exhibits an excellent fit with those principles of green chemistry4 which are concerned with process design.
Just as with all processes, solvent usage (section 7) is an important consideration in biocatalytic transformations. Enzymes function optimally in water, which is generally perceived as an advantage, but it can be a serious shortcoming if the organic substrate is only sparingly soluble in water. Moreover, some reactions such as (trans)esterifications and amidations cannot be conducted in water owing to equilibrium limitations and/or product hydrolysis. Hence, the long-standing interest in non-aqueous biocatalysis,138 which was triggered by the seminal paper of Zaks and Klibanov.128 Additional benefits of non-aqueous biocatalysis are easier product recovery from volatile organic solvents and elimination of microbial contamination. However, the use of volatile organic solvents in biocatalytic processes is subject to the same caveats as for organic syntheses in general (section 7). It should also be mentioned that if water is used as the solvent it still has to be treated to remove traces of organics before it can be discharged (and the complete E factor includes water).139 Hence, high substrate concentrations (preferably 10 wt% or more), or even suspensions of substrates, should be used in order to limit the amount of water. Moreover, highly polar substrates, such as carbohydrates, nucleosides and peptides are sparingly soluble in common organic solvents. There is a growing interest, therefore, in the use of neoteric solvents such as ionic liquids (ILs) and deep eutectic solvents (DESs) as reaction media for biocatalytic processes (see section 9).138
Biocatalytic processes can be performed with isolated enzymes or as whole cell biotransformations. Isolated enzymes have the advantage of not being contaminated with other enzymes present in the cell while the use of whole cells is less expensive as it avoids separation and purification of the enzyme. When growing microbial cells are used i.e. in fermentation processes, substantial amounts of biomass are generated as waste, which is generally easy to dispose of, e.g. as animal feed or can, in principle, be used as a source of energy for the process.
Natural enzymes are often highly selective catalysts but they have evolved over millions of years to be able to convert their natural substrates in high rates in vivo. It is perhaps not surprising therefore, that they are generally not sufficiently active or stable to be able to sustain a high activity and productivity with non-natural substrates under more challenging conditions in vitro, e.g. high substrate concentrations, non-aqueous media and so forth. Nonetheless, spectacular results have been obtained in some cases with wild-type enzymes, without the need for protein engineering. An early example is the enzymatic process for the synthesis of the key chiral intermediate in the manufacture of the antihypertensive drug, diltiazem, developed and commercialised by DSM-Andeno in the 1980s. It involved highly enantioselective hydrolysis of a chiral glycidate ester catalysed by Thermomyces lanuginosus lipase (E.C. 3.1.1.3), otherwise known as lipolase (Scheme 13a),140 an inexpensive, readily available enzyme used in a wide variety of industrial applications.141
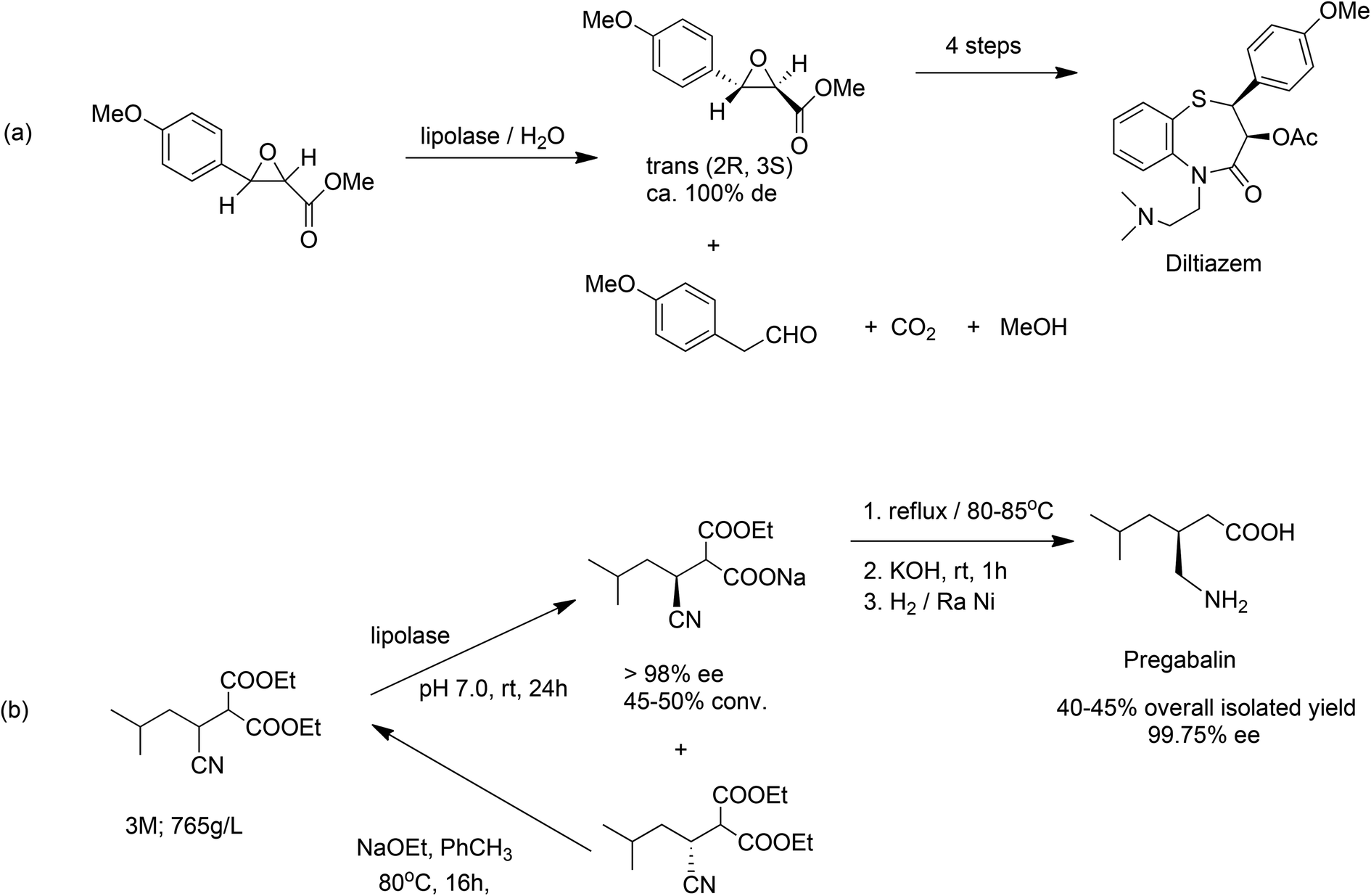 | ||
| Scheme 13 Enzymatic processes for diltiazem (a) and pregabalin. (b) Using lipolase. | ||
More recently, Pfizer developed an extremely effective chemoenzymatic process (Scheme 13b) for the manufacture of pregabalin,142 the active ingredient of the CNS drug Lyrica® using the same lipase. This second generation route afforded a dramatic improvement in process efficiency by setting the stereocentre early in the synthesis (the golden rule of chirotechnology127) and enabling the facile racemisation and re-use of the wrong enantiomer. The key enzymatic step was conducted at an impressive 3 M (765 g l−1) substrate concentration in a largely aqueous process with dramatically reduced organic solvent usage. Compared to the first generation manufacturing process, the new one afforded a higher yield and a five-fold reduction in the E factor from 86 to 17, and dispelled the notion that enzymatic reactions only work at high dilution.
The use of oxidoreductases143 in industrial organic synthesis has been gaining momentum in the last decade. In particular, ketoreductase (KRED) catalyzed enantioselective reduction of ketones to chiral alcohols, is a mature technology which is finding broad application in the pharmaceutical and fine chemical industries as an alternative to asymmetric hydrogenation catalyzed by noble metal complexes.144 The development of efficient cofactor regeneration systems together with enzyme optimisation using protein engineering techniques, has enabled the commercially viable application of KREDs at high substrate loadings.
A pertinent example is the Codexis process (Scheme 14a) for the manufacture of a key intermediate for atorvastatin, the active ingredient of the cholesterol lowering drug Lipitor.6 Ethyl-4-chloroacetoacetate undergoes highly enantioselective reduction catalysed by a ketoreductase (KRED) to afford the (S)-alcohol in 96% isolated yield and >99.5% ee. Cofactor regeneration is achieved with glucose and a NADP-dependent glucose dehydrogenase (GDH). In a second step, a halohydrin dehalogenase (HHDH) was employed to catalyse a nucleophilic substitution of chloride by cyanide using HCN at neutral pH and ambient temperature. All three enzymes were optimised by in vitro evolution using gene shuffling.145 The activity of the wild-type HHDH in the non-natural cyanation reaction was extremely low and was improved >2500 fold. The E factor for the overall process is 5.8 if water is excluded (2.3 for the reduction and 3.5 for the cyanation). If water is included the E factor is 18 (6.6 for reduction and 11.4 for cyanation). The main contributors to the E factor are solvent (butyl and ethyl acetate for extraction of the product in the first and second step, respectively) losses (51%), sodium gluconate (25%) and innocuous inorganic salts, NaCl and Na2SO4 (combined 22%). The three enzymes and the NADP cofactor account for <1% of the waste. Solvent recovery was 85% which probably can be further improved.
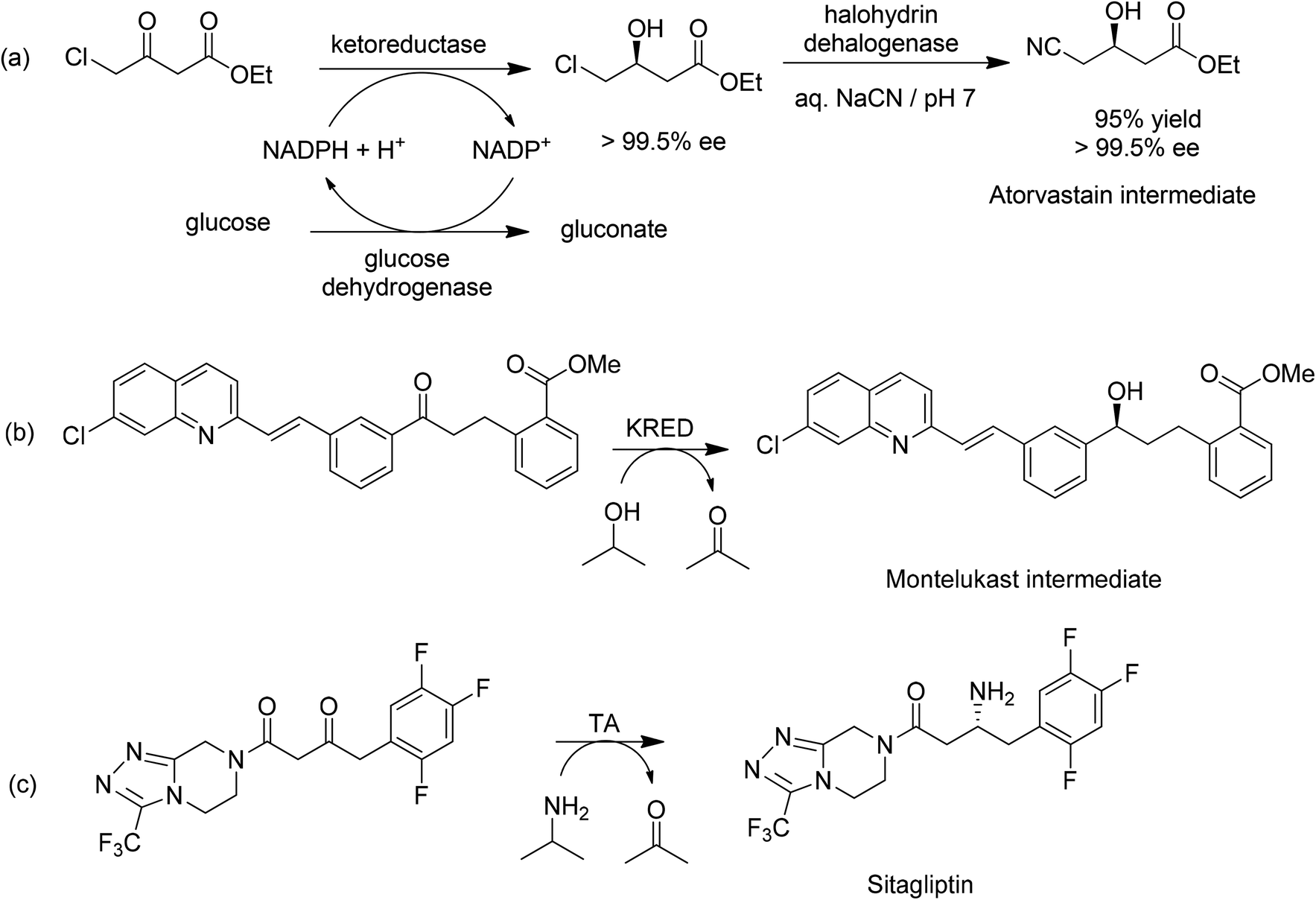 | ||
| Scheme 14 Biocatalytic reduction and reductive amination processes in API synthesis. | ||
Similarly, a biocatalytic process was developed146 for an intermediate for montelukast, the active ingredient of Singulair, an anti-asthmatic agent of Merck. It involved a KRED catalysed reduction of a prochiral ketone (Scheme 14b) as a suspension in a 4:1 mixture of isopropyl alcohol (for cofactor regeneration) and toluene. An enzyme was identified and its performance subsequently improved >1000 fold by in vitro evolution. The (S)-alcohol was obtained in >99.9% ee at a volumetric activity of >4 g L−1 h−1 and 100 g L−1 substrate. The biocatalytic process replaced a chemical process involving reduction by 1.8 eq. of a chiral borane in tetrahydrofuran at −25 °C and, hence, a much higher E factor.
Another striking example is the enzymatic synthesis of the antidiabetic, sitagliptin, involving an overall enantioselective reductive amination of a ketone using an (R)-transaminase-catalysed reaction with isopropylamine (Scheme 14c).147 Starting with an (R)-selective transaminase, which showed no activity towards the ketone substrate, in silico studies were used to identify what was needed to fit the ketone into the binding pocket of the enzyme. The amino residues surrounding the binding pocket were then engineered to provide the extra space, affording an enzyme with low activity which was further improved up to a commercially viable level using in vitro evolution. Under optimised conditions, 6 g l−1 of the best variant, in 50% aq. DMSO, converted 200 g l−1 of the ketone substrate in 24 h to sitagliptin in 92% yield and >99.95% ee. The biocatalytic process afforded sitagliptin with a 10 to 13% better yield, a 53% increase in productivity (kg per l per day), a 19% reduction in total waste, the elimination of all heavy metals, and a reduction in total manufacturing cost compared with the competing chemical process (a rhodium catalysed asymmetric hydrogenation of an enamine). Furthermore, the enzymatic reaction is run in multipurpose vessels, avoiding the need for specialised high-pressure hydrogenation equipment.
The above described processes constitute the tip of an iceberg. Biocatalytic processes are more environmentally attractive and more cost-effective than classical chemical processes and we expect, therefore, that more and more of the latter will be substituted by biocatalytic alternatives in the coming decade.
9. The bio-based economy and the metrics of biomass conversion
Although attention continues to be focused on waste minimisation and avoiding the use of toxic and/or hazardous reagents and solvents in chemicals manufacture, the last decade has witnessed a growing emphasis on the third element of green chemistry, namely the utilisation of renewable resources.148 Indeed, one of the great challenges of the twenty first century is to facilitate the transition from an unsustainable global economy largely based on non-renewable fossil resources – oil, coal and natural gas – to a sustainable one based on renewable resources. A carbon neutral, bio-based economy is envisaged in which liquid fuels, commodity chemicals, and novel materials, such as bioplastics, are produced in biorefineries.149 An additional green benefit could be derived from the substitution of existing products by inherently safer alternatives with reduced environmental footprints, as exemplified by biocompatible and biodegradable plastics.150 Surely, the ultimate in sustainable development must be to emulate nature by harnessing today's solar energy, rather than solar energy of the past laid down in geological reserves, for the synthesis of chemicals from carbon dioxide and water.A switch to renewable biomass as a feedstock in integrated biorefineries, employing green chemo- and biocatalytic processes,151 will afford an environmentally beneficial reduction in the carbon footprint of chemicals, liquid fuels and materials. However, the use of first generation biomass feedstocks, such as corn and edible oil seeds, is not perceived as a sustainable long-term option because it competes, directly or indirectly, with food production. The ideal scenario, from the viewpoint of a truly circular economy involves the valorisation of unavoidable waste biomass generated as agricultural residues, for example, sugar cane bagasse, corn stover, wheat straw and rice husks. Hundreds of millions of tons of this waste are produced annually on a global basis (Scheme 15).152 Recently, attention has focused on another feedstock for biorefineries: food supply chain waste (FSCW),153 most of which goes to landfill which has a negative value. Hence, the second-generation bio-based economy is founded on the full utilisation of agricultural biomass for the production of fuels and chemicals.
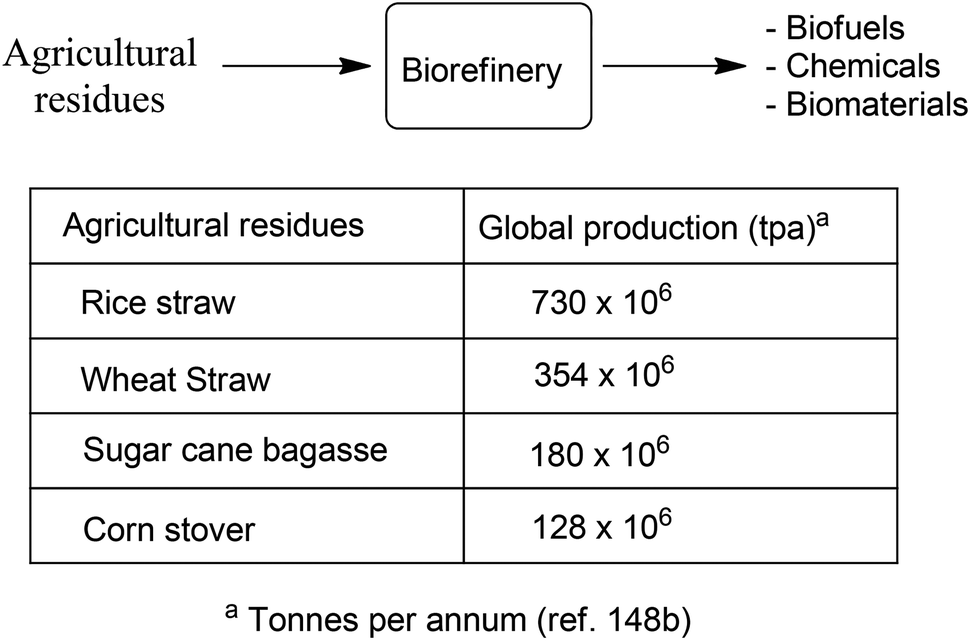 | ||
| Scheme 15 Conversion of waste lignocellulose in a biorefinery. | ||
This second-generation biomass consists primarily of lignocellulose derived from the cell walls of plants. It is available in enormous quantities but is much more difficult to process than first generation feedstocks such as sucrose and starch. Lignocellulose feedstocks consist of three major polymeric components: lignin (ca. 20%), cellulose (ca. 40%), and hemicellulose (ca. 25%) which have to be depolymerized and (partially) deoxygenated, in thermochemical or hydrolytic processes.154 An example of the former is gasification to a mixture of carbon monoxide and hydrogen (syn gas) which can be further processed using established catalytic technologies or fermentation155 to produce biofuels or platform chemicals. Interestingly, commercialisation of fermentation technology155d is targeting waste ‘syn gas’, which is available in large quantities, e.g. from steel mills, but could, in principle, also be produced from any organic waste.
Currently, the method of choice for hydrolytic conversion involves some form of pretreatment, such as a steam explosion, to open up the recalcitrant lignocellulose,156 followed by the addition of enzyme cocktails to catalyse the hydrolysis (saccharification) of the cellulose and hemicellulose to a mixture of C6 and C5 sugars.157 The remaining lignin is used to generate electricity but it would be more attractive to valorise it by (bio)catalytic conversion to commodity chemicals. The C6 and C5 sugars are subsequently converted to biofuels or platform chemicals, by either fermentation or chemocatalytic processes. Alternatively, hydrolysis and fermentation can be combined in a one-pot SSF (simultaneous saccharification and fermentation) process.158
The costs of both the pretreatment and the enzyme cocktail contribute significantly to the overall cost of second-generation bioethanol. The enzyme costs have decreased substantially over the last decade, and are still decreasing, as a result of optimisation of the production and properties of the cellulytic enzyme cocktail.159 Further cost reductions could probably be obtained by immobilisation of the enzyme(s) as, for example, cross-linked enzyme aggregates (CLEAs),160 thus enabling multiple recycling as opposed to the current single use, throw-away practice. However, in second-generation biofuels production the immobilised enzyme generally has to be separated from other suspended solids. This can be readily achieved on a large scale and in a cost-effective manner, using magnetic CLEAs and magnetic separation equipment commonly used in the mining industry.161
Currently, much attention is focused on the use of alternative reaction media138,162 such as ILs, and DESs, particularly if they are derived from renewable raw materials, for both saccharification of lignocellulose and further conversion of the substituent sugars to biofuels and commodity chemicals.
9.1. Carbohydrates to commodity chemicals
Two strategies are envisaged for conversion of C6 and C5 sugars to commodity chemicals: (i) conversion to ‘drop-in’ petroleum hydrocarbons which can be further processed employing established petrochemical technologies, and (ii) direct, redox-economic conversion to oxygenates as platform chemicals.There are various chemo- and biocatalytic strategies for the conversion of hexoses and pentoses to petroleum hydrocarbons (Scheme 16).163 One approach is to produce lower alcohols – ethanol, propanols and butanols – by fermentation and dehydrate them to the corresponding olefins, thereby providing a direct link into existing petrochemical supply chains. Indeed, the optimum use of bioethanol could well be as a platform chemical rather than as a biofuel.164 Similarly, 1-butanol and isobutanol can be produced efficiently by fermentation and dehydrated to 1-butene and isobutene, respectively. 2,3-Butane diol and 1,4-butane diol can also be produced by fermentation. They are interesting commodity chemicals themselves but they can also be dehydrated to butadiene.
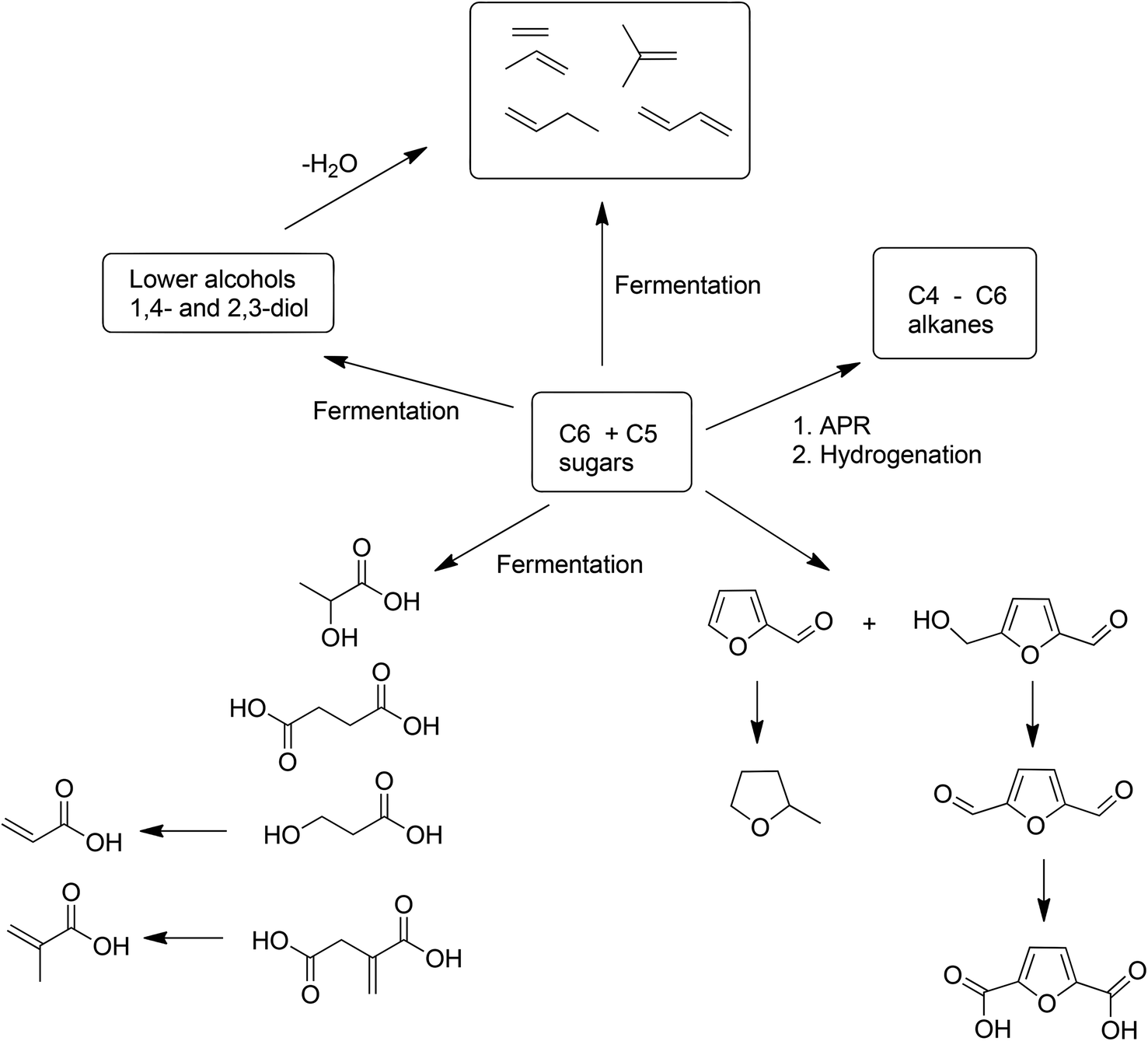 | ||
| Scheme 16 Conversion of carbohydrates to commodity chemicals and fuels. | ||
Alternatively, hydrocarbons can be produced directly by fermentation165 thereby circumventing the energy intensive separation of water miscible lower alcohols from the aqueous fermentation medium. Metabolic engineering is being used to re-engineer the isoprenoid pathway or fatty acid biosynthesis, in bacteria or yeast, to directly yield hydrocarbons, such as isobutene and isoprene. Alternatively, chemocatalytic conversion by so-called aqueous phase reforming (APR) over supported Pt or Pt–Re catalysts produces, inter alia, furfural, 5-hydroxymethyl furfural (HMF) and levulinic acid (LA), which can be hydrogenated in situ to a mixture of mainly C4–C6 alkanes.166
Similarly, direct conversion of hexoses and pentoses to commodity platform chemicals can be conducted using chemical or biological catalysis, or combinations thereof.167,168 Thanks to the significant advances in metabolic engineering and synthetic biology in the last two decades, a wide variety of oxygenates, including lower alcohols, diols and a variety of mono- and di-carboxylic acids can be produced directly, in a redox economic and cost effective manner, by fermentation. Citric acid and lactic acid are examples of first generation, large volume commodity chemicals produced by fermentation. There are many examples of second generation (di)carboxylic acids, such as succinic acid and acrylic acid, and diols which can be produced cost-effectively by fermentation (Scheme 16). However, the current low cost of crude oil and natural gas is not exactly conducive to the introduction of bio-based processes to the same products at this moment in time.
Alternatively, acid catalysed hydrolysis of pentoses and hexoses affords furfural169 and HMF, respectively. Furfural is already produced on an industrial scale and has broad applications. HMF, in contrast, has enormous potential170 as a feedstock for chemicals, polymers and biofuels but its production has not yet been reduced to commercial practice. HMF can be oxidised to 2,5-diformylfuran (DFF) and further to furan-2,5-dicarboxylic acid (FDCA), potential building blocks for polymers (Scheme 16). However, acid catalysed dehydration of hexoses to HMF in a cost-effective manner forms a significant challenge, mainly owing to the unstable nature of HMF towards further reaction, under the acidic reaction conditions, to levulinic acid.171
9.2. Metrics of commodity chemicals from biomass
Much attention has been focused in recent years on the sustainability of transportation fuels from renewable biomass and various metrics have been used to measure it.172 In contrast, much less attention has been devoted to assessing the sustainability of commodity chemicals from renewable biomass vs. non-renewable fossil resources. Saling and co-workers173 at BASF have used their earlier mentioned (see section 4) eco-efficiency analysis for a cradle-to-gate assessment of bio-based vs. petroleum-based routes to, for example, vitamin B2 (riboflavin). More recently, Patel and co-workers,174 building on earlier work of Fischer and co-workers,175 described a methodology for relatively quick assessment of the sustainability of processes in the laboratory phase, based on green chemistry principles, techno-economic analysis and some elements of LCA, reflecting the environmental impact of both the raw materials and the process. The method was used to compare bio-based vs. naphtha-based butadiene and was later extended to other early stage bio-based vs. petroleum-based products.176 A more comprehensive study of bio-based vs. naphtha-based butadiene, using a simplified life cycle thinking based on five indicators – cumulative energy demand, carbon footprint, water usage, a mid-point oriented analysis method and an economic index – was subsequently reported by Cavani and coworkers.177 They concluded that the direct conversion of (bio)ethanol to butadiene has a lower burden than the naphtha-based route and that future efforts should be focused on this route.The European Union COST Action CM0903 “Utilisation of Biomass for Sustainable Fuels and Chemicals” (UBIOCHEM) was launched in November 2009 with special emphasis on the utilisation of agricultural residues and non-edible or waste triglycerides. An important objective of UBIOCHEM was to shape a unified view and develop concise metrics for comparing different processes to sustainable fuels and platform chemicals from biomass. A Special Issue of Catalysis Today, “Sustainability metrics of chemicals from biomass” was devoted to this topic in 2015.178 The goal was to produce a concise set of sustainability metrics which would enable a relatively quick, cradle-to-gate comparison of fossil- vs. renewable biomass-based routes to commodity chemicals. It soon became apparent that mass-based metrics alone were not sufficient to differentiate as the competing processes often had comparable E factors. Four criteria were eventually selected: material and energy efficiency, land use and process economics.179 Material efficiency was defined as the total mass of useful products divided by the total mass of useful products + waste, i.e. it is 1/(E + 1). The definition of useful products in this context is debatable.
Seven commodity chemicals were chosen for the study: lactic acid, acrylonitrile, 1-butanol, 1,2-propane diol, succinic acid, isoprene and methionine. A preliminary conclusion was that some chemicals can already be produced from biomass with less energy input and even at lower cost compared to established petrochemical routes while others are currently more expensive and less energy efficient. Indeed, many bio-based routes are at the beginning of the learning curve and these concise metrics are useful in identifying bottlenecks and providing a basis for planning further optimisation.
Interestingly, Horvath and co-workers180 have proposed ‘ethanol equivalent’, a relatively simple metric, as a common currency for assessing the sustainability of biomass-based routes to fuels and chemicals. An ethanol equivalent is defined as the ‘mass of ethanol required to deliver the equivalent amount of energy from a given feedstock using energy equivalency or produce the equivalent mass of a carbon-based chemical using molar equivalency’. Since the ‘ethanol equivalent’ can be produced by well-known fermentation, the required mass of biomass feedstock, the land area and even the volume of water can be calculated. The calculations were based on the first generation corn-based bioethanol technology commercially practiced in the US and the stoichiometry shown in Scheme 17. Based on their calculations of ethanol equivalents, the authors concluded, inter alia, that replacement of the 387 × 106 tons of gasoline used in the US in 2008 is not a viable proposition by a long way. In contrast, the conversion of biomass to basic chemicals, such as ethylene, propylene and xylenes, could be a sustainable option in the near future, especially with second generation bioethanol from waste lignocellulose.
 | ||
| Scheme 17 Stoichiometry of carbon dioxide to ethanol via glucose. | ||
When the relevant processes have been demonstrated at an industrial scale full-blown sustainability assessments can be used to compare different process strategies. For example, Morales and coworkers181 carried out a sustainability assessment of technologies for the production of succinic acid by fermentation with metabolically engineered strains of E. coli, including isolation of the succinic acid from the fermentation broth. Technical, economic, environmental and process hazard aspects were considered. They showed that fermentation with strains active at acidic pH together with reactive extraction of the product provide the most environmentally competitive process, while strains with resistance to high sugar concentrations afforded the most economically attractive one. Succinic acid is currently produced mainly from n-butane via maleic anhydride. Substitution of this petrochemical succinic acid by bio-succinic acid would afford greenhouse gas savings of ca. 5 tonnes of CO2 per tonne of succinic acid. The authors noted that realisation of a high market share is dependent on a future decrease in total production costs and the product isolation step is responsible for 60–70% of the latter.
10. Conclusions and future outlook
Publication of the table of E factors twenty five years ago presented a challenge to the fine chemical and pharmaceutical industries to make the paradigm shift from a concept of process efficiency which was exclusively focused on chemical yield to one that is motivated by optimising resource efficiency and eliminating waste. The pharmaceutical industry in particular picked up the gauntlet and has made substantial progress in the integration of the principles of green chemistry and sustainability into the development and commercialisation of processes for the manufacture of APIs. The E factor concept played a pivotal role in driving the greening of the chemical and allied industries, and has been embraced by industry and academia world-wide as it provided a very simple means to measure the efficiency and sustainability of the chemical industry.Looking to the future, the displacement of archaic “stoichiometric” technologies by greener catalytic alternatives and the replacement of toxic and/or hazardous solvents and reagents with cleaner alternatives will continue to be important but it will be bolstered by two more drivers on the road to sustainability. First, the change from a fossil-based to a renewable bio-based manufacture of existing commodity chemicals and new materials which are biocompatible and biodegradable. Second, the transition from an unsustainable linear economy to a circular one which designs products and processes with conservation of resources and elimination of waste in mind, a truly green economy. In the words of William Ford Jr, “A good company delivers excellent products and services. A great company does all this and strives to make the world a better place”.
Notes and references
- R. A. Sheldon, Chem. Ind., 1992, 903–906 CAS.
- R. A. Sheldon, Green Chem., 2007, 9, 1273–1283 RSC.
- http://www.epa.gov/laws-regulations/summary-pollution-prevention-act .
- P. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, Oxford, 1998 Search PubMed.
- B. M. Trost, Science, 1991, 254, 1471–1477 CAS.
- S. K. Ma, J. Gruber, C. Davis, L. Newman, D. Gray, A. Wang, J. Grate, G. W. Huisman and R. A. Sheldon, Green Chem., 2010, 12, 81–86 RSC.
- F. Roschangar, R. A. Sheldon and C. H. Senanayake, Green Chem., 2015, 17, 752–768 RSC.
- (a) J. W. Hill and T. W. McCreary, Chemistry for Changing Times, Pearson, 14th edn, 2015, ch. 8 Search PubMed; (b) http://www.beyondbenign.org/k12education/highschool.html ; (c) http://nieuwescheikunde.nl/Publicaties/Lesmodulen/groenechemie/ ; (d) R. Hudson, D. Leaman, K. E. Kawamura, K. N. Esdale, S. Glaisher, A. Bishop and J. L. Katz, J. Chem. Educ., 2016, 93, 691–694 CrossRef CAS.
- Green Chemistry Metrics: Measuring and Monitoring Sustainable Processes, ed. A. Lapkin and D. J. C. Constable, Wiley-Blackwell, Chichester, 2008 Search PubMed; J. Andraos, The Algebra of Organic Synthesis: Green Metrics, Design Strategy, Route Selection and Optimization, CRC Press, Taylor and Francis Group, Boca Raton, 2012 Search PubMed.
- J. Andraos, Org. Process Res. Dev., 2005, 9, 149–163 CrossRef CAS.
- (a) A. D. Curzons, D. J. C. Constable, D. N. Mortimer and V. L. Cunningham, Green Chem., 2001, 3, 1–6 RSC; (b) D. J. C. Constable, A. D. Curzons and V. L. Cunningham, Green Chem., 2002, 4, 521–527 RSC.
- (a) C. Jiménez-González, C. S. Ponder, Q. B. Broxterman and J. Manley, Org. Process Res. Dev., 2011, 15, 912–917 CrossRef; (b) C. Jiménez-González, C. Ollech, W. Pyrz, D. Hughes, Q. B. Broxterman and N. Bhathela, Org. Process Res. Dev., 2013, 17, 239–246 CrossRef.
- P. J. Dunn, S. Galvin and K. Hettenbach, Green Chem., 2004, 6, 43–48 RSC.
- P. A. Wender, M. P. Croatt and B. Witulski, Tetrahedron, 2006, 62, 7505–7511 CrossRef CAS.
- R. A. Sheldon, CHEMTECH, 1994, 38–47 CAS.
- M. Eissen and J. O. Metzger, Chem. – Eur. J., 2002, 8, 3580–3585 CrossRef CAS.
- K. van Aken, L. Strekowski and L. Patiny, Beilstein J. Org. Chem., 2006, 2(3), 1–7 Search PubMed.
- (a) M. A. Curran, Curr. Opin. Chem. Eng., 2013, 2, 273–277 CrossRef; (b) Life Cycle Assessment Handbook; A Guide for Environmentally Sustainable Products, ed. M. A. Curran, Wiley, Hoboken, NJ, USA, 2012 Search PubMed; (c) J. G. Moretz-Sohn Monteiro, O. de Queiroz Fernandes Araujo and J. L. de Medeiros, Clean Technol. Environ. Policy, 2009, 11, 459–472 CrossRef; (d) G. Finnveden, M. Z. Hauschild, T. Ekvali, T. Guinée, R. Heijungs, S. Hellweg, A. Koehler, D. Pennington and S. Suh, J. Environ. Manage., 2009, 91, 1–21 CrossRef PubMed; (e) Background and Future Prospects in Life Cycle Assessment, ed. W. Klöpffer, Springer, Dordrecht, 2014 Search PubMed.
- See in: R. G. Hunt and W. E. Franklin, Int. J. Life Cycle Assess, 1996, 1, 4–7 CrossRef.
- J. A. Fava, A. Smerek, A. B. Heinrich and L. Morrison, in Background and Future Prospects in Life Cycle Assessment, ed. W. Klöpffer, Springer, Dordrecht, 2014, pp. 39–83 Search PubMed.
- (a) EN ISO 14040 Environmental Management, Life Cycle Assessment, Principles and Framework, International Organization for Standardization, Geneva, Switzerland, 2006 Search PubMed; (b) EN ISO 14044 Environmental Management, Life Cycle Assessment, Requirements and Guidelines, International Organization for Standardization, Geneva, Switzerland, 2006 Search PubMed.
- T. Graedel, Green Chem., 1999, 1, G126–G128 RSC.
- (a) P. T. Anastas and R. L. Lankey, Green Chem., 2000, 2, 289–295 RSC; (b) R. L. Lankey and P. T. Anastas, Ind. Eng. Chem. Res., 2002, 41, 4498–4502 CrossRef CAS; (c) X. Domenech, J. A. Ayllon and J. Peral, Environ. Sci. Technol., 2002, 36, 5517–5520 CrossRef CAS PubMed; (d) L. M. Gustafsson and P. Börjesson, Int. J. Life Cycle Assess., 2007, 12(3), 151–159 CAS; (e) L. M. Gilbertson, J. B. Zimmerman, D. L. Plata, J. E. Hutchison and P. T. Anastas, Chem. Soc. Rev., 2015, 44, 5758–5777 RSC.
- S. M. Mercer, J. Andraos and P. G. Jessop, J. Chem. Educ., 2012, 89, 215–220 CrossRef CAS.
- (a) M. A. Gonzalez and R. L. Smith, Environ. Prog., 2003, 22(4), 269–276 CrossRef CAS; (b) R. L. Smith, G. J. Ruiz-Mercado and M. A. Gonzalez, Comput. Chem. Eng., 2015, 81, 272–277 CrossRef CAS.
- C. Jiménez-González, A. D. Curzons, D. J. C. Constable and V. L. Cunningham, Int. J. Life Cycle Assess., 2004, 9(2), 114–121 CrossRef.
- A. D. Curzons, C. Jiménez-Gonzalez, A. L. Duncan, D. J. C. Constable and V. L. Cunningham, Int. J. Life Cycle Assess., 2007, 12(4), 272–280 CrossRef CAS.
- C. Jiménez-González and M. R. Overcash, Green Chem., 2014, 16, 3392–3400 RSC.
- (a) P. Saling, A. Kicherer, B. Dittrich-Krämer, R. Wittlinger, W. Zombik, I. Schmidt, W. Schrott and S. Schmidt, Int. J. Life Cycle Assess., 2002, 7(4), 203–218 CrossRef; (b) P. Saling, R. Maisch, M. Sylvani and N. König, Int. J. Life Cycle Assess., 2005, 10(5), 364–371 CrossRef; (c) A. Kicherer, S. Schaltegger, H. Tschochohei and B. Ferreira Bozo, Int. J. Life Cycle Assess., 2007, 12(7), 537–543 CAS; (d) R. Landsiedel and P. Saling, Int. J. Life Cycle Assess., 2002, 5, 261–268 CrossRef; (e) D. R. Shonnard, A. Kicherer and P. Saling, Environ. Sci. Technol., 2003, 37, 5340–5348 CrossRef CAS PubMed.
- L. Leseurre, C. Merea, S. Duprat de Paule and A. Pinchart, Green Chem., 2014, 16, 1139–1148 RSC.
- T. V. T. Phan, C. Gallardo and J. Mane, Green Chem., 2015, 17, 2846–2852 RSC.
- For a recent review see: L. M. Tufvesson, P. Tufvesson, J. M. Woodley and P. Börjesson, Int. J. Life Cycle Assess., 2013, 18, 431–444 CrossRef CAS.
- (a) G. Koller, U. Fischer and K. Hungerbühler, Ind. Eng. Chem. Res., 2000, 36, 960–972 CrossRef; (b) G. Wernet, S. Conradt, H. P. Isenring, C. Jiménez-González and K. Hungerbühler, Int. J. Life Cycle Assess., 2010, 15, 294–303 CrossRef CAS.
- D. Cespi, E. S. Beach, T. E. Swarr, F. Passarinin, I. Vassura, P. J. Dunn and P. T. Anastas, Green Chem., 2015, 17, 3390–3400 RSC.
- (a) G. Wernet, S. Papadokonstantakis, S. Hellweg and K. Hungerbühler, Green Chem., 2009, 11, 1826–1831 RSC; (b) G. Wernet, S. Hellweg, U. Fischer, S. Papadokonstantakis and K. Hungerbühler, Environ. Sci. Technol., 2008, 42, 6717–6722 CrossRef CAS; (c) G. Wernet, S. Hellweg and K. Hungerbühler, Int. J. Life Cycle Assess., 2012, 17, 720–728 CrossRef CAS.
- M. J. Eckelman, Green Chem., 2016, 18, 3257–3264 RSC.
- K. A. Hossain, F. I. Khan and K. Hawboldt, Ind. Eng. Chem. Res., 2007, 46, 8787–8795 CrossRef CAS.
- For an excellent tutorial review of LCA see: D. Kralisch, D. Ott and D. Gericke, Green Chem., 2015, 17, 123–145 RSC . See also: C. R. McElroy, A. Constantinou, L. C. Jones, L. Summerton and J. H. Clark, Green Chem., 2015, 17, 3111–1321 RSC.
- C. G. Brundtland, Our Common Future, The World Commission on Environmental Development, Oxford University Press, Oxford, 1987 Search PubMed.
- S. K. Sikdar, AIChE J., 2003, 49(8), 1928–1932 CrossRef CAS.
- E. Heinzle, D. Weirich, F. Brogli, V. H. Hoffmann, G. Koller, M. A. Verduyn and K. Hungerbühler, Ind. Eng. Chem. Res., 1998, 37, 3395–3407 CrossRef CAS.
- R. Dach, J. J. Song, F. Roschangar, W. Samstag and C. H. Senanayake, Org. Process Res. Dev., 2012, 16, 1697–1706 CrossRef CAS.
- T. E. Graedel, in Handbook of Green Chemistry and Technology, ed. J. Clark and D. J. Macquarrie, Wiley, New York, 2002, pp. 56–61 Search PubMed.
- See http://www.ipcc.ch/pdf/assessment-report/ar4/syr/ar4_syr_spm.pdf.
- J. M. Köhler, Green Process. Synth., 2014, 3, 33–45 Search PubMed.
- J. H. Clark, T. J. Farmer, L. Herrero-Davila and J. Sherwood, Green Chem., 2016, 18, 3914–3934 RSC.
- Communication from the Commission to the European Parliament, the Council, the European Economic and Social Committee and the Committee of the Regions, 2011, Roadmap to a Resource Efficient Europe, COM/2011/0571 final.
- B. Commoner, The Closing Circle: Nature, Man and Technology, Knopf, New York, 1971 Search PubMed.
- R. A. Sheldon, I. W. C. E. Arends and U. Hanefeld, Green Chemistry and Catalysis, Wiley-VCH, Weinheim, 2007 Search PubMed.
- (a) N. Z. Burns, P. S. Baran and R. W. Hoffmann, Angew. Chem., Int. Ed., 2009, 48, 2854–2867 CrossRef CAS PubMed; (b) J. M. Richter, Y. Ishihara, T. Masuda, B. W. Whitefield, T. Llamas, A. Pohjakallio and P. S. Baran, J. Am. Chem. Soc., 2008, 130, 17938–17954 CrossRef CAS PubMed.
- A. J. Mancuso, S.-L. Huang and D. Swern, J. Org. Chem., 1978, 12, 2480–2482 CrossRef CAS; A. J. Mancuso and D. Swern, Synthesis, 1981, 165–185 CrossRef.
- D. B. Dess and J. C. Martin, J. Org. Chem., 1983, 48, 4155–4156 CrossRef CAS.
- A. E. J. de Nooy, A. C. Besemer and H. van Bekkum, Synthesis, 1996, 1153–1174 CrossRef CAS.
- (a) A. Dijksman and I. W. C. E. Arends, Chem. Commun., 2000, 271–272 RSC; (b) A. Dijksman, I. W. C. E. Arends and R. A. Sheldon, Synlett, 2001, 102–104 CAS.
- K. Alfonsi, J. Colberg, P. J. Dunn, T. Fevig, S. Jennings, T. A. Johnson, H. P. Kleine, C. Knight, M. A. Nagy, D. A. Perry and M. Stefaniak, Green Chem., 2008, 10, 31–36 RSC.
- M. H. A. Janssen, J. F. Chesa Castellana, H. Jackman, P. J. Dunn and R. A. Sheldon, Green Chem., 2011, 13, 905–912 RSC.
- For a recent review see: R. A. Sheldon, Catal. Today, 2015, 247, 4–13 CrossRef CAS.
- G. J. ten Brink, I. W. C. E. Arends and R. A. Sheldon, Science, 2000, 287, 1636–1639 CrossRef CAS PubMed . See also: A. N. Campbell and S. S. Stahl, Acc. Chem. Res., 2012, 45, 851–863 CrossRef PubMed.
- (a) P. Gamez, I. W. C. E. Arends, J. Reedijk and R. A. Sheldon, Chem. Commun., 2003, 2414 RSC; (b) P. Gamez, I. W. C. E. Arends, R. A. Sheldon and J. Reedijk, Adv. Synth. Catal., 2004, 346, 805–811 CrossRef CAS; (c) A. Dijksman, I. W. C. E. Arends and R. A. Sheldon, Org. Biomol. Chem., 2003, 1, 3232–3237 RSC.
- M. Fabbrini, C. Galli, P. Gentili and D. Macchitella, Tetrahedron Lett., 2001, 42, 7551–7553 CrossRef CAS.
- I. W. C. E. Arends, Y. X. Li and R. A. Sheldon, Biocatal. Biotransform., 2006, 24, 443–448 CrossRef CAS; I. W. C. E. Arends, Y. X. Li, R. Ausan and R. A. Sheldon, Tetrahedron, 2006, 62, 6659–6665 CrossRef.
- V. Elango, M. A. Murhpy, B. L. Smith, K. G. Davenport, G. N. Mott and G. L. Moss, US Pat, 4981995, 1991, to Hoechst-Celanese Corp Search PubMed.
- (a) C. Torborg and M. Beller, Adv. Synth. Catal., 2009, 351, 3027–3043 CrossRef CAS; (b) A. Dumrath, C. Luebbe and M. Beller, in Palladium Catalyzed Coupling Reactions: Practical Aspects and Future Developments, ed. A. Molnar, Wiley-VCH, Weinheim, 2013, pp. 445–489 Search PubMed.
- (a) T. Mizoroki, K. Mori and A. Ozaki, Bull. Chem. Soc. Jpn., 1971, 44, 581 CrossRef CAS; (b) R. F. Heck and J. P. Nolley Jr., J. Org. Chem., 1972, 37, 2320–2322 CrossRef CAS.
- N. Miyaura, K. Yamada and A. Suzuki, Tetrahedron Lett., 1979, 27, 3437–3443 CrossRef.
- (a) A. S. Guram, R. A. Rennels and S. L. Buchwald, Angew. Chem., Int. Ed. Engl., 1995, 34, 1348–1350 CrossRef CAS; (b) J. Louie and J. F. Hartwig, Tetrahedron Lett., 1995, 36, 3609–3612 CrossRef CAS.
- (a) E. F. Lutz, J. Chem. Educ., 1986, 63, 202–204 CrossRef CAS; (b) B. Reuben and H. Wittcoff, J. Chem. Educ., 1988, 65, 605–607 CrossRef CAS.
- C. S. Higman, J. A. M. Lummiss and D. E. Fogg, Angew. Chem., Int. Ed., 2016, 55, 3552–3565 CrossRef CAS PubMed.
- T. Nicola, M. Brenner, K. Donsbach and P. Kreye, Org. Process Res. Dev., 2005, 9, 513–515 CrossRef CAS.
- (a) S. Chikkali and S. Mecking, Angew. Chem., Int. Ed., 2012, 51, 5802–5808 CrossRef CAS PubMed; (b) U. Biermann, U. Bornscheuer, M. A. R. Meier, J. Metzger and H. J. Schäfer, Angew. Chem., Int. Ed., 2011, 50, 3854–3871 CrossRef CAS PubMed.
- A. Ryback and M. A. R. Meier, Green Chem., 2007, 9, 1356–1361 RSC.
- M. J. Climent, A. Corma, S. Iborra, M. Mifsud and A. Velty, Green Chem., 2010, 12, 99–107 RSC.
- R. A. Sheldon, Green Chem., 2005, 7, 267–278 RSC.
- D. J. C. Constable, C. Jiménez-González and R. K. Henderson, Org. Process Res. Dev., 2007, 11, 133–137 CrossRef CAS.
- C. Jiménez-González, A. D. Curzons, D. J. C. Constable and V. L. Cunningham, Clean Technol. Environ. Policy, 2005, 7, 42–50 CrossRef.
- C. P. Ashcroft, P. J. Dunn, J. D. Hayler and A. S. Wells, Org. Process Res. Dev., 2015, 19, 740–747 CrossRef CAS.
- http://echa.europa.eu/candidate-list-table : the list is updated twice a year.
- (a) C.-W. Cho, T. P. T. Pham, Y.-C. Jeon and Y.-S. Yun, Green Chem., 2008, 10, 67–72 RSC; (b) A. S. Wells and V. T. Coombe, Org. Process Res. Dev., 2006, 10, 794–798 CrossRef CAS; (c) C. Pretti, C. Chiappe, D. Pieraccinin, M. Gregori, M. Abramon, G. Monni and L. Torre, Green Chem., 2006, 8, 238–240 RSC.
- A. D. Curzons, D. J. C. Constable and V. L. Cunningham, Clean Technol. Environ. Policy, 1999, 1, 82–90 CrossRef.
- C. Jiménez-González, A. D. Curzons, D. J. C. Constable and V. L. Cunningham, Clean Technol. Environ. Policy, 2005, 7, 42–50 CrossRef.
- R. K. Henderson, C. Jiménez-González, D. J. C. Constable, S. R. Alston, G. G. A. Inglis, G. Fisher, J. Sherwood, S. P. Binks and A. D. Curzons, Green Chem., 2011, 13, 854–862 RSC.
- C. M. Alder, J. D. Hayler, R. K. Henderson, A. M. Redman, L. Shukla, L. E. Shuster and H. F. Sneddon, Green Chem., 2016, 18, 3879–3890 RSC.
- D. Prat, O. Pardigon, H.-W. Flemming, S. Letestu, V. Ducandas, P. Isnard, E. Guntrum, T. Senac, S. Ruisseau, P. Cruciano and P. Hosek, Org. Process Res. Dev., 2013, 17, 1517–1525 CrossRef CAS.
- J. P. Adams, C. M. Alder, I. Andrews, A. M. Bullion, M. Campbell-Crawford, M. G. Darcy, J. D. Hayler, R. K. Henderson, C. A. Oare, I. Pendrak, A. M. Redman, L. E. Shuster, H. F. Sneddon and M. D. Walker, Green Chem., 2013, 15, 1542–1549 RSC.
- D. S. MacMillan, J. Murray, H. F. Sneddon, C. Jamieson and A. J. B. Watson, Green Chem., 2013, 15, 596–600 RSC.
- F. I. McGonagle, D. S. MacMillan, J. Murray, H. F. Sneddon, C. Jamieson and A. J. B. Watson, Green Chem., 2013, 15, 1159–1165 RSC.
- K. Skowerski, J. Biatecki, A. Tracz and T. K. Olszewski, Green Chem., 2014, 16, 1125–1130 RSC.
- D. Prat, J. Hayler and A. Wells, Green Chem., 2014, 16, 4546–4551 RSC . See also: H. E. Eastman, C. Jamieson and A. J. B. Watson, Aldrichimica. Acta, 2015, 48(2), 51–55 Search PubMed.
- M. Tobiszewski, S. Tsakovski, V. Simeonov, J. Namiesnik and F. Pena-Pereira, Green Chem., 2015, 17, 4773–4785 RSC.
- S. Hellweg, U. Fischer, M. Scheringer and K. Hungerbühler, Green Chem., 2004, 6, 418–427 RSC.
- G. P. Taber, D. M. Pfistere and J. C. Colberg, Org. Process Res. Dev., 2004, 8, 385–388 CrossRef CAS.
- A. G. Corrêa, M. W. Paixão and R. S. Schwab, Curr. Org. Synth., 2015, 12, 675–695 CrossRef.
- (a) Multiphase Homogeneous Catalysis, ed. B. Cornils, W. Herrmann, I. T. Horvath, W. Leitner, S. Mecking, H. Olivier-Bourbigou and D. Vogt, Wiley, Weinheim, 2005 Search PubMed; (b) W. Keim, Green Chem., 2003, 5(2), 105–111 RSC; (c) B. Driessen-Holscher, Adv. Catal., 1998, 42, 473–505 CAS.
- (a) G. Papadogianakis, R. A. Sheldon (Eds.), special issue on recent advances in catalysis in green aqueous media, Catal. Today, 2015, 247, 1–190; ; (b) C. J. Li, Chem. Rev., 1993, 93, 2023–2035 CrossRef CAS; (c) Metal-Catalyzed Reactions in Water, ed. P. H. Dixneuf and V. Cadierno, Wiley-VCH, Weinheim, 2013 Search PubMed.
- (a) B. Cornils, J. Mol. Catal. A: Chem., 1999, 143(1–3), 1–10 CrossRef CAS; (b) B. Cornils, Org. Process Res. Dev., 1998, 2(2), 121–127 CrossRef CAS; (c) A. Behr, M. Urschey and V. A. Brehme, Green Chem., 2003, 5, 198–204 RSC.
- C. W. Kohlpaintner, R. W. Fischer and B. Cornils, Appl. Catal., A, 2001, 221(1–2), 219–225 CrossRef CAS.
- (a) B. H. Lipshutz and S. Ghorai, Aldrichimica. Acta, 2008, 41, 59 CAS; B. H. Lipshutz, S. Ghorai, W. W. Y. Leong, B. R. Taft and D. V. Krogstad, J. Org. Chem., 2011, 76, 5061–5073 Search PubMed.
- (a) B. H. Lipshutz and S. Ghorai, Aldrichimica. Acta, 2012, 45, 3 CAS; (b) B. H. Lipshutz, S. Ghorai, A. R. Abela, R. Moser, T. Nishikata, C. Duplais, A. Krasovski, R. D. Gaston and R. C. Gadwood, J. Org. Chem., 2011, 76, 4379–4391 CrossRef CAS PubMed.
- P. Klumphu and B. H. Lipshutz, J. Org. Chem., 2014, 79, 888–900 CrossRef CAS PubMed.
- B. H. Lipshutz, N. A. Isley, J. C. Fennewald and E. D. Slack, Angew. Chem., Int. Ed., 2013, 52, 10952–10958 CrossRef CAS PubMed.
- B. H. Lipshutz and S. Ghorai, Green Chem., 2014, 16, 3660–3679 RSC.
- A. T. Straub, M. Otto, I. Usui and B. Breit, Adv. Synth. Catal., 2013, 355, 2071–2075 CrossRef CAS.
- (a) I. T. Horvath and J. Rabai, Science, 1994, 266, 72–75 CAS; (b) For a recent example see: J. Balogh, A. R. Hlil, H.-L. Su, Z. Xi, H. S. Bazzi and J. A. Gladysz, ChemCatChem, 2016, 8(1), 125–128 CrossRef CAS.
- For a tutorial review see: X. Han and M. Poliakoff, Chem. Soc. Rev., 2012, 41, 1428–1436 RSC.
- Catalysis in Ionic Liquids: From Catalyst Synthesis to Application, ed. C. Hardacre and V. I. Parvulescu, Royal Soc. Chem., Cambridge, 2014, vol. 15, RSC Catalysis Series Search PubMed.
- M. J. Muldoon, Dalton Trans., 2010, 39, 337–348 RSC.
- D. Coleman and N. Gathergood, Chem. Soc. Rev., 2010, 39, 600–637 RSC.
- (a) S. Stolte, M. Matzke, J. Arning, A. Böschen, W. R. Pitner, U. Welz-Biermann, B. Jastorff and J. Ranke, Green Chem., 2007, 9, 1170–1179 RSC; (b) R. F. M. Frade and C. A. M. Afonso, Hum. Exp. Toxicol., 2010, 29(12), 1038–1054 CrossRef CAS PubMed; (c) S. Bruzzone, C. Chiappe, S. E. Focardi, C. Pretti and M. Renzi, Chem. Eng. J., 2011, 175, 17–23 CrossRef CAS; (d) T. P. T. Pham, C.-W. Cho and Y.-S. Chun, Water Res., 2010, 44, 352–372 CrossRef CAS PubMed.
- M. Deetlefs and K. R. Seddon, Green Chem., 2010, 12, 17–30 RSC.
- M. Petkovic, K. R. Seddon, L. P. N. Rebelo and C. Silva Pereira, Chem. Soc. Rev., 2011, 40, 1383–1403 RSC.
- (a) G. Imperato, B. König and C. Chiappe, Eur. J. Org. Chem., 2007, 1049–1058 CrossRef CAS; (b) C. Chiappe, A. Marra and A. Mele, Top. Curr. Chem., 2010, 295, 177–195 CrossRef CAS PubMed; (c) K. Fukumoto, M. Yoshizawa and H. Ohno, J. Am. Chem. Soc., 2006, 127, 2398–2399 CrossRef PubMed; (d) X. Chen, X. Li, A. Hu and F. Wang, Tetrahedron: Asymmetry, 2008, 19, 1–14 CrossRef CAS.
- (a) Y. Fukaya, Y. Iizuka, K. Sekikawa and H. Ohno, Green Chem., 2007, 9, 1155–1157 RSC; (b) Y. Yu, X. Lu, Q. Zhou, K. Dong, H. Yao and S. Zhang, Chem. – Eur. J., 2008, 14, 11174–11182 CrossRef CAS PubMed.
- (a) M. Petkovic, J. L. Ferguson, H. Q. Nimal Gunaratne, R. Ferreira, M. C. Leitão, K. R. Seddon, L. P. N. Rebelo and C. S. Pereira, Green Chem., 2010, 12, 643–649 RSC; (b) X.-D. Hou, Q.-P. Liu, T. J. Smith, N. Li and M.-H. Zong, PLoS One, 2013, 8(3), e59145 CAS.
- T. L. Greaves and C. J. Drummond, Chem. Rev., 2008, 108, 206–237 CrossRef CAS PubMed.
- C. Pretti, C. Chiappe, I. Baldetti, S. Brunini, G. Monni and L. Intorre, Ecotoxicol. Environ. Saf., 2009, 72, 1170–1176 CrossRef CAS PubMed.
- L. Chen, M. Sharifzadeh, N. MacDowell, T. Welton, N. Shah and J. P. Hallett, Green Chem., 2014, 16, 3098–3106 RSC.
- S. Pavlovica, A. Ziemanis, E. Gzibovska, M. Klavins and P. Mekss, Green Sustainable Chem., 2011, 1, 103–110 CrossRef CAS.
- (a) A. P. Abbott, G. Capper, D. L. Davies, R. K. Rasheed and V. Tambyrajah, Chem. Commun., 2003, 70–71 RSC; (b) E. L. Smith, A. P. Abbott and K. S. Ryder, Chem. Rev., 2014, 114, 11060–11082 CrossRef CAS PubMed; (c) C. Ruβ and B. König, Green Chem., 2012, 14, 2969–2982 RSC; (d) Q. Zhang, K. De Oliveira Vigier, S. Royer and F. Jérôme, Chem. Soc. Rev., 2012, 41, 7108–7146 RSC.
- M. Francisco, A. van den Bruinhorst and M. C. Kroon, Angew. Chem., Int. Ed., 2013, 52, 3074–3085 CrossRef CAS PubMed.
- O. S. Hammond, D. T. Bowron and K. J. Edler, Green Chem., 2016, 18, 2736–2744 RSC.
- A. P. Abbott, R. C. Harris, K. S. Ryder, C. D'Agostino, L. F. Gladden and M. D. Mantle, Green Chem., 2011, 13, 82–90 RSC.
- (a) A. Hayyan, F. S. Mjalli, I. M. Al Nashef, Y. M. Al-Wahaibi, T. Al-Wahaibi and M. Ali Hashim, J. Mol. Liq., 2013, 178, 137–141 CrossRef CAS; (b) Z. Maugeri and P. Dominguez de Maria, RSC Adv., 2012, 2, 421–425 RSC.
- (a) Y. H. Choi, J. Van Spronsen, Y. Dai, M. Verberne, F. Hollmann, I. W. C. E. Arends, G.-J. Witkamp and R. Verpoorte, Plant Physiol., 2011, 156, 1701–1705 CrossRef CAS PubMed; (b) Y. Dai, J. Van Spronsen, G. J. Witkamp, R. Verpoorte and Y. H. Choi, Anal. Chim. Acta, 2013, 766, 61–68 CrossRef CAS PubMed.
- A. Paiva, R. Craveiro, I. Aroso, M. Martins, R. L. Reis and A. R. C. Duarte, ACS Sustainable Chem. Eng., 2014, 2, 1063–1071 CrossRef CAS.
- For a recent review of the use of DESs in organic synthesis see: D. A. Alonso, A. Baeza, R. Chinchilla, G. Guillena, I. M. Pastor and D. J. Ramon, Eur. J. Org. Chem., 2016, 612–632 CrossRef CAS.
- C. Vidal, J. Garcia-Alvarez, A. Hernan-Gomez, A. R. Kennedy and E. Hevia, Angew. Chem., Int. Ed., 2014, 53, 5969–5973 CrossRef CAS PubMed.
- R. A. Sheldon, Chirotechnology: the Industrial Synthesis of Optically Active Compounds, Marcel Dekker, New York, 1993 Search PubMed.
- A. Zaks and A. M. Klibanov, Science, 1984, 224, 1249–1251 CAS.
- R. Holt, Specialty Chem., 2013, 21–23 Search PubMed.
- (a) M. T. Reetz, J. Am. Chem. Soc., 2013, 135, 12480–12496 CrossRef CAS PubMed; (b) C. A. Tracewell and F. A. Arnold, Curr. Opin. Chem. Biol., 2009, 13, 3–9 CrossRef CAS PubMed; (c) N. J. Turner, Nat. Chem. Biol., 2009, 5, 567–573 CrossRef CAS PubMed; (d) M. T. Reetz, J. Org. Chem., 2009, 74, 5767–5778 CrossRef CAS PubMed; (e) S. Luetz, L. Giver and J. Lalonde, Biotechnol. Bioeng., 2008, 101, 647–653 CrossRef CAS PubMed; (f) J. M. Woodley, Curr. Opin. Chem. Biol., 2013, 17, 310–316 CrossRef CAS PubMed.
- (a) U. T. Bornscheuer, G. W. Huisman, R. J. Kazlauskas, S. Lutz, J. C. Moore and K. Robins, Nature, 2012, 485, 185–194 CrossRef CAS PubMed; (b) A. Illanes, A. Cauerhff, L. Wilson and G. R. Castro, Bioresour. Technol., 2012, 115, 48–57 CrossRef CAS PubMed.
- R. A. Sheldon and S. van Pelt, Chem. Soc. Rev., 2013, 42, 6223–6235 RSC.
- (a) Practical Methods for Biocatalysis and Biotransformations, ed. J. Whittall and P. W. Sutton, Wiley, Chichester, UK, 2009, vol. 1 Search PubMed; (b) Practical Methods for Biocatalysis and Biotransformations, ed. J. Whittall and P. W. Sutton, Wiley, Chichester, UK, 2012, vol. 2 Search PubMed; (c) Practical Methods for Biocatalysis and Biotransformations, ed. J. Whittall, P. W. Sutton and W. Kroutil, Chichester, UK, 2016, vol. 3 Search PubMed.
- (a) Green Biocatalysis, ed. R. N. Patel, Wiley, Hoboken, New Jersey, 2016 Search PubMed; (b) Biocatalysis for Green Chemistry and Chemical Process Development, ed. A. Tao and R. J. Kazlaukas, Wiley, Hoboken, New Jersey, 2011 Search PubMed; (c) J.-M. Choi, S.-S. Han and H.-S. Kim, Biotechnol. Adv., 2015, 13, 1443–1454 CrossRef PubMed.
- (a) G. W. Huisman and S. J. Collier, Curr. Opin. Chem. Biol., 2013, 17, 284–292 CrossRef CAS PubMed; (b) A. Tao and J. H. Xu, Curr. Opin. Chem. Biol., 2009, 13, 43–50 CrossRef PubMed; (c) G.-W. Zheng and J. H. Xu, Curr. Opin. Biotechnol., 2011, 22, 784–792 CrossRef CAS PubMed; (d) P. Hoyos, V. Pace, M. J. Hernaiz and A. R. Alcantara, Curr. Green Chem., 2014, 1(2), 155–181 CrossRef CAS; (e) Biocatalysis in the Pharmaceutical and Biotechnology Industries, ed. R. N. Patel, CRC Press, Boca Raton, Florida, 2007 Search PubMed.
- N. Turner and E. O'Reilly, Nat. Chem. Biol., 2013, 9, 285–288 CrossRef CAS PubMed.
- R. A. Sheldon, in Multi-Step Enzyme Catalysis: Biotransformations and Chemo-enzymatic Synthesis, ed. E. Garcia-Junceda, Wiley-VCH, Weinheim, 2008, pp. 109–135 Search PubMed.
- R. A. Sheldon, Chem. – Eur. J., 2016, 22, 12984–12999 CrossRef CAS PubMed and references therein.
- P. Dominguez de Maria and F. Hollmann, Front. Microbiol., 2015, 6, 1257 Search PubMed.
- (a) L. A. Hulshof and J. H. Roskam, Eur. Pat. Appl, EP343714A119891129, 1989 Search PubMed; (b) R. A. Sheldon, J. Chem. Technol. Biotechnol., 1996, 67, 1–14 CrossRef CAS.
- R. Fernandez-Lafuente, J. Mol. Catal. B: Enzym., 2010, 62, 197–212 CrossRef CAS.
- C. A. Martinez, S. Hu, Y. Dumond, J. Tao, P. Kellcher and L. Tully, Org. Process Res. Dev., 2008, 12, 392–398 CrossRef CAS.
- D. Gamenara, G. Seoane, P. Saénz Mendez and P. Dominguez de Maria, Redox Biocatalysis: Fundamentals and Applications, Wiley, Hoboken, NJ, 2012 Search PubMed.
- (a) Y. Ni and J.-H. Xu, Biotechnol. Adv., 2012, 30, 1279–1288 CrossRef CAS PubMed; (b) G. W. Huisman, J. Liang and A. Krebber, Curr. Opin. Chem. Biol., 2010, 14, 122–129 CrossRef CAS PubMed; (c) J. C. Moore, D. J. Pollard, B. Kosjek and P. N. Devine, Acc. Chem. Res., 2007, 40, 1412–1419 CrossRef CAS PubMed; (d) S. M. A. De Wildeman, T. Sonke, H. E. Schoemaker and O. May, Acc. Chem. Res., 2007, 40, 1260–1266 CrossRef CAS PubMed.
- R. J. Fox, C. S. Davis, E. C. Mundorff, L. M. Newman, V. Gavrilovic, S. K. Ma, L. M. Chung, C. Ching, S. Tam, S. Muley, J. Grate, J. Gruber, J. C. Whitman, R. A. Sheldon and G. W. Huisman, Nat. Biotechnol., 2007, 25, 338–344 CrossRef CAS PubMed.
- J. Liang, J. Lalonde, B. Borup, V. Mitchell, E. Mundorff, T. Na, D. A. Kochrekar, R. N. Cherat and G. G. Pai, Org. Process Res. Dev., 2010, 14, 193–198 CrossRef CAS.
- C. K. Savile, J. M. Janey, E. C. Mundorff, J. C. Moore, S. Tam, W. R. Jarvis, J. C. Colbeck, A. Krebber, F. J. Fleitz, J. Brands, P. N. Devine, G. W. Huisman and G. J. Hughes, Science, 2010, 329, 305 CrossRef CAS PubMed.
- R. A. Sheldon, Green Chem., 2014, 16, 950–963 RSC.
- (a) Catalytic Process Development for Renewable Materials, ed. P. Imhof and J. C. van der Waal, Wiley-VCH, Weinheim, 2013 Search PubMed; (b) S.-T. Yang, H. A. El-Enshasy and N. Thongchul, Bioprocessing Technologies Biorefinery for Sustainable Production of Fuels, Chemicals and Polymers, Wiley, Hoboken, 2013 Search PubMed; (c) P. Gallezot, Chem. Soc. Rev., 2012, 41, 1538–1558 RSC.
- E. de Jong, A. Higson, P. Walsh and M. Wellisch, Biofuels, Bioprod. Biorefin., 2012, 6, 606–624 CrossRef CAS.
- E. F. Sousa-Aguiar, L. Gorenstin Appel, P. Costa Zonetti, A. do Couto Fraga, A. Azevedo Bicudo and I. Fonseca, Catal. Today, 2014, 234, 13–23 CrossRef CAS.
- (a) C. O. Tuck, E. Perez, I. T. Horvath, R. A. Sheldon and M. Poliakoff, Science, 2012, 337, 695–699 CrossRef CAS PubMed; (b) J. K. Saini, R. Saini and L. Tewari, 3 Biotech., 2015, 5, 337–353 CrossRef.
- (a) L. A. Pfaltzgraff, M. De Bruyn, E. C. Cooper, V. Budarin and J. H. Clark, Green Chem., 2013, 15, 307–314 RSC; (b) C. S. Ki Lin, L. A. Pfaltzgraff, L. Herrero-Davila, E. B. Mubofu, S. Abderrahim, J. H. Clark, A. A. Koutinas, N. Kopsahelis, K. Stamatelatou, F. Dickson, S. Thankappan, Z. Mohamed, R. Brocklesby and R. Luque, Energy Environ. Sci., 2013, 6, 426–464 RSC.
- J. P. Lange, Biofuels, Bioprod. Biorefin., 2007, 1, 39–48 CrossRef CAS.
- (a) P. C. Munasinghe and S. K. Khanal, Bioresour. Technol., 2010, 101, 5013–5022 CrossRef CAS PubMed; (b) A. M. Henstra, J. Sipma, A. Rinzema and A. J. M. Stams, Curr. Opin. Biotechnol., 2007, 18, 200–206 CrossRef CAS PubMed; (c) J. Daniell, M. Köpke and S. D. Simpson, Energies, 2012, 5, 5372–5417 CrossRef CAS; (d) http://www.lanzatech.com .
- (a) P. Bajpai, Pretreatment of Lignocellulosic Biomass for Biofuel Production, Springer Briefs in Green Chemistry for Sustainability, Springer, Dordrecht, 2016 Search PubMed; (b) Green Biomass Pretreatment for Biofuels Production, Springer Briefs in Green Chemistry for Sustainability, ed. T. Gu, Springer, Dordrecht, 2013 Search PubMed.
- U. Bornscheuer, K. Buchholz and J. Seibel, Angew. Chem., Int. Ed., 2014, 53, 10876–10893 CrossRef CAS PubMed.
- M. M. Ishola, A. Jahandideh, B. Haidarian, T. Brandberg and M. J. Taherzadeh, Bioresour. Technol., 2013, 133, 68–73 CrossRef CAS PubMed.
- (a) A. Margeot, B. Hahn-Hagerdal, M. Edlund, R. Slade and F. Monot, Curr. Opin. Biotechnol., 2009, 20, 372–380 CrossRef CAS PubMed; (b) C. Ayrinhac, A. Margeot, N. Lopes Ferreira, F. B. Chaabane, F. Monot, G. Ravot, J.-M. Sonet and L. Fourage, Org. Process Res. Dev., 2011, 15, 275–278 CrossRef CAS.
- R. A. Sheldon, Appl. Microbiol. Biotechnol., 2011, 92, 467–477 CrossRef CAS PubMed.
- (a) R. A. Sheldon, M. J. Sorgedrager and B. Kondor, Non-leachable magnetic cross-linked enzyme aggregate, PCT Int. Appl, WO2012/023847A2, 2012, to CLEA Technologies Search PubMed; (b) A. Bhattacharya and B. I. Pleschke, Enzyme Microb. Technol., 2014, 61–62, 17–27 CrossRef CAS PubMed; (c) K. J. Khorshidi, H. Lenjannezhadian, M. Jamalan and M. Zeinali, J. Chem. Technol. Biotechnol., 2016, 91, 539–546 CrossRef.
- (a) Ionic Liquids in the Biorefinery Concept: Challenges and Perspectives, ed. R. Bogel-Lukasic, RSC Green Chemistry Series No. 36, Royal Society of Chemistry, 2016 Search PubMed; (b) Production of Biofuels and Chemicals with Ionic Liquids, ed. Z. Fang, R. L. Smith and X. Qi, Springer, Dordrecht, 2014 Search PubMed; (c) M. Francisco, A. van den Bruinhorst and M. C. Kroon, Green Chem., 2012, 14, 2153–2157 RSC.
- R. A. Sheldon, J. Mol. Catal. A: Chem., 2016, 422, 3–12 CrossRef CAS and references therein.
- (a) J. Rass-Hansen, H. Falsig, B. Jørgensen and C. H. Christensen, J. Chem. Technol. Biotechnol., 2007, 82, 329–333 CrossRef CAS; (b) J. M. R. Gallo, J. M. C. Bueno and U. Schuchardt, J. Braz. Chem. Soc., 2014, 25, 2229–2243 CAS; (c) J. A. Posada, A. D. Patel, A. Roes, K. Blok, A. P. C. Faaij and M. K. Patel, Bioresour. Technol., 2013, 135, 490–499 CrossRef CAS PubMed.
- N. Ladygina, E. G. Dedyukhina and M. B. Vainshtein, Process Biochem., 2006, 41, 1001–1014 CrossRef CAS.
- J. N. Chheda and J. A. Dumesic, Catal. Today, 2007, 123, 59–70 CrossRef CAS.
- R. Beerhuis, G. Rothenburg and N. R. Shiju, Green Chem., 2015, 17, 1341–1361 RSC.
- T. J. Schwartz, B. J. O'Neill, B. H. Shanks and J. A. Dumesic, ACS Catal., 2014, 4, 2060–2069 CrossRef CAS.
- J.-P. Lange, E. van der Heide, J. van Buijtenen and R. Price, ChemSusChem, 2012, 5, 150–166 CrossRef CAS PubMed.
- R.-J. van Putten, J. C. van der Waal, E. de Jong, C. B. Rasrendra, H. J. Heeres and J. G. de Vries, Chem. Rev., 2013, 113, 1499–1597 CrossRef CAS PubMed.
- S. P. Simeonov, J. A. S. Coelho and C. A. M. Afonso, ChemSusChem, 2013, 6, 997–1000 CrossRef CAS PubMed.
- P. R. D. Williams, D. Inman, A. Aden and G. A. Heath, Environ. Sci. Technol., 2009, 43, 4763–4775 CrossRef CAS PubMed.
- (a) C. A. Wall-Markowski, A. Kicherer and P. Saling, Environ. Prog., 2004, 23, 329–333 CrossRef CAS; (b) P. Saling, Appl. Microbiol. Biotechnol., 2005, 68, 1–8 CrossRef CAS PubMed.
- A. D. Patel, K. Meesters, H. den Uil, E. de Jong, K. Blok and M. K. Patel, Energy Environ. Sci., 2012, 5, 8430–8444 CAS.
- H. Sugiyama, U. Fischer, K. Hungerbühler and M. Hirao, AIChE J., 2008, 54, 1037–1053 CrossRef CAS.
- (a) A. D. Patel, K. Meesters, H. den Uil, E. de Jong, E. Worrell and M. K. Patel, ChemSusChem, 2013, 6, 1724–1736 CrossRef CAS PubMed; (b) A. D. Patel, S. Telalovic, J. H. Bitter, E. Worrell and M. K. Patel, Catal. Today, 2015, 239, 56–79 CrossRef CAS.
- D. Cespi, F. Passarini, I. Vassura and F. Cavani, Green Chem., 2016, 18, 1625–1638 RSC.
- R. A. Sheldon, J. P. M. Sanders and A. Marinas, Catal. Today, 2015, 239, 1–96 CrossRef CAS.
- R. A. Sheldon and J. P. M. Sanders, Catal. Today, 2015, 239, 3–6 CrossRef CAS.
- E. Cséfalvay, G. R. Akien, L. Qi and I. T. Horvath, Catal. Today, 2015, 239, 50–55 CrossRef.
- M. Morales, M. Ataman, S. Badr, S. Linster, J. Kourlimpinis, S. Papadokonstantanis, V. Hatzimanikatis and K. Hungerbühler, Energy Environ. Sci., 2016 10.1039/c0xx00000x.
| This journal is © The Royal Society of Chemistry 2017 |